 Sietse Q. Nagelkerke
Sietse Q. Nagelkerke Taco W. Kuijpers
Taco W. Kuijpers
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 21 January 2015
Sec. Primary Immunodeficiencies
Volume 5 - 2014 | https://doi.org/10.3389/fimmu.2014.00674
This article is part of the Research Topic Immunoglobulin therapy in the 21st century : the dark side of the moon View all 13 articles
Intravenous IgG (IVIg) contains polyclonal immunoglobulin G (IgG) from thousands of donors. It is administered at a low dose at regular intervals as antibody replacement therapy and at a higher dose as immunomodulatory treatment in various auto-immune or auto-inflammatory diseases. The working mechanism of immunomodulation is not well understood. Many different explanations have been given. During the last decade, we have focused on classical antibody binding via the Fc-domain of the IgG molecules to the common IgG receptors, i.e. the Fcγ receptors (FcγRs). Variation in the genes encoding human FcγRs determines function as well as expression among immune cells. As described here, NK cells and myeloid cells, including macrophages, can express different FcγR variants, depending on the individual’s genotype, copy number variation (CNV), and promoter polymorphisms. B-cells seem to only express the single inhibitory receptor. Although these inhibitory FcγRIIb receptors are also expressed by monocytes, macrophages, and only rarely by NK cells or neutrophils, their presence is unlikely to explain the immunomodulatory capacity of IVIg, nor does the sialylation of IgG. Direct IVIg effects at the level of the activating FcγRs, including the more recently described FcγRIIc, deserve renewed attention to describe IVIg-related immunomodulation.
Intravenous IgG (IVIg) is a blood product containing polyclonal immunoglobulin G (IgG) isolated and pooled from thousands of donors. IVIg is mainly used in two clinical situations. It is administered at a low dose at regular intervals as antibody replacement therapy in primary immunodeficiencies such as agammaglobulinemia and common variable immunodeficiency (CVID), or acquired immunodeficiencies where IgG plasma concentrations have become very low. In this context, suppletion essentially functions to provide the recipient with a repertoire of protective antibodies against a range of predominantly bacterial infections.
On the other hand, IVIg can also be used at a high dose as an immunomodulatory treatment in auto-immune or auto-inflammatory diseases, of which a handful are FDA- and EMEA-approved, including Kawasaki disease and immune thrombocytopenia (ITP). In this respect, IgG administration has also been used off-label for a growing number of additional diseases, including hematologic, dermatologic, and neuromuscular disorders. A list of indications for treatment with IVIg is provided in Table 1. Many but certainly not all of these diseases involve (auto)-antibody responses, questioning the actual working mechanism of IVIg under conditions where auto-antibodies have not yet been shown to be involved.

Table 1. Overview of indications for treatment with IVIg.
Even though IVIg is widely used and has proven to be an effective treatment for many diseases, the exact immunomodulatory mechanism(s) have remained elusive. Several mechanisms by which IVIg may exert its anti-inflammatory effects have been proposed over the past decades (1), listed in Table 2. These mechanisms are not necessarily mutually exclusive, and may act in concert to modulate the immune system. Furthermore, different mechanisms may be at work in the different diseases for which IVIg is administered. In this review, we describe the different theories that may explain the immunomodulatory effect of IVIg, with a special interest in the actions of IVIg in ITP, being the first disease for which the usefulness of IVIg as an immune-modulatory agent was discovered (2).

Table 2. Potential immunomodulatory mechanisms of IVIg.
Many of the theories aiming to explain the working mechanism involve Fc gamma receptors (FcγRs), which are the main receptors for IgG and therefore very likely to be involved in the working mechanisms of IVIg. Therefore, we start with an introduction of human FcγRs, followed by a description of the potential working mechanisms of IVIg, discussing how IVIg can shape immune responses by altering or interfering with FcγR expression and function.
FcγRs are receptors for (IgG), the most abundant of five classes of Ig. IgG consists of a Fab (fragment, antigen-binding) region, which determines specificity to specific antigens, and a constant region, which is the Fc (fragment, crystallizable) region, which (among other functions) mediates the effector functions of IgG, including the interactions with their major receptors, the FcγRs. These receptors are found on almost all immune cells (Table 3) and, upon binding of IgG, mediate a wide range of cellular responses, such as phagocytosis of IgG-opsonized microorganisms or immune complexes, antibody-dependent cellular cytotoxicity (ADCC), activation of the NADPH oxidase, and the release of cytokines.
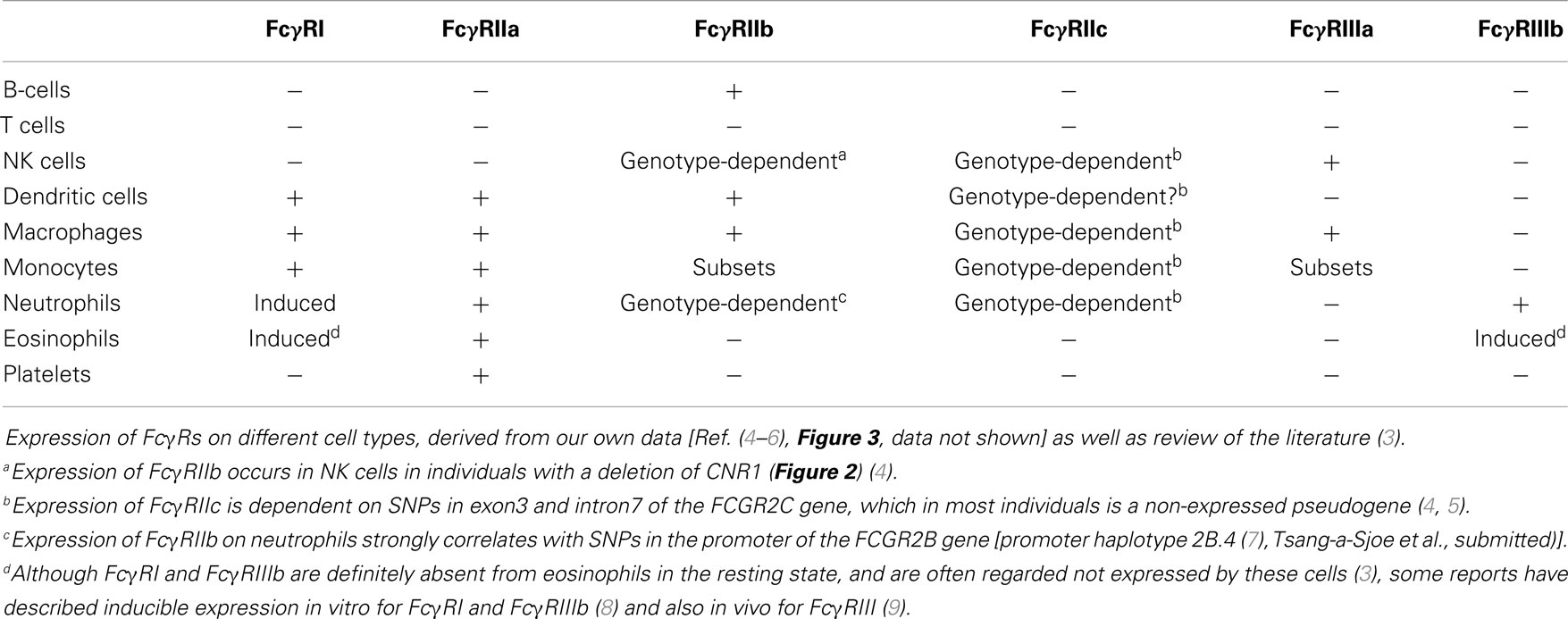
Table 3. Expression of FcγRs on different cell types.
Importantly, as compared to many innate pattern recognition receptors, human FcγRs are quite different from their murine counterparts in the sense that no clear orthologs can be assigned. As a result, human and murine FcγRs that share nomenclature and CD numbers actually have quite different protein structures, expression patterns, and Ig binding affinities (3). Thus, functional studies on mouse FcγRs can only provide very limited information for understanding the contributions of individual FcγRs to human disease.
Based on their affinity for monomeric IgG, FcγRs can be divided into the high-affinity FcγRI and the low-affinity FcγRII and FcγRIII (Figure 1). Signaling by FcγR is mediated by immunoreceptor tyrosine-based activating (ITAM) or inhibitory (ITIM) motifs that are present either in the cytoplasmic tail of the receptor itself or in non-covalently associated signaling adaptor proteins, such as the common γ-chain (FcRγ; see below). Aggregation of activating FcγR, i.e., those containing or associated with ITAMs, by binding of multivalent ligands, such as an opsonized pathogen or cancer cell, results in the phosphorylation of ITAM tyrosine residues by Src family protein tyrosine kinases (PTKs), and ultimately leads to activation of cellular responses (10). Aggregation of inhibitory FcγR, i.e., those containing ITIMs, also results in phosphorylation of tyrosine residues by Src family PTKs. In contrast to ITAMs, phosphorylated ITIMs serve as binding sites for phosphotyrosine phosphatases (PTPs), which dephosphorylate other proteins resulting in inhibition of activating pathways (11).
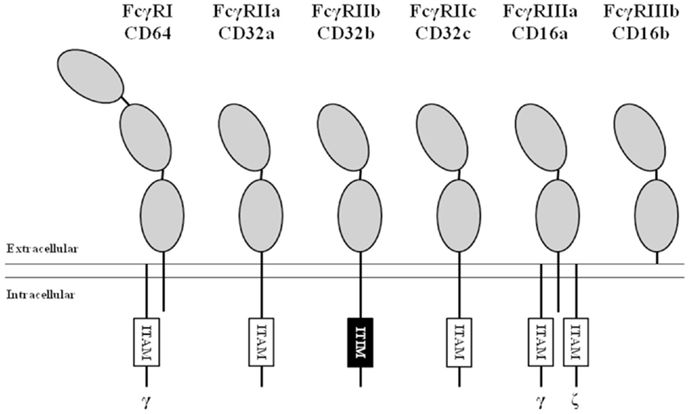
Figure 1. Nomenclature and structure of human FcγRs. All human FcγRs are transmembrane proteins, except the GPI-linked FcγRIIIb. The high-affinity FcγRI contains three extracellular (EC) domains (gray ellipses), the low-affinity FcγRs contain two. All isoforms of FcγRII contain either an immunoreceptor tyrosine-based activating (ITAM, white boxes) or inhibitory (ITIM, black box) motif in their α-chain. FcγRI and FcγRIIIa associate with ITAM-containing adaptor proteins such as the Fc receptor common gamma chain (indicated by “γ”) and the CD3 ζ-chain (indicated by “ζ”).
FcγRI (CD64) is a 72 kDa protein that has three extracellular (EC) Ig-like domains, involved in binding of IgG, a transmembrane (TM) domain and a short intracellular (IC) domain of 61 amino acids. The TM domain associates with the FcRγ-chain, an adaptor protein containing an ITAM, to induce signaling and maintain stable expression (12). FcγRI is constitutively expressed by monocytes, macrophages, and dendritic cells and its expression can be induced on neutrophils by stimulation with IFN-γ and/or G-CSF (13, 14). Although there are three genes with various transcripts (15, 16), it is generally believed that only one, the FCGR1A transcript, results in the expression of the classical FcγRIa (CD64).
FcγRII (CD32) is actually a collection of three highly homologous proteins, known as FcγRIIa, -b, and -c that all have a molecular mass of ~40 kDa. Their genes are located in one gene cluster at chromosome 1q23.3 (Figure 2). In contrast to FcγRI, the FcγRII proteins have only two IgG binding EC domains. On the other hand, the much larger intracellular domains of FcγRIIa, -b, and -c harbor intrinsic signaling motifs. In contrast to the activating FcγRIIa and -c, FcγRIIb contains an immunoreceptor tyrosine-based inhibitory motif (ITIM) (17). As no other FcγR contains or associates with proteins containing ITIMs, FcγRIIb is the only inhibitory FcγR (18).
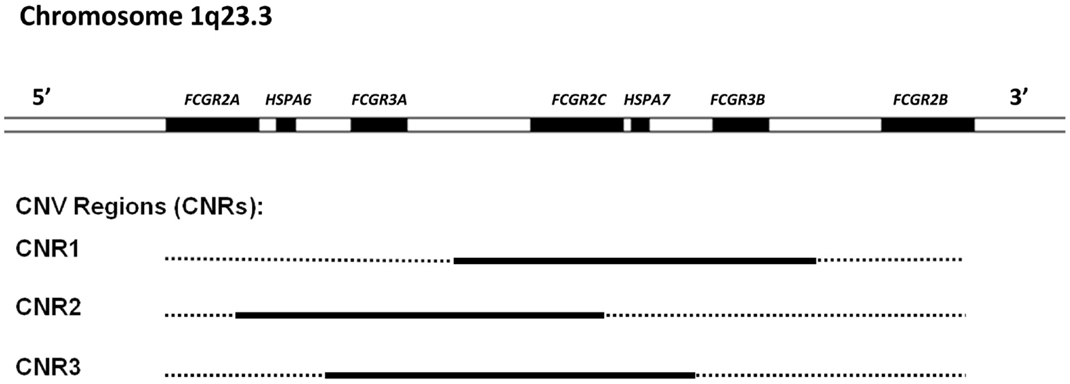
Figure 2. Overview of the low-affinity FcγR gene cluster and the corresponding CNV. Three combinations of FcγR genes have been shown to occur in duplication/deletion. Black lines indicate which genes are involved in CNV.
FcγRIIa is the most widely expressed isoform of FcγRII and is found on monocytes, macrophages, dendritic cells, neutrophils and platelets.
FcγRIIb is highly expressed on B-cells, where it constitutes the only surface-expressed FcγR. FcγRIIb is also expressed, albeit at much lower levels, on a subset of monocytes, on macrophages, and on dendritic cells. Expression of FcγRIIb can also be detected on neutrophils and NK cells, but only in individuals with certain genotypes [Ref. (4), Tsang-a-Sjoe et al., submitted].
FcγRIIc has long been considered not to be expressed at all, as its gene (FCGR2C) was thought to be a pseudogene (19, 20), and therefore, relatively little was known about the expression pattern of this receptor. In 1998, FcγRIIc was first found on NK cells of individuals with a particular haplotype of the receptor (21), but we now know that – apart from NK cells – it can also be expressed on neutrophils and monocytes in the individuals with the appropriate genotype (4, 5) (Figure 3A). As this activating FcγRIIc is expressed on circulating monocytes of some individuals, it may be expected that expression also occurs on (monocyte-derived) macrophages of these same individuals, but this has not been reported to date. Here, we show for the first time that monocyte-derived macrophages do indeed express FcγRIIc, at least when cultured in the presence of M-CSF (M2 phenotype), as shown by flowcytometry stainings with MoAb 2B6, which recognizes both FcγRIIb and FcγRIIc (Figure 3C). Although these data are difficult to interpret because of the (varying) presence of FcγRIIb on these cells, we can assume the mean difference in MFI between FCGR2C-ORF and FCGR2C-Stop donors to derive from FcγRIIc. Expression of FcγRIIc specifically was confirmed by qPCR of FCGR2C mRNA and a specific immunoprecipitation using a combination of MoAb 2B6 and a polyclonal antibody that binds FcγRIIc but not FcγRIIb (Figure 3C). Monocyte-derived macrophages differentiated with GM-CSF (M1 phenotype) do not seem to express FcγRIIc based on flowcytometry, although also in these cells, low levels of FCGR2C mRNA could be detected, therefore expression of FcγRIIc cannot be ruled out and may occur in later stages of differentiation of M1 macrophages. Recently, it was proposed that FcγRIIc can also be expressed on B-cells (22). However, evidence of expression of this receptor on B-cells on our own cohort of healthy donors was not confirmed on B-cells, irrespective their naïve or memory phenotype (Figure 3B; data not shown).
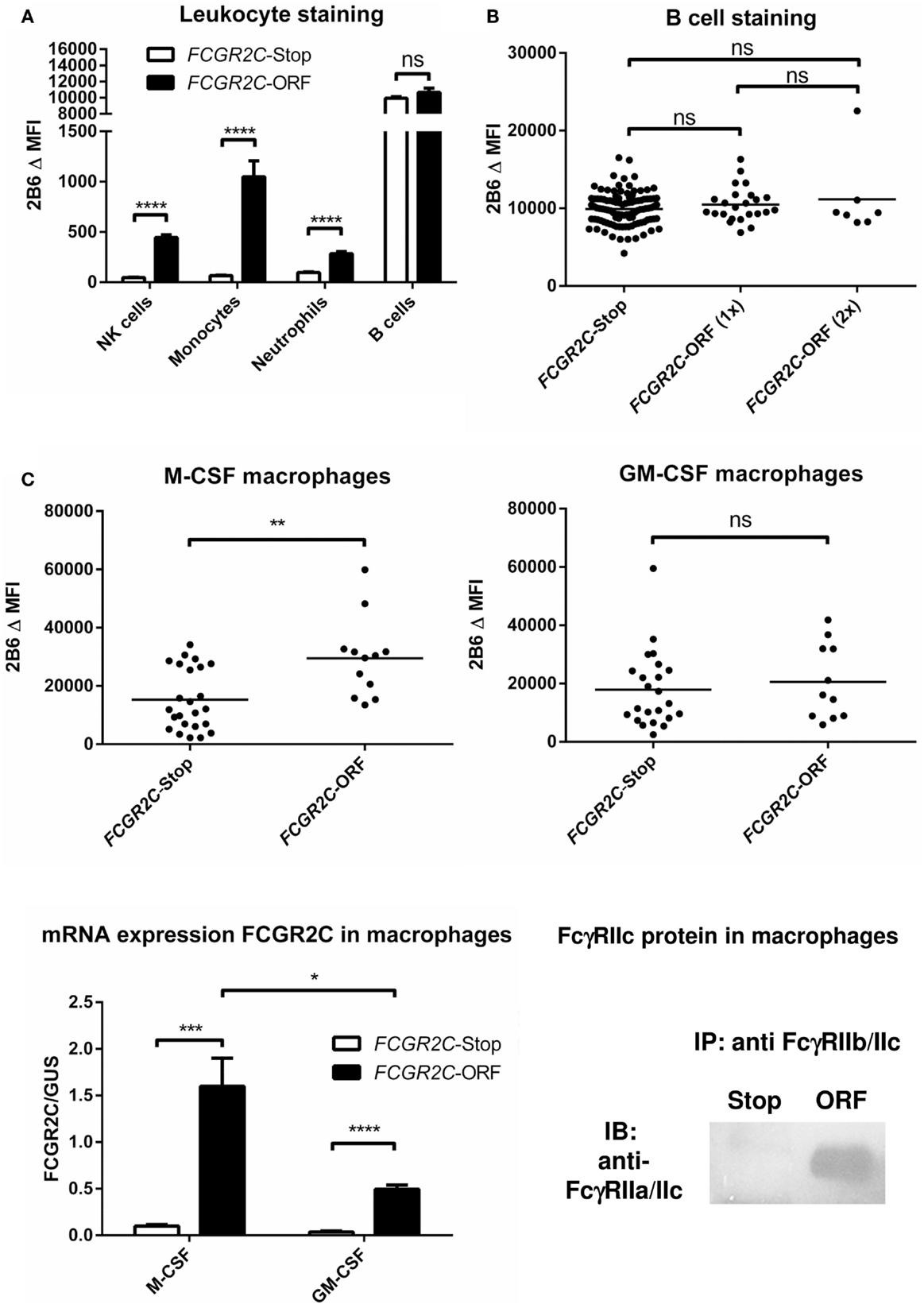
Figure 3. FcγRIIc expression on various cell types in healthy human subjects. (A) Expression of FcγRIIc and FcγRIIb on circulating leukocytes. Figure adapted from van der Heijden et al. (4), now including measurements from additional individuals. MoAb 2B6 recognizes an extracellular epitope of both FcγRIIb and FcγRIIc, but since FCGR2C-Stop individuals cannot express FcγRIIc, the difference in MFI between FCGR2C-Stop and FCGR2C-ORF individuals can be assumed to derive from expression of FcγRIIc. FCGR2C-Stop (individuals with zero copies of FCGR2C-ORF) n = 105; FCGR2C-ORF (including individuals with one and two copies of FCGR2C-ORF) n = 31. Means + SEM are shown. (B) Detailed analysis of MoAb 2B6 staining on circulating B-cells, showing individual measurements, reveals no evidence of expression of FcγRIIc on B-cells. FCGR2C-Stop n = 105; FCGR2C-ORF(1x), individuals with one copy of FCGR2C-ORF, n = 24; FCGR2C-ORF(2x), individuals with two copies of FCGR2C-ORF, n = 7. (C) Expression of FcγRIIc on macrophages. Upper panel: MoAb 2B6 staining on monocyte-derived macrophages cultured for 9 days from 36 healthy individuals, performed as previously described (79). FCGR2C-Stop: n = 24; FCGR2C-ORF, individuals with one (n = 11) or two (n = 1) copies of the FCGR2C-ORF allele. Data are shown for M-CSF (left) and GM-CSF (right) cultured macrophages. Lower left panel: detection of FCGR2C mRNA in monocyte-derived macrophages cultured for 9 days. qPCR with FCGR2C1-specific primers was performed as previously described (5), using cDNA from M-CSF monocyte-derived macrophages as a calibration curve, as described in (79). FCGR2C-ORF, individuals with 1 FCGR2C-ORF allele, n = 3; FCGR2C-Stop, individuals with zero copies of FCGR2C-ORF, n = 4. Means + SEM are shown. Lower right panel: immunoprecipitation of FcγRIIc from M-CSF monocyte-derived macrophages confirms expression of FcγRIIc. Experiment was performed essentially as described in Ref. (4), in this case using MoAb 2B6 to capture FcγRIIc (and FcγRIIb), followed by a specific staining for the intracellular part of FcγRIIc with a rabbit polyclonal antibody against the cytoplasmic tail shared by FcγRIIc and FcγRIIa (25), in macrophages from an individual with zero copies of FCGR2C-ORF (Stop), and an individual with one copy of FCGR2C-ORF (ORF). Data are representative of three independent experiments with different individuals. For reasons of simplicity, in this figure, individuals with the non-classical FCGR2C-ORF allele that is not expressed (4) [n = 6 for (A,B), were grouped with FCGR2C-stop individuals. Individuals with a deletion of CNR1 (FCGR2C and FCGR3B genes], which leads to ectopic expression of FcγRIIb on NK cells (4) (n = 14), were left out of the analysis of NK cells in (A). Statistical significance was tested by Mann–Whitney U test. ns (p > 0.05); *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Δ MFI: median fluorescence intensity of MoAb 2B6 minus median fluorescence intensity of isotype control. Some individuals were analyzed more than once at different time points with similar results, means are shown for these individuals.
FcγRIII (CD16), similarly to FcγRII, actually represents a collection of two genes, expressed within the gene cluster at chromosome 1q23.3, each encoding proteins with two EC Ig-like domains (Figures 1 and 2). Due to differences in glycosylation, their molecular masses are in the range of 50–80 kDa. FcγRIIIa is similar to FcγRI in its TM and IC domains. In monocytes and macrophages, this receptor associates with the FcRγ-chain, while in NK cells it associates with the CD3 ζ-chain to induce signaling (26–28). In contrast to FcγRI, association with these adaptor proteins is not only essential for maintaining stable expression but also for targeting the receptor to the cell membrane (28).
FcγRIIIb is a GPI-anchored protein, expressed only on neutrophils and eosinophils. As it does not have a TM domain, it cannot associate with FcRγ or the ζ-chain. Nonetheless, FcγRIIIb has been suggested to induce signaling, although the exact mechanism(s) is still unclear (29, 30), and current believe is that it functions mainly as a decoy receptor (31).
Recently, it has been proposed that a totally different class of receptors, the C-type lectins, may also bind the Fc-region of IVIg (32), and such receptors were termed “type II Fc receptors.” In humans, DC-SIGN was proposed to bind IgG with a sialic acid sugar moiety at the Asn297 N-linked glycosylation site of IgG. However, this interaction could not be reproduced by another group (33), which found no binding at all of IgG-Fc regions to DC-SIGN, regardless of the sialylation status. Therefore, with evidence for the interaction of IgG-Fc and DC-SIGN being currently marginal and not broadly supported in the literature, in our opinion, it remains to be seen whether DC-SIGN should indeed be classified as a true IgG-Fc receptor.
The genes encoding the classical FcγRs are highly polymorphic and functionally relevant genetic variations have been described for all low-affinity FcγRs (Table 4).

Table 4. Functionally relevant genetic variation in FcγR.
In FCGR2A, encoding for FcγRIIa, a single nucleotide polymorphism (SNP) was first noticed, which results in either a histidine or an arginine at position 131 (H131R) in the IgG binding domain (EC2) (34). FcγRIIa-H131 has a higher binding affinity for IgG1 and especially IgG2, as compared to FcγRIIa-R131, but binding to IgG3 and IgG4 is similar for both variants (38). Functionally, mononuclear cells from FcγRIIa-131HH individuals produce more IL-1beta when stimulated with IgG2 than FcγRIIa-131HR and -131RR individuals (39). Similarly, neutrophils from individuals homozygous for H131 (FcγRIIa-131HH) have been shown to have increased phagocytosis and degranulation in response to serum-opsonized bacteria and increased rosette formation and phagocytosis in presence of IgG3 anti-D sensitized erythrocytes when compared to FcγRIIa-131RR individuals (36, 40).
FCGR2B also exists in two allelic variants, encoding for FcγRIIb containing either an isoleucine or a threonine at position 232 in the TM domain (35). As this SNP (I232T) does not affect the IgG-binding EC domains, it has no influence on the binding affinity. However, its localization at the TM domain results in differences in downstream signaling and subsequent inhibition of FcγRI signaling in macrophages and BCR signaling in B-cells. In particular, I232 provides stronger inhibitory signaling than T232, and this is caused by the exclusion from lipid rafts of FcγRIIb-T232 (41, 42). As FcγRIIb is the only inhibitory FcγR, it has a central role in the regulation of immune responses. The loss-of-function FcγRIIb-T232 has been linked to susceptibility and/or severity of several auto-immune diseases, particularly SLE (43–45), and also in rheumatoid arthritis (RA) (46) and ITP (47).
Inter-individual variation in FcγRIIb is also found in expression patterns and levels. Similar to the I232T SNP, the important immune-regulatory role for FcγRIIb is also reflected in the observations of aberrant expression levels of FcγRIIb in SLE, RA, ITP, and chronic inflammatory demyelinating polyneuropathy (7, 48–51). As a result of a deletion in the FCGR locus that includes FCGR2C, FCGR3B and is called CNR1, FcγRIIb can surprisingly also be expressed on the surface of NK cells, where it is capable to inhibit killing of target cells in ADCC (4). Expression of FcγRIIb in other cells is hardly affected by this deletion. Furthermore, two SNPs in the proximal promoter of FCGR2B and FCGR2C, a guanine or cytosine at position −386 and a thymine or adenine at position −120, form four haplotypes of which one (−386G, −120A; 2B.3) has never been found in any individual thus far. In case of FCGR2B, the wildtype promoter (−386G, −120T; 2B.1) has a lower transcriptional activity than one of the other haplotypes (−386C, −120A; 2B.4) [Ref. (51); Tsang-a-Sjoe et al., submitted].
In case of FCGR2C, only the wildtype and one other promoter haplotype (−386C, −120T; 2B.2) are found. Moreover, the 2B.2 haplotype is linked to another polymorphism in FCGR2C (5). This other polymorphism, a SNP in exon 3, determines whether or not individuals can express FcγRIIc at all. This C > T mutation results in either an open-reading frame (FCGR2C-ORF, allele frequency ~10–15% in Caucasians) or a stop codon (FCGR2C-Stop) (5). Although expression on NK cells is low, it has been shown to be capable of inducing killing of target cells in a redirected ADCC assay (5). Classically, ORF/Stop genotyping of individuals is done based on this SNP alone. However, we have recently found that some individuals carry splice site mutations in intron 7 that introduce novel stop codons, leading to a loss of FcγRIIc expression (4), and genotyping FCGR2C should include these novel mutations to provide an accurate prediction for FcγRIIc expression.
The FcγRIIIa-encoding FCGR3A gene contains a SNP that results in either a valine or a phenylalanine at position 158 (V158F), located in the second EC domain (52). FcγRIIIa-V158 has a higher binding affinity for all human IgG classes compared to FcγRIIIa-F158 (38). In ADCC assays, NK cells from FcγRIIIa-V158 donors show increased killing of target cells that are opsonized with sub-saturating levels of Rituximab (53).
FcγRIIIb-encoding FCGR3B gene exists in three polymorphic variant proteins, NA1, NA2, and SH, which are also known as HNA-1a, -1b, and -1c, respectively (54, 55). FcγRIIIb-NA1 and -NA2 nucleotide sequences differ at five positions [G > C at nucleotide (nt) 141, C > T at nt 147, A > G at nt 227, G > A at nt 277, and G > A at nt 349], with four predicted amino acid differences (R36S, N65S, D82N, and V106I for NA1 and NA2, respectively). As a consequence, the NA2 variant has two additional N-linked glycosylation sites, compared to NA1. The SH variant is identical to NA2 at the five positions that distinguish NA1 from NA2, but differs from both variants at one additional position (C > A at nt 266), resulting in an A78D amino acid change that predicts a change in the tertiary structure of the protein. Additional complexity is added by the discovery of rare individuals carrying other mutations within this gene or different combinations of these nucleotide polymorphisms (37, 56), indicating that the NA1/NA2/SH typing is incomplete. While the binding affinities for IgG1 and IgG3 appear similar between the three variants (38), neutrophils from FcγRIIIb-NA1NA1 individuals bind and phagocytize IgG-opsonized bacteria and red blood cells more efficiently than those from FcγRIIIb-NA1NA2 and -NA2NA2 individuals (40, 57).
Besides being polymorphic, some of the low-affinity FCGR genes are subject to gene copy number variation (CNV). Although several large-scale studies on CNV have suggested that human FCGR2A and FCGR2B are candidate genes for CNV (58–61), our group has shown previously that this is not the case. In fact, CNV in the FCGR locus is restricted to FCGR2C, FCGR3A, and FCGR3B (24). It occurs in three different combinations: FCGR3A/FCGR2C (two possibilities with slightly different borders to the CNV region), and FCGR2C/FCGR3B (Figure 2).
Copy number variation translates into differences in expression levels of FcγRIIc (in case of FCGR2C-ORF), FcγRIIIa, and FcγRIIIb, with more gene copies leading to a higher receptor expression (and vice versa) (21, 62, 63). In case of FcγRIIIa, the level of expression on NK cells is, at least for 1 versus 2 copies, related to the level of killing of target cells in (redirected) ADCC assays (24). Increased expression of FcγRIIIb leads to higher binding and uptake of immune complexes (ICs) (64).
As is the case with polymorphic variants, CNV in FCGR genes is associated with several auto-immune diseases. Our group has previously shown that FCGR2C-ORF predisposes for ITP. The SNP in exon 3 causing an open-reading frame instead of a stop allele of FCGR2C, results in the expression of FcγRIIc and thus behaves as if it were CNV of FCGR2C-ORF. However, individuals can have an increased CNV at this locus of three FCGR2C-Stop alleles without increased risk since only the ORF allele was shown to predispose to ITP (5).
Although we could not find an association with various disease cohorts (24), an increased copy number of FCGR3A has been observed in anti-glomerular basement membrane antibody disease (anti-GBM disease) (65).
In contrast, a low copy number of FCGR3B has been shown to be a risk factor for SLE, even when linkage disequilibrium between FCGR3B CNV and FcγR SNPs that have previously been shown to be associated with SLE is taken into account (66–70). Similar associations have been reported for Sjögren’s syndrome (67), systemic sclerosis (71), and RA (72, 73), although other reports have shown no association with RA (67, 74).
Potential mechanisms can be divided into two categories, being dependent on either the Fc part or the Fab part of the IgG molecule. For some indications, such as ITP, clinical studies with human subjects have in fact already revealed what part of the IgG molecule is effective, as preparations with only Fab fragments of IVIg were not effective (75), whereas purified Fc fragments did have a good clinical effect (76). Thus, we know that at least for ITP, the immunomodulatory effect is Fc-mediated, although this may be different for other indications. Here, we focus most on Fc-mediated modes of action, and will briefly discuss Fab-mediated mechanisms.
Administration of IVIg greatly increases the total concentration of IgG in the recipients’ plasma and extracellular fluid, and with such an increase, more FcγRs may be bound by circulating non-complexed IgG, thereby saturating the FcγRs and making them less available for auto-antibodies in oligo- or polymeric complex with their (auto)antigen. The idea that especially the low-affinity FcγRs can be blocked by their monomeric ligand in vivo may at first hand seem surprising, but it has been shown in the past that also low-affinity receptors bind monomeric IgG (77), indicating that some “low-affinity” FcγRs are not so low-affinity, and maybe should be better named “medium-affinity,” especially in the case of FcγRIIa and FcγRIIIa (38). Greatly increasing the concentration of monomeric IgG above the normal plasma levels may shift the equilibrium toward a situation in which too many FcγRs are occupied for proper functioning – which may in part explain the immunomodulatory actions of IVIg under some of the conditions for which IVIg is used. Saturation of activating FcγRs was one of the first theories that was formulated to explain the working mechanism of IVIg (2), and this “classic” mechanism has for a long time been assumed as the most plausible explanation for the effect of IVIg in ITP (23, 78). Circumstantial evidence for this theory derives from observations that IgG preparations with increased affinity for FcγRs appear to have an increased effect (6, 79), and that in all diseases in which an immunomodulatory effect is wanted, high doses of IVIg are needed. Nevertheless, there is no formal proof for this concept, and although it has never been disproven, focus has shifted away from this theory as other explaining theories arose.
Over the past decade, the prevailing theory for the working mechanism of IVIg in most immunomodulatory situations has become that IVIg induces an upregulation of the inhibitory FcγRIIb on effector cells. More specifically, a fraction of IVIg, i.e., the IgGs containing a sialic acid sugar residue at the end of the N-linked glycosylation site at Asn297, would be responsible for this effect by binding to SIGNR1 (mouse), or its human ortholog DC-SIGN, inducing various signaling cascades ultimately leading to the upregulation FcγRIIb. This theory has recently been excellently reviewed in Ref. (80). However, the major problem with this theory is that many findings could not be reproduced by other research groups. For instance, we have recently found that FcγRIIb is not upregulated in human macrophages in response to IVIg, but nevertheless, these macrophages respond very well to IVIg treatment, being inhibited in phagocytosis (79). The role of FcγRIIb in ITP treatment by IVIg was also questioned in mouse studies (81). Similarly, we found that IgG-Fc sialylation was not important for the effect of IVIg on human macrophages (79), and many groups have recently published evidence that IgG-Fc sialylation of IVIg is not required for the immunomodulatory effects (82–85). As mentioned before, even the binding of sialylated IgG-Fc to DC-SIGN could not be reproduced (33). Furthermore, essentially all the evidence supporting this theory derives from murine studies, which may not be translated to the human situation, as mice and humans extensively differ in FcγR expression. Many of the murine studies describing this theory for instance use a model for arthritis, but IVIg has never proven to be a useful therapy in treating arthritic patients (86–88).
On the other hand, glycosylation may still be important, influencing the binding affinity IgG molecules to the various FcγRs. For instance, the binding affinity of FcγRIIIa is undoubtedly influenced by the level of fucosylation of the Fc-domain of IgG, a notion that may help to develop new, afucosylated IgG treatment options (89, 90). An important question will be whether the anti-inflammatory properties are directly influenced by afucosylated IgG or IgG otherwise modified in their glycosylation status.
FcRn is a receptor expressed by human endothelial cells to recycle plasma IgG, extending its half-life in the circulation (91, 92); saturating this “rescue-receptor” with a high dose of IVIg may shorten the half-life of all IgG including harmful auto-antibodies. Interestingly, for a number of diseases in which IVIg therapy is beneficial, plasmapheresis, aiming to remove pathogenic auto-antibodies by replacing the patients’ plasma with donor plasma is also a good option. This is for instance the case in Guillain Barre syndrome (93, 94). On the other hand, plasmapheresis is not effective in ITP (95, 96), and so apparently, rapid removal of auto-antibodies is not effective in ITP, suggesting that the effect of IVIg in ITP must be exerted in a different way.
Recent data have confirmed the expression of FcγRII isoforms, including FcγRIIb (97, 98) on dendritic cells (DCs), which may help to explain the subsequent steps in which inhibition of autoantibody release by B-cells, inhibition of T-helper (Th)1 and Th17 differentiation, and enhancement of CD4+FoxP3+ regulatory T cells (Treg), helps to modulate certain unwanted (auto)inflammatory responses. IVIg may be able to reset the balance at the level of DCs, involving not only the classical IgG receptors but also non-classical lectin-like surface molecules, as has been repeatedly proposed during the last decade (33, 99). We should emphasize that the relevance of such mechanisms and non-classical IgG receptors remain to be shown in humans for the IVIg-associated effects for immunomodulation.
A recent report showed an increased expression of type I interferon response genes in ITP patients, which was rapidly reduced in patients after receiving IVIg, leading to decreased expression of FcγRIII on monocytes, thereby altering the balance between activating and inhibiting FcγRs (100). The relevance of interferons in such responses is unclear, as interferon response genes have been found in various diseases, among which is SLE, and sometimes independent of clear-cut reaction to treatment and clinical response to therapy (101, 102).
Inhibition of the complement cascade by sequestering complement away from the deposited auto-antibodies as suggested in dermatomyositis (103). On the other hand, with the recent insight that IgG is only able to activate complement by means of generating hexamers, and not as single molecules or dimers (104), it is less likely that complement scavenging roles can realistically be involved in the anti-inflammatory IVIg-mediated effects.
One of the first explanations for the anti-inflammatory effect of IVIg was that there are anti-idiotypic antibodies present in the IVIg that neutralize the pathogenic auto-antibodies. This theory is often claimed but to our knowledge has only been proven to effectively explain the anti-inflammatory potential of IVIg in the case of neutralizing antibodies to coagulation factor VIII, which could be inhibited by anti-idiotypic antibodies in IVIg (105).
Apart from the known microbial antigen-specific binding properties, IgG preparations also contain neutralizing and clearance-enhancing antibodies that may switch a proinflammatory trigger into an anti-inflammatory condition. This suggests that healthy individuals from which plasma is collected and pooled for therapeutic IgG preparations already contain autoreactive “natural” antibodies at low levels in their blood. The infusion of such natural antibodies into the patient may be sufficient to reset certain diseases by the cross-reactive capacity of such natural “auto”antibodies (106–110).
Clearly, the different genetic FcγR variants may not only be a risk factor for the development of some auto-immune diseases but may possibly also influence the efficacy of treatment of these diseases by IVIg. Indeed, some SNPs can be overrepresented in Kawasaki disease (KD) patients that respond well to IVIg therapy, but not in the non-responders. Among KD patients, patients who respond well to IVIg have been reported to more often carry the promoter polymorphism 2B.4 in FCGR2B and the FcγRIIIb-NA1 when compared to non-responders (111, 112). In both these cases, the balance between activating and inhibitory receptor signaling is altered. A shift toward the inhibitory side of the balance increases the efficacy of IVIg treatment, while a shift toward the activating side shows the opposite effect. Connections of IVIg efficacy in KD and/or other auto-immune diseases with other polymorphisms or CNV in FcγR have not been found to date. Given the growing number of diseases in which IVIg therapy is successfully used and the number of possible working mechanisms that involve FcγR, it does not seem unlikely that more such connections exist. The ways in which IVIg may interact with FcγRs to exert its immunomodulatory actions are multiple, since many different FcγRs are expressed by different immune cells. An overview of the potential interactions is given in Figure 4. One special case is the FcγRIIc, discussed in more detail below.
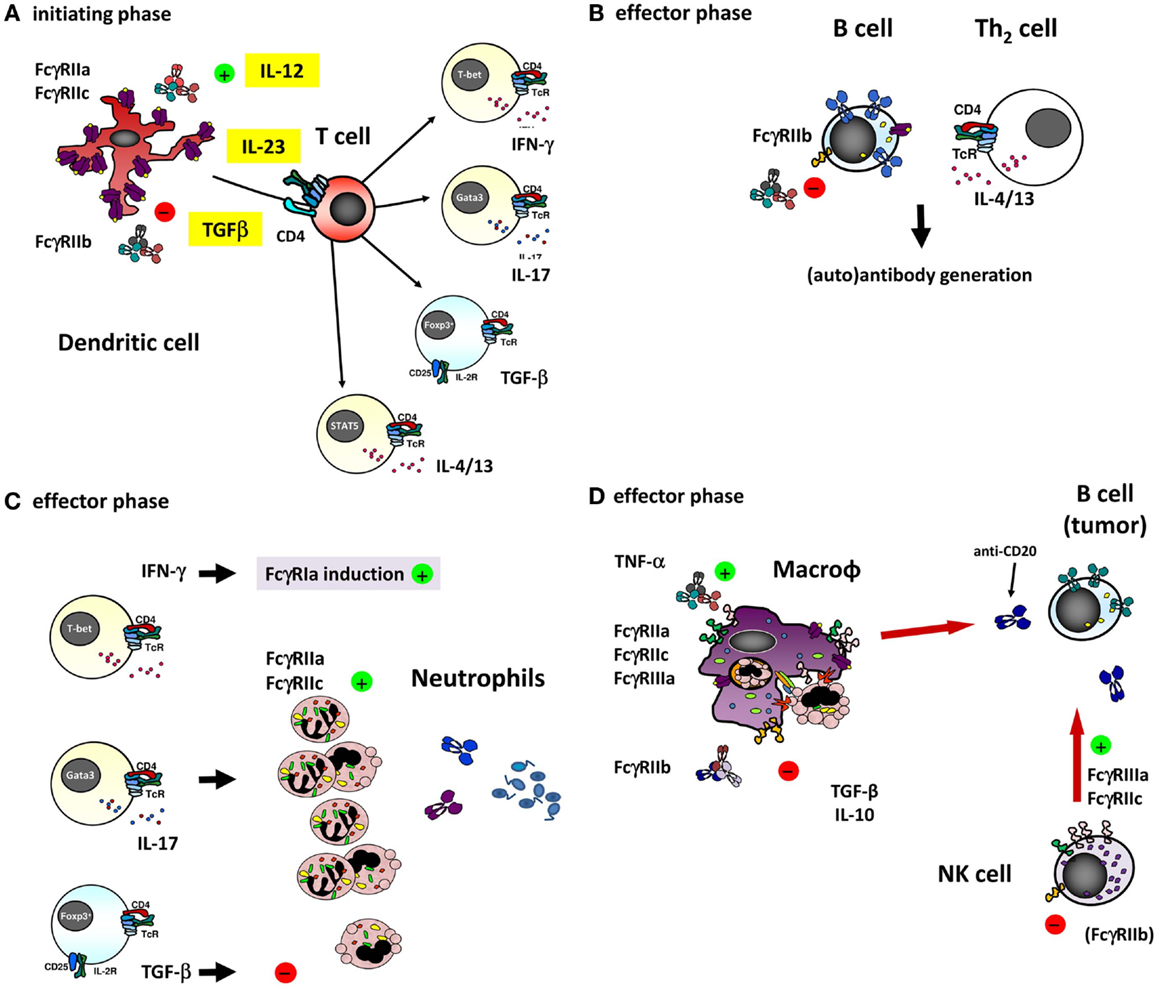
Figure 4. The different ways of how IVIg may influence an immune response involving (auto)-antibodies by interfering with the function of the different FcγRs on various cell types. (A) dendritic cells (DCs) play a major role in the initiating phase of an antibody response, and expression and function of different FcγRs on these cells may influence this response in several ways. For instance, ligation of FcγRIIa on DCs has been shown to influence the cytokine secretion by DCs in response to various TLR ligands, promoting Th17 responses (98). Furthermore, expression of FcγRIIa on myeloid DCs was recently shown to be downregulated by IVIg in an IL-33 dependent manner, which resulted in a Th2 cytokine response (97). As the co-stimulatory signals derived from DCs determine T cell function, they are ultimately important in shaping an antibody response, as well as the extent of generating a neutrophil-driven response. T cells themselves are crucial in determining the differentiation of most B-cells necessary for the antibody response, but are not known to express any classical FcγRs or other IgG receptors themselves, suggesting that they may be influenced by IVIg in indirect ways only. (B) Differentiation of B-cells into antibody producing cells is a major component of an (auto)-antibody response, and involves somatic hypermutation, class switch recombination and plasma cell differentiation. B-cells express high levels of FcγRIIb, and ligation of this receptor has been shown to dampen B-cell receptor signaling. Recently, it was proposed that also FcγRIIc is expressed on B-cells in some individuals, and may influence the immune response in vaccination settings, although it appears that the expression of FcγRIIc on B-cells is very limited if present at all, especially when compared to expression of FcγRIIb (see also Figure 3B). (C) Once an (auto)-antibody is formed, innate effector cells such as neutrophils are determinants of tissue damage at the site of deposited immune complexes by release of toxic components such as reactive oxygen species or proteases. Neutrophils express FcγRIIa and FcγRIIc in some individuals, which can contribute to tissue damage and inflammation, and may be influenced by IVIg. Following the afferent immunological phase, the effector phase of activated neutrophils may be in part driven by the presence of certain Th subsets. The “inflammation-promoting” Th17 activity may be counterbalanced by Tregs inducing immunosuppressive properties by TGF-β1 as indicated by expression of neutrophil IL-10 and IL-6, indoleamine 2,3-dioxygenase (IDO), heme oxygenase-1, and “suppressor of cytokine signaling-3” (SOCS3) (113), although the production of these cytokines by human neutrophils remains controversial. (D) In case of natural or therapeutic antibody responses against cellular targets, these targets can be eliminated by cells of the innate immune system, for instance through antibody-dependent phagocytosis by macrophages, or antibody-dependent cellular cytotoxicity by NK cells (or monocytes and neutrophils). These responses are mediated by FcγRs on these effector cells, which could be influenced by IVIg in a direct way, i.e., saturating activating FcγRs by binding to them, or in a more indirect way as a result of upregulation of the inhibitory FcγRIIb on effector macrophages (discussed in the text).
As mentioned above, a SNP in exon 3 of FCGR2C determines whether or not individuals can express FcγRIIc at all (5). Although expression on NK cells is low, it has been shown to be capable of inducing killing of target cells in a redirected ADCC assay (5). We could also detect FcγRIIc expression on neutrophils and monocytes in individuals with an ORF allele (Figure 3A) (4). When investigating surface FcγRIIc expression on monocyte-derived macrophages skewed to either M1 or M2 phenotype, the M-CSF-cultured cells were clearly expressing FcγRIIc (Figure 3C).
Surprisingly, a recent report also found FcγRIIc expression on B-cells (22). Upon transfection into a murine B-cell line, the co-ligation of FcγRIIc with the BCR resulted in enhanced and more sustained tyrosine phosphorylation of the key B-cells signaling components Syk and BLNK. In contrast, the engagement of FcγRIIb with the BCR and its activation caused a reduced level of Syk and BLNK phosphorylation. Antibodies generated upon immunization in this transgene mouse model were found to be enhanced, coinciding with a higher level of B-cell activation (22). In a cohort of about 300 individuals of unknown ethnicity the levels of Ab against a neoantigen (Anthrax protein) were tested (22). At the earliest time points in the vaccine study donors homozygous for the FCGR2C-ORF allele [i.e., two alleles (n = 11)] showed higher Ab levels at 4 weeks (p < 0.02) but not any longer at 8 weeks (22). The more common single-ORF donors were not included.
Although interesting, direct proof of FcγRIIc expression in human B-cells is lacking, as the protein was only specifically detected in EBV-transformed B-cell lines (22). Stainings with an antibody detecting both FcγRIIb and FcγRIIc showed a difference between Stop and ORF donors (22), suggesting some expression of FcγRIIc in primary and memory B-cells. However, we performed similar stainings in a much larger cohort and did not detect such differences. Hence, we must conclude that the expression of FcγRIIc on primary B-cells is at most marginal if present at all.
In fact, the myeloid expression on macrophages and DCs of FcγRIIc may be held responsible for the earlier peak in Ab generation in FCGR2C-ORF-positive donors. Not only the macrophage as effector mechanism in immune responses could be relevant for adaptive immunity including Ab generation but also the afferent part of adaptive responses may be involved, as may also be suggested for FcγRIIc-expressing human DCs in FCGR2C-ORF individuals. Thus, we hypothesize that FcγRIIc may be a subtle but relevant genetic factor in the fine balance between health and disease, including the way the immune response will shape the adaptive repertoire as indicated by the immunization studies mentioned above (22) (Figure 4, overview).
The Fc-gamma Receptors constitute the major receptors for human IgG. There may be low-affinity receptors with lectin-like binding properties that have been suggested to bind a fraction of IgG depending on IgG glycosylation, such as sialylation, but definite proof awaits further study. The beneficial effects of sialylated IVIg in mice are model-dependent, and evidence that sialylation of IgG plays a role in humans has not been generated thus far. Evidence for a prominent role of DC-SIGN in mediating the anti-inflammatory activity of IVIg in humans is also lacking. Thus, clinical application of sialic acid-enriched IVIg in humans is supposed not to be superior to conventional IVIg. In fact, the classically proposed mechanism of IVIg saturating the FcγRs still appears to be the most logical explanation for the immunomodulatory effects in at least some diseases for which it is indicated, for instance in ITP. However, different mechanisms may be at work in other inflammatory diseases for which IVIg is used, and it is not unlikely that different mechanisms act in concert. Fab-mediated IVIg actions may be relevant for some indications, but clinical studies have in the past ruled out Fab-mediated mechanisms to be important for ITP. Knowledge on whether the immunomodulatory effect of IVIg for a given indication is Fc- or Fab-mediated may become very important if alternative (i.e., not donor-derived) sources of IgG are to be used in the future. When the effects of IVIg are Fc-mediated, the polyclonal aspect of IVIg is clearly not important, and recombinant IgG preparations may suffice, which can then be specifically modified to enhance function. On the other hand, for indication in which the effects are Fab-mediated, the polyclonality is likely to be very important, and recombinant preparations can only be successful if the relevant clones can be identified and expanded for therapeutic IgG production. However, the results of the ITP studies preclude further clinical trials with Fab-only or Fc-only preparations for other indications, as this may withhold patients a currently effective therapy – which clearly is unethical. Hence, it will remain difficult to determine the relative importance of Fab and Fc for indications other than ITP in the human situation.
Although the sialylation of IgG-Fc appears not to be relevant for its immunomodulatory effect, the glycosylation status of IgG may still be important for its function, as the properties of IVIg preparations can for instance be dependent on the level of fucosylation, having effect on the binding affinity to FcγRs. Further studies will help to resolve the effects of the different glycosylation moieties of IgG-Fc on the interactions with the various receptors for IgG-Fc, and the relevance of these interactions for IVIg function. Finally, FcγRIIc is one novel activating IgG receptor that may add to tip the balance of immune responses, which needs further in-depth study, using proper detection methods to obtain evidence by genotyping and biochemistry.
Concluding, an enormous increase in insight has been generated during the last decade that may help to improve IgG therapy, either as supplement or anti-inflammatory approach. Further studies related to glycosylation may be highly relevant in this respect, but the immunomodulatory effects of IVIg seem not to be determined by the level of sialylation as studied in-depth over the last decade.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med (2012) 367:2015–25. doi:10.1056/NEJMra1009433
2. Imbach P, Barandun S, d’Apuzzo V, Baumgartner C, Hirt A, Morell A, et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet (1981) 1(8232):1228–31. doi:10.1016/S0140-6736(81)92400-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood (2012) 119:5640–9. doi:10.1182/blood-2012-01-380121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. van der Heijden J, Breunis WB, Geissler J, de Boer M, van den Berg TK, Kuijpers TW. Phenotypic variation in IgG receptors by nonclassical FCGR2C alleles. J Immunol (2012) 188:1318–24. doi:10.4049/jimmunol.1003945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Breunis WB, van Mirre E, Bruin M, Geissler J, de Boer M, Peters M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood (2008) 111:1029–38. doi:10.1182/blood-2007-03-079913
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Teeling JL, Jansen-Hendriks T, Kuijpers TW, de Haas M, van de Winkel JG, Hack CE, et al. Therapeutic efficacy of intravenous immunoglobulin preparations depends on the immunoglobulin G dimers: studies in experimental immune thrombocytopenia. Blood (2001) 98:1095–9. doi:10.1182/blood.V98.4.1095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Tackenberg B, Jelcic I, Baerenwaldt A, Oertel WH, Sommer N, Nimmerjahn F, et al. Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci U S A (2009) 106:4788–92. doi:10.1073/pnas.0807319106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Hartnell A, Kay AB, Wardlaw AJ. IFN-gamma induces expression of Fc gamma RIII (CD16) on human eosinophils. J Immunol (1992) 148(5):1471–8.
9. Davoine F, Lavigne S, Chakir J, Ferland C, Boulay ME, Laviolette M. Expression of FcgammaRIII (CD16) on human peripheral blood eosinophils increases in allergic conditions. J Allergy Clin Immunol (2002) 109:463–9. doi:10.1067/mai.2002.121952
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol (2010) 10:387–402. doi:10.1038/nri2765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Bezbradica JS, Medzhitov R. Role of ITAM signaling module in signal integration. Curr Opin Immunol (2012) 24(1):58–66. doi:10.1016/j.coi.2011.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. van Vugt MJ, Heijnen AF, Capel PJ, Park SY, Ra C, Saito T, et al. FcR gamma-chain is essential for both surface expression and function of human Fc gamma RI (CD64) in vivo. Blood (1996) 87:3593–9.
13. Cohen DM, Bhalla SC, Anaissie EJ, Hester JP, Savary CA, Rex JH. Effects of in vitro and in vivo cytokine treatment, leucapheresis and irradiation on the function of human neutrophils: implications for white blood cell transfusion therapy. Clin Lab Haematol (1997) 19:39–47. doi:10.1046/j.1365-2257.1997.00207.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Gericke GH, Ericson SG, Pan L, Mills LE, Guyre PM, Ely P. Mature polymorphonuclear leukocytes express high-affinity receptors for IgG (Fc gamma RI) after stimulation with granulocyte colony-stimulating factor (G-CSF). J Leukoc Biol (1995) 57:455–61.
15. Maresco DL, Chang E, Theil KS, Francke U, Anderson CL. The three genes of the human FCGR1 gene family encoding Fc gamma RI flank the centromere of chromosome 1 at 1p12 and 1q21. Cytogenet Cell Genet (1996) 73:157–63. doi:10.1159/000134330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Ernst LK, Duchemin AM, Miller KL, Anderson CL. Molecular characterization of six variant Fcgamma receptor class I (CD64) transcripts. Mol Immunol (1998) 35:943–54. doi:10.1016/S0161-5890(98)00079-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Van den Herik-Oudijk IE, Capel PJ, van der Bruggen T, van de Winkel JG. Identification of signaling motifs within human Fc gamma RIIa and Fc gamma RIIb isoforms. Blood (1995) 85:2202–11.
18. Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol (2010) 10:328–43. doi:10.1038/nri2762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Brooks DG, Qiu WQ, Luster AD, Ravetch JV. Structure and expression of human IgG FcRII(CD32). Functional heterogeneity is encoded by the alternatively spliced products of multiple genes. J Exp Med (1989) 170:1369–85. doi:10.1084/jem.170.4.1369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Warmerdam PA, Nabben NM, van de Graaf SA, van de Winkel JG, Capel PJ. The human low affinity immunoglobulin G Fc receptor IIC gene is a result of an unequal crossover event. J Biol Chem (1993) 268:7346–9.
21. Metes D, Ernst LK, Chambers WH, Sulica A, Herberman RB, Morel PA. Expression of functional CD32 molecules on human NK cells is determined by an allelic polymorphism of the FcgammaRIIC gene. Blood (1998) 91:2369–80.
22. Li X, Wu J, Ptacek T, Redden DT, Brown EE, Alarcon GS, et al. Allelic dependent expression of an activating Fc receptor n B cells enhances humoral immune responses. Sci Transl Med (2013) 5(216):216ra175. doi:10.1126/scitranslmed.3007097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Bussel J. Fc receptor blockade and immune thrombocytopenic purpura. Semin Hematol (2000) 37:261–6. doi:10.1053/shem.2000.8957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Breunis WB, van Mirre E, Geissler J, Laddach N, Wolbink G, van der Schoot E, et al. Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum Mutat (2009) 30:E640–50. doi:10.1002/humu.20997
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. van der Heijden J, Geissler J, van Mirre E, van Deuren M, van der Meer JW, Salama A, et al. A novel splice variant of FcγRIIa: a risk factor for anaphylaxis in patients with hypogammaglobulinemia. J Allergy Clin Immunol (2013) 131(5):1408–16. doi:10.1016/j.jaci.2013.02.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Masuda M, Roos D. Association of all three types of Fc gamma R (CD64, CD32, and CD16) with a gamma-chain homodimer in cultured human monocytes. J Immunol (1993) 151:7188–95.
27. Trinchieri G, Valiante N. Receptors for the Fc fragment of IgG on natural killer cells. Nat Immunol (1993) 12(4–5):218–34.
28. Park JG, Isaacs RE, Chien P, Schreiber AD. In the absence of other Fc receptors, Fc gamma RIIIA transmits a phagocytic signal that requires the cytoplasmic domain of its gamma subunit. J Clin Invest (1993) 92:1967–73. doi:10.1172/JCI116790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Edberg JC, Moon JJ, Chang DJ, Kimberly RP. Differential regulation of human neutrophil FcgammaRIIa (CD32) and FcgammaRIIIb (CD16)-induced Ca2+ transients. J Biol Chem (1998) 273:8071–9. doi:10.1074/jbc.273.14.8071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Fernandes MJ, Lachance G, Pare G, Rollet-Labelle E, Naccache PH. Signaling through CD16b in human neutrophils involves the Tec family of tyrosine kinases. J Leukoc Biol (2005) 78:524–32. doi:10.1189/jlb.0804479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcgamma receptors in dendritic cells and macrophages. Nat Rev Immunol (2014) 14:94–108. doi:10.1038/nri3582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol (2014) 15:707–16. doi:10.1038/ni.2939
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Yu XJ, Vasiljevic S, Mitchell DA, Crispin M, Scanlan CN. Dissecting the molecular mechanism of IVIg therapy: the interaction between serum IgG and DC-SIGN is independent of antibody glycoform or Fc domain. J Mol Biol (2013) 425:1253–8. doi:10.1016/j.jmb.2013.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Warmerdam PA, van de Winkel JG, Gosselin EJ, Capel PJ. Molecular basis for a polymorphism of human Fc gamma receptor II (CD32). J Exp Med (1990) 172:19–25. doi:10.1084/jem.172.1.19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Li X, Wu J, Carter RH, Edberg JC, Su K, Cooper GS, et al. A novel polymorphism in the Fcgamma receptor IIB (CD32B) transmembrane region alters receptor signaling. Arthritis Rheum (2003) 48:3242–52. doi:10.1002/art.11313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Nicu EA, van der Velden U, Everts V, van Winkelhoff AJ, Roos D, Loos BG. Hyper-reactive PMNs in FcgammaRIIa 131 H/H genotype periodontitis patients. J Clin Periodontol (2007) 34:938–45. doi:10.1111/j.1600-051X.2007.01136.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Yamamoto K, Sugita N, Kobayashi T, Okuda K, van de Winkel JG, Yoshie H. Evidence for a novel polymorphism affecting both N-linked glycosylation and ligand binding of the IgG receptor IIIB (CD16). Tissue Antigens (2001) 57:363–6. doi:10.1034/j.1399-0039.2001.057004363.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113:3716–25. doi:10.1182/blood-2008-09-179754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Yamamoto K, Kobayashi T, Sugita N, Tai H, Yoshie H. The FcgammaRIIa polymorphism influences production of interleukin-1 by mononuclear cells. Int J Immunogenet (2007) 34:369–72. doi:10.1111/j.1744-313X.2007.00701.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Bredius RG, Fijen CA, de Haas M, Kuijper EJ, Weening RS, van de Winkel JG, et al. Role of neutrophil Fc gamma RIIa (CD32) and Fc gamma RIIIb (CD16) polymorphic forms in phagocytosis of human IgG1- and IgG3-opsonized bacteria and erythrocytes. Immunology (1994) 83:624–30.
41. Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med (2005) 11:1056–8. doi:10.1038/nm1288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kono H, Kyogoku C, Suzuki T, Tsuchiya N, Honda H, Yamamoto K, et al. FcgammaRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet (2005) 14:2881–92. doi:10.1093/hmg/ddi320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Willcocks LC, Carr EJ, Niederer HA, Rayner TF, Williams TN, Yang W, et al. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc Natl Acad Sci U S A (2010) 107:7881–5. doi:10.1073/pnas.0915133107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Siriboonrit U, Tsuchiya N, Sirikong M, Kyogoku C, Bejrachandra S, Suthipinittharm P, et al. Association of Fcgamma receptor IIb and IIIb polymorphisms with susceptibility to systemic lupus erythematosus in Thais. Tissue Antigens (2003) 61:374–83. doi:10.1034/j.1399-0039.2003.00047.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, et al. Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum (2002) 46:1242–54. doi:10.1002/art.10257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Radstake TR, Franke B, Wenink MH, Nabbe KC, Coenen MJ, Welsing P, et al. The functional variant of the inhibitory Fcgamma receptor IIb (CD32B) is associated with the rate of radiologic joint damage and dendritic cell function in rheumatoid arthritis. Arthritis Rheum (2006) 54:3828–37. doi:10.1002/art.22275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bruin M, Bierings M, Uiterwaal C, Revesz T, Bode L, Wiesman ME, et al. Platelet count, previous infection and FCGR2B genotype predict development of chronic disease in newly diagnosed idiopathic thrombocytopenia in childhood: results of a prospective study. Br J Haematol (2004) 127:561–7. doi:10.1111/j.1365-2141.2004.05235.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, et al. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med (2006) 203:2157–64. doi:10.1084/jem.20051503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Carreno LJ, Pacheco R, Gutierrez MA, Jacobelli S, Kalergis AM. Disease activity in systemic lupus erythematosus is associated with an altered expression of low-affinity Fc gamma receptors and costimulatory molecules on dendritic cells. Immunology (2009) 128:334–41. doi:10.1111/j.1365-2567.2009.03138.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Wenink MH, Santegoets KC, Roelofs MF, Huijbens R, Koenen HJ, van Beek R, et al. The inhibitory Fc gamma IIb receptor dampens TLR4-mediated immune responses and is selectively up-regulated on dendritic cells from rheumatoid arthritis patients with quiescent disease. J Immunol (2009) 183:4509–20. doi:10.4049/jimmunol.0900153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Su K, Wu J, Edberg JC, Li X, Ferguson L, Cooper GS, et al. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. I. Regulatory FCGR2B polymorphisms and their association with systemic lupus erythematosus. J Immunol (2004) 172:7186–91. doi:10.4049/jimmunol.172.11.7186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest (1997) 100:1059–70. doi:10.1172/JCI119616
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, et al. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res (2004) 64:4664–9. doi:10.1158/0008-5472.CAN-03-2862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Ory PA, Clark MR, Kwoh EE, Clarkson SB, Goldstein IM. Sequences of complementary DNAs that encode the NA1 and NA2 forms of Fc receptor III on human neutrophils. J Clin Invest (1989) 84:1688–91. doi:10.1172/JCI114350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Bux J, Stein EL, Bierling P, Fromont P, Clay M, Stroncek D, et al. Characterization of a new alloantigen (SH) on the human neutrophil Fc gamma receptor IIIb. Blood (1997) 89:1027–34.
56. Matsuo K, Procter J, Stroncek D. Variations in genes encoding neutrophil antigens NA1 and NA2. Transfusion (2000) 40:645–53. doi:10.1046/j.1537-2995.2000.40060645.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Salmon JE, Edberg JC, Kimberly RP. Fc gamma receptor III on human neutrophils. Allelic variants have functionally distinct capacities. J Clin Invest (1990) 85:1287–95. doi:10.1172/JCI114566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. de Smith AJ, Tsalenko A, Sampas N, Scheffer A, Yamada NA, Tsang P, et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet (2007) 16:2783–94. doi:10.1093/hmg/ddm208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature (2006) 444:444–54. doi:10.1038/nature05329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Wong KK, de Leeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet (2007) 80:91–104. doi:10.1086/510560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nat Genet (2004) 36:949–51. doi:10.1038/ng1416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Koene HR, Kleijer M, Roos D, de Haas M, von dem Borne AE. Fc gamma RIIIB gene duplication: evidence for presence and expression of three distinct Fc gamma RIIIB genes in NA(1+,2+)SH(+) individuals. Blood (1998) 91:673–9.
63. de Haas M, Kleijer M, van Zwieten R, Roos D, von dem Borne AE. Neutrophil Fc gamma RIIIb deficiency, nature, and clinical consequences: a study of 21 individuals from 14 families. Blood (1995) 86:2403–13.
64. Willcocks LC, Lyons PA, Clatworthy MR, Robinson JI, Yang W, Newland SA, et al. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med (2008) 205:1573–82. doi:10.1084/jem.20072413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Zhou XJ, Lv JC, Bu DF, Yu L, Yang YR, Zhao J, et al. Copy number variation of FCGR3A rather than FCGR3B and FCGR2B is associated with susceptibility to anti-GBM disease. Int Immunol (2010) 22:45–51. doi:10.1093/intimm/dxp113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet (2007) 39:721–3. doi:10.1038/ng2046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Mamtani M, Anaya JM, He W, Ahuja SK. Association of copy number variation in the FCGR3B gene with risk of autoimmune diseases. Genes Immun (2010) 11:155–60. doi:10.1038/gene.2009.71
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Morris DL, Roberts AL, Witherden AS, Tarzi R, Barros P, Whittaker JC, et al. Evidence for both copy number and allelic (NA1/NA2) risk at the FCGR3B locus in systemic lupus erythematosus. Eur J Hum Genet (2010) 18:1027–31. doi:10.1038/ejhg.2010.56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Niederer HA, Willcocks LC, Rayner TF, Yang W, Lau YL, Williams TN, et al. Copy number, linkage disequilibrium and disease association in the FCGR locus. Hum Mol Genet (2010) 19:3282–94. doi:10.1093/hmg/ddq216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature (2006) 439:851–5. doi:10.1038/nature04489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. McKinney C, Broen JC, Vonk MC, Beretta L, Hesselstrand R, Hunzelmann N, et al. Evidence that deletion at FCGR3B is a risk factor for systemic sclerosis. Genes Immun (2012) 13:458–60. doi:10.1038/gene.2012.15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. McKinney C, Fanciulli M, Merriman ME, Phipps-Green A, Alizadeh BZ, Koeleman BP, et al. Association of variation in Fcgamma receptor 3B gene copy number with rheumatoid arthritis in Caucasian samples. Ann Rheum Dis (2010) 69:1711–6. doi:10.1136/ard.2009.123588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Graf SW, Lester S, Nossent JC, Hill CL, Proudman SM, Lee A, et al. Low copy number of the FCGR3B gene and rheumatoid arthritis: a case-control study and meta-analysis. Arthritis Res Ther (2012) 14:R28. doi:10.1186/ar3731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Marques RB, Thabet MM, White SJ, Houwing-Duistermaat JJ, Bakker AM, Hendriks GJ, et al. Genetic variation of the Fc gamma receptor 3B gene and association with rheumatoid arthritis. PLoS One (2010) 5(10):ii:e13173. doi:10.1371/journal.pone.0013173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Burdach SE, Evers KG, Geursen RG. Treatment of acute idiopathic thrombocytopenic purpura of childhood with intravenous immunoglobulin G: comparative efficacy of 7S and 5S preparations. J Pediatr (1986) 109:770–5. doi:10.1016/S0022-3476(86)80691-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Debré M, Bonnet MC, Fridman WH, Carosella E, Philippe N, Reinert P, et al. Infusion of Fc gamma fragments for treatment of children with acute immune thrombocytopenic purpura. Lancet (1993) 342:945–9. doi:10.1016/0140-6736(93)92000-J
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. van Mirre E, Teeling JL, van der Meer JW, Bleeker WK, Hack CE. Monomeric IgG in intravenous Ig preparations is a functional antagonist of FcgammaRII and FcgammaRIIIb. J Immunol (2004) 173:332–9. doi:10.4049/jimmunol.173.1.332
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Crow AR, Lazarus AH. Role of Fcgamma receptors in the pathogenesis and treatment of idiopathic thrombocytopenic purpura. J Pediatr Hematol Oncol (2003) 25(Suppl 1):S14–8. doi:10.1097/00043426-200312001-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Nagelkerke SQ, Dekkers G, Kustiawan I, van de Bovenkamp FS, Geissler J, Plomp R, et al. Inhibition of FcγR-mediated phagocytosis by IVIg is independent of IgG-Fc sialylation and FcγRIIb in human macrophages. Blood (2014) 124:3709–18. doi:10.1182/blood-2014-05-576835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol (2013) 13:176–89. doi:10.1038/nri3401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Leontyev D, Katsman Y, Branch DR. Mouse background and IVIG dosage are critical in establishing the role of inhibitory Fc gamma receptor for the amelioration of experimental ITP. Blood (2012) 119:5261–4. doi:10.1182/blood-2012-03-415695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Guhr T, Bloem J, Derksen NI, Wuhrer M, Koenderman AH, Aalberse RC, et al. Enrichment of sialylated IgG by lectin fractionation does not enhance the efficacy of immunoglobulin G in a murine model of immune thrombocytopenia. PLoS One (2011) 2011(6):e21246. doi:10.1371/journal.pone.0021246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Leontyev D, Katsman Y, Ma XZ, Miescher S, Käsermann F, Branch DR. Sialylation-independent mechanism involved in the amelioration of murine immune thrombocytopenia using intravenous gammaglobulin. Transfusion (2012) 52:1799–805. doi:10.1111/j.1537-2995.2011.03517.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Campbell IK, Miescher S, Branch DR, Mott PJ, Lazarus AH, Han D, et al. Therapeutic effect of IVIG on inflammatory arthritis in mice is dependent on the Fc portion and independent of sialylation or basophils. J Immunol (2014) 192:5031–8. doi:10.4049/jimmunol.1301611
85. Kasermann F, Boerema DJ, Ruegsegger M, Hofmann A, Wymann S, Zuercher AW, et al. Analysis and functional consequences of increased Fab-sialylation of intravenous immunoglobulin (IVIG) after lectin fractionation. PLoS One (2012) 7:e37243. doi:10.1371/journal.pone.0037243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Maksymowych WP, Aviña-Zubieta A, Luong M, Russell AS. High dose intravenous immunoglobulin (IVIg) in severe refractory rheumatoid arthritis: no evidence for efficacy. Clin Exp Rheumatol (1996) 14(6):657–60.
87. Kanik KS, Yarboro CH, Naparstek Y, Plotz PH, Wilder RL. Failure of low-dose intravenous immunoglobulin therapy to suppress disease activity in patients with treatment-refractory rheumatoid arthritis. Arthritis Rheum (1996) 39(6):1027–9. doi:10.1002/art.1780390621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. De Vita S, Ferraccioli GF, Di Poi E, Bartoli E, Bombardieri S. High dose intravenous immunoglobulin therapy for rheumatic diseases: clinical relevance and personal experience. Clin Exp Rheumatol (1996) 14(Suppl 15):S85–92.
89. Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, et al. Unique carbhydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A (2011) 108(31):12669–74. doi:10.1073/pnas.1108455108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Peipp M, Lammerts van Bueren JJ, Schneider-Merck T, Bleeker WW, Dechant M, Beyer T, et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood (2008) 112(6):2390–9. doi:10.1182/blood-2008-03-144600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol (2007) 7(9):715–25. doi:10.1038/nri2155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med (2003) 197(3):315–22. doi:10.1084/jem.20021829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Raphael JC, Chevret S, Hughes RA, Annane D. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev (2012) 7:CD001798. doi:10.1002/14651858.CD001798.pub2
94. Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev (2014) 9:CD002063. doi:10.1002/14651858.CD002063.pub5
95. Provan D, Stasi R, Newland AC, Blanchette VS, Bolton-Maggs P, Bussel JB, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood (2010) 115:168–86. doi:10.1182/blood-2009-06-225565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Masseau A, Guitton C, Bretonnière C, Renard B, Villers D, Hamidou M. Plasma exchanges as treatment of severe acute immune thrombocytopenic purpura. Rev Med Interne (2005) 26(10):824–6. doi:10.1016/j.revmed.2005.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Tjon AS, van Gent R, Jaadar H, van Hagen MP, Mancham S, van der Laan LJ, et al. Intravenous immunoglobulin treatment in humans suppresses dendritic cell function via stimulation of IL-4 and IL-13 production. J Immunol (2014) 192(12):5625–34. doi:10.4049/jimmunol.1301260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. den Dunnen J, Vogelpoel LT, Wypych T, Muller FJ, de Boer L, Kuijpers TW, et al. IgG opsonization of bacteria promotes Th17 responses via synergy between TLRs and FcγRIIa in human dendritic cells. Blood (2012) 120(1):112–21. doi:10.1182/blood-2011-12-399931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Massoud AH, Yona M, Xue D, Chouiali F, Alturaihi H, Ablona A, et al. Dendritic cell immunoreceptor: a novel receptor for intravenous immunoglobulin mediates induction of regulatory T cells. J Allergy Clin Immunol (2013) 133(3):853–63. doi:10.1016/j.jaci.2013.09.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Sehgal K, Guo X, Koduru S, Shah A, Lin A, Yan X, et al. Plasmacytoid dendritic cells, interferon signaling, and FcgammaR contribute to pathogenesis and therapeutic response in childhood immune thrombocytopenia. Sci Transl Med (2013) 5(193):193ra89. doi:10.1126/scitranslmed.3006277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci (2011) 1238:91–8. doi:10.1111/j.1749-6632.2011.06220.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet (2012) 44:1243–8. doi:10.1038/ng.2414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Basta M, Dalakas MC. High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J Clin Invest (1994) 94(5):1729–35. doi:10.1172/JCI117520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science (2014) 343(6176):1260–3. doi:10.1126/science.1248943
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Sultan Y, Kazatchkine MD, Nydegger U, Rossi F, Dietrich G, Algiman M. Intravenous immunoglobulin in the treatment of spontaneously acquired factor VIII:C inhibitors. Am J Med (1991) 91(5A):35S–9S. doi:10.1016/S0002-9343(91)80147-E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Abe Y, Horiuchi A, Miyake M, Kimura S. Anti-cytokine nature of natural human immunoglobulin: one possible mechanism of the clinical effect of intravenous immunoglobulin therapy. Immunol Rev (1994) 139:5–19. doi:10.1111/j.1600-065X.1994.tb00854.x
107. Ross C, Svenson M, Hansen MB, Vejlsgaard GL, Bendtzen K. High avidity IFN-neutralizing antibodies in pharmaceutically prepared human IgG. J Clin Invest (1995) 95(5):1974–8. doi:10.1172/JCI117881
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Svenson M, Hansen MB, Ross C, Diamant M, Rieneck K, Nielsen H, et al. Antibody to granulocyte-macrophage colony-stimulating factor is a dominant anti-cytokine activity in human IgG preparations. Blood (1998) 91(6):2054–61.
109. Reipert BM, Stellamor MT, Poell M, Ilas J, Sasgary M, Reipert S, et al. Variation of anti-Fas antibodies in different lots of intravenous immunoglobulin. Vox Sang (2008) 94(4):334–41. doi:10.1111/j.1423-0410.2008.001036.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Viard I, Wehrli P, Bullani R, Schneider P, Holler N, Salomon D, et al. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science (1998) 282(5388):490–3. doi:10.1126/science.282.5388.490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Shrestha S, Wiener HW, Olson AK, Edberg JC, Bowles NE, Patel H, et al. Functional FCGR2B gene variants influence intravenous immunoglobulin response in patients with Kawasaki disease. J Allergy Clin Immunol (2011) 128(3):677–80. doi:10.1016/j.jaci.2011.04.027
112. Shrestha S, Wiener H, Shendre A, Kaslow RA, Wu J, Olson A, et al. Role of activating FcgammaR gene polymorphisms in Kawasaki disease susceptibility and intravenous immunoglobulin response. Circ Cardiovasc Genet (2012) 5(3):309–16. doi:10.1161/CIRCGENETICS.111.962464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Lewkowicz N, Klink M, Mycko MP, Lewkowicz P. Neutrophil-CD4+CD25+ T regulatory cell interactions: a possible new mechanism of infectious tolerance. Immunobiology (2013) 218(4):455–64. doi:10.1016/j.imbio.2012.05.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: IgG, Fc gamma receptors, immunomodulation, IVIg, mechanisms of action
Citation: Nagelkerke SQ and Kuijpers TW (2015) Immunomodulation by IVIg and the role of Fc-gamma receptors: classic mechanisms of action after all? Front. Immunol. 5:674. doi: 10.3389/fimmu.2014.00674
Received: 24 October 2014; Accepted: 15 December 2014;
Published online: 21 January 2015.
Edited by:
Isabella Quinti, Sapienza University of Rome, ItalyReviewed by:
Antonio Condino-Neto, University of São Paulo, BrazilCopyright: © 2015 Nagelkerke and Kuijpers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taco W. Kuijpers, Department of Pediatric Hematology, Immunology and Infectious disease (KHI), Academic Medical Center (AMC), Room H7-230, Meibergdreef 9, Amsterdam 1105 AZ, Netherlands e-mail:dC53Lmt1aWpwZXJzQGFtYy51dmEubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.