 Avinash Sheshachalam
Avinash Sheshachalam Nutan Srivastava
Nutan Srivastava Troy Mitchell
Troy Mitchell Paige Lacy
Paige Lacy Gary Eitzen
Gary Eitzen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 September 2014
Sec. Molecular Innate Immunity
Volume 5 - 2014 | https://doi.org/10.3389/fimmu.2014.00448
This article is part of the Research Topic Secretion of Cytokines and Chemokines by Innate Immune Cells View all 12 articles
Neutrophils are part of a family of granulocytes that, together with eosinophils and basophils, play an essential role in innate immunity. Neutrophils are the most abundant circulating leukocytes and are vital for rapid immune responses, being recruited to sites of injury or infection within minutes, where they can act as specialized phagocytic cells. However, another prominent function of neutrophils is the release of pro-inflammatory compounds, including cytokines, chemokines, and digestive enzymes, which are stored in intracellular compartments and released through regulated exocytosis. Hence, an important feature that contributes to rapid immune responses is capacity of neutrophils to synthesize and store pre-formed pro-inflammatory mediators in specialized intracellular vesicles and thus no new synthesis is required. This review will focus on advancement in three topics relevant to neutrophil secretion. First, we will examine what is known about basal level pro-inflammatory mediator synthesis, trafficking, and storage in secretory compartments. Second, we will review recent advancements in the mechanisms that control vesicle mobilization and the release of pre-formed mediators. Third, we will examine the upregulation and de novo synthesis of pro-inflammatory mediators by neutrophils engaged at sites of infection.
Neutrophils are the most abundant leukocytes in blood, comprising 60–70% of all circulating white blood cells, and therefore, make up an essential part of both innate and adaptive immunity. Neutrophils are highly mobile and provide rapid responses to infection via phagocytosis of pathogens or release of potent pro-inflammatory mediators that chemically incapacitate pathogens and infected cells. Neutrophils are also recruited to the site of injury following trauma, resulting in an acute inflammatory response. Their short-life span allows for quick resolution of inflammation and can promote wound healing through stimulation of local tissue remodeling and chemoattraction of macrophage (1). Neutrophil immunity function depends on both the biogenesis of granules that store proteins with active antimicrobial activity, and on their ability to generate oxidative burst. Defects in either of these two processes results in severe immunodeficiency such as neutropenia, neutrophil-specific granule deficiencies, or chronic granulomatous disease when oxidative burst is lacking (2). In this review, we will consider the protein processing events that occur in neutrophils, resulting in their initial granulocytic morphology, and protein transitions that occur to activate neutrophil “attack” mode.
Mature neutrophils are differentiated from hematopoietic stem cells in the bone marrow in a process termed granulocytopoiesis or granulopoiesis (3). New neutrophils are produced daily in high numbers, up to 1011 in healthy individuals, which can increase several fold during infection (4). Consequently, neutrophil homeostasis is highly regulated to control their numbers. Granulocyte-colony-stimulating factor (G-CSF) is the principal cytokine regulating granulocytopoiesis. Lack of G-CSF production impairs granulocytopoiesis, resulting in neutropenia and severe immune deficiency (5, 6). Hematopoietic stem cell differentiation into granulocytes is regulated by the coordinated expression of three key myeloid transcription factors, GFI-1, PU.1, and members of the CCAAT enhancer binding protein family (C/EBPs). The balance between PU.1 and C/EBPα determines whether myeloblasts differentiate into granulocytes (high C/EBPα) or monocytes (high PU.1) (7).
The chemokines CXCL12 and CXCL2 and their neutrophil receptors, CXCR4 and CXCR2, respectively, regulate the release of neutrophils from the bone marrow. CXCR4–CXCL12 signaling promotes bone marrow retention, while CXCL2–CXCR2 promotes their release in what has been described as a tug of war model. The contribution of these chemokine signaling pathways to the regulation of neutrophil trafficking from bone marrow has been recently reviewed (8).
Neutrophils have been developed from human embryonic stem cells (hESCs) in culture. High yields were achieved by growing hESC in co-culture with semi-confluent OP9 stromal cells to stimulate hESC differentiation (9). The OP9 stromal cell line, which is derived from the osteopetrotic mouse, was specifically selected since they lack production of macrophage-colony stimulating factor (M-CSF) and thus support neutrophil rather than macrophage differentiation (10, 11). The procedure was further improved using a unique cytokine cocktail to yield highly functional neutrophils capable of chemotaxis, phagocytosis, oxidative burst, and bacterial killing (12, 13). Recently, protocols have been reported that differentiated neutrophils can be prepared from induced pluripotent stem cells (iPSCs) (14–16). These are significant advancements since the ability to obtain functional neutrophils from hESCs, iPSCs, and even patient-derived iPSCs may progress toward their eventual use in the treatment of hematopoietic disorders (17, 18).
Mature neutrophils emerge in the blood devoid of any proliferative capacity, but fully capable of launching an immune response. Ultrastructural images of mature neutrophils reveal a characteristic multi-lobed nucleus, very few mitochondria, a small Golgi structure, and a highly granular cytosol that is packed with vesicles (Figure 1A). The presence of different granule subsets is revealed by their varying size and intensity of stained with the peroxidase-reactive dye, 3,3′-diaminobenzidine (Figure 1B). Granule subsets are also distinguished by their protein content and their propensity for mobilization, which will be further discussed in following sections (19–21).
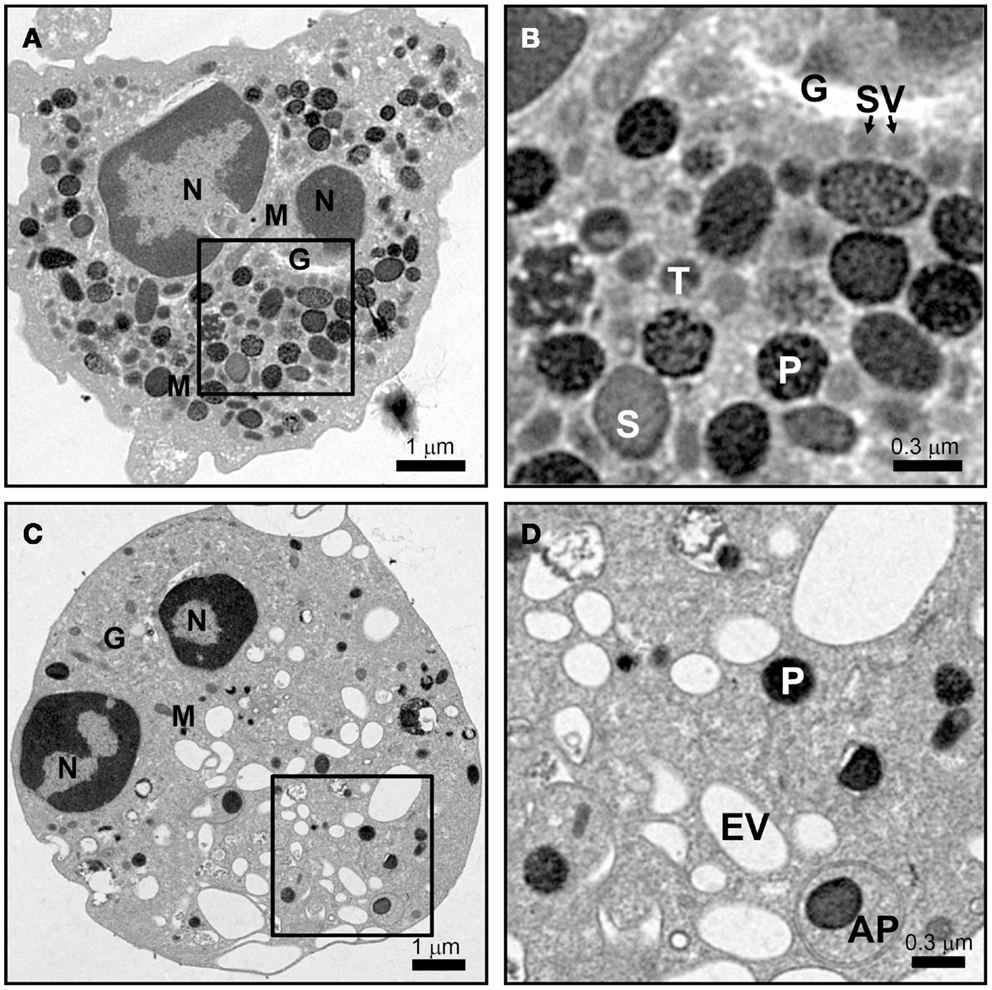
Figure 1. Neutrophil morphology as observed by electron microscopy. Neutrophils were isolated from human peripheral blood, and processed for electron microscopy after incubation for 15 min at 37°C with vehicle (A,B) or 10 μM cytochalasin B and 5 μM fMLF (C,D) as described in Ref. (22). (A,B) The cytosol of a resting cell is filled with vesicles, with primary granules (P) staining intensely dark with DAB, while secondary (S) and tertiary (T) granules show more translucent staining. Secretory vesicles (SV) are in close proximity to the Golgi complex (G). Few mitochondria (M) are observed. (C,D) After stimulation only a few dense primary granules (P) remain and many empty vesicles (EV) appear. A double membrane autophagosome (AP) was observed to form around a primary granule; however, the relevance of this observation is unknown. Scale (A,C) = 8 μm × 8 μm, 9,100× magnification; (B,D) = 2.4 μm × 2.4 μm, 27,600× magnification.
The difference in protein contents among neutrophil granule subsets is not driven by protein sorting. Instead, different granules are sequentially formed during neutrophil differentiation in what has been described as a targeting by timing model (23). According to this model, as different granule proteins are synthesized during different stages of neutrophil differentiation several granule subsets are generated (24–26). Neutrophils have at least three distinct granule subsets: (i) primary or azurophilic granules, which contain potent hydrolytic enzymes (e.g., elastase) and myeloperoxidases (MPO), (ii) secondary or specific granules, which contain high levels of the iron-binding protein lactoferrin, and (iii) tertiary or gelatinase granules, which contain matrix metalloproteinases (Figure 1B). Secondary and tertiary granules contain an overlapping set of proteins; however, all granules have distinctive buoyant densities and can be isolated by density gradient centrifugation (27). Recently, a fourth granule population has been described that was enriched in the microbial lectin ficolin-1. Ficolin-1 is found in tertiary granules; however, the authors found a second pool of ficolin-1-rich/gelatinase-poor granules with an elevated exocytosis propensity (21, 28). The importance of these granules may be to provide rapid release of pattern recognition molecules to activate the lectin complement pathway (28). Secretory vesicles (SV) also appear in density gradients that contain albumin indicative of their formation via endocytosis. SV can also be formed in mature neutrophils from a small Golgi structure (Figures 2A,B). These contain cytokines synthesized during immune activation indicative of an active biosynthetic pathway (29, 30), which will be discussed further in Section “Neutrophil De Novo Synthesis of Pro-Inflammatory Mediators.”
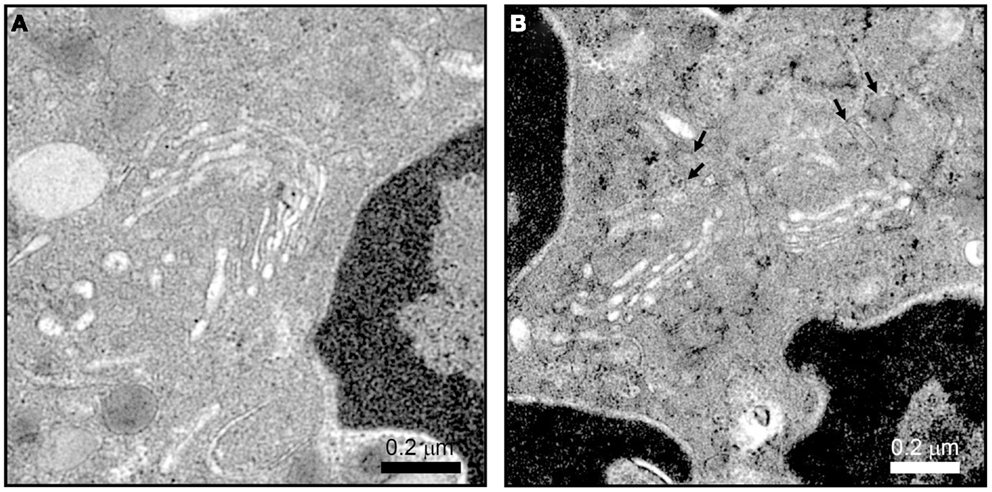
Figure 2. Neutrophil Golgi complex as observed by electron microscopy. Neutrophils processed for electron microscopy as described in Ref. (22) show a small stacked Golgi with an increase in electron dense vesicle (arrows) after stimulation. Conditions were incubation with vehicle (A) or 10 μM cytochalasin B and 5 μM fMLF (B) for 15 min at 37°C. Scale, 2.4 μm × 2.4 μm, 27,600×magnification.
Neutrophil granule proteins undergo a series of co- and post-translational processing and targeting events during sorting to their granule sub-compartment. Most, if not all, proteins packaged into neutrophil granules enter through the biosynthetic process as deduced by the presence of amino-terminal signal sequences for co-translational translocation into the endoplasmic reticulum (ER). The emergence of this “pre” sequence from the ribosome associates with the signal recognition particle, which drives co-translation into the lumen of the ER, followed by post-translational processing as proteins traverse the Golgi complex (e.g., protein maturation via glycosylation).
The precise mechanism by which proteins are targeted to neutrophil granules from the trans-Golgi network (TGN) is largely unknown, hampered by the fact that these cells are end-differentiated and do not synthesize granule proteins de novo. However, much information has been gained from analogous studies in other immune cells and the characterization of neutrophils from immunological disorders associated with sorting defects (31, 32). These studies confirm that neutrophils generate lysosome-like secretory granules both directly from the TGN and indirectly via a sorting endosomes (Figure 3). Post-TGN sorting is mediated by the multi-subunit adaptor protein complexes (APs) and the monomeric Golgi-localized γ-adaptin ear homology ARF binding (GGAs) protein. Both nucleate clathrin-coated vesicles and recruit luminal cargo for packaging into the vesicles (33). Three of the five known AP complexes function at the TGN and are likely involved in granule sorting since they have prominent roles in lysosomal sorting in other cell types (33, 34). AP-1 and GGAs direct the bulk sorting program from the TGN to sorting endosomes, while AP-3 and AP-4 shuttle cargo to granules from the sorting endosomes or directly from the TGN, bypassing the sorting endosomes (35). This distinction in pathways serves an important mechanistic role since soluble cargo utilizes receptors that require a dissociation step catalyzed in endosomes. This allows the receptor to be sorted back to the TGN while the cargo is transported into granules (Figure 3).
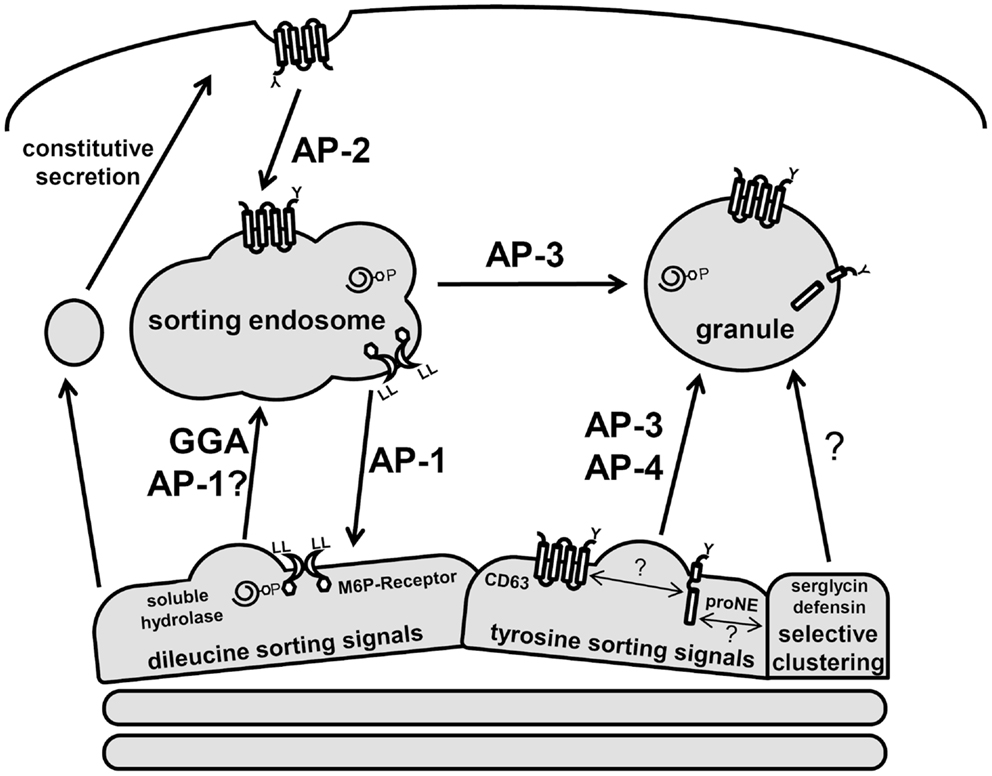
Figure 3. Schematic of neutrophil protein sorting pathways. Three prominent trafficking pathways for granule proteins are depicted: AP-1/GGA-dileucine based sorting, AP-3/AP-4-tyrosine-based sorting, and selective clustering-based sorting. Soluble hydrolases contain M6P signals and are sorted via the M6P-receptors, which contain dileucine signals. These complexes are shuttle to the sorting endosome through GGA and AP-1-dependent pathways. In the endosome, a drop in pH results in dissociation of the receptor–cargo complex with the retrieval of the M6P-receptor by an AP-1 dependent pathway. Serine proteases (neutrophil elastase, proNE) and trans-membrane proteins (CD63) contain tyrosine signals and are sorted directly to granules via AP-3/AP-4. Many granule proteins contain neither dileucine nor tyrosine signals and are thought to sort to granules via a clustering-based pathway since this occurs for many granule proteins that are highly concentrated. CD63 trafficking is unique as it can be found on several neutrophil membranes and may act to selectively retrieve granule proteins that escape normal trafficking via constitutive secretion.
The sorting of granule membrane proteins, including cargo receptors, depends on dileucine or tyrosine-based sorting signals present in carboxy-terminal cytosolic domains (35). The consensus dileucine sequences, DXXLL or [DE]XXXL[LI], and tyrosine sequence, [G]YXXØ (where X represents hydrophobic residues and Ø a bulky hydrophobic amino acid), interact with GGAs/AP-1 and AP-3/AP-4 complexes, respectively.
The majority of soluble hydrolases and granzymes are modified with mannose-6-phosphate (M6P) in the Golgi, which is recognized by M6P-receptors to facilitate their exit from the TGN (33). Unlike hydrolases and granzymes, many soluble granule proteases are sorted via M6P-independent mechanisms (31, 36). Targeting of the major serine protease, neutrophil elastase, as well as other proteases requires AP-3 (37). Neutrophil elastase contains a tyrosine on the cytosolic face of it trans-membrane pro-domain, which interacts with AP-3 presumably for its sorting (38). However, neutrophil elastase mutants that lack a pro-domain still get sorted to granules via a CD63-dependent mechanism (39). CD63, an abundant membrane protein of primary granules, is sorted by AP-3, and has been shown to play a role in granule protein quality control since it can be trafficked from many membranes to granules (40, 41). A recent study showed that CD63 and neutrophil elastase are present in a complex, highly indicative of a role for CD63 in the targeting of elastase to primary granules (42). Other serine proteases affected by CD63 granule sorting include proteinase-3 (39), and possibly the cathepsin family of proteases, which exhibit varying degrees of M6P-dependent/independent sorting (36, 43–45).
Neutrophils, as well as other granulocytes, may have acquired additional unique granule sorting mechanisms based on the fact that proteins in some granules can be tightly packed to the point of having a crystalline core. A selective aggregation of proteins destined for storage in granules would eliminate the need for distinct sorting motifs on each granule protein (24). Granule proteins, which are often cationic, may be clustered with serglycin, a major anionic proteoglycan of hematopoietic cells. Serglycin has been proposed to play a role in sorting and packing of several granule proteins including α-defensin and elastase (46, 47). Neutrophil elastase is absent from mature neutrophils in serglycin knock-out mice (48). Whether serglycin-induced clustering of granule proteins promotes granule localization via sorting or their retention in granules is still under investigation (49).
Most granule enzymes are synthesized as zymogens and undergo a proteolytic processing step that depends on the acidic pH levels within this lysosome-like compartment. The proteolytic cleavage of granule pro-domains converts these enzymes into their active forms and thus many granule proteins are maintained inactive until the proper compartment is reached. Some pro-domains have also been shown to have a role in sorting. The amino-terminal pro-peptide of pro-MPO has been shown to facilitate its targeting to primary granules (50). Unlike MPO, serine proteases (e.g., elastase and cathepsin G) are synthesized with both amino-terminal and carboxy-terminal pro-peptides that do not seem to be involved in their sorting (51).
Granule protein trafficking disorders often manifest as lysosomal storage disorders (LSDs), which are a large group of metabolic diseases that result from deficiencies in specific lysosomal enzymes or defects in lysosome biogenesis (31, 52). For instance, defects in the ability to generate M6P sorting signals in the Golgi result in the LSDs mucolipidosis II (also referred to as I-cell disease) and mucolipidosis III. Mutations in GlcNAc-phosphotransferase are believed to be the primary genetic defect in mucolipidosis. GlcNAc-phosphotransferase activity is absent in MLII and altered in MLIII (53). Studies of patients with mucolipidosis revealed neutropenia, with a major reduction in the number of neutrophils in MLII, but less severe in MLIII patients (53, 54). There have been no reports of neutrophil function studies from these patients, but presumably granules would lack most soluble hydrolases since these are sorted by the M6P pathway. However, studies of B lymphocytes showed that a portion of lysosomal enzymes were retained in granules with an unknown a back-up sorting event facilitating this partial sorting (36). The recent development of a mucolipidosis II mouse model might advance neutrophil functional characterizations (55, 56). These studies could reveal the prominence of M6P-independent sorting pathways in neutrophils and whether these can facilitate the partial retention of enzymes normally sorted by M6P.
Hermansky–Pudlak syndrome type 2 is also a LSD disorder that results from a mutation in AP3B1, which encodes a subunit of the AP-3 complex. The type 2 form includes immunodeficiency associated with neutropenia and partial albinism. The basis of this disease was first discovered as a similar autosomal recessive disease of dogs, canine cyclic hematopoiesis (38). The lack of AP-3 sorting resulted in reduced levels of neutrophil elastase and gelatinase in granules, but normal levels of other enzymes such as MPO and proteinase-3, which reside in the same granule fraction as elastase (57). This reveals the divergence in sorting pathways in neutrophils; AP-3 for neutrophil elastase versus GGA/AP-1 for MPO. Interestingly, AP-3 deficiency leads to congenital neutropenia in humans and cyclic neutropenia in dogs (38, 58), even though both are linked to the lack of neutrophil elastase sorting. Neutrophil elastase mutations cause a much more severe congenital neutropenia phenotype in humans (59). It has been suggested that increased apoptosis of myeloid precursors in patients carrying mutations in the elastase gene could lead to a maturation arrest of myelopoiesis and this triggers the more severe neutropenia phenotype (60).
Although neutrophil elastase is a soluble protein, there is evidence that in an intermediate stage of its processing it is a disulfide bonded trans-membrane protein (38). Upon enzymatic removal of the carboxy-terminal pro-domain, the protein maintains a trans-membrane conformation capable of associating with AP-3 complexes. Membrane-associated neutrophil elastase utilizes a tyrosine-based sorting signal that is located in the carboxy-terminal domain of the protein. Most elastase mutations associated with severe congenital neutropenia result in the removal of this sorting signal (38). Horwitz et al. have proposed that the localization of the enzyme in the lumen of granules or on the plasma membrane may regulate the differentiation pattern of myeloid progenitor cells into monocytes or granulocytes. This suggests that neutropenia may result from a defective differentiation switch between the monocytic and granulocytic lineage of myelopoiesis (61, 62). This is an appealing hypothesis that may account for the observation that granulocytes and monocytes reciprocally cycle in numbers during normal hematopoiesis and for the typical increase of monocyte blood counts observed in the majority of severe congenital neutropenia patients. Hence, not only granule sorting pathways are important for granule biogenesis but also in determining the population of cell types during hematopoiesis.
Three functions that involve protein processing occur in neutrophils when activated for an immune response: (1) neutrophil migration up chemotactic gradients to sites of infection, (2) destruction of pathogens or injured/infected cells through oxidative burst and phagocytosis, and (3) mobilization of granules and release of inflammatory mediators. In this section, we review these functions with a particular focus on the mobilization of granules and the release of pre-formed inflammatory mediators.
Upon activation, neutrophils migrate out of blood vessels following gradients of chemoattractants in a process known as chemotaxis. Neutrophils respond to a wide range of “neutrophil-active” chemoattractants, including chemokines and cytokines (CXCL8, IFNγ), complement (C5a), eicosanoids (LTB4), and pathogen-derived peptides such as formylated met–leu–phe (fMLF) (63, 64). Chemotaxis begins with neutrophil recruitment from circulation through physical interactions of neutrophil-specific adhesion molecules. The neutrophil adhesion cascade is the focus of recent reviews and therefore only briefly described here (65, 66). Neutrophils are initially tethered by selectin interactions, which can be upregulated on the surface of endothelial cells (E-selectin) and leukocytes (L-selectin). The lectin-like domains of selectins interact with sialylated carbohydrate groups present on surface proteins. This results in the characteristic rolling of neutrophils along the luminal side of the endothelium. Reduced velocity allows neutrophils to recognize chemokines on the surface of the endothelium, which activates integrins. Neutrophils will be released from low-affinity selectin interactions unless firm adhesive contacts are made between integrins and endothelial intracellular adhesion molecules (ICAMs). Integrin ligation triggers intracellular signaling for cytoskeletal rearrangement, which polarizes neutrophils and drives transmigration through the endothelium (diapedesis). Integrin signaling is essential as deficiencies result in leukocyte adhesion deficiency, which not only causes defects in chemotaxis and adhesion but also in phagocytosis and respiratory burst (2).
Regulation of the adhesion and chemotaxis requires the coordination of multiple protein signaling and processing events, which result in cytoskeletal rearrangements that generate cell polarity (67). The activation of distinct Rho proteins at the front (Rac1) and back (RhoA) of neutrophils create actin formations that drive movement (68). Rho proteins are central regulators of multiple intracellular processes, and it is interesting how signaling downstream from Rho has evolved to coordinate cytoskeletal remodeling in conjunction with other processes required for immune cell functions. These include many non-cytoskeletal-related functions such as regulation of gene transcription (69–72), calcium flux (73, 74), and oxidative burst (75–77).
Chemokines interact with G protein-coupled receptors (GPCRs) on the surface of neutrophils. One pathway activated by GPCRs is the lipid kinase pathway. PI3-kinase is activated, which converts phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] into phosphatidylinositol-3,4,5-triphosphate [PI(3,4,5)P3] at the plasma membrane. PI(3,4,5)P3 stimulates F-actin reorganization at the leading edge driving lamellipodia formation. The Rac GEFs, Vav1, and P-Rex1, bind PI(3,4,5)P3 via pleckstrin homology domains, activating Rac, which leads to the production of branched actin filaments at the leading edge (78–80). High levels of the phosphatase PTEN are present in the trailing edge of neutrophils, which consumes PI(3,4,5)P3. Thus, a gradient of PI(3,4,5)P3 is established toward the leading edge, which promotes polarization and directional motility (81). A recent report examined neutrophils derived from a conditional PTEN knock-out and showed it not only functions in polarization but also prioritizes chemoattractant signals. Normal neutrophils were shown to prefer bacterial chemoattractants over endogenous chemokines, but this preferential selection was lost in the PTEN knock-out (82).
Opposing roles for the two Rho proteins, RhoA and Cdc42, in the trailing edge (or uropod) were shown by examining neutrophils devoid of these proteins. In the absence of RhoA, neutrophil priming was unregulated, resulting in hyper-responsive activation and uncoordinated motility (83). Cdc42, on the other hand, was required specifically to maintain neutrophil polarity (84). WASp, a downstream Cdc42 effector, regulated polarity through the CD11b integrin, which recruits the microtubule end binding protein, EB1, to capture and stabilize microtubules at the uropod. This would allow the generation of “pushing” forces from the back of neutrophils. Cross-talk between Rho proteins has been an area of intense investigation (68) and these results show how Rho/Cdc42 signaling cross-talking can coordinately control neutrophil chemotaxis while at the same time minimize other immunity functions such as degranulation.
Neutrophil phagocytosis has been the focus of several reviews (85–87). For the purpose of this review, we will limit our consideration to the protein processes involved in NADPH oxidase assembly and how this is directed primarily to the phagosomal membrane.
The rapid immunological response of neutrophils not only depends partly on pre-formed granule proteins with antimicrobial activity but also on the ability to generate respiratory burst activity. Deficiency in respiratory burst results in a severe immunodeficiency called chronic granulomatous disease (2). Chronic granulomatous disease is linked to genetic abnormalities in any one of the five “phox” proteins, which comprise the NADPH oxidase complex. The small GTPase Rac is a sixth component that is physically part of the oxidase complex that regulates its function (75).
The NADPH oxidase complex generates reactive oxygen species (ROS) that are crucial for antimicrobial activity and antigen processing. However, excessive ROS production causes tissue damage and oxidative stress and therefore neutrophils must precisely control both the location and timing of NADPH oxidase activity. In unstimulated neutrophils, three oxidase subunits, p47-phox, p67-phox, and p40-phox, are cytosolic. Rac2 is also cytosolic, bound to its natural inhibitor RhoGDI. The remaining two subunits, gp91-phox and p22-phox, form the membrane-spanning catalytic core of cytochrome b558. Formation of the holoenzyme allows electrons to follow from NADPH bound at the cytosolic face of the complex, to FAD-bound cytochrome b558, which has two heme-bound oxygen groups that accept the electrons and produce superoxide.
The oxidase complex can be preferentially incorporated into the phagosome during pathogen endocytosis, which minimizes collateral tissue damage. However, residual oxidase complex at the plasma membrane or its activation in the absence of phagocytosis results in the release of ROS extracellularly. Three regulatory steps may facilitate the phagosomal-predominant spatial activation of the oxidase complex. p47-phox phosphorylation leads to a conformational change that exposes two amino-terminal SH3 domains, which interact with the proline-rich region of the membrane-bound p22-phox (88). Assembly that occurs on the plasma membrane is susceptible to in rapid dephosphorylation, resulting in unproductive oxidase assembly. Second is the regulation of the p40-phox subunit by PI3-kinase. This effect is mediated by the PX domain of p40-phox binding to PI(3)P, which results in an open conformation allowing NADPH access to the catalytic core (89, 90). PI(3)P is enriched on phagosome membranes, which facilitates the spatial restriction of oxidase activity to the phagosome rather than at the plasma membrane where the predominant phosphoinositol lipids are PI(4,5)P2 and PI(3,4,5)P3. A third regulatory step involves Rac. Rac is activated by guanine nucleotide exchange factors (GEFs) from the Vav or P-Rex families (78–80, 91). The function of these GEFs depend on membrane recruitment via increased levels of PI(3,4,5)P3 (92). However, this create a spatial paradox since PI(3,4,5)P3 is localized to the plasma membrane and not phagosomes. This is resolved by the oxidase complex showing preference for assembly with Rac2 over Rac1 (91, 93, 94). While Rac1 is predominantly localized to the plasma membrane, Rac2 has less affinity for the plasma membrane, likely due to fewer polybasic residues in its carboxy-terminus (95). This may result in spontaneous disassembly from the oxidase complex in the presence of high levels of PI(4,5)P2 and PI(3,4,5)P3, unless rapidly endocytosis occurs and the highly acidic phosphoinositides are metabolized.
Interestingly, the Rac GEF Vav has also been shown to affect oxidase assembly through a secondary affect involving the p40-phox subunit (96). This may involve other downstream effectors of Rac, which includes the serine kinase PAK1, which has been shown to phosphorylation p47-phox (97). In addition, RhoA has recently been shown to downregulate ROS production (98). The RhoA signaling pathway was linked to cytoskeletal remodeling, which would provide for an elegant way to downregulate oxidase activity during chemotaxis. Further studies of the convergence of multiple signaling pathways will be needed to reveal the full control of oxidase activity.
Neutrophils, upon stimulation undergo a series of immediate changes without the need for de novo synthesis of proteins (compare Figures 1A and 1B vs. 1C and 1D). Exocytosis, also known as degranulation in neutrophil, is the release pre-formed mediators from granules. Granule subsets are markedly different in their capacity for mobilization in response to stimulation (99, 100). Granules formed during the later stages of granulocytopoiesis are more prone to undergo exocytosis than granules formed during the earlier stages. A recent study reported exocytosis levels of 100% for SV, 38% for tertiary granules, 22% for secondary granules, and only 7% for primary granules after stimulation (28). The specific steps of exocytosis involve granule translocation toward a target membrane via actin remodeling and microtubule assembly, followed by tethering and docking through the sequential action of the core fusion machinery of Rab and SNARE proteins (Figure 4) (101, 102).
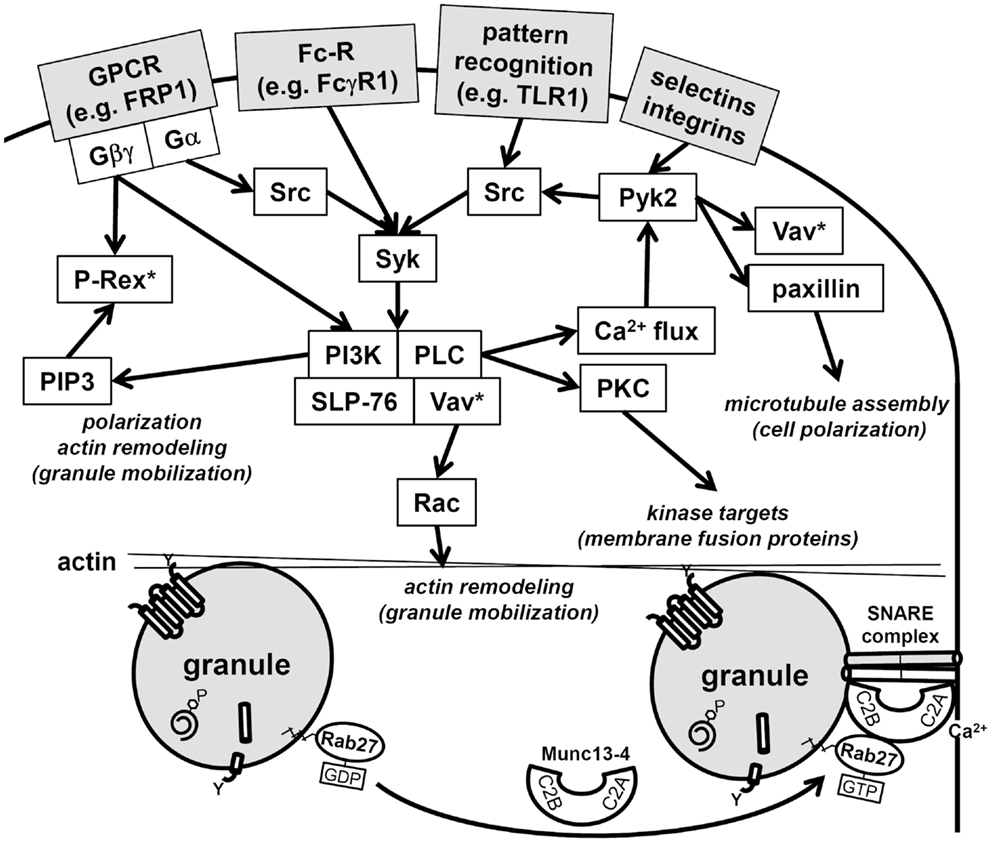
Figure 4. Schematic of neutrophil signaling pathways regulating degranulation. Two pathways that regulate granule mobilization are depicted: upstream kinase cascade and downstream fusion machinery. Activation of neutrophils through surface receptors triggers the activation of a kinase cascade. Central downstream effectors of these kinases target cytoskeletal remodeling, these include Vav, which activates Rac, paxillin, which facilitates microtubule polarization, and the generation of PI(3,4,5)P3, which facilitates polarization and actin remodeling. Note that Rac is activated at multiple points in the signaling pathways (Vav*, P-Rex*) and hence may be needed for several immune cell functions in addition to degranulation. Granule tethering is regulated by Rab27 recruitment of Munc13-4. Munc13-4 is a calcium-sensitive link between Rab function and the fusion machinery of SNAREs. The Munc13-4:SNARE interaction requires calcium flux and is targeted by PKC, which are activated in the upstream kinase cascade.
Neutrophil granule contents, which include MPO, elastase, lactoferrin, and matrix metalloproteinases, possess potent antimicrobial activity but are also highly cytotoxic. Therefore, their release is highly regulated by binary signals to minimize aberrant degranulation. The first of these binary signals is adhesion-dependent while the second involves activation of immune receptors by ligand interactions. The adhesion-dependent step in vivo involves β2-integrins (100), which can be reconstituted in vitro by adhesion to biological surfaces or the addition of actin depolymerizing agents (22, 103, 104). The adhesion-dependent signaling cascade for degranulation operates through the Src kinases Fgr and Hck. Neutrophils from double knock-out hck−/− fgr−/−, while still able to adhere to substrate, fail to undergo degranulation in response to tumor necrosis factor (TNF) (105). However, Src kinases are well known to be membrane proximal regulators of neutrophil degranulation during receptor-mediated activation, which comprises the second half of the binary signal (106, 107). For example, degranulation requires immune receptor–ligand interactions such as formylated peptides binding to GPCRs. This also triggers the activation of Src kinases, and hence it is curious why binary signals are needed that seemingly activate the same signaling cascade. Binary signals may be needed to reach an activation threshold for degranulation or perhaps distinct downstream pathways are activated. One downstream pathway specifically activated by the receptor-mediated kinase cascade is Ca2+ release from intracellular stores (107). Increasing concentrations of Ca2+ are responsible for the hierarchical release of neutrophil granules in the order of secretory vesicles > tertiary granules > secondary granules > primary granules (99).
Cross-talk between the binary degranulation signals makes it difficult to clearly define each signaling pathways. For example, recently a role for the highly abundant non-receptor tyrosine kinase, proline-rich kinase 2 (Pyk2), was described for degranulation (108). Pyk2 undergoes auto-phosphorylation after integrin ligation in calcium-dependent manner, allowing its association with the Src-family kinases. Furthermore, studies with pyk2−/− cells have shown its effect is mediated through paxillin and Vav, which are both phosphorylated by Pyk2 (108). Vav is a direct activator of Rac, which remodels actin to facilitate degranulation (78), while paxillin serves as an important scaffolding protein and has a direct effect on microtubule assembly, which may contribute to polarized granule motility (109). Thus, Pyk2 signaling may be central to the transduction of multiple upstream signals into downstream granule mobilization and degranulation (Figure 4).
Downstream events are regulated by distinct core fusion machinery that drives granule docking and exocytosis at specific target membranes. Initially, Rab proteins tether vesicles to target membranes, then SNARE proteins catalyze fusion (102). Neutrophil granules fuse with target membranes when v-SNARES pair with their corresponding t-SNARES. VAMP-2 is predominantly localized to SV and tertiary granules, whereas VAMP-7 is predominantly localized to primary granules (110, 111). It seems that the membrane density of VAMP-2 provides a functional role in mobilization, docking, and fusion to the plasma membrane, while VAMP-7 redirects vesicles to fuse predominantly with phagosomes (110, 112).
Numerous additional regulatory proteins interact with the core fusion machinery of Rabs and SNAREs. Among these, the Munc family of proteins is important regulators of SNARE complex formation (101). Munc13-4 (also known as UNC13D) is highly expressed in neutrophils and interacts with Rab27, which is the Rab that direct granules to dock at the plasma membrane. Munc13-4 tethers granules to the plasma membrane for exocytosis via two calcium-sensitive lipid bind C2 domains (Figure 4) (113). Neutrophil subfractionation revealed that high levels of Munc13-4 rapidly translocate from the cytosol to granules upon stimulation with a secretagogue like fMLF (114). In this study, Munc13-4 was shown to associate with secondary and tertiary granules and down-regulation of Munc13-4 using small interfering RNA decreased their exocytosis (114). Recently, STK24 and CCM-3, two regulators of neutrophil exocytosis, were shown to interact with Munc13-4 (115). Lack of either STK24 or CCM-3 inhibited degranulation; however, they were shown to play opposing roles in the regulation of Munc13-4. Whereas STK24 inhibited Munc13-4 function by binding its C2B domain, CCM-3 counteracted STK24-mediated C2B inhibition (115). The STK24/CCM-3 complex seems provide an important additional control mechanism to halt aberrant degranulation.
Neutrophils are well characterized for their ability to synthesize and secrete over 70 different cytokines, chemokines, and growth factors. Although several of these have been characterized at the mRNA level only, 11 of these show controversial data for human neutrophils (116). The trafficking pathways that govern the synthesis, storage, and release of these factors are poorly understood. Several studies using immunogold staining analysis of transmission electron microscopy have revealed that neutrophils store pre-formed cytokines in secretory granules. Transforming growth factor-α (TGFα) is stored as a pre-formed mediator in secretory granules that are peroxidase-negative, suggesting localization to secondary or tertiary granules (117). Similarly, TNF-α was found as a pre-formed mediator in cytoplasmic vesicles following immunogold staining (118). However, the characteristics of these cytoplasmic vesicles were not further elucidated in this study. Other studies investigating cytokine and chemokine expression in neutrophils have indicated that mature peripheral blood neutrophils possess pre-formed IL-6, IL-12, and CXCL2 in their SV or tertiary granules (119).
In neutrophils, CD63 and CD68 are abundant membrane-bound proteins in primary and secondary granules, and both of these possess the YXXΦ motif, which interacts with AP-3/AP-4 complexes. AP-3 traffics cargo from tubular endosomes (recycling endosomes) to late endosomes, lysosomes, and related organelles, while AP-4 traffics protein from the TGN to endosomes or directly to lysosomes (33, 35). This indicates that CD63 and CD68 may be engaged in trafficking to primary and secondary granules through recycling endosomes and late endosomes. Conversely, cytokines that are secreted by neutrophils, including IL-1α/β, IL-6, CXCL8, IL-12, TNF, IFNγ, CXCL2, TGFβ, MIP-1α/β, and VEGF do not possess adaptor motifs, suggesting that they do not directly bind to adaptors in order to enter sorting pathways for their trafficking. Based on the absence of adaptor motifs in cytokines, it is likely that cytokine trafficking is mainly determined by adaptor motifs present in membrane proteins such as CD63 and CD68, which direct cytokine cargo to appropriate granular and vesicular compartments in cells.
Lipopolysaccharide (LPS) is major TLR agonist that is shed from the outer membranes of Gram-negative bacteria, which cause a significant burden of disease by inducing respiratory infections, gastrointestinal disorders, sepsis, pneumonia, and many other transmissible infections. LPS stimulation results in substantial production of TNF, a potent pro-inflammatory cytokine with cytotoxic and pyrogenic effects. Neutrophils stimulated by LPS have the ability to release TNF, potentially through granule stores (118, 120). However, the mechanisms used to secrete cytokines and chemokines has not been thoroughly investigated in neutrophils although it is evident that SNAREs and other trafficking machinery are required for exocytosis of cytokine and chemokine-carrying granules (110, 111, 121, 122).
Granule exocytosis and release of their cytotoxic contents at the plasma membrane is most often an undesirable effect of inflammation. We have highlighted many of the protein processes involved in pro-inflammatory mediator synthesis and granule biogenesis. Neutrophils possess several mechanisms that promote fusion of granules with phagosomes to minimize the effect of neutrophil-induced co-lateral tissue damage. However, in many inflammatory disorders, such as acute lung injury, ischemia/reperfusion injury, severe asphyxic asthma, rheumatoid arthritis, and septic shock, excessive neutrophil degranulation is a common feature. Insight into neutrophil protein processes holds much promise in the future treatment of these diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
2. Schäffer AA, Klein C. Animal models of human granulocyte diseases. Hematol Oncol Clin North Am (2013) 27:129–48. doi:10.1016/j.hoc.2012.10.005
3. Borregaard N. Neutrophils, from marrow to microbes. Immunity (2010) 33:657–70. doi:10.1016/j.immuni.2010.11.011
4. Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood (1991) 78:2791–808.
5. Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood (1994) 84:1737–46.
6. Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity (1996) 5:491–501. doi:10.1016/S1074-7613(00)80504-X
7. Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene (2007) 26:6816–28. doi:10.1038/sj.onc.1210764
8. Day RB, Link DC. Regulation of neutrophil trafficking from the bone marrow. Cell Mol Life Sci (2012) 69:1415–23. doi:10.1007/s00018-011-0870-8
9. Lieber JG, Webb S, Suratt BT, Young SK, Johnson GL, Keller GM, et al. The in vitro production and characterization of neutrophils from embryonic stem cells. Blood (2004) 103:852–9. doi:10.1182/blood-2003-04-1030
10. Kodama H, Nose M, Niida S, Nishikawa S. Involvement of the c-kit receptor in the adhesion of hematopoietic stem cells to stromal cells. Exp Hematol (1994) 22:979–84.
11. Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature (1990) 345:442–4. doi:10.1038/345442a0
12. Yokoyama Y, Suzuki T, Sakata-Yanagimoto M, Kumano K, Higashi K, Takato T, et al. Derivation of functional mature neutrophils from human embryonic stem cells. Blood (2009) 113:6584–92. doi:10.1182/blood-2008-06-160838
13. Saeki K, Saeki K, Nakahara M, Matsuyama S, Nakamura N, Yogiashi Y, et al. A feeder-free and efficient production of functional neutrophils from human embryonic stem cells. Stem Cells (2009) 27:59–67. doi:10.1634/stemcells.2007-0980
14. Morishima T, Watanabe K, Niwa A, Fujino H, Matsubara H, Adachi S, et al. Neutrophil differentiation from human-induced pluripotent stem cells. J Cell Physiol (2011) 226:1283–91. doi:10.1002/jcp.22456
15. Mukherjee S, Santilli G, Blundell MP, Navarro S, Bueren JA, Thrasher AJ. Generation of functional neutrophils from a mouse model of X-linked chronic granulomatous disorder using induced pluripotent stem cells. PLoS One (2011) 6:e17565. doi:10.1371/journal.pone.0017565
16. Weinacht KG, Brauer PM, Felgentreff K, Devine A, Gennery AR, Giliani S, et al. The role of induced pluripotent stem cells in research and therapy of primary immunodeficiencies. Curr Opin Immunol (2012) 24:617–24. doi:10.1016/j.coi.2012.07.001
17. Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood (2011) 118:4599–608. doi:10.1182/blood-2011-02-335554
18. Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood (2011) 117:5561–72. doi:10.1182/blood-2010-12-328161
19. Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood (1997) 89:3503–21.
20. Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Proteomic analysis of human neutrophil granules. Mol Cell Proteomics (2005) 4:1503–21. doi:10.1074/mcp.M500143-MCP200
21. Rørvig S, Østergaard O, Heegaard NH, Borregaard N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. J Leukoc Biol (2013) 94:711–21. doi:10.1189/jlb.1212619
22. Mitchell T, Lo A, Logan MR, Lacy P, Eitzen G. Primary granule exocytosis in human neutrophils is regulated by Rac-dependent actin remodeling. Am J Physiol Cell Physiol (2008) 295:C1354–65. doi:10.1152/ajpcell.00239.2008
23. Le Cabec V, Cowland JB, Calafat J, Borregaard N. Targeting of proteins to granule subsets is determined by timing and not by sorting: the specific granule protein NGAL is localized to azurophil granules when expressed in HL-60 cells. Proc Natl Acad Sci U S A (1996) 93:6454–7. doi:10.1073/pnas.93.13.6454
24. Gullberg U, Andersson E, Garwicz D, Lindmark A, Olsson I. Biosynthesis, processing and sorting of neutrophil proteins: insight into neutrophil granule development. Eur J Haematol (1997) 58:137–53. doi:10.1111/j.1600-0609.1997.tb00940.x
25. Cowland JB, Borregaard N. The individual regulation of granule protein mRNA levels during neutrophil maturation explains the heterogeneity of neutrophil granules. J Leukoc Biol (1999) 66:989–95.
26. Theilgaard-Mönch K, Jacobsen LC, Borup R, Rasmussen T, Bjerregaard MD, Nielsen FC, et al. The transcriptional program of terminal granulocytic differentiation. Blood (2005) 105:1785–96. doi:10.1182/blood-2004-08-3346
27. Udby L, Borregaard N. Subcellular fractionation of human neutrophils and analysis of subcellular markers. Methods Mol Biol (2007) 412:35–56. doi:10.1007/978-1-59745-467-4_4
28. Rørvig S, Honore C, Larsson LI, Ohlsson S, Pedersen CC, Jacobsen LC, et al. Ficolin-1 is present in a highly mobilizable subset of human neutrophil granules and associates with the cell surface after stimulation with fMLP. J Leukoc Biol (2009) 86:1439–49. doi:10.1189/jlb.1008606
29. Lapinet JA, Scapini P, Calzetti F, Pérez O, Cassatella MA. Gene expression and production of tumor necrosis factor alpha, interleukin-1beta (IL-1beta), IL-8, macrophage inflammatory protein 1alpha (MIP-1alpha), MIP-1beta, and gamma interferon-inducible protein 10 by human neutrophils stimulated with group B meningococcal outer membrane vesicles. Infect Immun (2000) 68:6917–23. doi:10.1128/IAI.68.12.6917-6923.2000
30. Theilgaard-Mönch K, Knudsen S, Follin P, Borregaard N. The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol (2004) 172:7684–93. doi:10.4049/jimmunol.172.12.7684
31. Neufeld EF, Lim TW, Shapiro LJ. Inherited disorders of lysosomal metabolism. Annu Rev Biochem (1975) 44:357–76. doi:10.1146/annurev.bi.44.070175.002041
32. Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: the secrets of secretory lysosomes. Science (2004) 305:55–9. doi:10.1126/science.1095291
33. Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta (2009) 1793:605–14. doi:10.1016/j.bbamcr.2008.10.016
34. Hirst J, Barlow LD, Francisco GC, Sahlender DA, Seaman MN, Dacks JB, et al. The fifth adaptor protein complex. PLoS Biol (2011) 9:e1001170. doi:10.1371/journal.pbio.1001170
35. Nakatsu F, Ohno H. Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell Struct Funct (2003) 28:419–29. doi:10.1247/csf.28.419
36. Glickman JN, Kornfeld S. Mannose 6-phosphateindependent targeting of lysosomal enzymes in I-cell disease B lymphoblasts. J Cell Biol (1993) 123:99–108. doi:10.1083/jcb.123.1.99
37. Fontana S, Parolini S, Vermi W, Booth S, Gallo F, Donini M, et al. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood (2006) 2006(107):4857–64. doi:10.1182/blood-2005-11-4398
38. Benson KF, Li FQ, Person RE, Albani D, Duan Z, Wechsler J, et al. Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet (2003) 35:90–6. doi:10.1038/ng1224
39. Källquist L, Rosén H, Nordenfelt P, Calafat J, Janssen H, Persson AM, et al. Neutrophil elastase and proteinase 3 trafficking routes in myelomonocytic cells. Exp Cell Res (2010) 316:3182–96. doi:10.1016/j.yexcr.2010.08.016
40. Rous BA, Reaves BJ, Ihrke G, Briggs JA, Gray SR, Stephens DJ, et al. Role of adaptor complex AP-3 in targeting wild-type and mutated CD63 to lysosomes. Mol Biol Cell (2002) 13:1071–82. doi:10.1091/mbc.01-08-0409
41. Pols MS, Klumperman J. Trafficking and function of the tetraspanin CD63. Exp Cell Res (2009) 315:1584–92. doi:10.1016/j.yexcr.2008.09.020
42. Källquist L, Hansson M, Persson AM, Janssen H, Calafat J, Tapper H, et al. The tetraspanin CD63 is involved in granule targeting of neutrophil elastase. Blood (2008) 112:3444–54. doi:10.1182/blood-2007-10-116285
43. Garwicz D, Lindmark A, Gullberg U. Human cathepsin G lacking functional glycosylation site is proteolytically processed and targeted for storage in granules after transfection to the rat basophilic/mast cell line RBL or the murine myeloid cell line 32D. J Biol Chem (1995) 270:28413–8. doi:10.1074/jbc.270.47.28413
44. Steet R, Lee WS, Kornfeld S. Identification of the minimal lysosomal enzyme recognition domain in cathepsin D. J Biol Chem (2005) 280:33318–23. doi:10.1074/jbc.M505994200
45. Canuel M, Libin Y, Morales CR. The interactomics of sortilin: an ancient lysosomal receptor evolving new functions. Histol Histopathol (2009) 24:481–92.
46. Glenthøj A, Cowland JB, Heegaard NH, Larsen MT, Borregaard N. Serglycin participates in retention of α-defensin in granules during myelopoiesis. Blood (2011) 118:4440–8. doi:10.1182/blood-2011-06-362947
47. Lemansky P, Smolenova E, Wrocklage C, Hasilik A. Neutrophil elastase is associated with serglycin on its way to lysosomes in U937 cells. Cell Immunol (2007) 246:1–7. doi:10.1016/j.cellimm.2007.06.001
48. Niemann CU, Abrink M, Pejler G, Fischer RL, Christensen EI, Knight SD, et al. Neutrophil elastase depends on serglycin proteoglycan for localization in granules. Blood (2007) 109:4478–86. doi:10.1182/blood-2006-02-001719
49. Kolset SO, Tveit H. Serglycin – structure and biology. Cell Mol Life Sci (2008) 65:1073–85. doi:10.1007/s00018-007-7455-6
50. Gullberg U, Bengtsson N, Bülow E, Garwicz D, Lindmark A, Olsson I. Processing and targeting of granule proteins in human neutrophils. J Immunol Methods (1999) 232:201–10. doi:10.1016/S0022-1759(99)00177-5
51. Gullberg U, Lindmark A, Lindergren G, Persson A-M, Nilsson E, Olsson I. Carboxyl-terminal prodomain-deleted human leukocyte elastase and cathepsin G are efficiently targeted to granules and enzymatically activated in the rat basophilic/mast cell line RBL. J Biol Chem (1995) 270:12912–8. doi:10.1074/jbc.270.21.12912
52. Filocamo M, Morrone A. Lysosomal storage disorders: molecular basis and laboratory testing. Hum Genomics (2011) 5:156–69. doi:10.1186/1479-7364-5-3-156
53. Kudo M, Brem MS, Canfield WM. Mucolipidosis II (I-cell disease) and mucolipidosis IIIA (classical pseudo-hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase alpha/beta-subunits precursor gene. Am J Hum Genet (2006) 78:451–63. doi:10.1086/500849
54. Kumar TS, Scott JX, Raghupathy P, Moses PD. Mucolipidosis II (I-cell disease). J Postgrad Med (2005) 51:232–3.
55. Vogel P, Payne BJ, Read R, Lee WS, Gelfman CM, Kornfeld S. Comparative pathology of murine mucolipidosis types II and IIIC. Vet Pathol (2009) 46:313–24. doi:10.1354/vp.46-2-313
56. Kollmann K, Damme M, Markmann S, Morelle W, Schweizer M, Hermans-Borgmeyer I, et al. Lysosomal dysfunction causes neurodegeneration in mucolipidosis II “knock-in” mice. Brain (2012) 135:2661–75. doi:10.1093/brain/aws209
57. Jung J, Bohn G, Allroth A, Boztug K, Brandes G, Sandrock I, et al. Identification of a homozygous deletion in the AP3B1 gene causing Hermansky-Pudlak syndrome, type 2. Blood (2006) 108:362–9. doi:10.1182/blood-2005-11-4377
58. Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell (1999) 3:11–21. doi:10.1016/S1097-2765(00)80170-7
59. Horwitz MS, Duan Z, Korkmaz B, Lee HH, Mealiffe ME, Salipante SJ. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood (2007) 109:1817–24. doi:10.1182/blood-2006-08-019166
60. Duan Z, Li FQ, Wechsler J, Meade-White K, Williams K, Benson KF, et al. A novel notch protein, N2N, targeted by neutrophil elastase and implicated in hereditary neutropenia. Mol Cell Biol (2004) 24:58–70. doi:10.1128/MCB.24.1.58-70.2004
61. Horwitz M, Benson KF, Duan Z, Person RE, Wechsler J, Williams K, et al. Role of neutrophil elastase in bone marrow failure syndromes: molecular genetic revival of the chalone hypothesis. Curr Opin Hematol (2003) 10:49–54. doi:10.1097/00062752-200301000-00008
62. Horwitz M, Benson KF, Duan Z, Li FQ, Person RE. Hereditary neutropenia: dogs explain human neutrophil elastase mutations. Trends Mol Med (2004) 10:163–70. doi:10.1016/j.molmed.2004.02.002
63. Arancibia SA, Beltrán CJ, Aguirre IM, Silva P, Peralta AL, Malinarich F, et al. Toll-like receptors are key participants in innate immune responses. Biol Res (2007) 40:97–112. doi:10.4067/S0716-97602007000200001
64. Zeytun A, Chaudhary A, Pardington P, Cary R, Gupta G. Induction of cytokines and chemokines by toll-like receptor signaling: strategies for control of inflammation. Crit Rev Immunol (2010) 30:53–67. doi:10.1615/CritRevImmunol.v30.i1.40
65. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7:678–89. doi:10.1038/nri2156
66. Williams MR, Azcutia V, Newton G, Alcaide P, Luscinskas FW. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol (2011) 32:461–9. doi:10.1016/j.it.2011.06.009
67. Sánchez-Madrid F, Serrador JM. Bringing up the rear: defining the roles of the uropod. Nat Rev Mol Cell Biol (2009) 10:353–9. doi:10.1038/nrm2680
68. Burridge K, Doughman R. Front and back by Rho and Rac. Nat Cell Biol (2006) 8:781–2. doi:10.1038/ncb0806-781
69. Vojtek AB, Cooper JA. Rho family members: activators of MAP kinase cascades. Cell (1995) 82:527–9. doi:10.1016/0092-8674(95)90023-3
70. Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, et al. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell (1995) 81:1137–46. doi:10.1016/S0092-8674(05)80018-2
71. Perona R, Montaner S, Saniger L, Sánchez-Pérez I, Bravo R, Lacal JC. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev (1997) 11:463–75. doi:10.1101/gad.11.4.463
72. Jiang K, Zhong B, Ritchey C, Gilvary DL, Hong-Geller E, Wei S, et al. Regulation of Akt-dependent cell survival by Syk and Rac. Blood (2003) 101:236–44. doi:10.1182/blood-2002-04-1251
73. Hong-Geller E, Cerione RA. Cdc42 and Rac stimulate exocytosis of secretory granules by activating the IP(3)/calcium pathway in RBL-2H3 mast cells. J Cell Biol (2000) 148:481–94. doi:10.1083/jcb.148.3.481
74. Baier A, Ndoh VN, Lacy P, Eitzen G. Rac1 and Rac2 control distinct events during antigen-stimulated mast cell exocytosis. J Leukoc Biol (2014) 95:763–74. doi:10.1189/jlb.0513281
75. Knaus RG, Heyworth PG, Evans T, Curnutte JT, Bokoch GM. Regulation of phagocytic oxygen radical production by the GTP-binding protein Rac 2. Science (1991) 254:1512–5. doi:10.1126/science.1660188
76. Heyworth PG, Bohl BP, Bokoch GM, Curnutte JT. Rac translocates independently of the neutrophil NADPH oxidase components p47phox and p67phox. J Biol Chem (1994) 269:30749–52.
77. Diebold BA, Bokoch GM. Rho GTPases and the control of the oxidative burst in polymorphonuclear leukocytes. Curr Top Microbiol Immunol (2005) 291:91–111.
78. Kim C, Marchal CC, Penninger J, Dinauer MC. The hemopoietic Rho/Rac guanine nucleotide exchange factor Vav1 regulates N-formyl-methionyl-leucyl-phenylalanine-activated neutrophil functions. J Immunol (2003) 171:4425–30. doi:10.4049/jimmunol.171.8.4425
79. Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, et al. P-Rex1 regulates neutrophil function. Curr Biol (2005) 15:1867–73. doi:10.1016/j.cub.2005.09.050
80. Lawson CD, Donald S, Anderson KE, Patton DT, Welch HC. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine-dependent neutrophil responses. J Immunol (2011) 186:1467–76. doi:10.4049/jimmunol.1002738
81. Funamoto S, Meili R, Lee S, Parry L, Firtel RA. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell (2002) 109:611–23. doi:10.1016/S0092-8674(02)00755-9
82. Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, et al. PTEN functions to “prioritize” chemotactic cues and prevent “distraction” in migrating neutrophils. Nat Immunol (2008) 9:743–52. doi:10.1038/ni.1623
83. Jennings RT, Strengert M, Hayes P, El-Benna J, Brakebusch C, Kubica M, et al. RhoA determines disease progression by controlling neutrophil motility and restricting hyperresponsiveness. Blood (2014) 123:3635–45. doi:10.1182/blood-2014-02-557843
84. Kumar S, Xu J, Perkins C, Guo F, Snapper S, Finkelman FD, et al. Cdc42 regulates neutrophil migration via crosstalk between WASp, CD11b, and microtubules. Blood (2012) 120:3563–74. doi:10.1182/blood-2012-04-426981
85. Lee WL, Harrison RE, Grinstein S. Phagocytosis by neutrophils. Microbes Infect (2003) 5:1299–306. doi:10.1016/j.micinf.2003.09.014
86. Hallett MB, Dewitt S. Ironing out the wrinkles of neutrophil phagocytosis. Trends Cell Biol (2007) 17:209–14. doi:10.1016/j.tcb.2007.03.002
87. Nordenfelt P, Tapper H. Phagosome dynamics during phagocytosis by neutrophils. J Leukoc Biol (2011) 90:271–84. doi:10.1189/jlb.0810457
88. Peng G, Huang J, Boyd M, Kleinberg ME. Properties of phagocyte NADPH oxidase p47-phox mutants with unmasked SH3 (Src homology 3) domains: full reconstitution of oxidase activity in a semi-recombinant cell-free system lacking arachidonic acid. Biochem J (2003) 373(Pt 1):221–9. doi:10.1042/BJ20021629
89. Ellson CD, Gobert-Gosse S, Anderson KE, Davidson K, Erdjument-Bromage H, Tempst P, et al. PtdIns(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40(phox). Nat Cell Biol (2001) 3:679–82. doi:10.1038/35083076
90. Bissonnette SA, Glazier CM, Stewart MQ, Brown GE, Ellson CD, Yaffe MB. Phosphatidylinositol 3-phosphate-dependent and -independent functions of p40phox in activation of the neutrophil NADPH oxidase. J Biol Chem (2008) 283:2108–19. doi:10.1074/jbc.M706639200
91. Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol (2005) 15:1874–9. doi:10.1016/j.cub.2005.09.014
92. Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene (2013) 33(31):4021–35. doi:10.1038/onc.2013.362
93. Kim C, Dinauer MC. Rac2 is an essential regulator of neutrophil nicotinamide adenine dinucleotide phosphate oxidase activation in response to specific signaling pathways. J Immunol (2001) 166:1223–32. doi:10.4049/jimmunol.166.2.1223
94. Li S, Yamauchi A, Marchal CC, Molitoris JK, Quilliam LA, Dinauer MC. Chemoattractant-stimulated Rac activation in wild-type and Rac2-deficient murine neutrophils: preferential activation of Rac2 and Rac2 gene dosage effect on neutrophil functions. J Immunol (2002) 169:5043–51. doi:10.4049/jimmunol.169.9.5043
95. Yamauchi A, Marchal CC, Molitoris J, Pech N, Knaus U, Towe J, et al. Rac GTPase isoform-specific regulation of NADPH oxidase and chemotaxis in murine neutrophils in vivo. Role of the C-terminal polybasic domain. J Biol Chem (2005) 280:953–64. doi:10.1074/jbc.M408820200
96. Utomo A, Cullere X, Glogauer M, Swat W, Mayadas TN. Vav proteins in neutrophils are required for FcgammaR-mediated signaling to Rac GTPases and nicotinamide adenine dinucleotide phosphate oxidase component p40(phox). J Immunol (2006) 177:6388–97. doi:10.4049/jimmunol.177.9.6388
97. Roepstorff K, Rasmussen I, Sawada M, Cudre-Maroux C, Salmon P, Bokoch G, et al. Stimulus-dependent regulation of the phagocyte NADPH oxidase by a VAV1, Rac1, and PAK1 signaling axis. J Biol Chem (2008) 283:7983–93. doi:10.1074/jbc.M708281200
98. Filina JV, Gabdoulkhakova AG, Safronova VG. RhoA/ROCK downregulates FPR2-mediated NADPH oxidase activation in mouse bone marrow granulocytes. Cell Signal (2014) 26(10):2138–46. doi:10.1016/j.cellsig.2014.05.017
99. Sengeløv H, Kjeldsen L, Borregaard N. Control of exocytosis in early neutrophil activation. J Immunol (1993) 150:1535–43.
100. Sengeløv H, Follin P, Kjeldsen L, Lollike K, Dahlgren C, Borregaard N. Mobilization of granules and secretory vesicles during in vivo exudation of human neutrophils. J Immunol (1995) 154:4157–65.
101. Toonen RF, Verhage M. Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol (2003) 13:177–86. doi:10.1016/S0962-8924(03)00031-X
102. Lacy P, Eitzen G. Control of granule exocytosis in neutrophils. Front Biosci (2008) 13:5559–70. doi:10.2741/3099
103. Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest (1987) 80:1550–60. doi:10.1172/JCI113241
104. Han H, Roberts J, Lou O, Muller WA, Nathan N, Nathan C. Chemical inhibitors of TNF signal transduction in human neutrophils point to distinct steps in cell activation. J Leukoc Biol (2006) 79:147–54. doi:10.1189/jlb.0605308
105. Mócsai A, Ligeti E, Lowell CA, Berton G. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J Immunol (1999) 162:1120–6.
106. Fumagalli L, Zhang H, Baruzzi A, Lowell CA, Berton G. The Src family kinases Hck and Fgr regulate neutrophil responses to N-formyl-methionyl-leucyl-phenylalanine. J Immunol (2007) 178:3874–85. doi:10.4049/jimmunol.178.6.3874
107. Futosi K, Fodor S, Mócsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol (2013) 17:638–50. doi:10.1016/j.intimp.2013.11.010
108. Kamen LA, Schlessinger J, Lowell CA. Pyk2 is required for neutrophil degranulation and host defense responses to bacterial infection. J Immunol (2011) 186:1656–65. doi:10.4049/jimmunol.1002093
109. Efimov A, Schiefermeier N, Grigoriev I, Ohi R, Brown MC, Turner CE, et al. Paxillin-dependent stimulation of microtubule catastrophes at focal adhesion sites. J Cell Sci (2008) 121:196–204. doi:10.1242/jcs.012666
110. Mollinedo F, Calafat J, Janssen H, Martín-Martín B, Canchado J, Nabokina SM, et al. Combinatorial SNARE complexes modulate the secretion of cytoplasmic granules in human neutrophils. J Immunol (2006) 177:2831–41. doi:10.4049/jimmunol.177.5.2831
111. Logan MR, Lacy P, Odemuyiwa SO, Steward M, Davoine F, Kita H, et al. A critical role for vesicle-associated membrane protein-7 in exocytosis from human eosinophils and neutrophils. Allergy (2006) 61:777–84. doi:10.1111/j.1398-9995.2006.01089.x
112. Brumell JH, Volchuk A, Sengelov H, Borregaard N, Cieutat AM, Bainton DF, et al. Subcellular distribution of docking/fusion proteins in neutrophils, secretory cells with multiple exocytic compartments. J Immunol (1995) 155:5750–9.
113. Brzezinska AA, Johnson JL, Munafo DB, Crozat K, Beutler B, Kiosses WB, et al. The Rab27a effectors JFC1/Slp1 and Munc13-4 regulate exocytosis of neutrophil granules. Traffic (2008) 9:2151–64. doi:10.1111/j.1600-0854.2008.00838.x
114. Pivot-Pajot C, Varoqueaux F, de Saint Basile G, Bourgoin SG. Munc13-4 regulates granule secretion in human neutrophils. J Immunol (2008) 180:6786–97. doi:10.4049/jimmunol.180.10.6786
115. Zhang Y, Tang W, Zhang H, Niu X, Xu Y, Zhang J, et al. A network of interactions enables CCM3 and STK24 to coordinate UNC13D-driven vesicle exocytosis in neutrophils. Dev Cell (2013) 27:215–26. doi:10.1016/j.devcel.2013.09.021
116. Jaillon S, Galdiero MR, Del Prete D, Cassatella MA, Garlanda C, Mantovani A. Neutrophils in innate and adaptive immunity. Semin Immunopathol (2013) 35:377–94. doi:10.1007/s00281-013-0374-8
117. Calafat J, Janssen H, Ståhle-Bäckdahl M, Zuurbier AE, Knol EF, Egesten A. Human monocytes and neutrophils store transforming growth factor-alpha in a subpopulation of cytoplasmic granules. Blood (1997) 90:1255–66.
118. Beil WJ, Weller PF, Peppercorn MA, Galli SJ, Dvorak AM. Ultrastructural immunogold localization of subcellular sites of TNF-alpha in colonic Crohn’s disease. J Leukoc Biol (1995) 58:284–98.
119. Denkers E, Del Rio L, Bennouna S. Neutrophil production of IL-12 and other cytokines during microbial infection. In: Ma C, editor. The Neutrophil: An Emerging Regulator of Inflammatory and Immune Response. Basel: Karger (2003). p. 95–114.
120. Miskolci V, Ghosh CC, Rollins J, Romero C, Vu HY, Robinson S, et al. TNFalpha release from peripheral blood leukocytes depends on a CRM1-mediated nuclear export. Biochem Biophys Res Commun (2006) 351:354–60. doi:10.1016/j.bbrc.2006.10.045
121. Martín-Martín B, Nabokina SM, Blasi J, Lazo PA, Mollinedo F. Involvement of SNAP-23 and syntaxin 6 in human neutrophil exocytosis. Blood (2000) 96:2574–83.
122. Mollinedo F, Martín-Martín B, Calafat J, Nabokina SM, Lazo PA. Role of vesicle-associated membrane protein-2, through Q-soluble N-ethylmaleimide-sensitive factor attachment protein receptor/R-soluble N-ethylmaleimide-sensitive factor attachment protein receptor interaction, in the exocytosis of specific and tertiary granules of human neutrophils. J Immunol (2003) 170:1034–42. doi:10.4049/jimmunol.170.2.1034
Keywords: exocytosis, Rho GTPase, protein sorting, secretion, cytokine
Citation: Sheshachalam A, Srivastava N, Mitchell T, Lacy P and Eitzen G (2014) Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 5:448. doi: 10.3389/fimmu.2014.00448
Received: 17 July 2014; Accepted: 03 September 2014;
Published online: 19 September 2014.
Edited by:
Uday Kishore, Brunel University, UKReviewed by:
Janos G. Filep, University of Montreal, CanadaCopyright: © 2014 Sheshachalam, Srivastava, Mitchell, Lacy and Eitzen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gary Eitzen, Department of Cell Biology, University of Alberta, 514 Medical Sciences Building, Edmonton, AB T6G 2H7, Canada e-mail:Z2FyeS5laXR6ZW5AdWFsYmVydGEuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.