 Elena Tomasello
Elena Tomasello Emeline Pollet1,2,3†
Emeline Pollet1,2,3† Thien-Phong Vu Manh
Thien-Phong Vu Manh Gilles Uzé
Gilles Uzé Marc Dalod
Marc Dalod- 1UM2, Centre d’Immunologie de Marseille-Luminy (CIML), Aix-Marseille University, Marseille, France
- 2U1104, Institut National de la Santé et de la Recherche Médicale (INSERM), Marseille, France
- 3UMR7280, Centre National de la Recherche Scientifique (CNRS), Marseille, France
- 4UMR 5235, Centre National de la Recherche Scientifique (CNRS), University Montpellier II, Montpellier, France
Type I interferons (IFN-I) were identified over 50 years ago as cytokines critical for host defense against viral infections. IFN-I promote anti-viral defense through two main mechanisms. First, IFN-I directly reinforce or induce de novo in potentially all cells the expression of effector molecules of intrinsic anti-viral immunity. Second, IFN-I orchestrate innate and adaptive anti-viral immunity. However, IFN-I responses can be deleterious for the host in a number of circumstances, including secondary bacterial or fungal infections, several autoimmune diseases, and, paradoxically, certain chronic viral infections. We will review the proposed nature of protective versus deleterious IFN-I responses in selected diseases. Emphasis will be put on the potentially deleterious functions of IFN-I in human immunodeficiency virus type 1 (HIV-1) infection, and on the respective roles of IFN-I and IFN-III in promoting resolution of hepatitis C virus (HCV) infection. We will then discuss how the balance between beneficial versus deleterious IFN-I responses is modulated by several key parameters including (i) the subtypes and dose of IFN-I produced, (ii) the cell types affected by IFN-I, and (iii) the source and timing of IFN-I production. Finally, we will speculate how integration of this knowledge combined with advanced biochemical manipulation of the activity of the cytokines should allow designing innovative immunotherapeutic treatments in patients. Specifically, we will discuss how induction or blockade of specific IFN-I responses in targeted cell types could promote the beneficial functions of IFN-I and/or dampen their deleterious effects, in a manner adapted to each disease.
Introduction
Type I interferons (IFN-I) were the first cytokines discovered, over 50 years ago, based on their potent anti-viral effects (1, 2). IFN-I play a crucial, non-redundant role in vertebrate anti-viral defenses (3–5). IFN-I also mediate protective effects in other physiopathological contexts, including cancer (6) and multiple sclerosis (MS) (7). On the contrary, IFN-I responses can be deleterious in a number of circumstances, including bacterial or fungal infections (8–10), many autoimmune diseases (11), and, paradoxically, certain chronic viral infections (12–14). It is only recently that an integrated picture has emerged of the cellular and molecular mechanisms regulating the production of IFN-I and underlying their functions. Much knowledge was gained recently on another class of potent innate anti-viral interferons, the lambda, or type III IFNs (IFN-III). We will review knowledge on IFN-I/III (IFNs) and discuss how it could be harnessed to develop innovative therapeutic strategies aimed at surgically tuning IFN activity toward protective responses in a manner adapted to each disease. We will focus on IFN-α/β/λ because they are the best characterized IFNs and already used therapeutically. Recent reviews are covering information on other IFN-I subsets including IFN-ε, which is produced at mucosal sites and promotes local anti-viral defenses (15, 16).
Dendritic cells (DCs) are rare heterogeneous mononuclear phagocytes functionally characterized by their unique efficacy for antigen-specific activation of naïve T lymphocytes. DCs are sentinel cells of the immune system, able to sense and integrate a variety of danger input signals for delivery of output signals instructing the activation and functional polarization of effector immune cells. In mammals, five major DC subsets exist, which differ in their expression of innate immune recognition receptors (I2R2s) and in their functional specialization: monocyte-derived DCs (MoDCs), Langerhans cells, CD11b+ DCs, XCR1+ DCs, and plasmacytoid DCs (pDCs) (17). A recurrent theme of this review will be the intricate relationships between IFNs and DCs, since these cells can be a major source and/or target of these cytokines under various conditions.
The first section will synthesize current knowledge on IFN production and protective anti-viral functions. The I2R2s and downstream signaling pathways responsible for IFN-I production during viral infection will be listed. The roles of different cell types for this function will be discussed. The two main mechanisms through which IFN-I promote anti-viral defense will be reviewed, succinctly for direct anti-viral effects and in greater details for immunoregulatory functions.
The second section will focus on the detrimental functions of IFN-I. Selected diseases will be discussed to illustrate how different, and sometimes opposite, processes underlie deleterious IFN-I responses depending on the physiopathological contexts. IFN-I induction of unbridled inflammatory responses causing lethal tissue damage will be discussed as a major pathological mechanism during bacterial encounters secondary to influenza infection or in some autoimmune diseases. Inappropriate functional polarization of immune responses by IFN-I will be discussed as one potential cause for enhanced susceptibility to bacterial or fungal infections. The complex and disputed role of IFN-I in chronic viral infections will be reviewed, with emphasis on the physiopathology of the infections by human immunodeficiency virus type 1 (HIV-1) and human hepatitis C virus (HCV), with an outlook for the development of novel immunotherapeutic strategies to combine with anti-viral drugs.
The third section will recapitulate how the balance between beneficial versus deleterious IFN-I responses is modulated by several key parameters including (i) the source and timing of IFN-I production, (ii) the cell types affected by IFN-I, and (iii) the signaling pathways activated by IFN-I.
In the last section, we will speculate how integration of all the knowledge discussed before combined with advanced biochemical manipulation of the activity of the cytokines should allow designing innovative immunotherapeutic treatments, based on induction or blockade of specific IFN-I responses in targeted cell types. This “activity-by-targeting” concept is based on the design of novel “immuno-IFNs” consisting in covalent association between a mutated IFN-I with decreased affinity for its receptor and an antibody with high avidity for a molecule specifically expressed on target cell types (18). This design ensures lack of activity of the immuno-IFNs on all cell types but those targeted, contrary to previous strategies using IFNs with close to maximal potency that were still able to mediate strong off-target effects despite their coupling to cell-type specific antibodies and/or their local delivery.
General Concepts on IFN Production and Functions
How is the Production of IFN Controlled?
Type I interferons expression is not detectable under steady state conditions in vivo using classical methods such as gene expression analysis by RT-PCR or protein titration by ELISA or bioassays. However, mice deficient for the expression of the alpha chain of the IFN-I receptor (IFNAR1) harbor alteration in the ontogeny or functions of various cell types (19–26). Hence, extremely small or localized but functionally relevant quantities of IFN-I must be produced under steady state conditions (27). Indeed, the existence of steady state responses to IFN-I in various organs in vivo was demonstrated by using reporter mice expressing the firefly luciferase under the control of the promoter of Ifnb1 (28) or of Mx2 (29), a canonical IFN-I-stimulated gene (ISG). Steady state IFN-I responses are promoted by gut commensals (30). Early and transiently after many viral infections, large amounts of IFNs can be detected, in blood and spleen in the case of systemic infections or locally in the case of confined infections. IFN induction during viral infections results from the detection of specific danger signals by specialized I2R2s. This includes the detection of pathogen-associated molecular patterns as well as the sensing of stress signals or damage-associated molecular patterns (31, 32). Based on the nature and intracellular location of the danger signals that induce the production of the cytokines, the cellular sources of IFNs during viral infection can be classified in two main groups. Infected cells often contribute to IFN production as a response to their sensing of endogenous viral replication, or consecutive to the metabolic stress induced during massive translation of viral structural proteins, or as a result of plasma membrane perturbations upon viral entry. Specific subsets of uninfected cells can also significantly contribute to IFN production upon engulfment of material containing viral-derived nucleotide sequences and sensing of these molecules in endosomes by specific I2R2s. All sensing pathways leading to IFN induction converge on the activation of interferon response factors 3 or 7 (IRF3/7), which are the master transcription factors inducing IFN genes. Most cell types constitutively express IRF3 but not IRF7 or only at low levels. IRF7 expression requires IFN-I stimulation. IFN-β can directly be induced by IRF3. All but one of the IFN-α subtypes require IRF7 for their induction. Hence, IFN-β secretion promotes its own production and that of IFN-α in an autocrine manner (33, 34). This positive feedback loop strongly amplifies IFN production during viral infections, promoting fast and widespread induction of cell-intrinsic anti-viral defenses in uninfected cells to prevent virus dissemination. Other feedback loops tightly regulate IFN-I production positively or negatively. This section reviews different mechanisms controlling IFN production and how they could play different roles in host/virus interactions.
IFN production in infected cells is initiated by sensing of endogenous viral replication
Plasma membrane modifications occur upon virus entry which can induce IFN-I production and ISGs through a STING-dependent signaling. Infected cells can sense abnormal changes in the physical or biochemical properties of their plasma membrane upon virus entry, which can trigger their production of IFN-I (35, 36). This event depends on signaling by the endoplasmic reticulum (ER) – resident transmembrane protein stimulator of interferon genes (STING). Upon virus entry, STING translocates to the cytosol where it is activated by phosphatidylinositol 3-kinase (PI3K) and calcium-dependent pathways to initiate a signaling cascade leading to IRF3-dependent induction of IFN-I and ISGs (Figure 1) (31, 37).
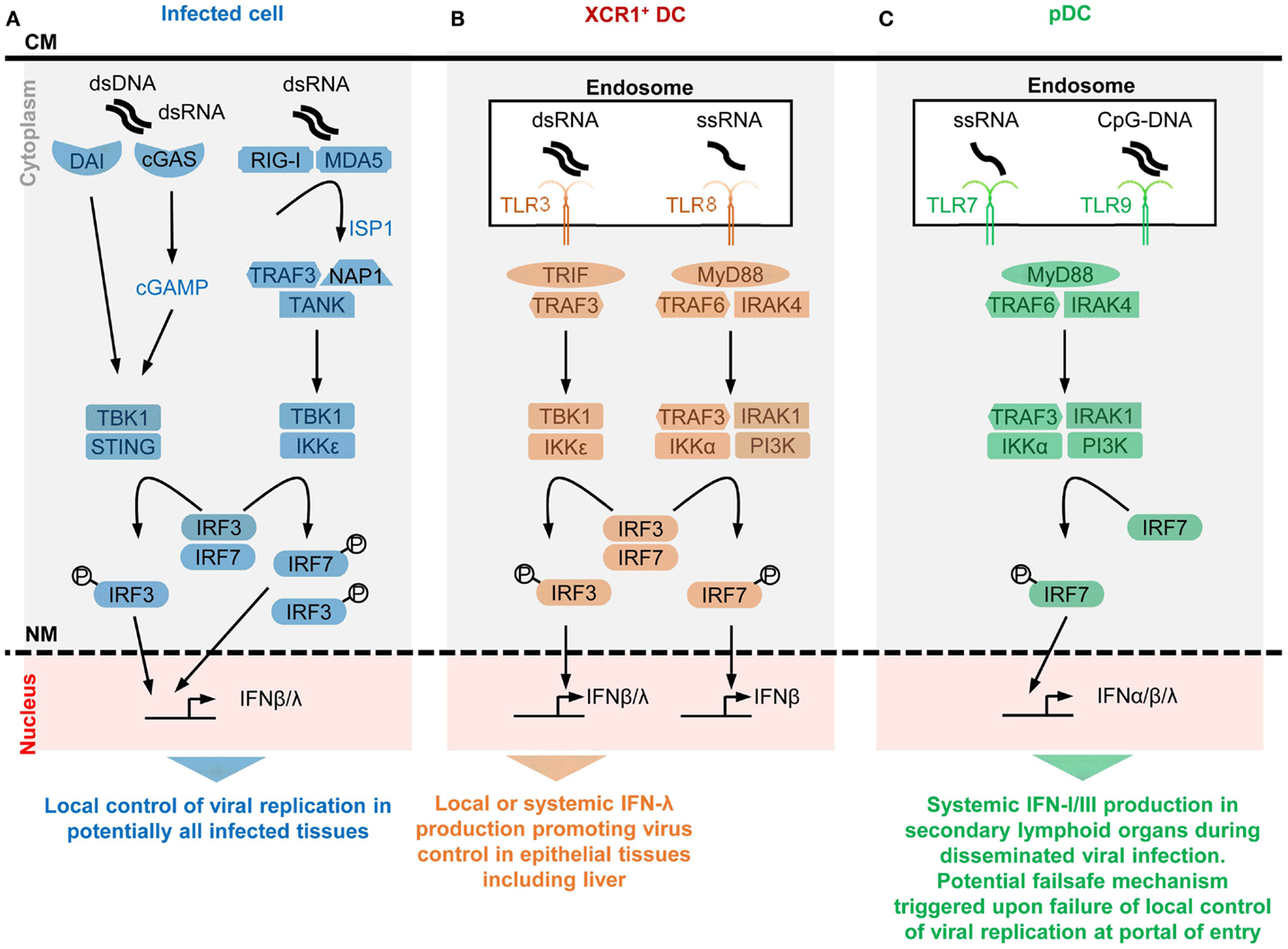
Figure 1. A simplified model of the potential contributions of selective sensors and cell types to IFN production during viral infections. Different innate immune recognition receptors are involved in sensing various types of viral nucleic acids in distinct categories of cells during viral infections, which may promote different types of anti-viral defenses. For each selected sensor shown, the types of viral nucleic acids recognized and the downstream signaling cascade induced are represented in a simplified, schematic manner. The potential specific role of each cell type in anti-viral defenses is also indicated at the bottom of each panel. (A) Potentially all types of infected cells can detect endogenous viral replication through cytosolic sensors triggering their local production of IFN-β/λ to control viral replication in an autocrine and paracrine fashion in infected tissues. (B) Uninfected XCR1+ DCs selectively produce high levels of IFN-λ and IFN-β upon engulfment of materials containing dsRNA and the consecutive triggering of TLR3 in endosomes. The receptor of IFN-λ is mostly expressed by epithelial cells. Hence, XCR1+ DCs might be involved in inducing local IFN responses in virally infected epithelial tissues. Since XCR1+ DCs are especially efficient at producing IFN-III upon HCV stimulation, they might contribute to local or systemic IFN production during infection with this virus, to promote IFN-λ-mediated protection of hepatocytes. Uninfected XCR1+ DCs and other uninfected cells may produce some IFN-β upon engulfment of materials containing ssRNA and the consecutive triggering of TLR8 in endosomes. The contribution of this pathway to anti-viral defense is not well understood yet, in part because mouse TLR8 is deficient for this function. (C) Uninfected pDCs selectively produce high levels of all subsets of IFNs upon engulfment of materials containing ssRNA or CpG DNA and the consecutive triggering of TLR7/9 in endosomes. However, pDCs seem to be activated for this function only in lymphoid tissues. Hence, pDC might contribute to systemic IFN production during blood-borne viral infections or as a failsafe mechanism activated upon abnormal widespread dissemination of a viral infection once it has escaped local confinement at its portal of entry. CM, cell membrane; NM, nuclear membrane.
Viral nucleotide sequences are sensed by dedicated I2R2s in the cytosol of infected cells, which induces IFN-I production. Some I2R2s are located in the cytosol and bind viral nucleotide sequences to induce IFN-I production in infected cells. These I2R2s are classified as cytosolic RNA or DNA sensors. Their specificity for particular nucleotide sequences or tertiary structures, their signaling pathways and their physiological significance have been recently reviewed (31, 32). Cytosolic RNA sensors encompass DExD/H helicases among which the retinoic-acid-inducible gene (RIG)-I-like receptors (RLRs) have been the most studied, namely RIG-I and melanoma differentiation associated gene 5 (MDA5). RIG-I recognizes RNA with a 5′-PPP or 5′-PP (38) (uncapped) moiety, or double-stranded RNA (dsRNA), both structures being present in viral, but not in cytosolic eukaryotic, RNA molecules. MDA5 might specifically recognize long dsRNA fragments. Both RIG-I and MDA5 contain a DexD/H box-containing RNA helicase domain, and 2 caspase recruitment domains (CARD1/2), which bind to mitochondrial anti-viral signaling protein (MAVS). RNA/RLR/STING molecular complexes initiate a signaling cascade leading to IRF3/7-dependent induction of IFNs (Figure 1). Other DExD/H helicases can promote IFN-I production in DCs, although their physiological roles for in vivo immune defenses against viral infections remain to be established (32). Cytosolic DNA sensors able to induce IFN-I (mostly IFN-β) and IFN-III encompass molecules belonging to different protein families, including DExD/H helicases, the inflammasome component IFN-γ-inducible protein 16 (IFI16), the Z-DNA binding protein 1 (ZBP1), and the cyclic GMP-AMP (cGAMP) synthase (cGAS) (31, 32). Most of the cytosolic DNA sensors activate STING and lead to IRF3/7- and NFκB-dependent induction of IFN-β and IFN-III. Many cell types express ZBP1 and are able to produce IFN-I upon triggering of this molecule, including macrophages, DCs, and fibroblasts following an HSV-1 infection (39, 40). Upon DNA binding, cGAS catalyzes the production of cGAMP. cGAS is critical for the detection of lentiviruses including HIV-1/2 (41, 42) and can contribute to sensing of, and protection against, other RNA viruses, including in vivo in mice (43). cGAMP also acts as a secreted second messenger signal alerting uninfected cells to directly induce their expression of intrinsic immune anti-viral defenses. The cGAS/STING/IRF3 signaling cascade and the IRF1 transcription factor are “master” inducers of cell-intrinsic immunity able to control the replication of most DNA and some RNA viruses at least in part independently of IFNs (43).
Viral hijacking of the protein synthesis apparatus of the host cell triggers ER overload, a stress, which synergizes with cytosolic sensing to promote IFN-I production. Infected cells become a factory for production of viral particles. Hijacking of the translation apparatus of the host cell for massive production of viral structural proteins leads to an overload of the capacity of the ER for correct folding of newly synthesized proteins. ER overload induces a homeostatic response of the cell, the unfolded protein response (UPR). UPR aims at restoring normal ER functions by inhibiting translation. UPR activation in infected cells contributes to prevent viral replication, including through inhibition of the production of viral proteins, promotion of IFN-I production, and induction of cell suicide (44).
IFN-I production in uninfected cells is initiated by endosomal sensing of viral nucleotide sequences derived from engulfed virions or infected cells
Toll-like receptors (TLRs) are among the first and best characterized I2R2s. TLRs are transmembrane proteins with a leucine-rich repeat extracellular domain involved in ligand recognition and an intracellular toll/interleukin-1 receptor domain essential for signaling (45). Among the nine TLRs conserved between mouse and human, TLR3, TLR7, TLR8, and TLR9 are located in endosomes where they can detect the abnormal presence of nucleic acids such as occurs upon endocytosis of virions or of virally infected cell material. TLR3 recognizes dsRNA, TLR7/8 ssRNA, and TLR9 DNA sequences containing unmethylated cytidine-phosphate-guanosine (CpG) motifs. TLR fine specificity and signaling pathways have been reviewed recently (32) and are summarized in Figure 1. We will discuss the expression patterns and functions of endosomal TLRs with regards to IFN production in uninfected specialized immune cell types, pDCs and XCR1+ DCs.
Selective expression of TLR7, TLR9, and IRF7 in pDCs endows them with a unique ability to produce very high amounts of all subtypes of IFNs upon virus stimulation irrespective of their own infection. Plasmacytoid DCs uniquely produce very large amounts of IFNs in response to in vitro stimulation with many viruses, without being infected (46). IFN-I mRNAs represent up to 40% of all mRNAs in pDCs at the peak of their activation (47). In vitro, upon exposure to influenza virus, herpes virus type 1, cytomegaloviruses, or vesicular stomatitis virus, individual pDCs produce 100–1000 times more IFNs than total PBMCs, monocytes, MoDCs, cDCs, neutrophils, and fibroblasts (47–52). However, in vitro, high molarity infection of cDCs with certain viruses unable to inhibit IFN-I production in their target cells can also induce massive IFN-β secretion (53). pDCs produce high levels of all subtypes of IFNs, contrary to many other cell types including infected cells, which often preferentially produce IFN-β (46, 47). In vivo, pDC depletion during systemic viral infections leads to over 95% decrease of IFN-I production, while the total number of pDCs producing IFN-I (<100,000 in one mouse) is much lower than the total number of infected cells (54–59). This shows that in vivo also individual activated pDCs produce much more IFN-I/III than most other cell types, including virus-infected cells. The professional IFN-producing function of pDCs largely results from their high constitutive and selective expression of IRF7, TLR7, and TLR9 (Figure 1). These molecules are pre-associated in ready-to-signal complexes located in specialized endosomes specific to pDCs (60, 61). pDCs must also be equipped for efficient sensing and up-take of virions and virus-infected cells. The corresponding cell surface I2R2s remain to be identified.
Selective expression of TLR3 in XCR1+ DC endows them with a unique ability to produce very high amounts of IFN-β and IFN-III upon stimulation with dsRNA or HCV irrespective of their own infection. XCR1+ DCs are very potent for antigen-specific activation of CD8+ T cells, in particular through cross-presentation of exogenous antigens that they have captured from other cells and processed for association with class I major complex histocompatibility (MHC-I) molecules (62). In mice, XCR1+ DCs are crucial for the initiation of protective adaptive immune responses against tumors and a variety of viruses (63). Mouse and human XCR1+ DCs constitutively and selectively express high levels of TLR3 (Figure 1). They produce large amounts of IFN-III and IFN-β upon stimulation with a synthetic mimetic of dsRNA, Polyinosinic:polycytidylic acid (PolyI:C) (64, 65). Human XCR1+ DCs uniquely respond to stimulation with HCV by producing large amounts of IFN-III in a TLR3-dependent manner (66, 67), irrespective of their own infection.
Positive and negative feedback loops regulating IFN-I production
Positive feedback loops. In addition to IRF7 induction, other positive feedback mechanisms exist to amplify the production of IFNs rapidly after initiation of a viral infection as illustrated by the following selected examples. IFNs induce the expression of many cytosolic RNA/DNA sensors and of TLR7. This broadens the spectrum of host’s cell types able to detect endogenous viral replication for IFN induction. Induction of OASL by IFNs in human cells enforces RIG-I signaling, counteracting viral immune evasion genes interfering with this sensing pathway (68). The IFN-inducible ribonuclease L (RNaseL) generates viral and cellular RNA degradation products, which engage RLRs for amplification of IFN production (69, 70). The IFN-inducible Protein kinase R (PKR) stabilizes IFN-I mRNA (71).
Negative feedback loops. To prevent unbridled responses deleterious for the host, IFN activity must be tightly controlled including during viral infections. Several negative feedback loops exist to terminate IFN production, after anti-viral defenses have been activated. The ISG ubiquitin specific peptidase 18 (USP18) binds to IFNAR2, preventing it from recruiting signal transducer and activator of transcription 1 (STAT1). IFNs induce the expression of TAM receptor tyrosine kinases in DCs, monocytes, and macrophages. TAM receptors associate and signal in part through IFNAR1. They activate the suppressors of cytokine signaling-1/3 (SOCS-1/3). SOCS inhibit TLR and RLR signaling, thereby terminating IFN production (72). TAM receptor ligands, Gas6 and ProS, bind phosphatidylserine on dying cells and are produced by activated DCs and monocytes/macrophages. Thus, IFN induction of TAM inhibitory receptors on uninfected phagocytic immune cells could limit their propensity to produce the cytokines upon engulfment of dying virally infected cells. IFNs induce Tetherin on most cell types. pDCs express a receptor for Tetherin, leukocyte immunoglobulin-like receptor, subfamily A (with TM domain), member 4 (LILRA4). LILRA4 triggering on pDCs inhibits their production of IFN-I. Hence, through LILRA4 engagement by Tetherin, pDCs can monitor their efficacy at inducing an anti-viral gene expression program in neighboring cells through IFNs, and timely terminate their IFN production.
How positive and negative feedback loops integrate in time and space to promote optimal kinetics and intensity of IFN production in order to efficiently control viral infection without causing severe immunopathology is not completely understood. Positive feedback loops may occur very rapidly after initiation of viral infection to allow rapid secretion of high levels of the cytokines for fast and strong induction of anti-viral cell-intrinsic immunity. Negative feedback loops occur likely later to terminate the response and thus avoid chronicity of cytokine production and its ensuing deleterious effects.
What are the respective roles of infected versus uninfected cells in IFN production during viral infections?
IFN production by infected cells serves as first line of defense to block viral replication at his portal of entry in the body, while IFN production by uninfected pDCs might constitute a failsafe mechanism activated only when viral infection gets systemic. pDCs do not constitute the major source of IFN production upon local infections by several viruses in the lung or in the female reproductive tract. pDCs are dispensable for resistance against these infections (56, 73, 74). During pulmonary infection by Newcastle disease virus (NDV), IFN-I are produced locally in the lungs mainly by infected alveolar macrophages. Lung pDCs do not express the cytokines (73). Selective depletion of lung alveolar macrophages leads to systemic dissemination of NDV, and to a strong activation of pDCs for IFN-I production specifically in the spleen. Even in the case of systemic viral infections such as caused by intravenous injection of NDV or intraperitoneal injection of mouse cytomegalovirus (MCMV), pDC IFN production is confined to the spleen. It is not detected in other organs even those with high viral replication (59, 73). Hence, splenic pDCs are especially prone to high level IFN production upon systemic acute viral infections. pDCs located in non-lymphoid organs, in particular mucosal barrier tissues, appear to be inhibited for IFN production. Thus, IFN production by infected cells serves as first line of defense to block virus replication at its portal of entry in the body. IFN production by uninfected pDCs might constitute a failsafe mechanism mainly activated in the spleen when viral infection gets systemic (75). Under these conditions, to promote health over disease, the benefits for the host of producing high circulating levels of IFNs in order to induce widespread cell-intrinsic anti-viral defenses might prevail over the deleterious effects that this could cause on certain cell types or tissues. Indeed, pDCs are required for protection against HSV-2 and HSV-1 in mice only in systemic but not local infections (56). This observation is consistent with the crucial role of pDCs for protection of mice against systemic infection by mouse hepatitis virus (MHV), a fast replicating coronavirus (55). Conflicting results have been obtained on the role of pDCs during intranasal influenza infection (74, 76–78). A possible explanation is that pDC IFN production contributes to resistance to highly pathogenic influenza strains that might systemically spread from the lung early after infection, even if at low levels. Another intriguing observation is that IFNs are critical for host resistance to MCMV and that pDCs are the major source of IFNs in this infection model but are dispensable for virus control (54). Studies are ongoing to understand this apparent paradox. Patients bearing genetic mutations disrupting endosomal TLR signaling do not appear to suffer from life-threatening viral infections (79, 80), contrary to patients impaired in IFNAR signaling (4, 81). A notable exception is the specific susceptibility to severe herpes virus encephalitis in individuals’ deficient for TLR3 signaling (82, 83). However, contrary to extracellular TLR, endosomal TLR have evolved under strong purifying selection in human beings (84). Hence, while pDCs and endosomal TLR might have been required for protection of our species against viral infections in the past, this appears not to be the case anymore perhaps due to improved social, hygiene, and health care in modern society (75).
IFN production by uninfected pDCs or XCR1+ DCs might promote protection against viruses able to interfere with the signaling pathways inducing cytokine production in infected cells. Attesting to the importance of IFNs for anti-viral defense in vertebrates, many mammalian viruses encode immune evasion genes specifically inhibiting the production of IFNs in infected cells (39, 85). pDCs or XCR1+ DCs might be essential for IFN-dependent host protection against these viruses, because they are spared from the intracellular functions of viral immune evasion genes (75). To the best of our knowledge, MCMV does not encode for immune evasion genes inhibiting IFN production. However, MCMV manipulates IFN-I responses through specific inhibition of STAT1 functions in infected cells. Thus, pDCs might be dispensable for resistance against systemic MCMV infection due to sufficient levels of IFN production by infected cells locally in all infected tissues. Hepatocyte responses to IFN-III appear to play a critical role in human resistance to HCV. In infected hepatocytes, HCV induces the expression of cellular microRNAs binding to IFN-III mRNA and leading to its degradation. Uninfected XCR1+ DCs produce high levels of IFN-III in vitro upon HCV stimulation (66, 67). Hence, during acute HCV infection in vivo, XCR1+ DC may be a strong and early source of IFN-III not subjected to virus immune evasion strategies, therefore, contributing to protect naturally resistant individuals.
Altruistic suicide of subcapsular sinus macrophages in secondary lymphoid organs promotes strong IFN responses to control viral dissemination. In secondary lymphoid organs, a subset of macrophages is critical for the clearance of viruses from the lymph (86). These macrophages are located on viral entry routes, near to subcapsular sinuses where the afferent lymph drained from non-lymphoid tissues flows. Contrary to other subsets of macrophages, subcapsular sinus macrophages are highly susceptible to viral infection, because they constitutively express only low levels of effector molecules of cell-intrinsic anti-viral immunity and because their responses to IFNs are inhibited by their high constitutive expression of USP18. Subcapsular sinus macrophages rapidly become infected by viruses incoming from the lymph and produce large amounts of IFNs. This altruistic suicide prevents virus dissemination to other adjacent cell types and promotes the induction of innate and adaptive anti-viral immunity (87).
How are IFNs Promoting Anti-Viral Immunity?
IFN direct anti-viral effector functions: induction of effector molecules of cell-intrinsic anti-viral immunity
Upon instruction by IFNs, cells express a wide variety of viral restriction factors, whose combined action blocks pathogen invasion by interfering with the different stages of viral life cycle (Figure 2A). This has been extensively reviewed recently (88) and will only be described succinctly here. Virus fusion with host cell membrane can be blocked by Cholesterol-25-hydrolase (CH25H) that inhibits sterol biosynthesis. Some viruses enter cells by escaping from endosomes/lysosomes, which can be blocked by interferon inducible transmembrane (IFITM) proteins. Virus uncoating can be blocked by tripartite motif (TRIM) proteins, such as TRIM5α, which bind to HIV-1 capsid thus promoting its degradation, and by Myxoma resistance GTPases, MX1, and MX2, which efficiently trap viral structural proteins at an early stage following virus entry into the cell. MX1 inhibits a number of viruses, including influenza virus through sequestration of its nucleocapsid. MX2 associates with host cyclophilin A and HIV-1 capsid protein. Virion assembly can be blocked at transcriptional, translational, and post-translational levels. The adenosine deaminase acting on RNA 1 (ADAR1) and the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) deaminases induce viral RNA destabilization and hypermutation (89, 90). The sterile alpha motif and histidine-aspartic domain (HD) containing protein 1 (SAMHD1) blocks reverse transcription by hydrolyzing dNTPs (91). ADAR1, APOBEC, and SAMHD1 functions have been mainly studied in infections by HIV-1 and other retroviruses. The 2′,5′-oligoadenylate synthase (OAS) proteins, the IFN-induced proteins with tetratricopeptide repeats (IFIT), and PKR inhibit viral and host protein translation by using complementary mechanisms (88). The major post-translation modification induced by IFNs is the binding of the ubiquitin-like modifier ISG15 to several viral and host proteins, a process called ISGylation. Most of the known ISGylated proteins are targeted for degradation, with few exceptions that are on the contrary stabilized like IRF3 (88). Finally, the egress and budding of virions of many enveloped viruses can be inhibited by Tetherin or by Viperin (88).
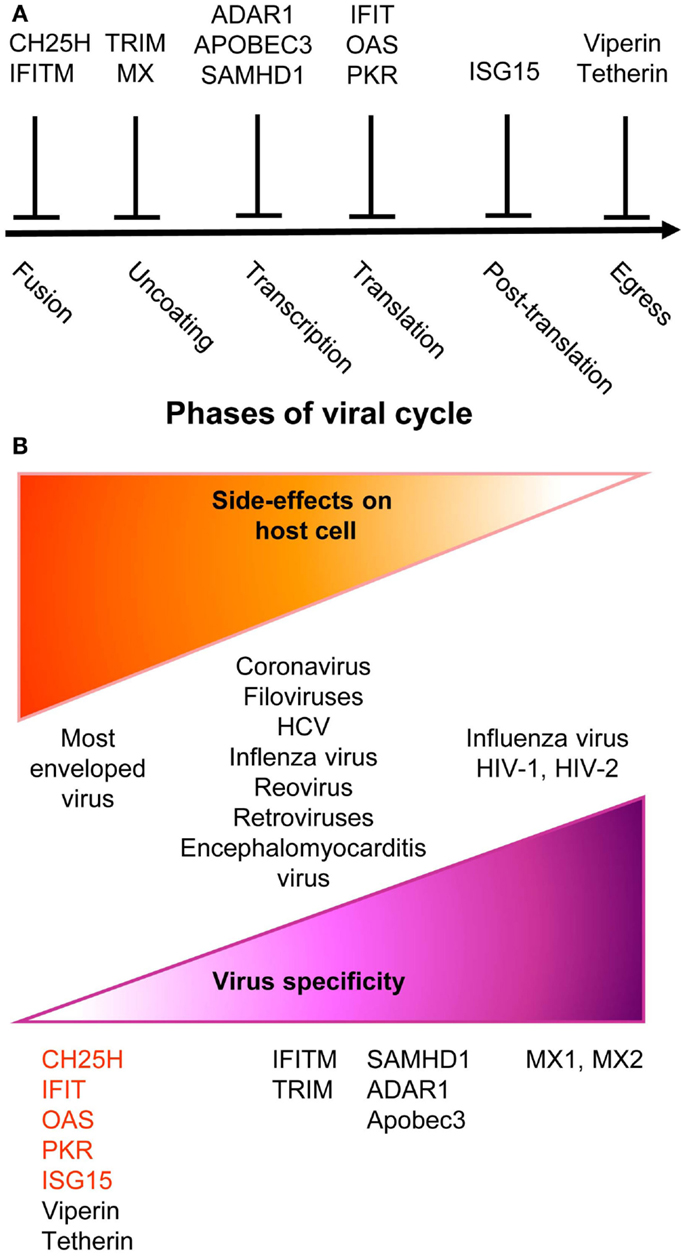
Figure 2. A simplified and schematic view of the modes of action and specificities of ISG acting as direct anti-viral restriction factors. (A) Different IFN-inducible restriction factors can block viral replication in infected cells in a cell-intrinsic manner at different stages of the viral life cycle. (B) Viral specificity (purple) might be inversely correlated to the breadth of side effects on host cells (orange).
Many anti-viral ISGs have been functionally characterized only recently, largely thanks to large-scale screening approaches. They display a variable degree of viral specificity (43, 92) that might inversely relate to the extent of their side effects on host cells (Figure 2B). Anti-viral effectors acting on a broad spectrum of viruses often target key metabolic pathways that are also crucial for host cell functions. This is the case for the control of cholesterol metabolism by CH25H (93) or of protein translation by PKR, OAS, or IFITs (88). Other anti-viral restriction factors such as MX2 may specifically target one molecule of a very restricted set of viruses with no apparent side effects on host cells. Some anti-viral ISGs target specific functions critical for only a restricted array of viruses and might similarly exert side effects only on a subset of host cell types. For example, SAMHD1 inhibits retrovirus replication through dNTP depletion, which might more specifically affect proliferating host cells. Hence, the infected host must balance the intensity, breadth, and location of ISG induction to circumvent viral replication while preventing life-threatening damages to vital cell types or tissues. One of the mechanisms contributing to this balance is translational control of the expression of ISGs, especially those with pro-apoptotic or anti-proliferative functions (94). While many anti-viral ISGs are transcriptionally activated in most IFN-stimulated cells, their translation can be specifically blocked in uninfected cells by cellular microRNA. This inhibition is relieved upon cell infection through negative regulation of the function of the RNA-induced silencing complex. Hence, IFN stimulation of uninfected cells prepares them for rapid and strong induction of cell-intrinsic anti-viral defenses upon viral infection while avoiding their unnecessary exposure to the toxic effects of certain ISGs.
Further knowledge on the functions and the dynamic regulation of ISGs is essential to develop novel therapeutic strategies against viral infections aiming at modulating IFN responses to promote their protective anti-viral cell-intrinsic functions over their deleterious toxic effects. A better understanding of the immunoregulatory effects of IFNs will also help.
IFN orchestration of anti-viral responses of both innate and adaptive immune cells
Type I interferon can modulate the functions of a broad spectrum of immune cells (Figure 3A). We will review this knowledge, focusing on the functions of DCs, NK cells, T cells, and B cells, since they are involved in the control of most viral infections. We will discuss the hypothesis that DCs play a central role in IFN-I orchestration of innate and adaptive immunity for the induction of optimal anti-viral defenses (Figure 3).
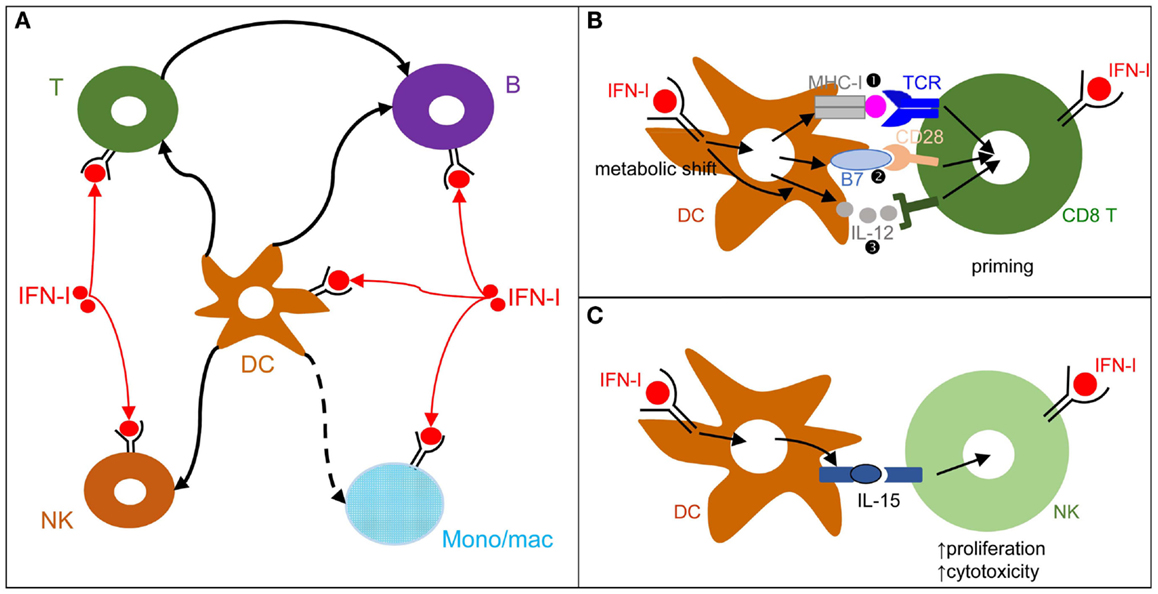
Figure 3. DCs play a central role in IFN-I orchestration of innate and adaptive immune responses. (A) IFN-I exert cell-intrinsic as well as indirect effects on a variety of immune cell populations. DC responses to IFN-I play a major role in promoting protective activation and functional polarization of other innate and adaptive immune cells, not only during viral infections but also in other physiopathological situations including cancer. (B) DC cell-intrinsic responses to IFN-I endow them to deliver appropriate signals for T cell priming and functional polarization. IFN-I can modulate all three types of signals delivered by DC to T cells: MHC-I/antigenic peptide complexes ( ), co-stimulation (
), co-stimulation ( ), and cytokines (
), and cytokines ( ). This depends both on IFN-I-dependent transcriptional induction in DC of some of the corresponding genes and on IFN-I-dependent metabolic reprogramming of DC. (C) DC cell-intrinsic responses to IFN-I endow them to deliver appropriate signals, in particular IL-15 trans-presentation, for NK cell activation. See main text for further details.
). This depends both on IFN-I-dependent transcriptional induction in DC of some of the corresponding genes and on IFN-I-dependent metabolic reprogramming of DC. (C) DC cell-intrinsic responses to IFN-I endow them to deliver appropriate signals, in particular IL-15 trans-presentation, for NK cell activation. See main text for further details.
During viral infections and cancer immunosurveillance, IFN-I constitute one of the most important input signal acting on DCs to promote their delivery of appropriate output signals to T cells, B cells, and NK cells for protective immunity (Figure 3A). DCs deliver three types of signals to activate and functionally polarize T cells. Signal 1 is the triggering of the T cell receptor by viral peptide-MHC complexes. Signal 2 is the triggering of activating T cell co-stimulation receptors such as CD28 or CD27 by the CD80/86 and CD70 co-stimulation molecules expressed on DCs. Signal 3 corresponds to cytokines, which can promote T cell proliferation and acquisition of specific effector functions. Under steady state conditions, most DCs are in an immature state characterized by low level expression of MHC-II (signal 1) and co-stimulation molecules (signal 2) and by the lack of production of T cell-activating cytokines (signal 3). Upon activation, including early after viral infections in vivo, DCs up-regulate their expression of signal 1 and activating signal 2 and secrete T cell-activating cytokines. This process is called DC maturation. Gene expression profiling of DCs stimulated by microbial stimuli identified a core set of genes up-regulated in mature DCs irrespective of the stimulus they receive, irrespective of the subset they belong to, and conserved across evolution (95). Most of these genes are induced during DC maturation in part through cell-intrinsic IFN-I signaling (95). Consistently, cell-intrinsic IFNAR signaling in DCs is required in many circumstances for the induction of protective immunity, including efficient CD8 T cell responses during viral infection or tumor development (96–98), Th1 responses upon PolyI:C injection independently of IL-12 or IFN-γ effects (99, 100), as well as follicular helper T cell and humoral responses (101, 102). Mechanistically, IFN-I promote DC immunogenicity for efficient T cell activation through a variety of effects (Figure 3B). It drives DC up-regulation of signal 2 in vivo during viral infections (103) and boosts their capacity to cross-present antigens for increased delivery of signal 1 to CD8 T cells (96–98). It shapes their delivery of activating signal 3, in particular inducing IL-15 and promoting or inhibiting IL-12 depending on experimental conditions (58, 104). Finally, it is necessary to induce their metabolic shift from mitochondrial oxidative phosphorylation to aerobic glycolysis, which fuels the increased needs in energy and the expansion of the intracellular organelles required for the production and proper intracellular routing of the signal 1 and 2 proteins (100, 105). Selective inactivation of IFNAR on cDCs compromises mouse resistance to MCMV and MHV infections (103, 106). In contrast, IFNAR expression is not required on NK cells for protection against MCMV and on pDCs, T cells, and B cells for early control of MHV replication (103, 106). Although cell-intrinsic IFN-I signaling in NK cells can promote their activation (107) (Figure 3A), IFN-I-induced IL-15 trans-presentation by DCs plays a more prominent role for this function in many conditions including in vivo during MCMV infection (103, 108) (Figure 3C).
Cell-intrinsic IFN-I signaling in CD4 T cells (109), CD8 T cells (110, 111), and B cells (112) can also contribute to their efficient activation and functional polarization (Figure 3). This depends on experimental settings. CD8 T cell-intrinsic IFN-I responses are crucial for mounting efficient cytotoxic CD8 T cell responses against LCMV but are less critical against Vaccinia virus and vesicular stomatitis virus (110, 113, 114). Mechanistically, cell-intrinsic IFN-I signaling in CD8 T cells can promote their survival during their antigen-induced proliferation (110). Cell-intrinsic signaling in DCs and CD8 T cells may act in a synergistic manner. Indeed, conditional inactivation of IFN-I responsiveness was required to occur simultaneously in both of these two cell types to dramatically affect CD8 T cell expansion upon vaccination with a modified Ankara vaccinia virus (115).
In summary, IFN-I generally play a crucial, beneficial, role in immune defenses against viral infections, both through the induction of cell-intrinsic anti-viral defenses and through the orchestration of innate and adaptive immunity. However, if these responses are not properly regulated, they can contribute to diseases as we will now discuss.
Different, and Sometimes Opposite, Processes Underlie Deleterious IFN-I Responses Depending on the Physiopathological Contexts
Deleterious Effects Resulting from the Induction of Unbridled Inflammatory Responses Causing Severe Tissue Damage, as Exemplified in Autoimmune Diseases
A frequent side effect of IFN-I treatment against cancer or chronic viral infections is the induction of autoimmune reactions. Consistently, ISG expression is a hallmark of many spontaneous systemic or tissue-specific autoimmune diseases, including systemic lupus erythematosous (SLE), Sjogren’s syndrome, psoriasis, and other skin disorders (11). The dysregulation of IFN-I responses observed in patients with these autoimmune diseases likely results from both genetic and environmental factors. Genome-wide association studies show that polymorphisms in genes involved in IFN-I responses strongly correlate with increased susceptibility to many autoimmune diseases (11). Diverse environmental factors can also contribute to the onset of autoimmune diseases. Microbial infections often precede first clinical manifestations of autoimmune diseases. Whether infections (116) and/or alterations in the commensal microbiota of the affected barrier tissues (117, 118) are the cause or rather the consequence of autoimmunity is still matter of debate. Infection- or dysbiosis-induced tissue damages and unbridled IFN-I responses can contribute to initiate autoimmune reactions. Gender is another prominent factor affecting susceptibility to autoimmune diseases. Women are more prone to autoimmunity, which may result from endocrine regulation of IFN-I responses. pDC IFN-I production is enhanced in human and mouse females, due at least in part to cell-intrinsic enhancement of TLR7/9 responses by the female hormone estradiol (119). In autoimmune diseases, different mechanisms could operate to initiate the dysregulation of immune responses leading to a vicious circle of reciprocal activation between innate IFN-I responses and adaptive self-reactive lymphocyte responses (Figure 4). Adaptive immune cells are educated to spare “self.” This occurs through negative selection of potentially autoimmune B and T cells during their development in the bone marrow or thymus, respectively, a process called central tolerance. Self-reactive B or T cells that have escaped this pruning can be either deleted or functionally inactivated once they have egressed in secondary lymphoid organs or non-lymphoid tissues, a process called peripheral tolerance. In some individuals, polymorphisms in genes involved in the promotion of central or peripheral tolerance lead to a higher number, diversity, and/or responsiveness of self-reactive lymphocytes in the periphery, in particular of B cells secreting anti-DNA or anti-RNP antibodies (120, 121). Mammalian DNA or RNA are poor inducers of pDC IFN-I induction under normal conditions. However, pre-existing anti-DNA or anti-RNP autoantibodies can break this innate tolerance of pDC. Indeed, antibodies binding to self nucleic acids can protect them from degradation and compact them into nanoparticles that are very effective for the induction of IFN-I in pDC (Figure 4). DNA-containing immune complexes (ICs) are frequently found in the serum of SLE patients (SLE-ICs) and can activate pDC IFN-I production (122). In turn, pDC IFN-I activate cDCs, monocytes (123), and B cells, leading to a vicious circle of reciprocal activation between DCs and self-reactive lymphocytes and to the exacerbation of autoimmune responses (Figure 4). Certain infections or dysbiosis of the commensal microbiota of the affected barrier tissues could promote chronic production of host amphiphatic peptides able to combine with eukaryotic DNA or RNA, likely released from dying cells, thus forming pDC-activating nanoparticles. Indeed, in psoriatic skin, both a high expression of LL37 and a massive infiltration of pDCs is observed (124) (Figure 4). Hence, to treat many autoimmune diseases, novel therapeutic strategies could be designed to target dysregulated pDC IFN-I production or B cell activation by IFN-I.
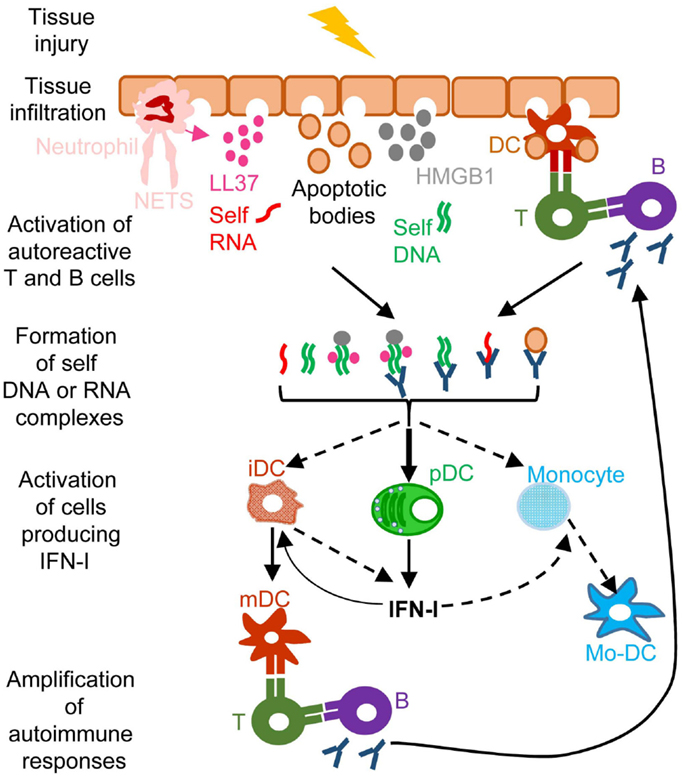
Figure 4. A simplified model of the deleterious role of IFN-I in several autoimmune diseases. When exposed to different kinds of injuries (microbial infection, commensal microbiota dysbiosis, chemical or physical insults), healthy tissues can undergo cell damage and death. These events induce the release of apoptotic bodies encompassing self RNA or DNA. Neutrophil recruitment and activation in inflamed tissues can also constitute a potent source of self nucleic acids, through the release of neutrophil extracellular traps (NET). Self RNA or DNA can associate with cationic peptides (e.g., LL37) as shown in psoriatic patients or with inflammatory molecules (e.g., high mobility group box 1, HMGB1) to generate nanoparticles that are extremely efficient for IFN-I production by pDC and eventually other cell types. pDC can also be efficiently activated for IFN-I production by immune complexes (ICs) generated by the association between self nucleic acids and auto-antibodies as frequently found in the serum of systemic lupus erythematosus patients. IFN-I promote the differentiation and/or the maturation of antigen-presenting cells, in particular different subsets of DC. Activated DC can then present self-antigens for activation of auto-reactive T CD4+ cells, including follicular helper lymphocytes, which in turn activate auto-reactive B cells for auto-antibody secretion, leading to a vicious circle of reciprocal activation between innate and auto-reactive adaptive immune cells. iDC, immature DC; mDC, mature DC; Mo-DC, monocyte-derived DC. See main text for further details.
Deleterious Responses Resulting from Inappropriate Functional Polarization of Immune Responses, as Exemplified in Failure to Control Secondary Bacterial or Fungal Infections
One of the most common complications of primary infections by many respiratory viruses, in particular influenza virus, is a life-threatening pneumonia due to secondary pulmonary infections by bacteria, such as Streptococcus pneumoniae, Staphylococcus aureus, or Haemophilus influenza (125, 126). These pathologies affect especially infants, elderly, and immunocompromised patients. Retrospective studies indicate that secondary bacterial pneumonia was highly recurrent in lung tissues isolated from patients who died during last century influenza pandemics, independently of antibiotic availability (127, 128). Influenza virus induces high IFN-I responses in human beings and mice. In both hosts, secondary bacterial infections are lethal only when they occur in a limited time window following primary viral infection (3–7 days), around the peak of IFN-I responses, before complete virus clearance. Mouse models of viral/bacterial coinfections are being used to dissect disease mechanisms (129). IFNAR1-deficient mice appear more resistant to secondary pulmonary bacterial infections, showing that IFN-I responsiveness contributes to disease (130). Similarly, after lymphochoriomeningitis virus (LCMV) infection, wild-type but not IFNAR1-deficient mice are more susceptible to LPS-induced septic shock (131). Several mechanisms may contribute to the detrimental role of IFN-I in secondary bacterial infections (Figure 5). Early during viral infection, IFN-I decrease the host ability to control bacterial replication, by dominantly polarizing immune responses toward anti-viral functions, simultaneously inhibiting the development of the types of immune responses required for protection against most bacterial infections. IFN-I can inhibit the production of chemokines required for the recruitment to the respiratory tract of antibacterial effector innate immune cells, in particular neutrophils or monocytes/macrophages (132, 133) (Figure 5). Depending on the experimental models used, IFN-I can on the contrary induce a CCR2-dependant recruitment of classical monocytes (134). In infected tissue, IFN-I might skew the functional polarization of resident or infiltrating monocytic cells toward immunosuppression, because it does limit their antibacterial functions by inhibiting their IL-1 production (135–137) while it might promote their production of IL-10 and nitric oxygen intermediates. The exact nature of infiltrating monocytic cells is not clear and could correspond to activated classical monocytes, MoDCs, monocyte-derived macrophages, or myeloid-derived suppressor cells (MDSCs). The boundaries between these putatively different cell types are currently ill-defined (138). These cells could fuel local replication of monocyte/macrophage-tropic bacteria (134), be immunosuppressive (139) or contribute to local immunopathology (140). The role of IFN-I on monocytes/macrophages is complex and will require further investigations to determine when it is protective versus deleterious and what the underlying mechanisms are. Depending on the context, IFN-I can either promote or inhibit the induction of Th1 cytokines such as IL-12 and IFN-γ, and myeloid cell responses to IFN-γ (10, 141-143). IFN-I can also polarize CD4 T cell responses toward Th1 at the expense of Th17, while the Th17-type cytokines IL-17A and IL-22 are required for host defense against pulmonary bacteria by inducing the production of anti-microbial peptides and of tissue repair molecules (Figure 5) (141–143). IFN-I may not only affect host resistance to bacterial infection, but also host tolerance, i.e., the ability of the host to tolerate a given burden of pathogen without undergoing excessive tissue damages (143, 144). Hence, to counter IFN-I deleterious effects during secondary bacterial infections, it will be important to better delineate the respective contribution of lung tissue tolerance modulation and of immune-mediated resistance weakening.
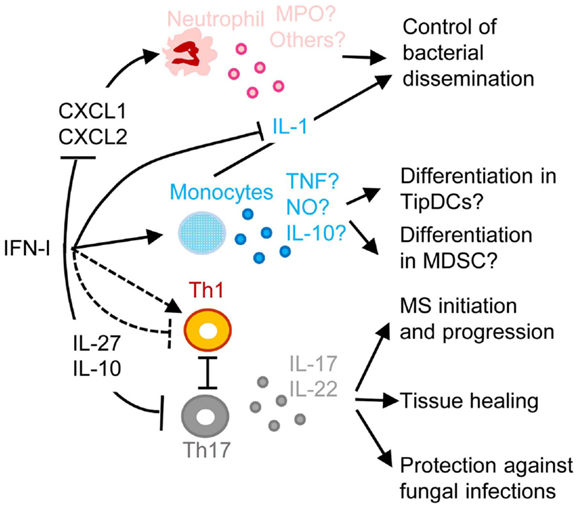
Figure 5. A simplified model of the deleterious role of IFN-I in secondary pulmonary bacterial infections or in fungal infections and of their protective role in multiple sclerosis (MS). IFN-I-dependent signals can block CXCL1 and CXCL2 production, thus inhibiting the recruitment and activation of neutrophils in inflamed tissues, hampering their protective functions against secondary pulmonary bacterial infections or fungal infections. IFN-I-dependent signals can enhance CCL2 production, promoting the recruitment in inflamed tissues of CCR2+ monocytes that can potentially differentiate into TipDC or into MDSC. This can contribute to disease either through enforcing T cell activation leading to immunopathology or on the contrary through suppressing anti-microbial immune defenses. IFN-I can either inhibit or promote Th1 responses, which in the latter case occurs at the expense of Th17 responses thus compromising the production of IL-17 and IL-22, which are respectively required for control of microbial replication and for tissue healing. IFN-I-induced IL-10 and IL-27 can directly inhibit Th17. In MS, inhibition of Th17 functions may contribute to the protective effects of IFN-β therapy. MPO, myeloperoxidase; TNF, tumor necrosis factor alpha; NO, nitric oxide. See main text for other abbreviations and further details.
Another well documented example of deleterious effects of IFN-I due to their inappropriate functional polarization of immune responses is the enhanced susceptibility to fungal infections of patients with genetically determined hyperactive IFN-I responses, as exemplified in the hereditary disease Chronic Mucocutaneous Candidiadis (CMC) (Figure 5) (145). Patients with CMC have a significant deficit in Th17 CD4 T cells, at least in part as a consequence of altered responsiveness to IL-6 or IL-21. Several STAT1 mutations were identified in patients with autosomal dominant CMC. Gain-of-function STAT1 mutations were found to hard wire CD4 T cell responses to cytokines toward STAT1 signaling, compromising their STAT3-dependent ability to produce IL-17 upon IL-6 or IL-21 stimulation. This was associated to induction of a global IFN-I transcriptomic signature in blood (145). Deleterious IFN-I effects on immunity to Candida might not only occur in CMC patients but also in other types of individuals upon secondary fungal infections occurring shortly after a primary viral infection, likewise to the situation discussed above for secondary bacterial infections. Indeed, PolyIC induced IFN-I abrogate innate immunity to systemic candidiasis in mice (146), and IFNAR-deficient mice can be more resistant to Candida infection under certain experimental settings (147). However, the role of IFN-I in the modulation of the ability of immunocompetent hosts to control fungal infection is disputed (148, 149).
The inhibition of Th17 responses by IFN-I could be protective in at least one important human pathology, MS (Figure 5). MS represents a striking exception to the previously discussed detrimental role of IFN-I in autoimmune diseases. Indeed, a large proportion of MS patients have low serum IFN-I activity and low ISG levels. These MS patients present a significant reduction of MS relapse upon IFN-β administration (150). The underlying mechanisms are not yet completely unraveled. However, in the experimental autoimmune encephalitis mouse model of MS, Th17 responses bear a major contribution to nervous system damages and are inhibited by the IL-10 and IL-27 induced upon IFN-I administration (151).
In summary, IFN-I responses can be deleterious in autoimmunity by promoting a vicious circle of reciprocal activation between innate immune cells and auto-reactive CD4 T or B lymphocytes. IFN-I responses can also be deleterious upon secondary bacterial or fungal infections in the lung or the kidneys occurring shortly after a primary viral infection, by compromising the recruitment of anti-microbial innate effector cells and/or by preventing the proper functional polarization of immune responses. We will now discuss how IFN-I responses can also compromise host immune defenses against certain viruses and promote chronic infections.
Deleterious Responses Resulting from the Induction of Immunosuppression, as Exemplified in Chronic LCMV Infection
Different LCMV strains such as Armstrong and clone-13 (Cl13), respectively, lead to acute versus chronic infections in mice. A hallmark of chronic LCMV infection is the loss of the proliferative potential and effector functions of anti-viral CD8 T cells, a process called exhaustion. Exhausted CD8 T cells are characterized by a high expression of the inhibitory receptors PD-1, CTLA4, and LAG-3 (152). In vivo blockade of these inhibitory receptors can reverse T cell exhaustion and allow resolution of the chronic infection (152). IFN-I and ISGs are induced early after infection with all strains of LCMV, albeit to lower levels with those leading to chronic infection. This early IFN-I production is critical to limit viral replication (3). In models of acute infection, IFN-I responses rapidly return to normal, undetectable, levels, before viral replication is completely controlled. In contrast, ISG induction is maintained in chronic infection, including the expression of PD-1 ligands on APCs and of the immunosuppressive IL-10 cytokine, consistent with a prolonged expression of IFN-I albeit at low levels (13, 14). In vivo neutralization of IFN-I by antibody administration promoted resolution of chronic LCMV Cl13 infection, allowing the restoration of functional anti-viral CD8 T cell responses at least in part through CD4 T cell- and IFN-γ-dependent mechanisms (13, 14). During persistent LCMV Cl13 infection, chronic low level IFN-I production polarizes CD4 T cell responses toward T follicular helper (Tfh) rather than Th1 functions. Thus, chronic IFN-I responses promote enhanced anti-viral B cell responses but facilitate CD8 T cell exhaustion due to deficient CD4 T cell help, therefore contributing to host failure to prevent chronic infection (153). Strikingly, establishment of chronic infection by LCMV Cl13 could also be prevented by early administration of two shots of a high dose of exogenous IFN-I, at days 2 and 5 post-LCMV inoculation. This treatment allowed viral clearance by rescuing anti-viral CD8 T cell from exhaustion (154). Altogether, these studies show that the timing and duration of IFN-I production during viral infections is critical in determining how this response will impact the balance between the virus and the host. An early and robust but transient production of IFN-I promotes strong induction of cell-intrinsic viral restriction mechanisms as well as adequate polarization of adaptive anti-viral immune responses, which combined effects lead to viral clearance. In contrast, if the production of IFN-I is too low and/or too late, both viral replication and low IFN-I responses become chronic, their combined action leading to induction of immunosuppressive effects and to inadequate functional polarization of CD4 T cells. This results in CD8 T cell exhaustion and maintenance of chronic infection. Chronic viral replication and CD8 T cell exhaustion is also a hallmark of HIV-1 infection. We will now discuss the complex and disputed role of IFN-I in this disease.
The Complex and Disputed Role of IFN-I in HIV-1 Infection
Both in HIV-1 infection and in its most relevant animal model, infection of non-human primates with simian immunodeficiency virus (SIV), disease progression after the acute phase of the infection is associated with high and chronic expression of ISGs while IFN-I production is inconsistently detected (155–157). In contrast, the individuals that do not progress toward disease despite persistent high viral loads show much lower immune activation, in particular low ISG expression, after the acute phase of the infection (158–161). Hence, chronic low levels of IFN-I are associated to disease progression independently of the level of viral replication. Therefore, an outstanding question still open for a better understanding of the physiopathology of HIV-1 infection is whether chronic IFN-I responses are merely a marker of progression, or whether they are implicated in driving disease development. In addition to mechanisms similar to those uncovered in the mouse model of chronic LCMV infection, during HIV-1 infection other effects of IFN-I could promote a vicious circle of reciprocal activation between chronic viral replication and sustained, deleterious immune responses (Figure 6). Very early after HIV-1 infection, in most individuals, IFN-I production might be too weak or too late to induce a combination of cell-intrinsic defense mechanisms and of immune responses efficient enough to prevent later establishment of chronic infection. On the contrary, as demonstrated in the case of the mouse model of LCMV infection, IFN-I responses could favor CD8 T cell exhaustion, either by direct cell-intrinsic effects on CD8 T cells (Figure 6, ) or by contributing to deprive them from CD4 T cell help (Figure 6, ). Several effects of IFN-I might compromise anti-HIV-1 Th1 responses or more generally contribute to the global depletion of CD4 T cells. These mechanisms include functional polarization of anti-HIV-1 CD4 T cells toward Tfh rather than Th1 responses, CXCL10 production leading to enhance recruitment of memory CD4 T cells to the sites of viral replication where they fuel chronic viral replication with new HIV-1 target cells (Figure 6, to  ), direct pro-apoptotic and anti-proliferative effects on CD4 T cells (Figure 6,
), direct pro-apoptotic and anti-proliferative effects on CD4 T cells (Figure 6,  ), as well as TRAIL induction on pDCs licensing them for killing CD4 T cells irrespective of their infection (Figure 6,
), as well as TRAIL induction on pDCs licensing them for killing CD4 T cells irrespective of their infection (Figure 6,  ) (162, 163). Altogether, these mechanisms entertain chronic viral replication and continuous depletion of CD4 T cells, leading to the dramatically enhanced susceptibility to opportunistic infections (Figure 6,
) (162, 163). Altogether, these mechanisms entertain chronic viral replication and continuous depletion of CD4 T cells, leading to the dramatically enhanced susceptibility to opportunistic infections (Figure 6,  ) characteristic of the acquired immunodeficiency syndrome (AIDS) (Figure 6,
) characteristic of the acquired immunodeficiency syndrome (AIDS) (Figure 6, 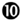 ). Other lines of evidences have been reported to support a deleterious role of pDC activation during HIV-1 infection. Women undergo faster HIV-1 disease progression than men with similar viral loads, which may result in part from the highest IFN-I production of women’s pDCs including in response to HIV-1 stimulation (164). pDC recruitment and activation in the vaginal mucosa of female macaques early after local SIV inoculation contribute to attract and activate CD4 T cells, which can then be infected and promote virus dissemination from its portal of entry (165). However, in vivo blockade of pDC IFN-α production by administration of TLR7/9-antagonistic oligonucleotides early after SIV infection of macaques did not decrease T lymphocyte activation, which suggests that additional sources of IFN-I likely contribute to the immune dysfunction observed in SIV/HIV-1 infections. Targeting dysregulated IFN-I responses during HIV-1 infection might represent an interesting adjuvant therapeutic strategy to highly active antiretroviral treatments. Administration of IFN-I in the non-pathogenic SIV infection model of sooty mangabeys was not sufficient to switch it into a pathogenic model. No CD4 T cell depletion ensued, no hyperactivation of immune responses were observed. Viral loads were even significantly decreased. However, this could be consistent with the positive impact of early and high dose IFN-I administration in chronic LCMV infection (154). Indeed, during the review process of this manuscript, it was reported that, early during primary SIV infection in the pathogenic rhesus macaque model, a high dose injection of IFN-I was protective while neutralization of endogenous IFN-I was deleterious. In contrast, in the same animal model, prolonged IFN-I administration accelerated disease development in the chronic stage of the infection (166). In mice with a humanized immune system, pDC depletion strongly decreased ISG induction and enhanced viral replication both in the acute and chronic phases of HIV-1 infection. However, pDC depletion during chronic infection decreased infection-induced T cell apoptosis and increased T cell numbers in lymphoid organs (167). These results further emphasize the dual role of IFN-I and pDCs in the physiopathology of HIV-1 infection. A strong and transient production of IFN-I early after infection benefits the host by lowering the set-point of viral replication during chronic infection. Sustained production of low levels of IFN-I during chronic infection contributes to immune dysregulation and CD4 T cell depletion. Further studies will be necessary to examine whether complementing standard-of-use antiretroviral drugs with pDC depletion, IFN-I neutralization, or selective inhibition of T cell responses to IFN-I could yield additional benefits to chemotherapy in non-human primates during chronic SIV infection.
). Other lines of evidences have been reported to support a deleterious role of pDC activation during HIV-1 infection. Women undergo faster HIV-1 disease progression than men with similar viral loads, which may result in part from the highest IFN-I production of women’s pDCs including in response to HIV-1 stimulation (164). pDC recruitment and activation in the vaginal mucosa of female macaques early after local SIV inoculation contribute to attract and activate CD4 T cells, which can then be infected and promote virus dissemination from its portal of entry (165). However, in vivo blockade of pDC IFN-α production by administration of TLR7/9-antagonistic oligonucleotides early after SIV infection of macaques did not decrease T lymphocyte activation, which suggests that additional sources of IFN-I likely contribute to the immune dysfunction observed in SIV/HIV-1 infections. Targeting dysregulated IFN-I responses during HIV-1 infection might represent an interesting adjuvant therapeutic strategy to highly active antiretroviral treatments. Administration of IFN-I in the non-pathogenic SIV infection model of sooty mangabeys was not sufficient to switch it into a pathogenic model. No CD4 T cell depletion ensued, no hyperactivation of immune responses were observed. Viral loads were even significantly decreased. However, this could be consistent with the positive impact of early and high dose IFN-I administration in chronic LCMV infection (154). Indeed, during the review process of this manuscript, it was reported that, early during primary SIV infection in the pathogenic rhesus macaque model, a high dose injection of IFN-I was protective while neutralization of endogenous IFN-I was deleterious. In contrast, in the same animal model, prolonged IFN-I administration accelerated disease development in the chronic stage of the infection (166). In mice with a humanized immune system, pDC depletion strongly decreased ISG induction and enhanced viral replication both in the acute and chronic phases of HIV-1 infection. However, pDC depletion during chronic infection decreased infection-induced T cell apoptosis and increased T cell numbers in lymphoid organs (167). These results further emphasize the dual role of IFN-I and pDCs in the physiopathology of HIV-1 infection. A strong and transient production of IFN-I early after infection benefits the host by lowering the set-point of viral replication during chronic infection. Sustained production of low levels of IFN-I during chronic infection contributes to immune dysregulation and CD4 T cell depletion. Further studies will be necessary to examine whether complementing standard-of-use antiretroviral drugs with pDC depletion, IFN-I neutralization, or selective inhibition of T cell responses to IFN-I could yield additional benefits to chemotherapy in non-human primates during chronic SIV infection.
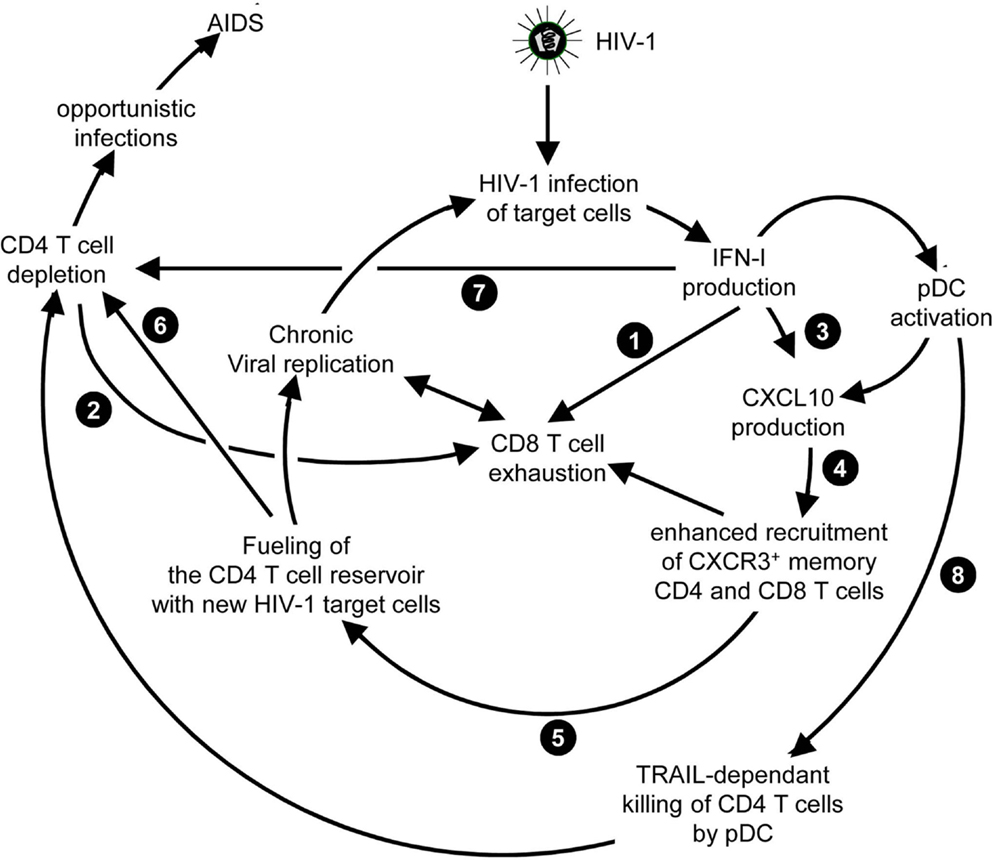
Figure 6. Potential mechanisms through which chronic, low level IFN-I production might promote disease progression in HIV-1 infection. High and sustained expression of ISG in blood and lymphoid organs is a hallmark of progressive infection with immunodeficiency viruses both in human beings and in non-human primates, irrespective of the levels of viral replication. Several mechanisms summarized here have been proposed to explain how chronic, low level IFN-I production might promote disease progression in HIV-1 infection. These mechanisms include direct () and indirect () promotion of the exhaustion of anti-viral CD8 T cell responses, as well as direct () and indirect (-to-, and ) promotion of CD4 T cell depletion with a proposed central role of pDC in this deleterious process. Altogether, these mechanisms may sustain a vicious circle of reciprocal activation between chronic viral replication and deleterious immune responses, driving the progressive depletion of all CD4 T cells ultimately causing the enhanced susceptibility to opportunistic infections characteristic of the acquired immunodeficiency syndrome (AIDS). See main text for further details.
IFN-I administration has been used for many years to treat another human chronic viral disease, HCV infection. Roughly, half of the patients do not show sustained virological responses (SVR). The treatment causes severe side effects in many individuals. New chemotherapeutic drugs very potent at blocking HCV replication in vivo have recently become available. Hence, whether IFN-I administration still constitutes a viable treatment against chronic HCV infection is being questioned (168, 169). We will now discuss this issue.
IFN-I Treatment of Chronic HCV Infection: Balancing Benefits for Virus Control with Side Effects Strongly Affecting Patient’s Quality of Life
Chronic HCV infection is the main cause of liver cirrhosis and hepatocellular carcinoma. There is currently no vaccine against HCV. The most common therapy for chronic HCV patients is the administration of recombinant pegylated IFN-α (Peg-IFN-α) combined with the anti-viral drug ribavirin. However, because of IFNAR pleiotropic expression, IFN-α administration induces severe side effects including flu-like syndrome, fever, fatigue, myalgia, and nervous depression (Figure 7A) (170). Moreover, only about half of treated patients harbor SVR (171). Prior-to-treatment high hepatic ISG expression is a negative predictor of SVR upon Peg-IFN-α therapy. High ISG expression in untreated patients likely results from chronic but low IFN-I production triggered by persistent HCV replication. Indeed, hepatocytes from non-responder patients were found to be infected at a greater frequency and to exhibit dampened antiviral and cell death responses (172). What the cellular sources of IFN-I production are and why they persist only in non-responder patients still remain to be established. In chronic HCV infection, cytotoxic effector lymphocytes may contribute to the development of hepatocarcinoma by causing low level but sustained hepatocyte death and renewal. In contrast, local production of IFN-γ in the liver by NK and T lymphocytes could promote resistance to disease through non-cytolytic control of viral replication. As discussed previously for LCMV and HIV-1, low chronic production of endogenous IFN-I in HCV patients could compromise both innate and adaptive anti-viral immune responses. Chronic exposure to IFN-I could dampen the ability of NK and CD8 T cells to produce IFN-γ (173, 174) and promote CD8 T cell exhaustion (175). It could also induce an antagonist form of CXCL10, a chemokine required for recruitment to the liver of anti-viral NK and CD8 T cell effectors (176). It may also polarize monocytes toward immunosuppressive functions (177). Therefore, better understanding IFN-I effects in HCV infection is critical to improve care of both responders and non-responder patients to Peg-IFN-α. For responder patients, the issue is to modify the treatment to favor beneficial antiviral and immunoactivating effects over side effects strongly affecting patient’s quality of life (Figure 7A). This might be achieved by specific delivery of IFN-I to targeted cell types as discussed later. For non-responder patients, the issue is to understand the mechanisms underlying treatment failure to determine whether alternative therapies could be designed (Figure 7A).
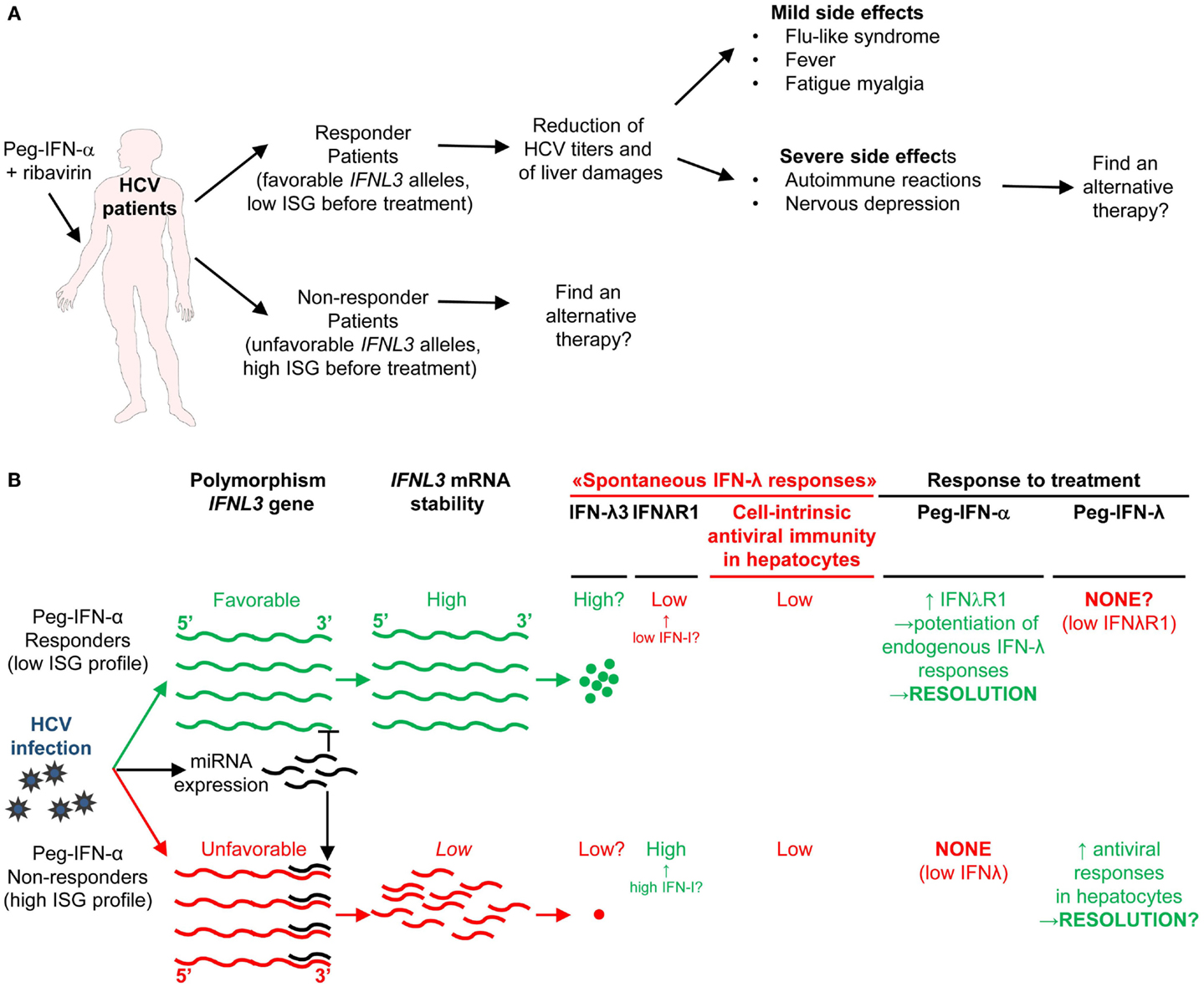
Figure 7. A novel hypothetical model attempting to explain the respective roles of IFN-I and IFN-III in HCV infection. (A) Classification of patients suffering from chronic HCV infection and treated with PEG-IFN-α in non-responders and responders, and identification of IFNL3 (IL28B) gene polymorphisms as the best predictors for treatment response. The side-effects induced by PEG-IFN-α treatment are also listed since they can severely affect the patient’s quality of life and lead to treatment failure due lack of compliance or suicide. Hence, new approaches are needed to promote beneficial over deleterious effects of IFN-I administration in chronic HCV infection. (B) Proposal of a new hypothesis explaining the relationships between endogenous ISG levels in patients prior to treatment, IFNL3 gene polymorphism, endogenous expression of IFN-I, IFN-λ, and IFNλR1, and responsiveness to IFN-I administration. Efficient control of HCV infection may depend on hepatocyte response to IFN-λ rather than IFN-α. Upon HCV infection, the virus induces the expression of host miRNA able to bind the 3′ UTR of IFNL3 mRNA to promote their degradation. The favorable IFNL3 allele associated with responsiveness to PEG-IFN-α treatment may allow endogenous expression of sufficient levels of IFNL3 for efficient induction of cell-intrinsic anti-viral defenses in hepatocytes. This process is, however, hampered by the limited expression of the receptor for this cytokine (IFNλR1) in these patients. PEG-IFN-α treatment might promote resolution of the infection by inducing IFNλR1 in these patients, potentiating their response to their endogenous production of IFNL3. In the patients that do not respond to PEG-IFN-α treatment, endogenous levels of IFNL3 are insufficient for efficient induction of cell-intrinsic anti-viral defenses in hepatocytes, due to the degradation of the corresponding mRNA in infected hepatocytes. In these patient’s hepatocytes, however, IFNλR1 is already expressed to high levels prior to treatment due to their high endogenous IFN-I responses. Administration of exogenous IFN-λ might cure these patients. See main text for further details.
Genome-wide association studies identified various single nucleotide polymorphisms (SNP) in the gene encoding IL-28B/IFN-λ3, one of the IFN-III, as well in its 5′ and 3′ non-coding regions (178–181). One SNP, called rs12979860, is located 3 kb upstream of the IFNL3 gene. Patients harboring the CC genotype have a favorable prognosis to IFN-I treatment. Patients with the TT genotype are at high risk of treatment failure (178, 179). In Europeans, the favorable CC genotype is the major, most common, IFNL3 allele. The unfavorable TT SNP is the minor allele. The frequency of these alleles is reversed in Africans. The favorable allele allows escape of IFNL3 mRNA from degradation by cellular microRNA induced upon HCV infection (181).
Until recently, IFNL3 genotypes and hepatic ISG expression were considered as independent predictors of response to Peg-IFN-α treatment in HCV patients (171). Here, we propose a potential explanation, which integrates both factors in a relatively simple model (Figure 7B). Our main hypothesis is that efficient control of HCV infection depends on hepatocyte response to IFN-λ rather than IFN-α. This is supported by reports that IFN-λ induces a stronger and more sustained ISG expression in hepatocyte cell lines in vitro (182), and that PolyI:C-induced control of HCV replication in humanized liver in chimeric mice is correlated to the induction of IFN-λ but not IFN-I in human hepatocytes (183). Responder patients harboring the favorable IFNL3 allele preventing the degradation of the corresponding RNA in infected cells might express significant levels of endogenous IFN-λ3, although this is disputed. However, they express only low levels of IFN-λR1, which limits IFNλ3 efficiency (Figure 7B) (184). How these patients benefit from Peg-IFN-α treatment could be that it induces IFN-λR1 expression on hepatocytes thus boosting endogenous IFN-λ3 effects (184). In contrast, high ISG-expressing non-responder patients harboring the unfavorable IFNL3 allele might not express enough IFN-λ3 for virus control. However, they do express IFN-λR1 as a result of their endogenous production of IFN-I. Hence, Peg-IFN-α might be ineffective in these patients because they already express IFN-λR1 but fail to produce endogenous IFN-λ3 due to the degradation of its mRNA in infected hepatocytes (Figure 7B). These patients may be good candidates for Peg-IFN-λ therapy, currently undergoing clinical development. Since the expression of IFN-λR1 is mainly restricted to epithelial cells, melanocytes, and hepatocytes, some of the side effects related to IFN-I treatment might be strongly attenuated in Peg-IFN-λ therapy. However, as IFN-I are key to induce anti-viral immune responses, it will be critical to determine whether, beside viral clearance, Peg-IFN-λ therapy can also induce long-term immune protection against HCV.
Cellular and Molecular Mechanisms Determining the Beneficial Versus Deleterious Outcome of IFN-I Effects
Existence of Different Pathways Downstream of IFNAR Signaling
IFN-I transduce intracellular signals through a single receptor, IFNAR, but via a multitude of downstream signaling pathways. The Janus activated kinase (JAK)/STAT pathway was the first to be identified (185). IFNAR is composed of two distinct subunits, IFNAR1 and IFNAR2, which are constitutively associated with members of the JAK family, tyrosine kinase 2 (TYK2) and JAK1, respectively (186). The binding of IFN-I to their receptor leads to the phosphorylation of JAK1 and TYK2, which in turn induce the phosphorylation and activation of the STAT proteins (186).
Different STAT complexes can form upon triggering of IFNAR (Figure 8). A transcriptional complex that forms in most conditions of IFN-I stimulation and induces the expression of many molecules of cell-intrinsic anti-viral immunity is interferon-stimulated gene factor 3 (ISGF3), a heterotrimer composed of pSTAT1, pSTAT2, and IRF9 (187) (Figure 8). Following its translocation into the nucleus, ISGF3 binds to ISRE regulatory sequences in target genes. Many molecules playing a key role in the function of innate or adaptive immune leukocytes are also induced by ISGF3, including CD80, CD86, or IL-15 in DC, and Granzyme B in NK cells. ISGF3 is generally composed of STAT1 phosphorylated on Tyr701 and Ser727 and of STAT2 phosphorylated on Tyr689. However, alternative ISGF3 complexes have been described in various contexts which could participate to the diversity of IFN-I effects (188).
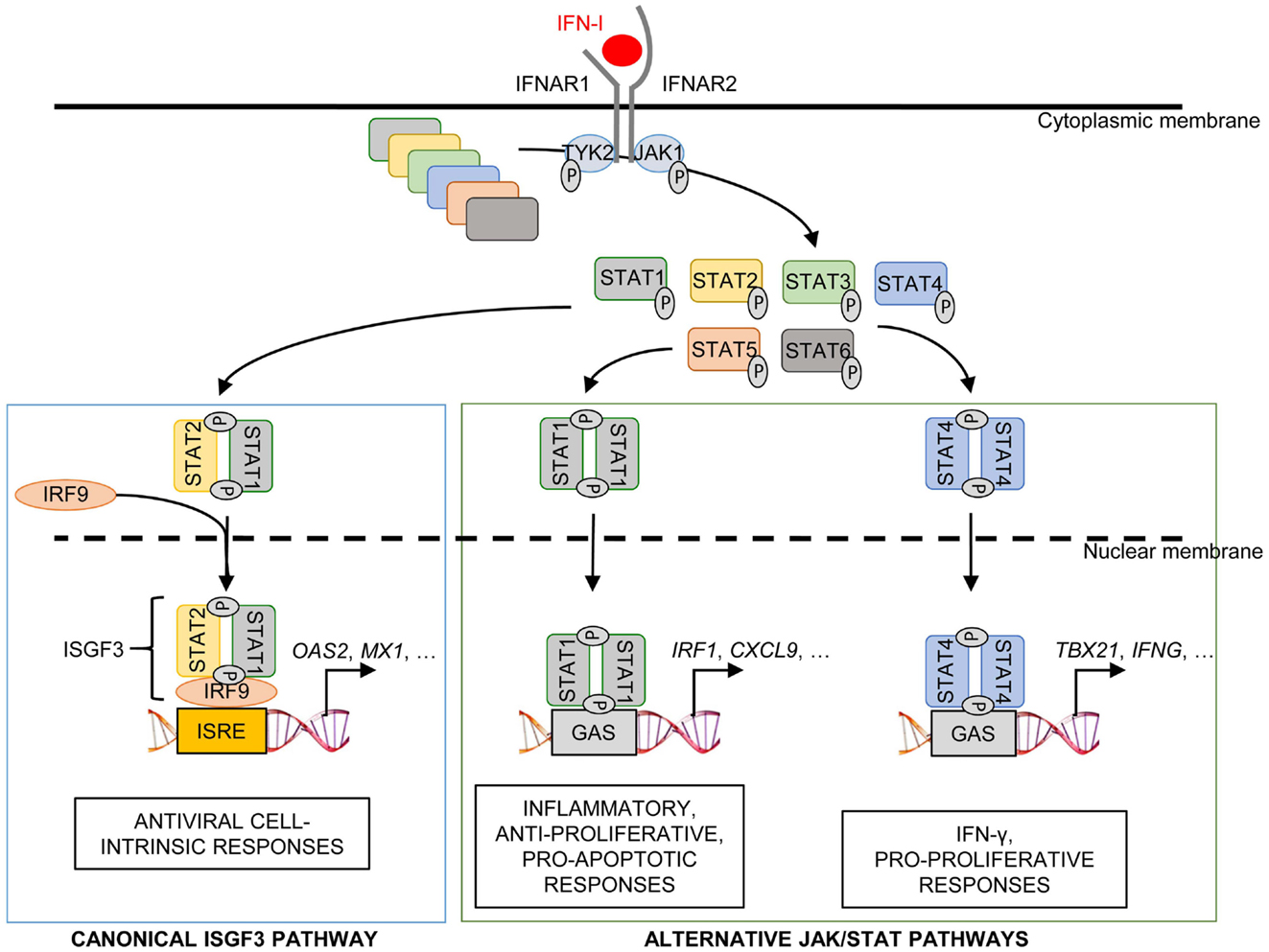
Figure 8. Schematic representation of the ISGF3 and alternative JAK/STAT signaling pathways induced by IFN-I. The receptor for IFN-I, IFNAR, is composed of two chains, IFNAR1 and IFNAR2, which are respectively associated with the JAK family kinases TYK2 and JAK1. IFN-I binding to IFNAR triggers the phosphorylation of TYK2 and JAK1, which in turn phosphorylate a variety of STAT proteins. Activated STATs are able to form complexes, as homo- or hetero-dimers. The heterodimer STAT1-STAT2 binds to a third partner, IFN-regulatory factor 9 (IRF9), in order to form the ISGF3 complex. This complex translocates into the nucleus and binds to specific regulatory sequences, IFN-stimulated response elements (ISRE), to activate the expression of many interferon-stimulated genes (ISGs). In particular, ISGF3 induces most, if not all, of the ISGs encoding effector molecules of cell-intrinsic anti-viral defenses such as OAS or MX1. Alternative JAK/STAT pathways include the formation of STAT1 or STAT4 homodimers, which may drive different functional responses to IFN-I. STAT1 homodimers bind to IFNγ-activated sequences (GAS) in the promoter of certain ISGs, which may promote inflammatory, anti-proliferative, and pro-apoptotic responses. STAT4 homodimers also bind to GAS but promote IFN-γ production and pro-proliferative responses.
The pSTAT1 homodimer also plays a prominent role in cell-intrinsic IFN-I-dependent gene induction. It binds IFNγ-activated sequences (GAS) and controls the expression of many pro-inflammatory molecules (187). pSTAT1 homodimers can form upon stimulation with either IFN-I or IFN-γ. Many GAS-regulated genes can be induced by either cytokines.
Depending on cell types, JAK signaling downstream of IFNAR can lead to the activation of virtually all STAT proteins and to their combinatorial association into a variety of complexes with different affinities for specific GAS elements (189–191) (Figure 8). This diversity contributes to IFN-I induction of different transcriptional programs in distinct cell types (39). STAT complex formation depends in part on the relative abundance of STAT molecules in the cell (192). While STAT1, STAT2, STAT3, and STAT5 can be activated in most cell populations, STAT4 and STAT6 are mainly activated in lymphocytes (193). For example, quiescent NK cells express more STAT4 than STAT1, leading to constitutive association of IFNAR to STAT4 in these cells. Hence, quiescent NK cells mount pSTAT4 homodimer-dependent responses to IFN-I stimulation, including IFN-γ production and T-bet-driven proliferation (Figure 8) (194, 195). Changes in STAT levels can also occur upon the differentiation/activation of a given cell type and lead to a shift in its functional response to the cytokines (196). Upon activation, NK cells decrease their expression of STAT4 and increase that of STAT1, shifting their IFN-I response from STAT4-dependent in a quiescent state to STAT1-dependent in pre-activated cells. This translates into opposite IFN-I effects on IFN-γ production and proliferation for quiescent versus pre-activated NK cells (194). However, this outcome can be modulated by simultaneous exposure to other cytokines such as IL-15 or IL-12/18. A reverse STAT1-to-STAT4 shift occurs in DC during their maturation, shifting their functional responses from inhibition to activation of IL-12 production in response to combined stimulations with IFN-I and CD40L (197). This enables mature DC to efficiently activate CD8 T cells. Other yet unknown mechanisms control the formation of different STAT complexes in distinct cell types. The nature and dynamics of the signaling pathways triggered by IFN-α or -β were evaluated in bulk cultures of human blood leukocytes using flow cytometry (191) or high throughput mass cytometry (190). A diversity of phosphorylation patterns of STAT1/3/5 was observed upon IFN-I stimulation. IFN-α activation induced phosphorylation of STAT1, STAT3, and STAT5 in most cell types, peaking at 15 min (190). IFN-β-induced STAT1 phosphorylation was found to be poor in B cells as compared to monocytes and T cells (191). However, the underlying mechanism remains to be identified since B cells did not express lower amount of IFNAR2 or STAT1 or enhanced levels of the inhibitory SOCS1 molecule. The high STAT1 activation in monocytes led to their induction of IFN-I-dependent pro-apoptotic genes while this was not the case in B cells. These results strikingly differ from those obtained in the other study upon IFN-α stimulation, where STAT1 phosphorylation was on the contrary lower in CD14+ monocytes and was prolonged in B cells and NK cells (190). The differences between these two studies might have resulted from the use of different subsets and doses of IFN-I. In any case, both studies consistently reported that CD4 T cells showed the highest activation of STAT5. All CD4 T cells but not all CD8 T cells activated STAT5 and for a longer time (190). IFN-β activation of STAT3 was delayed in CD4 T cells and B cells as compared to CD8 T cells and monocytes (191). Different STAT complexes may lead to distinct transcriptional programs linked to different biological functions (Figure 8). More systematic studies are needed to understand this complexity. Besides changing STAT levels between cell types or activation states, the processes controlling differential formation of STAT complexes downstream of IFNAR triggering remain to be identified.
In addition to JAK/STAT signaling, other pathways can be activated downstream of IFNAR, including those involving the phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinases (MAPK), and the CRK adaptor molecules (39, 198). This leads to the activation of other transcription factors such as IRF, NF-κB, or PU.1, which contribute to orchestrate cell responses to the cytokines by regulating both distinct and overlapping sets of genes as compared to STAT (199, 200).
In summary, IFNAR signals through a remarkable diversity of pathways, including but not limited to diverse combinations and kinetics of STAT phosphorylations. This explains at least in part the diversity of IFN-I effects, including their induction of opposite responses depending on the physiopathological contexts and/or the nature of the principal responding cell types (200, 201). IFN-III induce the same signaling pathways as IFN-I, although they engage a different heterodimeric receptor, composed of the IL-28RA and IL-10RB chains and preferentially expressed on epithelial cells including hepatocytes.
The Different IFN-I Subtypes have Different Affinities for Their Receptor Leading to Their Induction of Distinct Signaling
In mice and human beings, numerous IFN-I subtypes exist. Functional and population genetic analyses showed that these IFN-I subtypes significantly differ in their functions (202–207). Hence, one of most extraordinary feature of IFN-I biology is how IFN-I subtypes can elicit so many pleiotropic and diverse functions by interacting with the same receptor complex (208).
Both IFNAR1 and IFNAR2 are required for the initiation of IFN-I-dependent signals, as mice deficient in either one are highly susceptible to viral infections (3, 5). The assembling of the IFN-receptor ternary complex is a two-step process. First, a binary complex is formed by the binding of one side of the IFN molecule to IFNAR2. Then, a single binary complex interacts with IFNAR1 via the other side of the IFN molecule. The stability of the ternary complex will be determined in part by the association and dissociation kinetics between the cytokine and the two receptor chains, as well as by IFNAR expression levels since the cell surface concentrations of the receptor subunits are relatively low. Hence, both the affinity of IFN-I subsets for IFNAR and the amounts of IFN-I, IFNAR1, and IFNAR2 will regulate their biological effects (Figure 9A) (209, 210). Cell membrane density of IFNAR1 and IFNAR2 is also involved in differential IFN-β- versus IFN-α-induced functional activities, such as anti-proliferative function (211). A variety of cell-intrinsic parameters can also impact the lifetime of the IFN-receptor ternary complex, such as the rate of endocytosis/degradation/recycling of signaling complexes, and negative ISG regulators such as USP18 that decrease the affinity of IFN-IFNAR1 binding (203, 212).
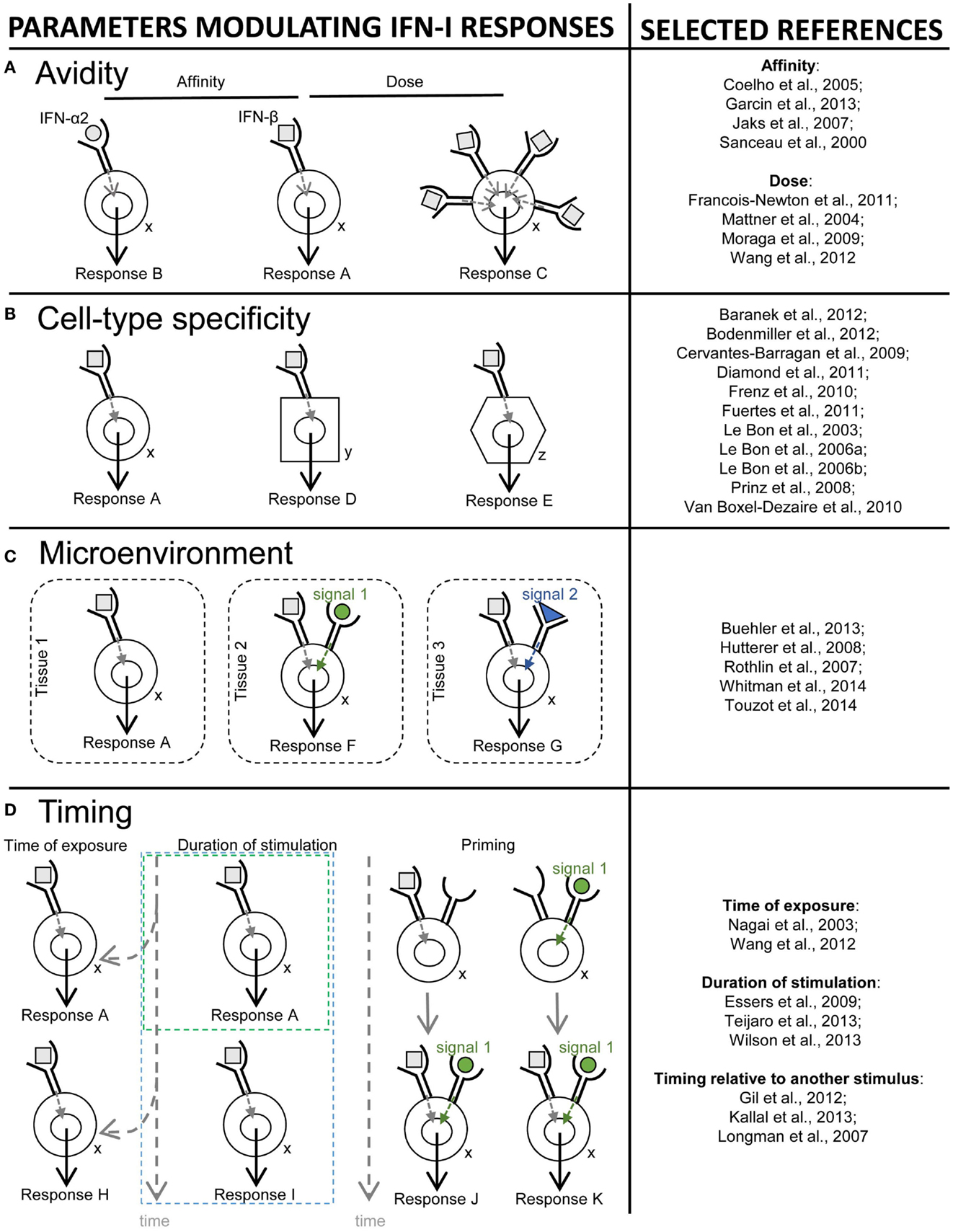
Figure 9. Schematic illustration of different mechanisms controlling the diversity of IFN-I effects. Different parameters that contribute to promote and control the diversity of IFN-I responses are depicted on the left side of the figure. References of papers illustrating each mechanism are given on the right side of the figure. (A) Avidity (a combination of affinity and dose). For example, the affinity of IFN-β for IFNAR1 is 100-times higher than that of IFN-α2, and IFNβ is much more potent in inhibiting cellular proliferation or monocyte differentiation into osteoclasts (response A), while both IFN-I subtypes are equipotent in establishing an anti-viral state (response B). The same subset of IFN-I can also exert different biological effects at low versus high doses. For example, low, but not high, doses of IFN-β protect BALB/c mice from progressive cutaneous and fatal visceral disease after Leishmania major infection. (B) Cell type specificity. Mouse DC but not NK cells are strong responders to IFN-I, and cell-intrinsic responses to IFN-I are critical in DC but not in some other cell types for immune defenses against viral infections or tumors. (C) Tissue microenvironment. The response of a given cell type to a given dose of a specific subset of IFN-I can also be modulated by the microenvironment of the cell. For example, in cancer, protective IFN-I effects on infiltrating DC or other immune cells might be dampened by inhibitors of IFNAR signaling locally produced by the tumor, such as ligands of the TAM receptor tyrosine kinases. (D) Timing. Differences in the time and duration of exposure to IFN-I can also determine distinct functional outcomes. For example, during viral infections, early and transient high levels of IFN-I promote protective DC and T cell responses, while delayed, chronic and low level IFN-I production compromises host immune defenses and promotes chronic viral infections. Within a given cell type, the outcome of IFN-I stimulation also depends on time of exposure to these cytokines relative to other modulatory signals (timing relative to other stimuli). For example, in naïve CD8 T cells, TCR signaling prior to IFN-I stimulation leads to increased expression of STAT4 and promotes IFN-γ production and proliferation, while IFN-I stimulation prior to TCR triggering leads to STAT1-dependent anti-proliferative and pro-apoptotic effects.
Based on a definition of a prototypic cytokine-receptor binding module and by analogy with the EPO receptor system, IFN-I subtypes were originally postulated to form ternary complexes of differing architectures, resulting in distinct geometry and assembling of intracellular signaling components (213). Experimental evidence rejected this hypothesis. Rather, the differential activities of IFN-I subtypes are determined by the stability of the ligand/receptor ternary complex (207, 212). Differential affinities of the IFN-I subtypes for IFNAR1 and IFNAR2 extracellular domains generate subtype-specific signaling cascades and biological outcomes (Figure 9A) (210, 214). Crystal structure of ternary IFN-I/IFNAR1/IFNAR2 complex illuminated the biochemical complexity of IFN-I interaction with their cognate receptors (215). The main conformational features of IFN-I/IFNAR1/IFNAR2 ternary complexes are conserved among the different IFN-I, but are quite different from the other cytokine receptors (214, 215). In the formation of the binary IFN-I/IFNAR2 complex, IFN-I ligand discrimination resides on differential energetics during the interaction of anchor points with IFNAR2, shared by all IFN-I, as well as on key amino acid substitution among IFN-I subtypes (215). IFNAR1 then performs major conformational changes to interact with IFN-I associated in the binary complex, thus displaying an optimized functional plasticity (215). These differences in the chemistry of IFN-I subtype interaction with IFNAR2 and IFNAR1 thus explain the different affinities of IFN-α versus IFN-β within ternary complex and their differential activities (210).
Cell-Intrinsic Responses of Distinct Cell Types Differentially Contribute to IFN-I Effects in Various Physiopathological Conditions
The functions regulated by IFN-I strongly depend on the main responding cell types (Figure 9B). This has been studied in vitro by examining the functional consequences of the stimulation of different cell types with IFN-I, and in vivo by determining the contribution of cell-intrinsic IFN-I responses of different cell types to resistance or susceptibility to various diseases. An emerging concept is the central role of DC responses to IFN-I for induction of protective immunity against viral infections or tumors (Figure 3). The development of mutant mice allowing conditional genetic inactivation of Ifnar1 in a cell-type specific manner using the Cre-lox system (216) has been instrumental in accelerating our understanding of how different cell types respond to IFN-I in vivo and what their respective contribution is to protective or deleterious IFN-I responses. This has been investigated most extensively in viral infections (106, 111, 112, 115, 217) but also in cancer (97, 98), bacterial infections (218), autoimmunity (216, 219), sepsis (220), or inflammatory diseases (221). Efforts are being pursued to better understand which cell types respond to IFN-I in a manner promoting protective versus deleterious effects in different physiopathological settings. That knowledge will considerably help to develop novel strategies to modulate IFN-I functions for promoting health over disease. The development of mutant mice allowing conditional genetic inactivation of Stat1, Stat3, and Stat5 (222–226) will help better understanding how different signaling pathways in different cell types determine the outcome of IFN-I response in vivo in various conditions. This knowledge might lead to the development of strategies aiming at targeting a given cell type with a specific subset of IFN-I, or in the presence of antagonists of certain signaling pathways, to surgically tune IFN-I responses in vivo toward the most desirable outcome.
The Anatomical Location and Timing of Cell Exposure to IFN-I Might also be Major Parameters Controlling the Effects of the Cytokines
The formation of specific STAT complexes is a highly dynamic process. It depends not only on the cell type but also on its specific state at the time it sees IFN-I. Hence, major parameters controlling the effects of IFN-I in a given cell type also include its microenvironment (Figure 9C) and the timing of its exposure to the cytokines both in terms of duration of the stimulation and of previous activation history (Figure 9D).
The TAM receptor ligand Gas6 is expressed within tumor cells in various solid cancers (227, 228). Elevated Gas6 expression is of bad prognosis in different cancers (228, 229). In a mouse model of ovarian cancer, early during tumorigenesis tumor-infiltrating DCs were found to be immunogenic and promote antitumor immunity, but they were later altered in the course of tumor development to acquire immunosuppressive properties beneficial to the tumor (230). One may thus hypothesize that expression of TAM soluble ligands in certain tumors and of TAM receptors on tumor-infiltrating DCs might contribute to dampen DC response to IFN-I and therefore facilitate their polarization by the tumor microenvironment into immunosuppressive cells (Figure 9C).
Acute versus chronic exposure to IFN-I can lead to strikingly opposite effects on a given cell type (13, 14, 231). In addition to duration, the time when a cell is exposed to IFN-I can also dramatically impact its functional response, depending on its previous activation history (Figure 9D). In vitro stimulation of DCs with IFN-β can lead to opposite outcomes depending whether it occurs simultaneously to, or after, TNFα-induced maturation. IFN-β polarizes DCs toward Th1 induction in the former case, and toward IL-10-secreting T cells in the latter case. These opposite effects result at least in part from the differential expression of IL-12/18 by DCs (232). Similarly, IFN-I effect on the functional polarization of CD4 T cells is strongly modulated by the other cytokines present in the lymphocyte microenvironment at the same time (233). IFN-I can also mediate opposite effects on CD8 T cells depending whether it occurs before or after cognate engagement of the T cell receptor. Indeed, while CD8 T cells have the potential to respond to IFN-I by inducing both STAT1- and STAT4-dependent genes, this depends upon their activation history. Naïve CD8 T cells respond mostly to IFN-I through STAT1 signaling, leading to the inhibition of their proliferation and eventually to the induction of their apoptosis. However, cognate triggering of the T cell receptor causes a decrease in STAT1 and an increase in STAT4 expression in CD8 T cells. This leads to a shift of their IFN-I response from STAT1-to-STAT4 signaling, resulting in the promotion of their proliferation and IFN-γ production. During LCMV infection, this mechanism promotes STAT4-dependant expansion of anti-viral CD8 T cells, but STAT1-dependant inhibition of naïve CD8 T cell proliferation (234).
Innovative Biochemical Engineering Approaches to Tune IFN-I Effects
Since the late 70s the clinical potential of IFN-I for the treatment of patients suffering of viral infection or cancer diseases has been widely acknowledged (235). Today, this expectation is tempered because IFN-I treatment can induce severe side effects and sufficient doses cannot be administered in patients. Therefore, there is a strong need to create tuned IFN molecules devoid of side effects. Based on our current understanding of IFN-I responses as reviewed above, many parameters could be tuned individually or in a combined manner to modulate IFN-I activity to promote their beneficial effects over the deleterious ones in a number of diseases. These parameters include modifying the affinity of IFN-I for its receptor, playing with the local quantity/concentration of IFN-I and with the duration of its delivery, and modulating the nature of the cells that are responding to IFN-I. We will discuss here novel strategies being developed to deliver IFN-I to, or block IFN-I responsiveness of, a specific target cell type in vivo (Figure 10).
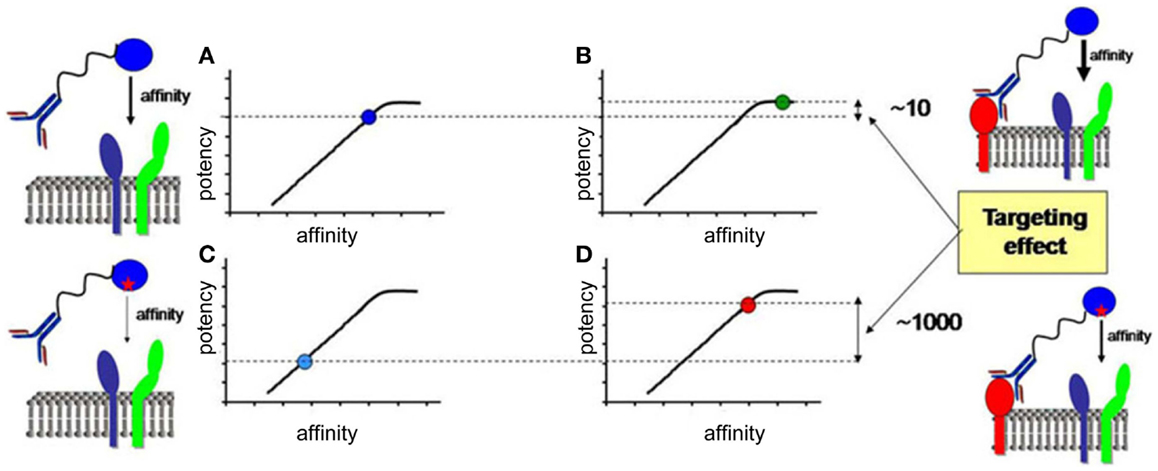
Figure 10. Strategy for high efficiency cell type-specific targeting of cytokine activity. (A) Most cytokines have evolved to exhibit optimized specific activities. (B) When the antibody moiety of a classical immunocytokine binds its cellular target, the ensuing increase of cytokine-receptor avidity translates into a modest increase of cytokine potency. (C,D) By introducing a mutation that decreases the affinity of the cytokine for its receptor, the activity of the mutated immunocytokine is now focused with a very high efficiency on target cells.
Modulating IFN-I Functions by Altering Their Binding to Their Receptor through Advanced Biochemical Engineering
If IFN-I-induced side effects are a consequence of the pleiotropic nature of IFN-I, and if the bioactivities mediating deleterious effects have some degree of independence from those mediating beneficial effects, one could mutate the IFN-I molecules in order to skew their activity toward a desired bioactivity. Indeed, introducing key mutation in IFN-α2 allowed increasing its affinity to IFNAR1 by a factor of 100. Accordingly, this IFN-α2 mutant is 100-times more potent in inhibiting cell proliferation, but as potent as WT IFN-α2 in inducing an anti-viral state (236–238). Hence, it is possible to tune IFN activity by modifying its binding to IFNAR. However, translating such an approach for the design of molecules for clinical application is severely hampered by the poor understanding we have on the IFN-I bioactivities mediating the side effects. Furthermore, we are far from having established the list of bioactivities that could be differentially modulated by changing the stability of the IFN-I/IFNAR complex. We know more about the cell types that mediate beneficial versus deleterious IFN responses in various diseases. Hence, we will now discuss strategies aimed at focusing IFN activity to specific cell types to promote health over disease.
Cell Specific Targeting of Exogenous IFN-I
Several strategies have been developed to specifically target IFNs on tumor cells, tumor-infiltrated immune cells or infected tissues. These strategies include intra-lesional injection (239, 240), adenoviral-mediated gene transfer (241–243), engineered tumor-infiltrating monocytes (244), and fusion of IFNs with a cleavable protecting shell (245). Another strategy to increase cytokine accumulation within the tumor or infected tissue is antibody-mediated targeting of cytokine delivery, where a cytokine moiety is fused to an antibody directed against a specific cell surface marker (Figure 10). The fusion molecule retains both antigen-binding and IFN-I bioactivities, and is enriched at the targeted site upon in vivo injection (246–249). When targeted to human CD20, IFN-I inhibited the proliferation of lymphoma cells engrafted in immunodeficient mice (250). An IFN-I targeted to a tumor antigen can also amplify the therapeutic effect of the antibody by acting on tumor-infiltrated DCs, thus increasing antigen cross-presentation and antitumor cytotoxic T cell responses (249). On non-targeted cells, the antibody conjugation negatively impacts IFN-I potency, but only modestly (18, 248, 251) (Figure 10A). Fusion molecules generally retain full IFN-I biological activity on the cells expressing the antibody target (Figure 10B). Hence, this difference only leads to a modest ratio between the IFN-I specific activity measured on target and non-target cells (Figure 10B). Such a targeting efficiency is definitely too low to reduce the toxic effect of IFN-I administration, because it will not specifically focus IFN-I activities on “beneficial cells” without stimulating “deleterious cells.”
The engineering of immuno-IFN-I must be improved to reach the very high targeting efficacy required to significantly diminish the treatment side effects. We recently reported an innovative strategy reaching this goal (18). It is based on the postulate that the antibody moiety of an immuno-IFN-I stabilizes the IFN-I/receptor-complex by avidity. It also takes into account the fact that the biological potency of an IFN-I is proportional to the stability of the IFN-I/receptor complex up to a certain threshold beyond which increasing the stability does not increase its potency (238, 252). IFN-α2 and IFN-β are used in most immuno-IFN-I studies. They have evolved to retain close to maximal potency. Hence, their targeting by an antibody that only provides a modest gain in terms of biological potency. However, it is expected that decreasing the affinity of the IFN-I for its receptor, by introducing a mutation, would increase the targeting effect of the antibody (Figures 10C,D). This is indeed the case. Using an IFN-I with a single point mutation that dramatically decreases its affinity for IFNAR2 (Figure 10C) allows engineering immuno-IFNs that are up to 1000-fold more potent on cells expressing the antibody target (Figure 10D). The three log targeting efficiency of these novel types of immuno-IFNs is found for various activities measured in vitro or in vivo when delivered in mice. If the toxic side effect experienced by the patients treated with IFN-I is due to systemic IFN-I activity, this targeting technology may find considerable clinical applications since such engineered immuno-IFNs are virtually inactive while “en route” and are activated only after binding of the fused antibody to the desired target. It remains to define the useful targets according to pathologies, for example, tumor cells themselves and professional cross-presenting XCR1+ DCs for cancer (97, 98, 249), or hepatocytes for chronic HCV infection.
Cell Specific Blockade of Endogenous IFN-I
To treat autoimmune diseases, novel therapeutics targeting IFN-I have been developed, including two IFN-α-neutralizing monoclonal antibodies currently in clinical trials (Sifalimumab and Rontalizumab) (253, 254). However, long-term systemic neutralization of IFN-I activity may increase susceptibility to viral infection and tumor development. Alternative strategies are needed to specifically inhibit IFN-I deleterious effects in these diseases without globally compromising IFN-I anti-viral and anti-tumoral functions. The sequential nature of the assembling of the IFN-I/receptor complex opens the possibility to design IFN-I antagonists specifically targeting the cell subsets responsible for IFN-I deleterious effects.
An IFN-α2 carrying a single amino acid substitution that blocks the IFN-I/IFNAR1 interaction engages IFNAR2 in a complex, which cannot bind IFNAR1 (255). Since the binary IFN-I/IFNAR2 complex is devoid of any IFN-I activity, such mutant behaves as a potent IFN-I antagonist. When linked to an antibody specific for a cell surface marker, the antagonistic activity of the mutant IFN-I should be significantly reinforced specifically on the cells expressing the target. Hence, it should be possible to design and construct targeted antagonists that inhibit responsiveness to endogenous IFN-I specifically on the cell subsets on which the cytokines act to promote autoimmunity or severe side effects, leaving the other cells fully responsive. For example, in chronic HCV patients treated with Peg-IFN-α, one of the most deleterious side effects is nervous depression, which might be prevented by co-administration of an IFN-I antagonist specifically targeting neurons or other cells of the central nervous system.
Conclusion
In the last decade, several major technological breakthroughs and the generation of novel animal models have remarkably advanced our understanding of the mode of action of IFNs. In vitro high throughput screening allowed systematically studying the functions of ISGs by ectopic expression or knock-down. Advance biophysical investigation of the interactions between IFN-I and the IFN-I receptor allowed to rigorously investigate the mechanistic basis for the differential bioactivities of IFN-I subtypes. The analyses of the responses of different cell types to IFNs or to viral infection, in vitro but also in vivo in various pathologies, demonstrated that IFN-I often mediate beneficial versus deleterious roles by acting on different cell types. From integrative analysis of these data, a picture is now emerging suggesting that it will be possible to segregate protective from deleterious IFN-I effects, based (i) on their differential induction depending on IFN-I subsets or on the magnitude/timing of IFN-I production, (ii) on their conditioning in different tissues, (iii) or on their occurrence in different cell types. Hence, innovative immunotherapeutic treatments are being designed to tune IFN-I activity toward desired effects in order to promote health over disease in a manner adapted to each physiopathological condition. In particular, a proof-of-concept has been made in vitro that it will be possible to target IFN-I activity on given cell types or tissues to administer to patients sufficiently high doses of the cytokine at the site of interest while limiting unwanted effects in other tissues or cell types. The next steps will be to demonstrate efficacy of this strategy in vivo in preclinical animal models. Importantly, to foster the development of these innovative immunotherapies, major efforts are still warranted to continue delineating which cell types are mainly responsible for the protective versus deleterious effects of IFN-I in different diseases. Combining high throughput technologies and systems biology approaches will also advance our understanding of the molecular mechanisms dynamically controlling IFN-I responses in health and diseases, which should reveal potentially novel therapeutic targets.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The studies performed in the laboratories are supported by funding from INSERM, CNRS, Aix-Marseille University, the LabEx MabImprove, and the European Community’s Seventh Framework Programme FP7/2007–2013 (grant agreement 223608 for Gilles Uzé, European Research Council Starting Grant Agreement number 281225 for Marc Dalod including salary support to Emeline Pollet and Thien-Phong Vu Manh). We thank past and present laboratory members for their contribution to studies on DCs or IFNs. We apologize for not quoting certain studies because of space limitations.
References
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci (1957) 147(927):258–67. doi: 10.1098/rspb.1957.0049
2. Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. Some properties of interferon. Proc R Soc Lond B Biol Sci (1957) 147(927):268–73. doi:10.1098/rspb.1957.0049
3. Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science (1994) 264(5167):1918–21. doi:10.1126/science.8009221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev (2008) 226:29–40. doi:10.1111/j.1600-065X.2008.00698.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Fenner JE, Starr R, Cornish AL, Zhang JG, Metcalf D, Schreiber RD, et al. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol (2006) 7(1):33–9. doi:10.1038/ni1287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases – elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi:10.1016/j.coi.2014.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Reder AT, Feng X. How type I interferons work in multiple sclerosis and other diseases: some unexpected mechanisms. J Interferon Cytokine Res (2014) 34(8):589–99. doi:10.1089/jir.2013.0158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Malireddi RK, Kanneganti TD. Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front Cell Infect Microbiol (2013) 3:77. doi:10.3389/fcimb.2013.00077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Furuya Y, Steiner D, Metzger DW. Does Type I interferon limit protective neutrophil responses during pulmonary Francisella Tularensis infection? Front Immunol (2014) 5:355. doi:10.3389/fimmu.2014.00355
10. Lenz LL, Eshleman EM. Type I interferons in bacterial infections: taming of myeloid cells and possible implications for autoimmunity. Front Immunol (2014) 5:431. doi:10.3389/fimmu.2014.00431
11. Di Domizio J, Cao W. Fueling autoimmunity: type I interferon in autoimmune diseases. Expert Rev Clin Immunol (2013) 9(3):201–10. doi:10.1586/eci.12.106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Herbeuval JP, Shearer GM. HIV-1 immunopathogenesis: how good interferon turns bad. Clin Immunol (2007) 123(2):121–8. doi:10.1016/j.clim.2006.09.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340(6129):207–11. doi:10.1126/science.1235214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340(6129):202–7. doi:10.1126/science.1235208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Wijesundara DK, Xi Y, Ranasinghe C. Unraveling the convoluted biological roles of type I interferons in infection and immunity: a way forward for therapeutics and vaccine design. Front Immunol (2014) 5:412. doi:10.3389/fimmu.2014.00412
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi:10.1111/j.0105-2896.2004.00204.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Dalod M, Chelbi R, Malissen B, Lawrence T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J (2014) 33(10):1104–16. doi:10.1002/embj.201488027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Garcin G, Paul F, Staufenbiel M, Bordat Y, Van der Heyden J, Wilmes S, et al. High efficiency cell-specific targeting of cytokine activity. Nat Commun (2014) 5:3016. doi:10.1038/ncomms4016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Hsu AC, Parsons K, Barr I, Lowther S, Middleton D, Hansbro PM, et al. Critical role of constitutive type I interferon response in bronchial epithelial cell to influenza infection. PLoS One (2012) 7(3):e32947. doi:10.1371/journal.pone.0032947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Tsugawa Y, Kato H, Fujita T, Shimotohno K, Hijikata M. Critical role of interferon-alpha constitutively produced in human hepatocytes in response to RNA virus infection. PLoS One (2014) 9(2):e89869. doi:10.1371/journal.pone.0089869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest (2010) 120(4):1151–64. doi:10.1172/JCI37223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Chen HM, Tanaka N, Mitani Y, Oda E, Nozawa H, Chen JZ, et al. Critical role for constitutive type I interferon signaling in the prevention of cellular transformation. Cancer Sci (2009) 100(3):449–56. doi:10.1111/j.1349-7006.2008.01051.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Kim S, Kaiser V, Beier E, Bechheim M, Guenthner-Biller M, Ablasser A, et al. Self-priming determines high type I IFN production by plasmacytoid dendritic cells. Eur J Immunol (2014) 44(3):807–18. doi:10.1002/eji.201343806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Otero DC, Baker DP, David M. IRF7-dependent IFN-beta production in response to RANKL promotes medullary thymic epithelial cell development. J Immunol (2013) 190(7):3289–98. doi:10.4049/jimmunol.1203086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Li HS, Gelbard A, Martinez GJ, Esashi E, Zhang H, Nguyen-Jackson H, et al. Cell-intrinsic role for IFN-alpha-STAT1 signals in regulating murine Peyer patch plasmacytoid dendritic cells and conditioning an inflammatory response. Blood (2011) 118(14):3879–89. doi:10.1182/blood-2011-04-349761
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Zietara N, Lyszkiewicz M, Gekara N, Puchalka J, Dos Santos VA, Hunt CR, et al. Absence of IFN-beta impairs antigen presentation capacity of splenic dendritic cells via down-regulation of heat shock protein 70. J Immunol (2009) 183(2):1099–109. doi:10.4049/jimmunol.0803214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol (2001) 2(5):378–86. doi:10.1038/35073080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol (2009) 183(5):3229–36. doi:10.4049/jimmunol.0804277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Pulverer JE, Rand U, Lienenklaus S, Kugel D, Zietara N, Kochs G, et al. Temporal and spatial resolution of type I and III interferon responses in vivo. J Virol (2010) 84(17):8626–38. doi:10.1128/JVI.00303-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity (2012) 37(1):171–86. doi:10.1016/j.immuni.2012.05.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity (2013) 38(5):870–80. doi:10.1016/j.immuni.2013.05.004
32. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity (2013) 38(5):855–69. doi:10.1016/j.immuni.2013.05.007
33. Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J (1998) 17(22):6660–9. doi:10.1093/emboj/17.22.6660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett (1998) 441(1):106–10. doi:10.1016/S0014-5793(98)01514-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol (2012) 13(8):737–43. doi:10.1038/ni.2350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Hare D, Mossman KL. Novel paradigms of innate immune sensing of viral infections. Cytokine (2013) 63(3):219–24. doi:10.1016/j.cyto.2013.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Olagnier D, Hiscott J. Breaking the barrier: membrane fusion triggers innate antiviral immunity. Nat Immunol (2012) 13(8):713–5. doi:10.1038/ni.2373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature (2014). doi:10.1038/nature13590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Bonjardim CA, Ferreira PC, Kroon EG. Interferons: signaling, antiviral and viral evasion. Immunol Lett (2009) 122(1):1–11. doi:10.1016/j.imlet.2008.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Furr SR, Chauhan VS, Moerdyk-Schauwecker MJ, Marriott I. A role for DNA-dependent activator of interferon regulatory factor in the recognition of herpes simplex virus type 1 by glial cells. J Neuroinflammation (2011) 8:99. doi:10.1186/1742-2094-8-99
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339(6121):786–91. doi:10.1126/science.1232458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity (2013) 39(6):1132–42. doi:10.1016/j.immuni.2013.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, et al. Virgin and C. M. rice: pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature (2014) 505(7485):691–5. doi:10.1038/nature12862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Claudio N, Dalet A, Gatti E, Pierre P. Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J (2013) 32(9):1214–24. doi:10.1038/emboj.2013.80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Severa M, Fitzgerald KA. TLR-mediated activation of type I IFN during antiviral immune responses: fighting the battle to win the war. Curr Top Microbiol Immunol (2007) 316:167–92.
46. Baranek T, Zucchini N, Dalod M. Plasmacytoid dendritic cells and the control of herpesvirus infections. Viruses (2009) 1(3):383–419. doi:10.3390/v1030383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood (2006) 107(6):2423–31. doi:10.1182/blood-2005-07-2709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med (1999) 5(8):919–23. doi:10.1038/11360
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science (1999) 284(5421):1835–7. doi:10.1126/science.284.5421.1835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol (2001) 2(12):1144–50. doi:10.1038/ni736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Barchet W, Cella M, Odermatt B, Asselin-Paturel C, Colonna M, Kalinke U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J Exp Med (2002) 195(4):507–16. doi:10.1084/jem.20011666
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity (2004) 21(1):107–19. doi:10.1016/j.immuni.2004.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, et al. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature (2003) 424(6946):324–8. doi:10.1038/nature01783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity (2010) 33(6):955–66. doi:10.1016/j.immuni.2010.11.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Cervantes-Barragan L, Zust R, Weber F, Spiegel M, Lang KS, Akira S, et al. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood (2007) 109(3):1131–7. doi:10.1182/blood-2006-05-023770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Swiecki M, Wang Y, Gilfillan S, Colonna M. Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog (2013) 9(10):e1003728. doi:10.1371/journal.ppat.1003728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, et al. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med (2003) 197(7):885–98. doi:10.1084/jem.20021522
58. Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, et al. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med (2002) 195(4):517–28. doi:10.1084/jem.20011672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Zucchini N, Bessou G, Robbins SH, Chasson L, Raper A, Crocker PR, et al. Individual plasmacytoid dendritic cells are major contributors to the production of multiple innate cytokines in an organ-specific manner during viral infection. Int Immunol (2008) 20(1):45–56. doi:10.1093/intimm/dxm119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi:10.1016/j.cell.2010.01.022
61. Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity (2010) 32(3):305–15. doi:10.1016/j.immuni.2010.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Alexandre YO, Cocita CD, Ghilas S, Dalod M. Deciphering the role of DC subsets in MCMV infection to better understand immune protection against viral infections. Front Microbiol (2014) 5:378. doi:10.3389/fmicb.2014.00378
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31:563–604. doi:10.1146/annurev-immunol-020711-074950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Lauterbach H, Bathke B, Gilles S, Traidl-Hoffmann C, Luber CA, Fejer G, et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med (2010) 207(12):2703–17. doi:10.1084/jem.20092720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Balan S, Ollion V, Colletti N, Chelbi R, Montanana-Sanchis F, Liu H, et al. Human XCR1+ dendritic cells derived in vitro from CD34+ progenitors closely resemble blood dendritic cells, including their adjuvant responsiveness, contrary to monocyte-derived dendritic cells. J Immunol (2014) 193(4):1622–35. doi:10.4049/jimmunol.1401243
66. Zhang S, Kodys K, Li K, Szabo G. Human type 2 myeloid dendritic cells produce interferon-lambda and amplify interferon-alpha in response to hepatitis C virus infection. Gastroenterology (2013) 144(2):414–425e7. doi:10.1053/j.gastro.2012.10.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Yoshio S, Kanto T, Kuroda S, Matsubara T, Higashitani K, Kakita N, et al. Human blood dendritic cell antigen 3 (BDCA3)(+) dendritic cells are a potent producer of interferon-lambda in response to hepatitis C virus. Hepatology (2013) 57(5):1705–15. doi:10.1002/hep.26182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Zhu J, Zhang Y, Ghosh A, Cuevas RA, Forero A, Dhar J, et al. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity (2014) 40(6):936–48. doi:10.1016/j.immuni.2014.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Luthra P, Sun D, Silverman RH, He B. Activation of IFN-β expression by a viral mRNA through RNase L and MDA5. Proc Natl Acad Sci USA (2011) 108(5):2118–23. doi:10.1073/pnas.1012409108
70. Malathi K, Saito T, Crochet N, Barton DJ, Gale M Jr, Silverman RH. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA (2010) 16(11):2108–19. doi:10.1261/rna.2244210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe (2010) 7(5):354–61. doi:10.1016/j.chom.2010.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell (2007) 131(6):1124–36. doi:10.1016/j.cell.2007.10.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, et al. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity (2007) 27(2):240–52. doi:10.1016/j.immuni.2007.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS Jr, Bakaletz LO, et al. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J Virol (2007) 81(18):9790–800. doi:10.1128/JVI.00530-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Crozat K, Vivier E, Dalod M. Crosstalk between components of the innate immune system: promoting anti-microbial defenses and avoiding immunopathologies. Immunol Rev (2009) 227(1):129–49. doi:10.1111/j.1600-065X.2008.00736.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Kaminski MM, Ohnemus A, Cornitescu M, Staeheli P. Plasmacytoid dendritic cells and Toll-like receptor 7-dependent signalling promote efficient protection of mice against highly virulent influenza A virus. J Gen Virol (2012) 93(Pt 3):555–9. doi:10.1099/vir.0.039065-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Wolf AI, Buehler D, Hensley SE, Cavanagh LL, Wherry EJ, Kastner P, et al. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J Immunol (2009) 182(2):871–9. doi:10.4049/jimmunol.182.2.871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J Exp Med (2008) 205(7):1621–34. doi:10.1084/jem.20071365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Yang K, Puel A, Zhang S, Eidenschenk C, Ku CL, Casrouge A, et al. Human TLR-7-, -8-, and -9-mediated induction of IFN-alpha/beta and -lambda Is IRAK-4 dependent and redundant for protective immunity to viruses. Immunity (2005) 23(5):465–78. doi:10.1016/j.immuni.2005.09.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321(5889):691–6. doi:10.1126/science.1158298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet (2003) 33(3):388–91. doi:10.1038/ng1097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med (2011) 208(10):2083–98. doi:10.1084/jem.20101568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Sancho-Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest (2011) 121(12):4889–902. doi:10.1172/JCI59259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, et al. Evolutionary dynamics of human toll-like receptors and their different contributions to host defense. PLoS Genet (2009) 5(7):e1000562. doi:10.1371/journal.pgen.1000562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science (2006) 312(5775):879–82. doi:10.1126/science.1125676
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, et al. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature (2010) 465(7301):1079–83. doi:10.1038/nature09118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang DE, Trilling M, et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol (2012) 13(1):51–7. doi:10.1038/ni.2169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi:10.1146/annurev-immunol-032713-120231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Pfaller CK, Li Z, George CX, Samuel CE. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr Opin Immunol (2011) 23(5):573–82. doi:10.1016/j.coi.2011.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, et al. DNA deamination mediates innate immunity to retroviral infection. Cell (2003) 113(6):803–9. doi:10.1016/S0092-8674(03)00423-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Sze A, Olagnier D, Lin R, van Grevenynghe J, Hiscott J. SAMHD1 host restriction factor: a link with innate immune sensing of retrovirus infection. J Mol Biol (2013) 425(24):4981–94. doi:10.1016/j.jmb.2013.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature (2011) 472(7344):481–5. doi:10.1038/nature09907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Blanc M, Hsieh WY, Robertson KA, Kropp KA, Forster T, Shui G, et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity (2013) 38(1):106–18. doi:10.1016/j.immuni.2012.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, Cox JE, et al. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe (2013) 14(4):435–45. doi:10.1016/j.chom.2013.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Vu Manh TP, Alexandre Y, Baranek T, Crozat K, Dalod M. Plasmacytoid, conventional, and monocyte-derived dendritic cells undergo a profound and convergent genetic reprogramming during their maturation. Eur J Immunol (2013) 43(7):1706–15. doi:10.1002/eji.201243106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol (2003) 4(10):1009–15. doi:10.1038/ni978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med (2011) 208(10):1989–2003. doi:10.1084/jem.20101158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med (2011) 208(10):2005–16. doi:10.1084/jem.20101159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med (2009) 206(7):1589–602. doi:10.1084/jem.20090247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol (2014) 12(1):e1001759. doi:10.1371/journal.pbio.1001759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Le Bon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, Tough DF. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity (2001) 14(4):461–70. doi:10.1016/S1074-7613(01)00126-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson-Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity (2009) 31(3):491–501. doi:10.1016/j.immuni.2009.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Baranek T, Manh TP, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe (2012) 12(4):571–84. doi:10.1016/j.chom.2012.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, et al. A type I interferon autocrine-paracrine loop is involved in toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med (2005) 201(9):1435–46. doi:10.1084/jem.20041964
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15(4):323–32. doi:10.1038/ni.2833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Cervantes-Barragan L, Kalinke U, Zust R, Konig M, Reizis B, Lopez-Macias C, et al. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J Immunol (2009) 182(2):1099–106. doi:10.4049/jimmunol.182.2.1099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Martinez J, Huang X, Yang Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J Immunol (2008) 180(3):1592–7. doi:10.4049/jimmunol.180.3.1592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26(4):503–17. doi:10.1016/j.immuni.2007.03.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting edge: the direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol (2006) 176(6):3315–9. doi:10.4049/jimmunol.176.6.3315
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med (2005) 202(5):637–50. doi:10.1084/jem.20050821
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, et al. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol (2006) 176(8):4682–9. doi:10.4049/jimmunol.176.8.4682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol (2006) 176(4):2074–8. doi:10.4049/jimmunol.176.8.4682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol (2006) 176(8):4525–9. doi:10.4049/jimmunol.176.8.4525
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol (2006) 177(3):1746–54. doi:10.4049/jimmunol.177.3.1746
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Frenz T, Waibler Z, Hofmann J, Hamdorf M, Lantermann M, Reizis B, et al. Concomitant type I IFN receptor-triggering of T cells and of DC is required to promote maximal modified vaccinia virus Ankara-induced T-cell expansion. Eur J Immunol (2010) 40(10):2769–77. doi:10.1002/eji.201040453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Munz C, Lunemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol (2009) 9(4):246–58. doi:10.1038/nri2527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Fry L, Baker BS, Powles AV, Fahlen A, Engstrand L. Is chronic plaque psoriasis triggered by microbiota in the skin? Br J Dermatol (2013) 169(1):47–52. doi:10.1111/bjd.12322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Nibali L, Henderson B, Sadiq ST, Donos N. Genetic dysbiosis: the role of microbial insults in chronic inflammatory diseases. J Oral Microbiol (2014) 6:22962. doi:10.3402/jom.v6.22962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Seillet C, Laffont S, Tremollieres F, Rouquie N, Ribot C, Arnal JF, et al. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood (2012) 119(2):454–64. doi:10.1182/blood-2011-08-371831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Kinnunen T, Chamberlain N, Morbach H, Choi J, Kim S, Craft J, et al. Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood (2013) 121(9):1595–603. doi:10.1182/blood-2012-09-457465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest (2013) 123(10):4283–93. doi:10.1172/JCI69854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest (2005) 115(2):407–17. doi:10.1172/JCI200523025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Rodriguez-Pla A, Patel P, Maecker HT, Rossello-Urgell J, Baldwin N, Bennett L, et al. IFN priming is necessary but not sufficient to turn on a migratory dendritic cell program in lupus monocytes. J Immunol (2014) 192(12):5586–98. doi:10.4049/jimmunol.1301319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature (2007) 449(7162):564–9. doi:10.1038/nature06116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol (2008) 3:499–522. doi:10.1146/annurev.pathmechdis.3.121806.154316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Beadling C, Slifka MK. How do viral infections predispose patients to bacterial infections? Curr Opin Infect Dis (2004) 17(3):185–91. doi:10.1097/00001432-200406000-00003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis (2008) 198(7):962–70. doi:10.1086/591708
128. Kuiken T, Taubenberger JK. Pathology of human influenza revisited. Vaccine (2008) 26(Suppl 4):D59–66. doi:10.1016/j.vaccine.2008.07.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. J Immunol (2013) 191(5):2047–52. doi:10.4049/jimmunol.1301152
130. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207(10):2053–63. doi:10.1084/jem.20101664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Doughty L, Nguyen K, Durbin J, Biron C. A role for IFN-alpha beta in virus infection-induced sensitization to endotoxin. J Immunol (2001) 166(4):2658–64. doi:10.4049/jimmunol.166.4.2658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest (2009) 119(7):1910–20. doi:10.1172/JCI35412
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest (2011) 121(9):3657–65. doi:10.1172/JCI57762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest (2010) 120(5):1674–82. doi:10.1172/JCI40817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35(6):1023–34. doi:10.1016/j.immuni.2011.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511(7507):99–103. doi:10.1038/nature13489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science (2014) 345(6197):679–84. doi:10.1126/science.1254790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi:10.1038/nri3712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Daley-Bauer LP, Wynn GM, Mocarski ES. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity (2012) 37(1):122–33. doi:10.1016/j.immuni.2012.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Aldridge JR Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, et al. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci U S A (2009) 106(13):5306–11. doi:10.1073/pnas.0900655106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol (2011) 186(3):1666–74. doi:10.4049/jimmunol.1002194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, et al. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J Infect Dis (2014) 209(6):865–75. doi:10.1093/infdis/jit527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, et al. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science (2013) 340(6137):1230–4. doi:10.1126/science.1233632
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science (2012) 335(6071):936–41. doi:10.1126/science.1214935
145. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med (2011) 208(8):1635–48. doi:10.1084/jem.20110958
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34(2):213–23. doi:10.1016/j.immuni.2011.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, et al. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog (2012) 8(7):e1002811. doi:10.1371/journal.ppat.1002811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Biondo C, Midiri A, Gambuzza M, Gerace E, Falduto M, Galbo R, et al. IFN-alpha/beta signaling is required for polarization of cytokine responses toward a protective type 1 pattern during experimental cryptococcosis. J Immunol (2008) 181(1):566–73. doi:10.4049/jimmunol.181.1.566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. del Fresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, et al. Interferon-beta production via dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity (2013) 38(6):1176–86. doi:10.1016/j.immuni.2013.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Reder AT, Feng X. Aberrant type I interferon regulation in autoimmunity: opposite directions in MS and SLE, Shaped by Evolution and Body Ecology. Front Immunol (2013) 4:281. doi:10.3389/fimmu.2013.00281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Axtell RC, Raman C. Janus-like effects of type I interferon in autoimmune diseases. Immunol Rev (2012) 248(1):23–35. doi:10.1111/j.1600-065X.2012.01131.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. doi:10.1038/ni.1679
153. Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, et al. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci U S A (2014) 111(20):7409–14. doi:10.1073/pnas.1401662111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, Gilfillan S, et al. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe (2012) 11(6):631–42. doi:10.1016/j.chom.2012.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Hyrcza MD, Kovacs C, Loutfy M, Halpenny R, Heisler L, Yang S, et al. Distinct transcriptional profiles in ex vivo CD4+ and CD8+ T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J Virol (2007) 81(7):3477–86. doi:10.1128/JVI.01552-06
156. Sedaghat AR, German J, Teslovich TM, Cofrancesco J Jr, Jie CC, Talbot CC Jr, et al. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. J Virol (2008) 82(4):1870–83. doi:10.1128/JVI.02228-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Smith AJ, Li Q, Wietgrefe SW, Schacker TW, Reilly CS, Haase AT. Host genes associated with HIV-1 replication in lymphatic tissue. J Immunol (2010) 185(9):5417–24. doi:10.4049/jimmunol.1002197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest (2009) 119(12):3556–72. doi:10.1172/JCI40115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest (2009) 119(12):3544–55. doi:10.1172/JCI40093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Harris LD, Tabb B, Sodora DL, Paiardini M, Klatt NR, Douek DC, et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J Virol (2010) 84(15):7886–91. doi:10.1128/JVI.02612-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Rotger M, Dalmau J, Rauch A, McLaren P, Bosinger SE, Martinez R, et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J Clin Invest (2011) 121(6):2391–400. doi:10.1172/JCI45235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Hardy AW, Graham DR, Shearer GM, Herbeuval JP. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by toll-like receptor 7-induced IFN-alpha. Proc Natl Acad Sci USA (2007) 104(44):17453–8. doi:10.1073/pnas.0707244104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Stary G, Klein I, Kohlhofer S, Koszik F, Scherzer T, Mullauer L, et al. Plasmacytoid dendritic cells express TRAIL and induce CD4+ T-cell apoptosis in HIV-1 viremic patients. Blood (2009) 114(18):3854–63. doi:10.1182/blood-2009-04-217927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, et al. Sex differences in the toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med (2009) 15(8):955–9. doi:10.1038/nm.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature (2009) 458(7241):1034–8. doi:10.1038/nature07831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature (2014) 511(7511):601–5. doi:10.1038/nature13554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Li G, Cheng M, Nunoya J, Cheng L, Guo H, Yu H, et al. Plasmacytoid dendritic cells suppress HIV-1 replication but contribute to HIV-1 induced immunopathogenesis in humanized mice. PLoS Pathog (2014) 10(7):e1004291. doi:10.1371/journal.ppat.1004291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Wendt A, Adhoute X, Castellani P, Oules V, Ansaldi C, Benali S, et al. Chronic hepatitis C: future treatment. Clin Pharmacol (2014) 6:1–17. doi:10.2147/CPAA.S30338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Baugh JM, Garcia-Rivera JA, Gallay PA. Host-targeting agents in the treatment of hepatitis C: a beginning and an end? Antiviral Res (2013) 100(2):555–61. doi:10.1016/j.antiviral.2013.09.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem (2007) 282(28):20047–51. doi:10.1074/jbc.R700004200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity (2014) 40(1):13–24. doi:10.1016/j.immuni.2013.12.010
172. Sheahan T, Imanaka N, Marukian S, Dorner M, Liu P, Ploss A, et al. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe (2014) 15(2):190–202. doi:10.1016/j.chom.2014.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Miyagi T, Takehara T, Nishio K, Shimizu S, Kohga K, Li W, et al. Altered interferon-alpha-signaling in natural killer cells from patients with chronic hepatitis C virus infection. J Hepatol (2010) 53(3):424–30. doi:10.1016/j.jhep.2010.03.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Ahlenstiel G, Titerence RH, Koh C, Edlich B, Feld JJ, Rotman Y, et al. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology (2010) 138(1):e1–2. doi:10.1053/j.gastro.2009.08.066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog (2010) 6(6):e1000947. doi:10.1371/journal.ppat.1000947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Casrouge A, Decalf J, Ahloulay M, Lababidi C, Mansour H, Vallet-Pichard A, et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest (2011) 121(1):308–17. doi:10.1172/JCI40594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Hartigan-O’Connor DJ, Lin D, Ryan JC, Shvachko VA, Cozen ML, Segal MR, et al. Monocyte activation by interferon alpha is associated with failure to achieve a sustained virologic response after treatment for hepatitis C virus infection. J Infect Dis (2014) 209(10):1602–12. doi:10.1093/infdis/jit801
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature (2009) 461(7265):798–801. doi:10.1038/nature08463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature (2009) 461(7262):399–401. doi:10.1038/nature08309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet (2013) 45(2):164–71. doi:10.1038/ng.2521
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. McFarland AP, Horner SM, Jarret A, Joslyn RC, Bindewald E, Shapiro BA, et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol (2014) 15(1):72–9. doi:10.1038/ni.2758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Kohli A, Zhang X, Yang J, Russell RS, Donnelly RP, Sheikh F, et al. Distinct and overlapping genomic profiles and antiviral effects of Interferon-lambda and -alpha on HCV-infected and noninfected hepatoma cells. J Viral Hepat (2012) 19(12):843–53. doi:10.1111/j.1365-2893.2012.01610.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Nakagawa S, Hirata Y, Kameyama T, Tokunaga Y, Nishito Y, Hirabayashi K, et al. Targeted induction of interferon-lambda in humanized chimeric mouse liver abrogates hepatotropic virus infection. PLoS One (2013) 8(3):e59611. doi:10.1371/journal.pone.0059611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Duong FH, Trincucci G, Boldanova T, Calabrese D, Campana B, Krol I, et al. IFN-lambda receptor 1 expression is induced in chronic hepatitis C and correlates with the IFN-lambda3 genotype and with nonresponsiveness to IFN-alpha therapies. J Exp Med (2014) 211(5):857–68. doi:10.1084/jem.20131557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem (1998) 67:227–64. doi:10.1146/annurev.biochem.67.1.227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264(5164):1415–21. doi:10.1126/science.8197455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi:10.1038/nri3581
188. Fink K, Grandvaux N. STAT2 and IRF9: beyond ISGF3. JAKSTAT (2013) 2(4):e27521. doi:10.4161/jkst.27521
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science (2011) 332(6030):687–96. doi:10.1126/science.1198704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Bodenmiller B, Zunder ER, Finck R, Chen TJ, Savig ES, Bruggner RV, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol (2012) 30(9):858–67. doi:10.1038/nbt.2317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. van Boxel-Dezaire AH, Zula JA, Xu Y, Ransohoff RM, Jacobberger JW, Stark GR. Major differences in the responses of primary human leukocyte subsets to IFN-beta. J Immunol (2010) 185(10):5888–99. doi:10.4049/jimmunol.0902314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, et al. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science (2002) 297(5589):2063–6. doi:10.1126/science.1074900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol (2012) 12(2):125–35. doi:10.1038/nri3133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J Exp Med (2007) 204(10):2383–96. doi:10.1084/jem.20070401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Mack EA, Kallal LE, Demers DA, Biron CA. Type 1 interferon induction of natural killer cell gamma interferon production for defense during lymphocytic choriomeningitis virus infection. MBio (2011) 2(4):e00169–11. doi:10.1128/mBio.00169-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Kallal LE, Biron CA. Changing partners at the dance: variations in STAT concentrations for shaping cytokine function and immune responses to viral infections. JAKSTAT (2013) 2(1):e23504. doi:10.4161/jkst.23504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Longman RS, Braun D, Pellegrini S, Rice CM, Darnell RB, Albert ML. Dendritic-cell maturation alters intracellular signaling networks, enabling differential effects of IFN-alpha/beta on antigen cross-presentation. Blood (2007) 109(3):1113–22. doi:10.1182/blood-2006-05-023465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5(5):375–86. doi:10.1038/nri1604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25(3):349–60. doi:10.1016/j.immuni.2006.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity (2006) 25(3):361–72. doi:10.1016/j.immuni.2006.08.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Kalvakolanu DV. Alternate interferon signaling pathways. Pharmacol Ther (2003) 100(1):1–29. doi:10.1016/S0163-7258(03)00070-6
202. Sanceau J, Hiscott J, Delattre O, Wietzerbin J. IFN-beta induces serine phosphorylation of Stat-1 in Ewing’s sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene (2000) 19(30):3372–83. doi:10.1038/sj.onc.1203670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Francois-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS One (2011) 6(7):e22200. doi:10.1371/journal.pone.0022200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Coelho LF, Magno de Freitas Almeida G, Mennechet FJ, Blangy A, Uze G. Interferon-alpha and -beta differentially regulate osteoclastogenesis: role of differential induction of chemokine CXCL11 expression. Proc Natl Acad Sci U S A (2005) 102(33):11917–22. doi:10.1073/pnas.0502188102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Garcin G, Bordat Y, Chuchana P, Monneron D, Law HK, Piehler J, et al. Differential activity of type I interferon subtypes for dendritic cell differentiation. PLoS One (2013) 8(3):e58465. doi:10.1371/journal.pone.0058465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Manry J, Laval G, Patin E, Fornarino S, Itan Y, Fumagalli M, et al. Evolutionary genetic dissection of human interferons. J Exp Med (2011) 208(13):2747–59. doi:10.1084/jem.20111680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Uze G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol (2007) 316:71–95.
208. Berry CM, Hertzog PJ, Mangan NE. Interferons as biomarkers and effectors: lessons learned from animal models. Biomark Med (2012) 6(2):159–76. doi:10.2217/BMM.12.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Mattner J, Wandersee-Steinhauser A, Pahl A, Rollinghoff M, Majeau GR, Hochman PS, et al. Protection against progressive leishmaniasis by IFN-beta. J Immunol (2004) 172(12):7574–82. doi:10.4049/jimmunol.172.12.7574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Levin D, Schneider WM, Hoffmann HH, Yarden G, Busetto AG, Manor O, et al. Multifaceted activities of type I interferon are revealed by a receptor antagonist. Sci Signal (2014) 7(327):ra50. doi:10.1126/scisignal.2004998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Moraga I, Harari D, Schreiber G, Uze G, Pellegrini S. Receptor density is key to the alpha2/beta interferon differential activities. Mol Cell Biol (2009) 29(17):4778–87. doi:10.1128/MCB.01808-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Piehler J, Thomas C, Garcia KC, Schreiber G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev (2012) 250(1):317–34. doi:10.1111/imr.12001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Mogensen KE, Lewerenz M, Reboul J, Lutfalla G, Uze G. The type I interferon receptor: structure, function, and evolution of a family business. J Interferon Cytokine Res (1999) 19(10):1069–98. doi:10.1089/107999099313019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Jaks E, Gavutis M, Uze G, Martal J, Piehler J. Differential receptor subunit affinities of type I interferons govern differential signal activation. J Mol Biol (2007) 366(2):525–39. doi:10.1016/j.jmb.2006.11.053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell (2011) 146(4):621–32. doi:10.1016/j.cell.2011.06.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity (2008) 28(5):675–86. doi:10.1016/j.immuni.2008.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, et al. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity (2014) 40(6):961–73. doi:10.1016/j.immuni.2014.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. Kunz S, Oberle K, Sander A, Bogdan C, Schleicher U. Lymphadenopathy in a novel mouse model of Bartonella-induced cat scratch disease results from lymphocyte immigration and proliferation and is regulated by interferon-alpha/beta. Am J Pathol (2008) 172(4):1005–18. doi:10.2353/ajpath.2008.070591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
219. Dann A, Poeck H, Croxford AL, Gaupp S, Kierdorf K, Knust M, et al. Cytosolic RIG-I-like helicases act as negative regulators of sterile inflammation in the CNS. Nat Neurosci (2012) 15(1):98–106. doi:10.1038/nn.2964
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Schwandt T, Schumak B, Gielen GH, Jungerkes F, Schmidbauer P, Klocke K, et al. Expression of type I interferon by splenic macrophages suppresses adaptive immunity during sepsis. EMBO J (2012) 31(1):201–13. doi:10.1038/emboj.2011.380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Goossens P, Gijbels MJ, Zernecke A, Eijgelaar W, Vergouwe MN, van der Made I, et al. Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab (2010) 12(2):142–53. doi:10.1016/j.cmet.2010.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol (1998) 161(9):4652–60.
223. Wallner B, Leitner NR, Vielnascher RM, Kernbauer E, Kolbe T, Karaghiosoff M, et al. Generation of mice with a conditional Stat1 null allele. Transgenic Res (2012) 21(1):217–24. doi:10.1007/s11248-011-9519-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Kernbauer E, Maier V, Stoiber D, Strobl B, Schneckenleithner C, Sexl V, et al. Conditional Stat1 ablation reveals the importance of interferon signaling for immunity to Listeria monocytogenes infection. PLoS Pathog (2012) 8(6):e1002763. doi:10.1371/journal.ppat.1002763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
225. Klover PJ, Muller WJ, Robinson GW, Pfeiffer RM, Yamaji D, Hennighausen L. Loss of STAT1 from mouse mammary epithelium results in an increased Neu-induced tumor burden. Neoplasia (2010) 12(11):899–905.
226. Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol (2004) 24(18):8037–47. doi:10.1128/MCB.24.18.8037-8047.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Buehler M, Tse B, Leboucq A, Jacob F, Caduff R, Fink D, et al. Meta-analysis of microarray data identifies GAS6 expression as an independent predictor of poor survival in ovarian cancer. Biomed Res Int (2013) 2013:238284. doi:10.1155/2013/238284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Hutterer M, Knyazev P, Abate A, Reschke M, Maier H, Stefanova N, et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin Cancer Res (2008) 14(1):130–8. doi:10.1158/1078-0432.CCR-07-0862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
229. Whitman SP, Kohlschmidt J, Maharry K, Volinia S, Mrozek K, Nicolet D, et al. GAS6 expression identifies high-risk adult AML patients: potential implications for therapy. Leukemia (2014) 28(6):1252–8. doi:10.1038/leu.2013.371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
230. Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med (2012) 209(3):495–506. doi:10.1084/jem.20111413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
231. Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature (2009) 458(7240):904–8. doi:10.1038/nature07815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Nagai T, Devergne O, Mueller TF, Perkins DL, van Seventer JM, van Seventer GA. Timing of IFN-beta exposure during human dendritic cell maturation and naive Th cell stimulation has contrasting effects on Th1 subset generation: a role for IFN-beta-mediated regulation of IL-12 family cytokines and IL-18 in naive Th cell differentiation. J Immunol (2003) 171(10):5233–43. doi:10.4049/jimmunol.171.10.5233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Touzot M, Grandclaudon M, Cappuccio A, Satoh T, Martinez-Cingolani C, Servant N, et al. Combinatorial flexibility of cytokine function during human T helper cell differentiation. Nat Commun (2014) 5:3987. doi:10.1038/ncomms4987
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Gil MP, Ploquin MJ, Watford WT, Lee SH, Kim K, Wang X, et al. Regulating type 1 IFN effects in CD8 T cells during viral infections: changing STAT4 and STAT1 expression for function. Blood (2012) 120(18):3718–28. doi:10.1182/blood-2012-05-428672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
235. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov (2007) 6(12):975–90. doi:10.1038/nrd2422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
236. Jaitin DA, Roisman LC, Jaks E, Gavutis M, Piehler J, Van der Heyden J, et al. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol (2006) 26(5):1888–97. doi:10.1128/MCB.26.5.1888-1897.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
237. Kalie E, Jaitin DA, Abramovich R, Schreiber G. An interferon alpha2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J Biol Chem (2007) 282(15):11602–11. doi:10.1074/jbc.M610115200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
238. Kalie E, Jaitin DA, Podoplelova Y, Piehler J, Schreiber G. The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J Biol Chem (2008) 283(47):32925–36. doi:10.1074/jbc.M806019200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
239. Cozzio A, Kempf W, Schmid-Meyer R, Gilliet M, Michaelis S, Scharer L, et al. Intra-lesional low-dose interferon alpha2a therapy for primary cutaneous marginal zone B-cell lymphoma. Leuk Lymphoma (2006) 47(5):865–9. doi:10.1080/10428190500399698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
240. Dubrot J, Palazon A, Alfaro C, Azpilikueta A, Ochoa MC, Rouzaut A, et al. Intratumoral injection of interferon-alpha and systemic delivery of agonist anti-CD137 monoclonal antibodies synergize for immunotherapy. Int J Cancer (2011) 128(1):105–18. doi:10.1002/ijc.25333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
241. Armstrong L, Davydova J, Brown E, Han J, Yamamoto M, Vickers SM. Delivery of interferon alpha using a novel Cox2-controlled adenovirus for pancreatic cancer therapy. Surgery (2012) 152(1):114–22. doi:10.1016/j.surg.2012.02.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
242. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res (2011) 71(7):2488–96. doi:10.1158/0008-5472.CAN-10-2820
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
243. Sterman DH, Haas A, Moon E, Recio A, Schwed D, Vachani A, et al. A trial of intrapleural adenoviral-mediated interferon-alpha2b gene transfer for malignant pleural mesothelioma. Am J Respir Crit Care Med (2011) 184(12):1395–9. doi:10.1164/rccm.201103-0554CR
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
244. De Palma M, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell (2008) 14(4):299–311. doi:10.1016/j.ccr.2008.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
245. Adams G, Vessillier S, Dreja H, Chernajovsky Y. Targeting cytokines to inflammation sites. Nat Biotechnol (2003) 21(11):1314–20. doi:10.1038/nbt888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
246. Coulstock E, Sosabowski J, Ovecka M, Prince R, Goodall L, Mudd C, et al. Liver-targeting of interferon-alpha with tissue-specific domain antibodies. PLoS One (2013) 8(2):e57263. doi:10.1371/journal.pone.0057263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
247. Frey K, Zivanovic A, Schwager K, Neri D. Antibody-based targeting of interferon-alpha to the tumor neovasculature: a critical evaluation. Integr Biol (Camb) (2011) 3(4):468–78. doi:10.1039/c0ib00099j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
248. Huang TH, Chintalacharuvu KR, Morrison SL. Targeting IFN-alpha to B cell lymphoma by a tumor-specific antibody elicits potent antitumor activities. J Immunol (2007) 179(10):6881–8. doi:10.4049/jimmunol.179.10.6881
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
249. Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, et al. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell (2014) 25(1):37–48. doi:10.1016/j.ccr.2013.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
250. Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood (2010) 115(14):2864–71. doi:10.1182/blood-2009-10-250555
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
251. Ji C, Sastry KS, Tiefenthaler G, Cano J, Tang T, Ho ZZ, et al. Targeted delivery of interferon-alpha to hepatitis B virus-infected cells using T-cell receptor-like antibodies. Hepatology (2012) 56(6):2027–38. doi:10.1002/hep.25875
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
252. Slutzki M, Jaitin DA, Yehezkel TB, Schreiber G. Variations in the unstructured C-terminal tail of interferons contribute to differential receptor binding and biological activity. J Mol Biol (2006) 360(5):1019–30. doi:10.1016/j.jmb.2006.05.069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
253. McBride JM, Jiang J, Abbas AR, Morimoto A, Li J, Maciuca R, et al. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum (2012) 64(11):3666–76. doi:10.1002/art.34632
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
254. Merrill JT, Wallace DJ, Petri M, Kirou KA, Yao Y, White WI, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann Rheum Dis (2011) 70(11):1905–13. doi:10.1136/ard.2010.144485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
255. Pan M, Kalie E, Scaglione BJ, Raveche ES, Schreiber G, Langer JA. Mutation of the IFNAR-1 receptor binding site of human IFN-alpha2 generates type I IFN competitive antagonists. Biochemistry (2008) 47(46):12018–27. doi:10.1021/bi801588g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: type I interferons, dendritic cells, chronic viral infections, immunotherapy, bioengineering
Citation: Tomasello E, Pollet E, Vu Manh T-P, Uzé G and Dalod M (2014) Harnessing mechanistic knowledge on beneficial versus deleterious IFN-I effects to design innovative immunotherapies targeting cytokine activity to specific cell types. Front. Immunol. 5:526. doi: 10.3389/fimmu.2014.00526
Received: 23 June 2014; Accepted: 07 October 2014;
Published online: 30 October 2014.
Edited by:
Yoichi Furuya, Albany Medical College, USAReviewed by:
Christine Anne Biron, Brown University, USAAlan Chen-Yu Hsu, The University of Newcastle, Australia
Danushka Kumara Wijesundara, The Basil Hetzel Institute, Australia
Copyright: © 2014 Tomasello, Pollet, Vu Manh, Uzé and Dalod. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc Dalod, Centre d’Immunologie de Marseille-Luminy, Parc scientifique et technologique de Luminy, case 906, F-13288 Marseille Cedex 09, France e-mail:ZGFsb2RAY2ltbC51bml2LW1ycy5mcg==
†Elena Tomasello, Emeline Pollet and Thien-Phong Vu Manh have contributed equally to this work.