 Matthew C. Morris
Matthew C. Morris Elizabeth A. Gilliam
Elizabeth A. Gilliam Liwu Li
Liwu Li- 1Department of Biological Sciences, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA
- 2Virginia Tech Carillion School of Medicine and Research Institute, Roanoke, VA, USA
Monocytes and macrophages play pivotal roles in inflammation and homeostasis. Recent studies suggest that dynamic programing of macrophages and monocytes may give rise to distinct “memory” states. Lipopolysaccharide (LPS), a classical pattern recognition molecule, dynamically programs innate immune responses. Emerging studies have revealed complex dynamics of cellular responses to LPS, with high doses causing acute, resolving inflammation, while lower doses are associated with low-grade and chronic non-resolving inflammation. These phenomena hint at dynamic complexities of intra-cellular signaling circuits downstream of the Toll-like receptor 4 (TLR4). In this review, we examine pathological effects of varying LPS doses with respect to the dynamics of innate immune responses and key molecular regulatory circuits responsible for these effects.
Current Dogma and Limitations with Regard to LPS Signaling in Innate Immunity
Lipopolysaccharide (LPS) is a ubiquitous molecule found on the surface of Gram-negative bacteria and is recognized by innate immune cells in humans. Slightly elevated levels of LPS persist in humans with chronic diseases and lifestyles that involve chronic smoking and drinking (1–7). Through a better understanding of how inflammation plays a role in the development of chronic disease, it is possible to devise better treatments to prevent or mitigate their debilitating effects. Currently, it is believed that low-grade inflammation plays a significant role in slowing and preventing normal healing processes from occurring, leading to chronic diseases including heart disease, diabetes, reduced wound healing, and even Parkinson’s disease and rheumatoid arthritis (RA) (2, 8–13).
Lipopolysaccharide challenge is known to induce a refractory state in cells, whereupon subsequent challenge, even with a high dose of LPS, is characterized by less robust induction of pro-inflammatory cytokines and increased production of anti-inflammatory cytokines, a state known as endotoxin tolerance (14–16). The duration of exposure has also been implicated in different immune responses (16). Pretreatment with a very low dose of endotoxin (in the picograms/milliliter range), in contrast, has an opposite effect, potentiating or “priming” the pro-inflammatory response to subsequent endotoxin challenge. This phenomenon is referred to as the Shwartzman-like reaction (17). We and others have documented the priming response to very low-dose LPS in vitro, where it results in augmented expression of pro-inflammatory cytokines such as IL-6 and tumor necrosis factor α (TNFα), and in vivo, where mice pretreated with super-low-dose LPS exhibit increased mortality in response to challenge with a higher dose (18, 19). Endotoxin priming and tolerance have both been well documented, though the molecular mechanisms governing the decision between one response and the other have not been well defined. Regardless, the “decision” must be made at the time of the primary challenge: since the secondary stimulus can be delivered at precisely the same dosage and for the same duration, the differences in the response cannot originate with the secondary challenge. The difference between priming and tolerance must therefore be in the response to the preparatory dose, and it is here that a detailed examination of the dynamics of the macrophage response to LPS would be most fruitful.
The first events in the immune response to LPS occur outside of the cell. LPS must first come into contact with the LPS-binding protein (LBP). The LPS–LBP complex can then be recognized by TLR4, acting in conjunction with MD-2 and cluster of differentiation 14 (CD14) (20). Once this recognition has occurred, the TLR4 signaling cascade can commence.
Upon ligation of TLR4 by LPS, signaling can proceed through an MyD88-dependent or MyD88-independent pathway. The intra-cellular portion of TLR4 contains a Toll/IL-1R homology (TIR) domain, by which it is enabled to interact with a family of related proteins and adaptor molecules, most prominently MyD88 and TIR-domain-containing adaptor protein inducing interferon-β (TRIF) (21). TLR4 is unique for its ability to signal through both MyD88 and TRIF, as the other TLRs are limited exclusively to either MyD88-dependent or TRIF-dependent signaling (22). Recruitment of MyD88 to TLR4 is followed by a signaling cascade involving the interleukin-1-receptor-associated kinases or interleukin-receptor-associated kinase (IRAKs). There are currently four known IRAKs, among which IRAK-1, -2, and -4 play positive roles in signal transduction, while IRAK-M (also known as IRAK-3) acts to suppress TLR signaling (23). The MyD88-dependent pathway of TLR signaling culminates in the activation of mitogen-activated protein kinases (MAPK) and NFκB, with subsequent induction of pro-inflammatory genes (24). Figure 1 presents an overview of prominent mediators of TLR4 signaling.
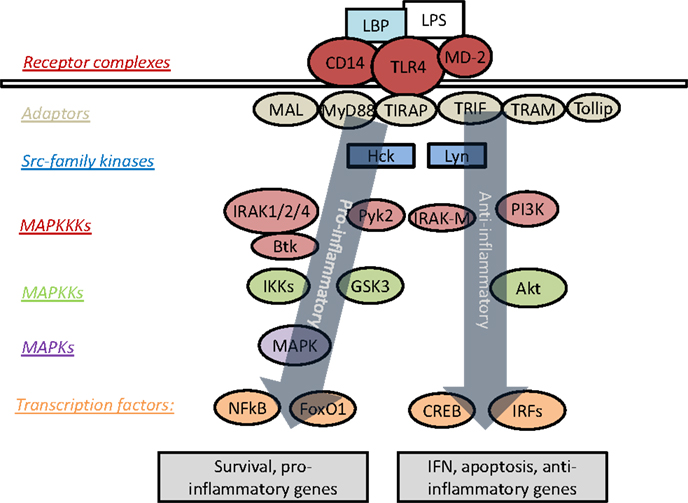
Figure 1. Overview of signaling cascades engaged by TLR4. LPS triggers recruitment of various adaptor molecules that may recruit a myriad of downstream kinases and transcription factors, resulting in the activation of both pro- and anti-inflammatory responses. However, the dynamic modulation and coordination of these complex events are not well understood.
Signaling through TRIF usually requires endocytosis of TLR4 (25). This endocytosis in turn requires CD14, as interference with CD14–TLR4–LBP interactions prevents effective TLR4 internalization (26, 27). Interestingly, Watanabe et al. demonstrated that TRIF-dependent signaling can be activated in the absence of CD14 (28), but this required the direct delivery of LPS to the interior of the cell, suggesting even more strongly that TRIF-dependent signaling requires signaling by TLR4 within the confines of the cytoplasmic membrane. A single amino acid mutation in TLR3 is sufficient to induce signaling through MyD88, rather than TRIF, indicating that the two pathways are closely related (29). Once recruited, TRIF in turn activates interferon regulatory factor 3 through TBK1, and signaling proceeds through PI3K, ultimately resulting in the activation of interferon-β (IFNβ) and related genes (14). LPS preconditioning has TRIF-dependent protective effects with respect to the ischemic injury associated with stroke (30), and deletion of TRIF exacerbates allergic dermatitis in mice (31). Taken together, these results point to a broadly anti-inflammatory role for TRIF-dependent signaling in addition to its established pro-inflammatory effects downstream of TLR4 activation.
Emerging Concept of Innate Programing and Memory
Past studies have largely treated the MyD88-dependent and TRIF-dependent pathways in isolation, with little attention paid to the possibility of cross-talk between them. However, there are intriguing indications that such cross-talk does occur. IRAK-1, a pivotal actor in MyD88-dependent signaling, plays a suppressive role in TRIF-dependent TLR3 signaling (32), and MyD88 is important for the suppression of TRIF-mediated apoptosis (33). The Src-family tyrosine kinases (SFK) also play a role in the differential regulation of pro- and anti-inflammatory effects downstream of TLR stimulation (34), including direct involvement in MyD88-dependent NFκB activation (35). The phenomena of endotoxin priming and tolerance indicate that the pro- and anti-inflammatory responses to TLR4 stimulation are not wholly independent; rather, the activation of one must affect the other somehow. These emerging studies hint at a novel concept of “innate immune programing” and “memory.” Based on the mutually inhibitory cross-talks among these pathways, innate leukocytes may be skewed to distinct phenotypes and retain certain memory states, such as M1, M2, and other intermediate states (36). However, the mechanisms responsible for this potential memory are not well understood. Our future efforts will be dedicated to the review of potential leads that may help reveal the underlying mechanisms.
The major mediators of cross-talk between MyD88- and TRIF-dependent TLR4 signaling appear to be the SFKs and phosphatidylinositol-3-kinase (PI3K), which act at different “layers” downstream of the receptors to integrate signals from the different pathways. The SFKs are engaged by tumor-necrosis-factor-receptor-associated factor 6 (TRAF6) (37), and activated within minutes of LPS stimulation, along with the Syk kinase Pyk2 and Bruton’s tyrosine kinase (Btk), a member of the Tec family (38). The SFK Lyn has been studied chiefly in B cells, due to the spontaneous appearance of a lupus-like B-cell-mediated autoimmune disease in Lyn-deficient mice (39). Lyn activates PI3K through B-cell adaptor for PI3K (BCAP) in B cells (40), but DC-specific deletion of Lyn still causes hyperactive MyD88 signaling and B cell-mediated autoimmunity (41), pointing to a role for Lyn in myeloid cells. Knockout of MyD88 either globally or conditionally in B cells or dendritic cells counteracts the autoimmune symptoms characteristic of Lyn deficiency (42, 43). BCAP itself is active in myeloid cells as well (44), indicating that the network is not limited to lymphoid cells. In mast cells, Lyn is necessary for TLR4-dependent NFκB and MAPK activation (45), and further contributes to the activation of Btk (46), a TIR-domain-containing molecule, which promotes LPS-induced NFκB activation in macrophages (47). Btk also drives MAPK-dependent TNFα production in response to TLR2 and TLR4 stimulation of myeloid cells (48).
Inhibition of Pyk2 ameliorates the symptoms of LPS-induced lung injury (49), and it also promotes MyD88-dependent signaling and NFκB activation (35). Together, these findings point toward a generally pro-inflammatory role for Pyk2, but the discovery that PI3K inhibitors suppress Pyk2 activity (50) indicates that Pyk2 is involved in both pathways. Downstream of PI3K, Pyk2 may act in part to modulate the inflammatory suppression driven by PI3K/Akt signaling.
The need for further study of the regulation of pro- and anti-inflammatory responses to TLR4 stimulation is clear. Since the “switch” between endotoxin priming and tolerance appears to depend on the dosage of the first challenge, the dynamics of that response must be investigated with the aim of determining what conditions lead to the activation of one pathway or another. The network motifs active here are key to the understanding of the LPS response. There exists a role for epigenetics in the broad reprograming of macrophages and endotoxin tolerance (51–53), but the response to single dosages over a matter of hours is likely to be regulated by faster dynamic molecular mechanisms, as discussed below in further detail.
Competitive Circuitry Governing Innate Programing of Leukocytes by LPS
A growing body of literature suggests that a competitive network may be responsible for the decision between a predominantly pro- or anti-inflammatory response to LPS, with PI3K, Akt, and the cAMP-response-element-binding-protein (CREB) acting in opposition to glycogen synthase kinase 3 (GSK3) and forkhead box O1 (FoxO1). BCAP, as mentioned above, is crucial for TLR-dependent PI3K activation in myeloid cells and the ensuing suppression of inflammation (44). PI3K dampens NFκB activation by means of phosphoinositide-dependent kinase-1, which suppresses TRAF6 activity and is necessary for the LPS-induced activation of Akt and ERK (54). PI3K also activates Akt in response to mammalian target of rapamycin (mTOR), competing with MAPK/p38/JNK signaling (55). Inhibition of PI3K leads to increased production of IL-6 and TNFα in response to TLR2 stimulation of macrophages (56), and activation of PI3K results in deactivation of FoxO1, preventing it from promoting TLR4 signaling (57). Overall, PI3K is important for negative feedback and control of TLR signaling, acting to counteract both NFκB and MAPK, two of the main transcription factors responsible for pro-inflammatory gene transcription in response to LPS.
Akt exerts its anti-inflammatory effects through NFκB and MAPK signaling, as well as its activation of CREB. In non-canonical NFκB signaling, the processing of p100–p52 requires Akt, paving the way for increased activity of RelB (58), a suppressive NFκB family member. Activation of Akt through the mTOR–PI3K pathway both counteracts MAPK signaling and activates CREB (59), at the same time directly inactivating FoxO1 (55). Quercetin treatment activates Akt in multiple cell types, leading to decreased activity of FoxO1 in pancreatic islets (60), and ameliorating the inflammatory response of adipocytes to TNFα (61). Akt activation has also been shown to correlate with suppression of FoxO1 in HEK293 cells (62). Inhibition of JAK3 leads to decreased activity of both Akt and CREB, and this loss of activity correlates with an augmented pro-inflammatory response to LPS (63). The role of Akt, then, seems to be to mediate the anti-inflammatory effects of PI3K, in large part by suppressing FoxO1 and activating CREB.
GSK3 has been implicated in many inflammatory signaling pathways. It directly suppresses genes with CREB binding sites (64). Inhibition of GSK3 increases IL-10 production and decreases IL-12 in response to LPS in monocytes (65), which, in light of the importance of IL-10 to the anti-inflammatory effects of PI3K (66), points strongly to GSK3 as an actor in opposition to PI3K. GSK3 suppresses IFNβ induction by LPS (67), indicating a suppressive effect on TRIF-dependent signaling. GSK3 inactivates CREB directly (68, 69), and IFNγ activates GSK3 and suppresses CREB (70), indicating that the pro-inflammatory effects of IFNγ may be due in large part to its effects on this sub-network.
The opposing effects of PI3K activation and GSK3 activation have been described in multiple cell types. They have opposing effects on the LPS response in DC (66), and in H2O2-induced apoptosis in neurons (71). Inactivation of GSK3β is important in inflammatory resolution and is associated with a blunted pro-inflammatory response to LPS (72). In macrophages, PI3K–Akt signaling directly opposes GSK3 activity during the LPS response, with GSK3α knockdown potentiating the effects of IL-10 while CREB knockdown reduces them. Furthermore, the pro-inflammatory effects of PI3K inhibition can be counteracted by treatment with IL-10 (73), another indicator that IL-10 is a downstream effector of PI3K. The PI3K-dependent increase in IL-10 production is due to its inactivation of GSK3 (67, 74), and direct activation of PI3K–Akt results in inhibitory phosphorylation of GSK3 (75). Knockout of the mTOR signaling molecule rictor prevents Akt from inactivating GSK3 upon TLR4 stimulation, correlating with increased FoxO1 activity and pro-inflammatory gene expression (76). Taken together, these findings constitute a strong body of evidence that the anti-inflammatory PI3K/Akt/CREB signaling axis acts by suppressing GSK3/FoxO1, and that this competition is the lynchpin of the primarily pro- or anti-inflammatory characteristics of the dynamic LPS response (Figure 2). We recently reported that super-low-dose LPS selectively activates GSK3 and JNK while suppressing Akt and ERK (77). This may explain the mild skewing of pro-inflammatory responses by low-grade endotoxemia in mice and humans. In contrast, high-dose LPS can induce robust activation of all MAPKs that include p38, JNK, and ERK, as well as PI3K/Akt (77), which may lead to the robust yet transient resolving inflammation followed by anti-inflammatory tolerance associated with high-dose endotoxin challenge. With regard to upstream signaling network, IRAK-1 is responsible for the effects of super-low-dose and high-dose LPS (Figure 2) (77, 78).
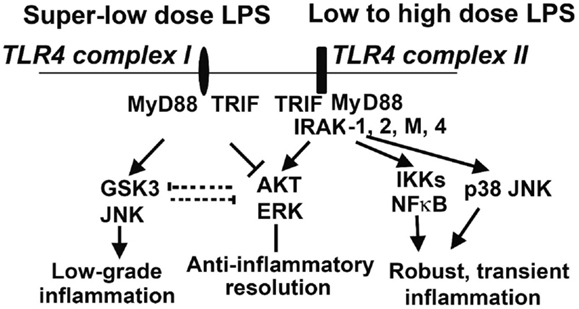
Figure 2. Competitive molecular circuits potentially responsible for the dynamic programing of innate leukocytes. Varying doses of LPS may engage unique TLR4 complexes that may cause intra-cellular pathway switching, leading to either low-grade non-resolving inflammation, or acute resolving inflammation. Super-low-dose LPS selectively activates GSK3 and JNK, while suppressing AKT and ERK. These two branches may also have mutually inhibitory interactions, further fine-tuning cellular inflammatory states. Low- to high-dose LPS potently induces NFκB and MAP kinases, leading to robust inflammatory reactions. On the other hand, low- to high-dose LPS could also trigger the activation of AKT and ERK, which may serve to dampen inflammatory responses. IRAK-1 appears to be critical for both the low-grade inflammation triggered by super-low-dose LPS and the anti-inflammatory responses (e.g., IL-10 expression through ERK activation) activated by high-dose LPS.
Pathological Effects of Varying Dosages of Endotoxin
Chronic diseases currently affect large proportions of the US population where currently one in three adults is obese and almost one in five children between 6 and 19 years are also considered obese (79–82). In 2005, the CDC estimated that one in two Americans suffered from at least one chronic disease, such as arthritis, greatly decreasing their quality of life and participation in daily activities (83).
Lipopolysaccharide is a ubiquitous molecule found on the surface of Gram-negative bacteria and is recognized by innate immune cells in humans. Slightly elevated levels of LPS persist in humans with chronic diseases and lifestyles that involve chronic smoking and drinking (1–7). Elevated circulating endotoxin may program host leukocytes into a low-grade “memory” state, and contribute to the pathogenesis of diverse diseases that may include atherosclerosis, diabetes, reduced wound healing, Parkinson’s disease, and RA (2, 8–13). Indeed, recent studies suggest that the lower circulating levels of endotoxin may lead to enhanced pathogenesis of atherosclerosis (84, 85). Elderly people tend to have elevated circulating endotoxin associated with neurological disease (86, 87). On the other hand, slightly elevated endotoxin in vivo may offer protection toward ischemia reperfusion injuries (88–91). Varying dosages of endotoxin may also affect the function of dendritic cells, and alter vaccine efficacies (92).
To put these dynamic pathological and physiological responses in perspective, we have simplified these contrasting profiles of acute and persistent inflammation in Figure 3. A normal inflammatory response comprises an early, pro-inflammatory phase, in which microbicidal functions predominate, and a secondary, anti-inflammatory phase, where wound healing occurs and inflammatory cells leave the area of damage (72, 93, 94). In chronic disease, the pro-inflammatory phase fails to resolve, leading to a persistent state of low-grade inflammation. This leads to changes in mucosal barriers and commensal bacteria that line the gastrointestinal tract. As a result, these individuals tend to have slightly elevated levels of LPS (1–100 pg/ml) circulating in their blood (1, 2, 95–100). However, while inflammatory processes for high doses of LPS (>10 ng/ml) have been intensively studied for its role in septic shock (101, 102), much less is known about the immunological response to subclinical doses of LPS. Typically, the activation of the Toll-like receptor-4 (TLR4) complex leads to the activation of nuclear factor κB (NFκB) where it initiates the transcription of genes encoding inflammatory cytokines (103–108). These inflammatory cytokines are responsible for the recruitment of neutrophils, natural killer cells, and antigen-presenting cells to the site of the infection (103). Once the infection is cleared, other cytokines such as IL-10 and transforming growth factor β (TGFβ) combine with apoptosis of pro-inflammatory cells such as neutrophils to resolve inflammation and restore homeostasis (103). Additionally, macrophages can enter a state of endotoxin tolerance wherein they suppress their expression of pro-inflammatory mediators to prevent excessive inflammation (109, 110). Suppression at multiple levels including inhibitor of κB, phosphatidylinositol-3-kinase (PI3K), MAP kinase phosphatases, and the inactivation of IRAK-1 helps prevent inflammation in the absence of danger signals (54, 56, 111).
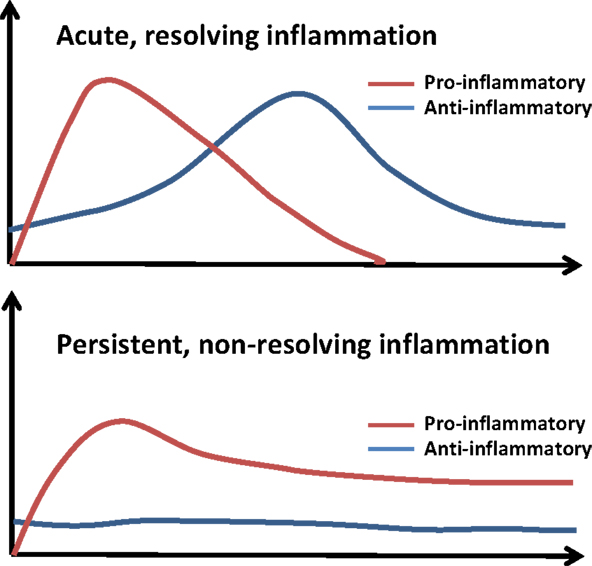
Figure 3. The course of acute and persistent inflammatory responses. The upper panel depicts the trajectory of a typical inflammatory response. An initial pro-inflammatory phase characterized by recruitment of neutrophils and production of cytokines such as IL-6 and TNFα is followed by resolution, with compensatory induction of anti-inflammatory cytokines (e.g., IL-10, TGFβ). Non-resolving and persistent inflammation is mild but unopposed by anti-inflammatory mechanisms.
It is important to note that “innate programing” and “memory” may not be limited exclusively to endotoxin responses. Rather, recent studies have indicated that other microbial products such as beta-glucan can analogously “train” or program host innate immunity, and led to improved host responses toward controlling infection (112–114).
Conclusion
Remaining questions include the plasticity of this network, and the persistence of the programing for which it is responsible. Can the character of the LPS response be easily altered by pharmacological intervention aimed at activating or inhibiting different components of this signaling nexus? Once the character of the response has been established, will the system reset itself? Answers to these questions will significantly advance the understanding of TLR signaling in particular and the behavior of innate immune cells in general. Some efforts have been made to apply large-scale systematic methods to the study of this system (115, 116), but a great deal of work remains to be done, particularly with respect to the network herein described. There is a growing appreciation for plasticity and memory in macrophages, with a movement away from strict classifications of macrophage populations along lines of classical/alternative activation to more flexible schemes of classification based on dedication to a variety of different functions (36, 117). It is likely that further examination of this and other myeloid signaling networks will accelerate this. Innate immune “memory” is not a function of dedicated cell types as in lymphoid cells but rather a characteristic intrinsic to individual cells, whereby signals percolating through a network change its state in such a way as to influence its responses to subsequent stimuli. Such “memory” is therefore likely to be an important characteristic of many different cell types, particularly those responding to many different stimuli through interlocking networks of receptors and signaling cascades (neurons, in particular, come readily to mind). Innate immune cell populations may come to be seen as temporary workers, dedicated to their functions less strongly than has hitherto been supposed. Increasing appreciation for this plasticity will open broad new vistas for both the theoretical understanding of innate immunity and the treatment of associated diseases. Further studies aimed at the unique characteristics of innate memory and the underlying mechanisms are urgently needed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in this laboratory is supported by NIH grant R01 HL115835 to Liwu Li.
Abbreviations
BCAP, B-cell adaptor for PI3K; Btk, Bruton’s tyrosine kinase; CD14, cluster of differentiation 14; CREB, cAMP response-element-binding protein; C/EBPδ, CCAAT-enhancer-binding protein δ; EPC, endothelial progenitor cell; FoxO1, forkhead box O1; GBP-1, guanylate-binding protein 1; GSK3, glycogen synthase kinase 3; IFN, interferon; IRAK, interleukin-receptor-associated kinase; LBP, lipid-binding protein; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NFκB, nuclear factor κ of activated B cells; PI3K, phosphatidylinositol-3-kinase; RA, rheumatoid arthritis; SFK, Src-family kinase; TIR, Toll/IL-1R homology; TGFβ, transforming growth factor β; TNFα, tumor necrosis factor α; TRAF6, tumor-necrosis-factor-receptor-associated factor 6; TRIF, TIR-domain-containing adaptor protein inducing interferon-β.
References
1. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes (2008) 57(6):1470–81. doi: 10.2337/db07-1403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev (2010) 31(6):817–44. doi:10.1210/er.2009-0030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Maachi M, Pieroni L, Bruckert E, Jardel C, Fellahi S, Hainque B. Systemic low-grade inflammation is related to both circulating and adipose tissue TNFalpha, leptin and IL-6 levels in obese women. Int J Obes Relat Metab Disord (2004) 28(8):993–7. doi:10.1038/sj.ijo.0802718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Sun L, Yu Z, Ye X, Zou S, Li H, Yu D. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care (2010) 33(9):1925–32. doi:10.2337/dc10-0340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes (2010) 59(1):172–81. doi:10.2337/db09-0367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Terawaki H, Yokoyama K, Yamada Y, Maruyama Y, Iida R, Hanaoka K. Low-grade endotoxemia contributes to chronic inflammation in hemodialysis patients: examination with a novel lipopolysaccharide detection method. Ther Apher Dial (2010) 14(5):477–82. doi:10.1111/j.1744-9987.2010.00815.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Laugerette F, Vors C, Geloen A, Chauvin MA, Soulage C, Lambert-Porcheron S. Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. J Nutr Biochem (2011) 22(1):53–9. doi:10.1016/j.jnutbio.2009.11.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Moreno-Navarrete JM, Manco M, Ibanez J, Garcia-Fuentes E, Ortega F, Gorostiaga E. Metabolic endotoxemia and saturated fat contribute to circulating NGAL concentrations in subjects with insulin resistance. Int J Obes (2010) 34(2):240–9. doi:10.1038/ijo.2009.242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res (2010) 107(1):56–65. doi:10.1161/CIRCRESAHA.110.218420
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia (2007) 55(5):453–62. doi:10.1002/glia.20467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol (2007) 49(21):2129–38. doi:10.1016/j.jacc.2007.02.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res Rev (2011) 10(3):319–29. doi:10.1016/j.arr.2010.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Sturm R. The effects of obesity, smoking, and drinking on medical problems and costs. Health Aff (2002) 21(2):245–53. doi:10.1377/hlthaff.21.2.245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol (2009) 30(10):475–87. doi:10.1016/j.it.2009.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Yoza BK, McCall CE. Facultative heterochromatin formation at the IL-1 beta promoter in LPS tolerance and sepsis. Cytokine (2011) 53(2):145–52. doi:10.1016/j.cyto.2010.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Vaknin I, Blinder L, Wang L, Gazit R, Shapira E, Genina O. A common pathway mediated through Toll-like receptors leads to T- and natural killer-cell immunosuppression. Blood (2008) 111(3):1437–47. doi:10.1182/blood-2007-07-100404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Heremans H, Van Damme J, Dillen C, Dijkmans R, Billiau A. Interferon gamma, a mediator of lethal lipopolysaccharide-induced Shwartzman-like shock reactions in mice. J Exp Med (1990) 171(6):1853–69. doi:10.1084/jem.171.6.1853
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Rocksén D, Koch B, Sandström T, Bucht A. Lung effects during a generalized Shwartzman reaction and therapeutic intervention with dexamethasone or vitamin E. Shock (2004) 22(5):482–90. doi:10.1097/01.shk.0000142254.38630.36
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Deng H, Maitra U, Morris M, Li L. Molecular mechanism responsible for the priming of macrophage activation. J Biol Chem (2013) 288(6):3897–906. doi:10.1074/jbc.M112.424390
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Park BS, Lee JO. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med (2013) 45:e66. doi:10.1038/emm.2013.97
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Watters TM, Kenny EF, O’Neill LA. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol (2007) 85(6):411–9. doi:10.1038/sj.icb.7100095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of Toll-like receptor-mediated immune responses. Nat Rev Immunol (2005) 5(6):446–58. doi:10.1038/nri1630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell (2003) 11(2):293–302. doi:10.1016/S1097-2765(03)00053-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Deguchi A, Tomita T, Omori T, Komatsu A, Ohto U, Takahashi S. Serum amyloid A3 binds MD-2 to activate p38 and NF-kappaB pathways in a MyD88-dependent manner. J Immunol (2013) 191(4):1856–64. doi:10.4049/jimmunol.1201996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell (2011) 147(4):868–80. doi:10.1016/j.cell.2011.09.051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Martin TR, Mongovin SM, Tobias PS, Mathison JC, Moriarty AM, Leturcq DJ. The CD14 differentiation antigen mediates the development of endotoxin responsiveness during differentiation of mononuclear phagocytes. J Leukoc Biol (1994) 56(1):1–9.
27. Kitchens RL, Munford RS. CD14-dependent internalization of bacterial lipopolysaccharide (LPS) is strongly influenced by LPS aggregation but not by cellular responses to LPS. J Immunol (1998) 160(4):1920–8.
28. Watanabe S, Kumazawa Y, Inoue J. Liposomal lipopolysaccharide initiates TRIF-dependent signaling pathway independent of CD14. PLoS One (2013) 8(4):e60078. doi:10.1371/journal.pone.0060078
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Verstak B, Arnot CJ, Gay NJ. An alanine-to-proline mutation in the BB-loop of TLR3 Toll/IL-1R domain switches signalling adaptor specificity from TRIF to MyD88. J Immunol (2013) 191(12):6101–9. doi:10.4049/jimmunol.1300849
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Vartanian KB, Stevens SL, Marsh BJ, Williams-Karnesky R, Lessov NS, Stenzel-Poore MP. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation (2011) 8:140. doi:10.1186/1742-2094-8-140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Brandt EB, Gibson AM, Bass S, Rydyznski C, Khurana Hershey GK. Exacerbation of allergen-induced eczema in TLR4- and TRIF-deficient mice. J Immunol (2013) 191(7):3519–25. doi:10.4049/jimmunol.1300789
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Bruni D, Sebastia J, Dunne S, Schroder M, Butler MP. A novel IRAK1-IKKepsilon signaling axis limits the activation of TAK1-IKKbeta downstream of TLR3. J Immunol (2013) 190(6):2844–56. doi:10.4049/jimmunol.1202042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Novikova L, Czymmeck N, Deuretzbacher A, Buck F, Richter K, Weber AN. Cell death triggered by Yersinia enterocolitica identifies processing of the proinflammatory signal adapter MyD88 as a general event in the execution of apoptosis. J Immunol (2014) 192(3):1209–19. doi:10.4049/jimmunol.1203464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Lowell CA. Src-family and Syk kinases in activating and inhibitory pathways in innate immune cells: signaling cross talk. Cold Spring Harb Perspect Biol (2011) 3(3):a002352. doi:10.1101/cshperspect.a002352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Xi CX, Xiong F, Zhou Z, Mei L, Xiong WC. PYK2 interacts with MyD88 and regulates MyD88-mediated NF-kappaB activation in macrophages. J Leukoc Biol (2010) 87(3):415–23. doi:10.1189/jlb.0309125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8(12):958–69. doi:10.1038/nri2448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Liu A, Gong P, Hyun SW, Wang KZ, Cates EA, Perkins D. TRAF6 protein couples Toll-like receptor 4 signaling to Src family kinase activation and opening of paracellular pathway in human lung microvascular endothelia. J Biol Chem (2012) 287(20):16132–45. doi:10.1074/jbc.M111.310102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Smolinska MJ, Page TH, Urbaniak AM, Mutch BE, Horwood NJ. Hck tyrosine kinase regulates TLR4-induced TNF and IL-6 production via AP-1. J Immunol (2011) 187(11):6043–51. doi:10.4049/jimmunol.1100967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Scapini P, Hu Y, Chu CL, Migone TS, Defranco AL, Cassatella MA. Myeloid cells, BAFF, and IFN-gamma establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J Exp Med (2010) 207(8):1757–73. doi:10.1084/jem.20100086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Inabe K. Tyrosine phosphorylation of B-cell adaptor for phosphoinositide 3-kinase is required for Akt activation in response to CD19 engagement. Blood (2002) 99(2):584–9. doi:10.1182/blood.V99.2.584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA. Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U S A (2013) 110(35):E3311–20. doi:10.1073/pnas.1300617110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Hua Z, Gross AJ, Lamagna C, Ramos-Hernandez N, Scapini P, Ji M. Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Immunol (2014) 192(3):875–85. doi:10.4049/jimmunol.1300683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Lamagna C, Hu Y, DeFranco AL, Lowell CA. B cell-specific loss of Lyn kinase leads to autoimmunity. J Immunol (2014) 192(3):919–28. doi:10.4049/jimmunol.1301979
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Ni M, MacFarlane AW IV, Toft M, Lowell CA, Campbell KS, Hamerman JA. B-cell adaptor for PI3K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3K. Proc Natl Acad Sci U S A (2012) 109(1):267–72. doi:10.1073/pnas.1111957108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Avila M, Martinez-Juarez A, Ibarra-Sanchez A, Gonzalez-Espinosa C. Lyn kinase controls TLR4-dependent IKK and MAPK activation modulating the activity of TRAF-6/TAK-1 protein complex in mast cells. Innate Immun (2012) 18(4):648–60. doi:10.1177/1753425911435265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Hong H, Kitaura J, Xiao W, Horejsi V, Ra C, Lowell CA. The Src family kinase Hck regulates mast cell activation by suppressing an inhibitory Src family kinase Lyn. Blood (2007) 110(7):2511–9. doi:10.1182/blood-2007-01-066092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem (2003) 278(28):26258–64. doi:10.1074/jbc.M301484200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Horwood NJ, Page TH, McDaid JP, Palmer CD, Campbell J, Mahon T. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol (2006) 176(6):3635–41. doi:10.4049/jimmunol.176.6.3635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Duan Y, Learoyd J, Meliton AY, Leff AR, Zhu X. Inhibition of Pyk2 blocks lung inflammation and injury in a mouse model of acute lung injury. Respir Res (2012) 13:4. doi:10.1186/1465-9921-13-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Cain RJ, Vanhaesebroeck B, Ridley AJ. Different PI 3-kinase inhibitors have distinct effects on endothelial permeability and leukocyte transmigration. Int J Biochem Cell Biol (2012) 44(11):1929–36. doi:10.1016/j.biocel.2012.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood (2009) 114(15):3244–54. doi:10.1182/blood-2009-04-217620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Lyn-Kew K, Rich E, Zeng X, Wen H, Kunkel SL, Newstead MW. IRAK-M regulates chromatin remodeling in lung macrophages during experimental sepsis. PLoS One (2010) 5(6):e11145. doi:10.1371/journal.pone.0011145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Shanmugam MK, Sethi G. Role of epigenetics in inflammation-associated diseases. Subcell Biochem (2013) 61:627–57. doi:10.1007/978-94-007-4525-4_27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Chaurasia B, Mauer J, Koch L, Goldau J, Kock AS, Bruning JC. Phosphoinositide-dependent kinase 1 provides negative feedback inhibition to Toll-like receptor-mediated NF-kappaB activation in macrophages. Mol Cell Biol (2010) 30(17):4354–66. doi:10.1128/MCB.00069-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci (2011) 36(6):320–8. doi:10.1016/j.tibs.2011.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Medina EA, Morris IR, Berton MT. Phosphatidylinositol 3-kinase activation attenuates the TLR2-mediated macrophage proinflammatory cytokine response to Francisella tularensis live vaccine strain. J Immunol (2010) 185(12):7562–72. doi:10.4049/jimmunol.0903790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Fan W, Morinaga H, Kim JJ, Bae E, Spann NJ, Heinz S. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J (2010) 29(24):4223–36. doi:10.1038/emboj.2010.268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Gustin JA, Korgaonkar CK, Pincheira R, Li Q, Donner DB. Akt regulates basal and induced processing of NF-kappaB2 (p100) to p52. J Biol Chem (2006) 281(24):16473–81. doi:10.1074/jbc.M507373200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Rastogi R, Jiang Z, Ahmad N, Rosati R, Liu Y, Beuret L. Rapamycin induces mitogen-activated protein (MAP) kinase phosphatase-1 (MKP-1) expression through activation of protein kinase B and mitogen-activated protein kinase kinase pathways. J Biol Chem (2013) 288(47):33966–77. doi:10.1074/jbc.M113.492702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Li JM, Wang W, Fan CY, Wang MX, Zhang X, Hu QH. Quercetin preserves beta – cell mass and function in fructose-induced hyperinsulinemia through modulating pancreatic Akt/FoxO1 activation. Evid Based Complement Alternat Med (2013) 2013:303902. doi:10.1155/2013/303902
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Chuang CC, Martinez K, Xie G, Kennedy A, Bumrungpert A, Overman A. Quercetin is equally or more effective than resveratrol in attenuating tumor necrosis factor-{alpha}-mediated inflammation and insulin resistance in primary human adipocytes. Am J Clin Nutr (2010) 92(6):1511–21. doi:10.3945/ajcn.2010.29807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Kim DH, Park CH, Park D, Choi YJ, Park MH, Chung KW. Ginsenoside Rc modulates Akt/FoxO1 pathways and suppresses oxidative stress. Arch Pharm Res (2014) 37(6):813–20. doi:10.1007/s12272-013-0223-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Wang H, Brown J, Gao S, Liang S, Jotwani R, Zhou H. The role of JAK-3 in regulating TLR-mediated inflammatory cytokine production in innate immune cells. J Immunol (2013) 191(3):1164–74. doi:10.4049/jimmunol.1203084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Tullai JW, Chen J, Schaffer ME, Kamenetsky E, Kasif S, Cooper GM. Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. J Biol Chem (2007) 282(13):9482–91. doi:10.1074/jbc.M700067200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol (2005) 6(8):777–84. doi:10.1038/ni1221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Ohtani M, Nagai S, Kondo S, Mizuno S, Nakamura K, Tanabe M. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood (2008) 112(3):635–43. doi:10.1182/blood-2008-02-137430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Wang H, Garcia CA, Rehani K, Cekic C, Alard P, Kinane DF. IFN-beta production by TLR4-stimulated innate immune cells is negatively regulated by GSK3-beta. J Immunol (2008) 181(10):6797–802. doi:10.4049/jimmunol.181.10.6797
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Woodgett JR, Ohashi PS. GSK3: an in-Toll-erant protein kinase? Nat Immunol (2005) 6(8):751–2. doi:10.1038/ni0805-751
69. Hu X, Chen J, Wang L, Ivashkiv LB. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol (2007) 82(2):237–43. doi:10.1189/jlb.1206763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity (2006) 24(5):563–74. doi:10.1016/j.immuni.2006.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Koh SH, Kim SH, Kwon H, Kim JG, Kim JH, Yang KH. Phosphatidylinositol-3 kinase/Akt and GSK-3 mediated cytoprotective effect of epigallocatechin gallate on oxidative stress-injured neuronal-differentiated N18D3 cells. Neurotoxicology (2004) 25(5):793–802. doi:10.1016/j.neuro.2004.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Das A, Ganesh K, Khanna S, Sen CK, Roy S. Engulfment of apoptotic cells by macrophages: a role of microRNA-21 in the resolution of wound inflammation. J Immunol (2014) 192(3):1120–9. doi:10.4049/jimmunol.1300613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Antoniv TT, Ivashkiv LB. Interleukin-10-induced gene expression and suppressive function are selectively modulated by the PI3K-Akt-GSK3 pathway. Immunology (2011) 132(4):567–77. doi:10.1111/j.1365-2567.2010.03402.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Ren F, Duan Z, Cheng Q, Shen X, Gao F, Bai L. Inhibition of glycogen synthase kinase 3 beta ameliorates liver ischemia reperfusion injury by way of an interleukin-10-mediated immune regulatory mechanism. Hepatology (2011) 54(2):687–96. doi:10.1002/hep.24419
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Lochhead PA, Coghlan M, Rice SQ, Sutherland C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes (2001) 50(5):937–46. doi:10.2337/diabetes.50.5.937
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Brown J, Wang H, Suttles J, Graves DT, Martin M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FoxO1. J Biol Chem (2011) 286(52):44295–305. doi:10.1074/jbc.M111.258053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Maitra U, Deng H, Glaros T, Baker B, Capelluto DG, Li Z. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol (2012) 189(2):1014–23. doi:10.4049/jimmunol.1200857
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Huang Y, Li T, Sane DC, Li L. IRAK1 serves as a novel regulator essential for lipopolysaccharide-induced interleukin-10 gene expression. J Biol Chem (2004) 279(49):51697–703. doi:10.1074/jbc.M410369200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Wu SY, Green A. Projection of Chronic Illness Prevalence and Cost Inflation. Santa Monica, CA: RAND Health (2000).
80. Ogden CL, Carroll MD, Flegal KM. Obesity Among Adults in the United States – No Change Since 2003–2004. NCHS Data Brief. Hyattsville, MD: Centers for Disease Control and Prevention (2007).
81. Ogden CL, Carroll MD, Flegal KM. High body mass index for age among US children and adolescents, 2003-2006. JAMA (2008) 299(20):2401–5. doi:10.1001/jama.299.20.2401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Anderson G. Chronic Conditions: Making the Case for Ongoing Care. Partnership for Solutions. Baltimore, MD: Johns Hopkins University (2004).
83. Hootman J, Bolen J, Helmick C, Langmaid G. Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation – United States, 2003-2005. MMWR Morb Mortal Wkly Rep (2006) 55(40):1089–92.
84. Stoll LL, Denning GM, Weintraub NL. Endotoxin, TLR4 signaling and vascular inflammation: potential therapeutic targets in cardiovascular disease. Curr Pharm Des (2006) 12(32):4229–45. doi:10.2174/138161206778743501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Stoll LL, Denning GM, Weintraub NL. Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler Thromb Vasc Biol (2004) 24(12):2227–36. doi:10.1161/01.ATV.0000147534.69062.dc
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Pang Y, Campbell L, Zheng B, Fan L, Cai Z, Rhodes P. Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development. Neuroscience (2010) 166(2):464–75. doi:10.1016/j.neuroscience.2009.12.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Herber DL, Maloney JL, Roth LM, Freeman MJ, Morgan D, Gordon MN. Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia (2006) 53(4):382–91. doi:10.1002/glia.20272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Chao W, Shen Y, Zhu X, Zhao H, Novikov M, Schmidt U. Lipopolysaccharide improves cardiomyocyte survival and function after serum deprivation. J Biol Chem (2005) 280(23):21997–2005. doi:10.1074/jbc.M413676200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Venkataraman R, Kellum JA, Song M, Fink MP. Resuscitation with Ringer’s ethyl pyruvate solution prolongs survival and modulates plasma cytokine and nitrite/nitrate concentrations in a rat model of lipopolysaccharide-induced shock. Shock (2002) 18(6):507–12. doi:10.1097/00024382-200212000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Downey JS, Han J. Cellular activation mechanisms in septic shock. Front Biosci (1998) 3:d468–76.
91. Meng X, Ao L, Brown JM, Meldrum DR, Sheridan BC, Cain BS. LPS induces late cardiac functional protection against ischemia independent of cardiac and circulating TNF-alpha. Am J Physiol (1997) 273(4 Pt 2):H1894–902.
92. Yadav R, Zammit DJ, Lefrancois L, Vella AT. Effects of LPS-mediated bystander activation in the innate immune system. J Leukoc Biol (2006) 80(6):1251–61. doi:10.1189/jlb.0406253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Cohen HB, Mosser DM. Extrinsic and intrinsic control of macrophage inflammatory responses. J Leukoc Biol (2013) 94(5):913–9. doi:10.1189/jlb.0413236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Nagaraja S, Wallqvist A, Reifman J, Mitrophanov AY. Computational approach to characterize causative factors and molecular indicators of chronic wound inflammation. J Immunol (2014) 192(4):1824–34. doi:10.4049/jimmunol.1302481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Goto T, Eden S, Nordenstam G, Sundh V, Svanborg-Eden C, Mattsby-Baltzer I. Endotoxin levels in sera of elderly individuals. Clin Diagn Lab Immunol (1994) 1(6):684–8.
96. Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One (2008) 3(6):e2516. doi:10.1371/journal.pone.0002516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Szeto CC, Kwan BC, Chow KM, Lai KB, Chung KY, Leung CB. Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin J Am Soc Nephrol (2008) 3(2):431–6. doi:10.2215/CJN.03600807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck study. J Am Coll Cardiol (1999) 34(7):1975–81. doi:10.1016/S0735-1097(99)00448-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology (2009) 50(2):638–44. doi:10.1002/hep.23009
100. Lira FS, Rosa JC, Pimentel GD, Souza HA, Caperuto EC, Carnevali LC Jr. Endotoxin levels correlate positively with a sedentary lifestyle and negatively with highly trained subjects. Lipids Health Dis (2010) 9:82. doi:10.1186/1476-511X-9-82
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Schlegel N, Baumer Y, Drenckhahn D, Waschke J. Lipopolysaccharide-induced endothelial barrier breakdown is cyclic adenosine monophosphate dependent in vivo and in vitro. Crit Care Med (2009) 37(5):1735–43. doi:10.1097/CCM.0b013e31819deb6a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Scheifele DW, Olsen EM, Pendray MR. Endotoxinemia and thrombocytopenia during neonatal necrotizing enterocolitis. Am J Clin Pathol (1985) 83(2):227–9.
103. Kawai T, Akira S. TLR signaling. Semin Immunol (2007) 19(1):24–32. doi:10.1016/j.smim.2006.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med (2007) 13(11):460–9. doi:10.1016/j.molmed.2007.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell (1998) 2(2):253–8. doi:10.1016/S1097-2765(00)80136-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol (2010) 10(5):365–76. doi:10.1038/nri2748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Li L, Jacinto R, Yoza B, McCall CE. Distinct post-receptor alterations generate gene- and signal-selective adaptation and cross-adaptation of TLR4 and TLR2 in human leukocytes. J Endotoxin Res (2003) 9(1):39–44. doi:10.1177/09680519030090010401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Adib-Conquy M, Cavaillon JM. Gamma interferon and granulocyte/monocyte colony-stimulating factor prevent endotoxin tolerance in human monocytes by promoting interleukin-1 receptor-associated kinase expression and its association to MyD88 and not by modulating TLR4 expression. J Biol Chem (2002) 277(31):27927–34. doi:10.1074/jbc.M200705200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med (2002) 30(1 Suppl):S64–73. doi:10.1097/00003246-200201001-00009
110. Li L, Cousart S, Hu J, McCall CE. Characterization of interleukin-1 receptor-associated kinase in normal and endotoxin-tolerant cells. J Biol Chem (2000) 275(30):23340–5. doi:10.1074/jbc.M001950200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Piao W, Song C, Chen H, Diaz MA, Wahl LM, Fitzgerald KA. Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-beta-dependent pathways and increases expression of negative regulators of TLR signaling. J Leukoc Biol (2009) 86(4):863–75. doi:10.1189/jlb.0309189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Kopanakis K, Tzepi IM, Pistiki A, Carrer DP, Netea MG, Georgitsi M. Pre-treatment with low-dose endotoxin prolongs survival from experimental lethal endotoxic shock: benefit for lethal peritonitis by Escherichia coli. Cytokine (2013) 62(3):382–8. doi:10.1016/j.cyto.2013.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Quintin J, Saeed S, Martens JH, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12(2):223–32. doi:10.1016/j.chom.2012.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe (2011) 9(5):355–61. doi:10.1016/j.chom.2011.04.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Roach JC. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature (2006) 441(7090):173–8. doi:10.1038/nature04768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Oda K, Kitano H. A comprehensive map of the Toll-like receptor signaling network. Mol Syst Biol (2006) 2(2006):0015. doi:10.1038/msb4100057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11(11):723–37. doi:10.1038/nri3073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: innate programing, endotoxin, priming and tolerance, systems dynamics, acute and chronic inflammation
Citation: Morris MC, Gilliam EA and Li L (2015) Innate immune programing by endotoxin and its pathological consequences. Front. Immunol. 5:680. doi: 10.3389/fimmu.2014.00680
Received: 14 October 2014; Accepted: 16 December 2014;
Published online: 06 January 2015.
Edited by:
Ulrich Blank, Paris Diderot University – Paris 7, FranceReviewed by:
Carlo Pucillo, University of Udine, ItalyAshley Mansell, Monash Institute of Medical Research, Australia
Copyright: © 2015 Morris, Gilliam and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liwu Li, Department of Biological Sciences, Virginia Polytechnic Institute and State University, 970 Washington Street SW, Blacksburg, VA 24061, USA e-mail:bHdsaUB2dC5lZHU=