
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol. , 15 September 2014
Sec. Primary Immunodeficiencies
Volume 5 - 2014 | https://doi.org/10.3389/fimmu.2014.00415
This article is part of the Research Topic Exploiting New Technologies for Immune System Evaluation in Primary Immune Deficiencies View all 10 articles
 Rohan Ameratunga1,2*
Rohan Ameratunga1,2* Maia Brewerton3
Maia Brewerton3 Charlotte Slade3
Charlotte Slade3 Anthony Jordan2David Gillis4Richard Steele1Wikke Koopmans1
Anthony Jordan2David Gillis4Richard Steele1Wikke Koopmans1 See-Tarn Woon1
See-Tarn Woon1
Common variable immunodeficiency disorders (CVIDs) are the most frequent symptomatic primary immune deficiency condition in adults. The genetic basis for the condition is not known and no single clinical feature or laboratory test can establish the diagnosis; it has been a diagnosis of exclusion. In areas of uncertainty, diagnostic criteria can provide valuable clinical information. Here, we compare the revised European society of immune deficiencies (ESID) registry (2014) criteria with the diagnostic criteria of Ameratunga et al. (2013) and the original ESID/pan American group for immune deficiency (ESID/PAGID 1999) criteria. The ESID/PAGID (1999) criteria either require absent isohemagglutinins or impaired vaccine responses to establish the diagnosis in patients with primary hypogammaglobulinemia. Although commonly encountered, infective and autoimmune sequelae of CVID were not part of the original ESID/PAGID (1999) criteria. Also excluded were a series of characteristic laboratory and histological abnormalities, which are useful when making the diagnosis. The diagnostic criteria of Ameratunga et al. (2013) for CVID are based on these markers. The revised ESID registry (2014) criteria for CVID require the presence of symptoms as well as laboratory abnormalities to establish the diagnosis. Once validated, criteria for CVID will improve diagnostic precision and will result in more equitable and judicious use of intravenous or subcutaneous immunoglobulin therapy.
Common variable immunodeficiency disorders (CVIDs) are the most frequent symptomatic primary immune deficiency disorder in adults. While most patients suffer recurrent infections (1–5), there is also an increased risk of autoimmune disorders and malignancy because of immune dysregulation. CVID is likely to represent a heterogeneous group of polygenic disorders (6).
Although some patients have symptoms dating back to early childhood (7), the hallmark of CVID is primary hypogammaglobulinemia, which is a consequence of late onset antibody failure (LOAF). The majority of adult patients with CVID have IgG levels below 5 g/l (5). Most patients also have reduced or undetectable IgA and/or IgM levels (1, 8). Other laboratory features may include reduced switched memory B cells and/or increased numbers of CD21 low B cells in the periphery (9). A subgroup of patients with severe T-cell defects, originally included within the spectrum of CVID, are now defined separately as late onset combined immune deficiency (LOCID) (10).
In addition to the laboratory features outlined above, many patients have characteristic histological lesions including a sarcoidosis-like granulomatous disorder, nodular lymphoid hyperplasia of the gut, nodular regenerative hyperplasia of the liver, or lymphoid interstitial pneumonitis of the lungs (11–16). Absence of plasma cells in gastrointestinal biopsies is another characteristic finding in the majority of CVID patients (17–19).
It can be very difficult to determine which patients are suffering from CVID and require intravenous immunoglobulin (IVIG) or subcutaneous immunoglobulin (SCIG) replacement. IVIG/SCIG treatment can substantially improve both quality of life (20) and longevity in patients with CVID. This underscores the importance of accurate diagnostic criteria for CVID (21).
Here, we compare the original (1999) European society of immune deficiencies (ESID)/pan American group for immune deficiency (PAGID) criteria, the Ameratunga et al. (2013) criteria and the revised ESID registry (2014) criteria (Tables 1–3). We have previously described in detail the evidence base for the Ameratunga et al. (2013) criteria (21). It is necessary to provide an abbreviated version of these criteria here to allow comparison with the revised ESID registry (2014) criteria. We have devoted similar space to each of these criteria.

Table 1. The original ESID/PAGID (1999) criteria for probable and possible CVID.

Table 2. New diagnostic criteria (Ameratunga et al., 2013) for CVID.

Table 3. Revised ESID (2014) diagnostic criteria for CVID.
According to the ESID/PAGID (1999) criteria, CVID is a diagnosis of exclusion (Table 1) (33). Patients were required to have an IgG level below 7–8 g/l [2 SD below the mean, or more accurately, below the 97.5th percentile, as immunoglobulin levels are not normally distributed (34)], as well as impaired vaccine challenge responses or absent isohemagglutinins and other secondary causes of hypogammaglobulinemia were to be excluded. The criteria did not include the characteristic histological features of the disorder or clinical sequelae resulting from the immune system failure (ISF).
Since that time, there have been major advances in the understanding of CVID. The majority of CVID patients have impaired memory B cell function with a reduction in switched memory B cells (9). In the last decade, there have been several genetic discoveries in patients with CVID-like conditions. Rare patients with monogenic defects of CD19, CD20, CD21, CD81, and ICOS have been identified (35–38). If identified by molecular diagnostic studies (39), these patients are, however, no longer classified as having CVID and are removed from further consideration of the disorder (40, 41). Genetic alterations from genome wide association studies, including copy number variations (42) and sequence variations in genes such as TACI, BAFF receptor, and MSH5 may predispose to CVID. Mutations of TACI, BAFF receptor, and MSH5 are also found in healthy individuals, but at lower frequency (28, 31, 43).
The ESID/PAGID (1999) criteria require IgG levels to be below 2 SD of the mean (Table 1). This means that 2.5% of the general population would meet this criterion (23). There is general agreement with the third ESID/PAGID (1999) criterion that other secondary causes of hypogammaglobulinemia including drug-induced disorders need to be excluded (22, 44, 45).
Perhaps the greatest difficulty with the ESID/PAGID (1999) criteria is the requirement for poor responses to vaccines. The ESID/PAGID (1999) criteria do not specify which vaccines should be used and there are significant variations in vaccine protocols in different studies (46, 47). Therefore, patients with trivial hypogammaglobulinemia with mildly impaired diphtheria antibody responses could be classified as having CVID. Poor responses to the diphtheria vaccine are common, even in normal persons, particularly with increasing age (23).
It is likely many patients with CVID have already generated vaccine-specific memory B cells following childhood immunization prior to LOAF. Therefore, assessing booster responses to childhood vaccines may be diagnostically misleading. This may explain why a significant minority of patients with presumed CVID have protective responses to tetanus toxoid and Pneumovax® (48, 49). It is also debatable if the response to highly immunogenic proteins such as tetanus toxoid, administered with adjuvant, is a valid and reliable predictor of a protective response to pathogens in vivo (21). Specific concerns about using vaccines to assess the immune response are shown in Table 4.

Table 4. Difficulties interpreting vaccine responses in CVID.
The use of neoantigens such as rabies vaccine, typhoid vaccine, and experimental vaccines such as ψX174 to assess LOAF may be more predictive of an immune defect as patients are unlikely to have previously encountered these antigens (54). However, there have been concerns about risks associated with the rabies vaccine (54) and the typhoid vaccine is not yet widely used. The ψx174 vaccine has not been registered by the FDA and cannot be used in routine clinical practice (67).
The difficulty with diagnosis is illustrated in a recent study in which a new category of idiopathic primary hypogammaglobulinemia was proposed for symptomatic patients who did not meet the ESID/PAGID (1999) criteria for CVID (68). In spite of not meeting the ESID/PAGID (1999) criteria, many of these patients were treated with immunoglobulin. There is thus a discord between diagnosis and treatment in many patients with hypogammaglobulinemia/CVID.
The ESID/PAGID (1999) criteria do not specify the need for symptoms to establish the diagnosis. Therefore, important clinical manifestations and complications may not be obvious from different parts of the world, when using these criteria. This was illustrated in recent CVID studies, where there were wide variations in complications leading to different clinical phenotypes as well as bronchiectasis in countries within the European Union as well as the United States (4, 7, 69).
It is accepted that these criteria are relatively simple and could be used to diagnose patients in developing countries. The major difficulty is the interpretation of vaccine responses (Table 4), which is a pivotal component of these criteria. We are also concerned that the application of relatively simple criteria to a complex set of disorders will result in inaccuracies, as evidenced by the need for a new category of disorders, idiopathic primary hypogammaglobulinemia, described above. We are thus left with criteria that can be difficult to measure and interpret. Several eminent authors have expressed concern about the need for revised diagnostic criteria for CVID (70, 71).
We recently proposed new diagnostic criteria for CVID (Table 2) (21). Treatment recommendations are closely linked to these diagnostic criteria (Figure 1). The Ameratunga et al. (2013) criteria emphasize both the clinical and laboratory sequelae of ISF in these patients (72).
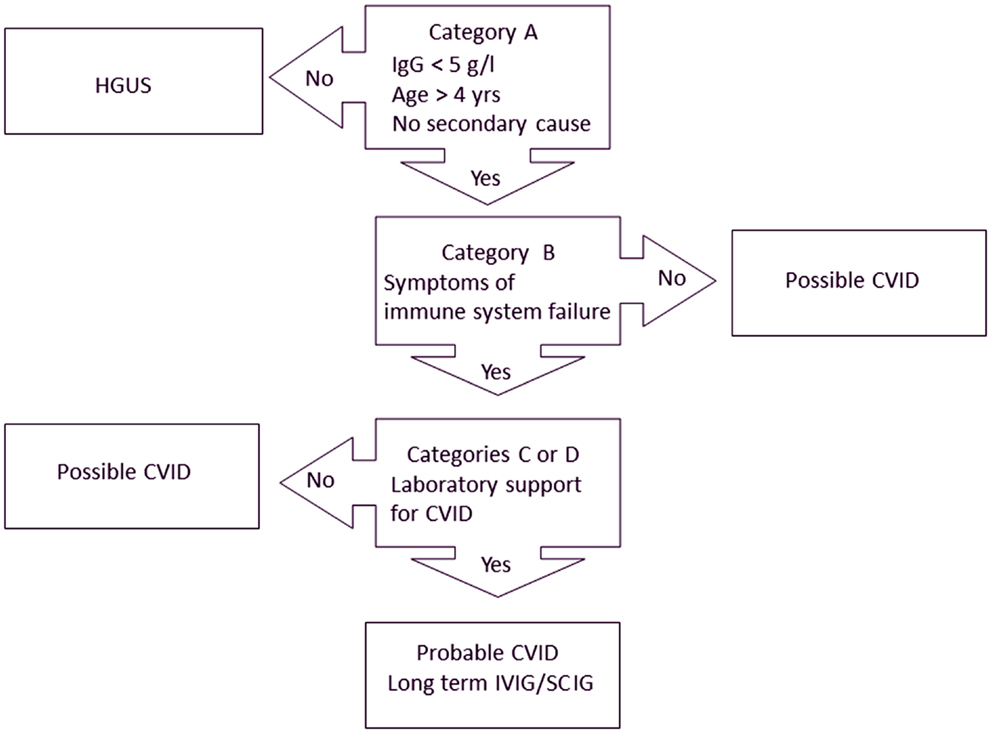
Figure 1. Treatment algorithm for CVID (21). Patients must meet all major criteria in category A for consideration of CVID. Category B confirms the presence of symptoms indicating immune system failure (ISF). To have probable CVID, patients must also have supportive laboratory evidence of immune system dysfunction (category C) or characteristic histological lesions of CVID (category D). Patients with mild hypogammaglobulinemia (IgG > 5 g/l) are termed hypogammaglobulinemia of uncertain significance (HGUS). Patients meeting category A criteria but not other criteria are deemed to have possible CVID. Most patients with probable CVID are likely to require IVIG/SCIG. Some patients with possible CVID who have profound hypogammaglobulinemia will require IVIG/SCIG but most patients with HGUS are unlikely to need IVIG/SCIG replacement.
In order to qualify as having probable CVID, patients must meet criteria in categories A, B, and C or D (Table 2). It is important for these criteria to be applied sequentially, as each subsequent category confers increasing specificity (Figure 1). A threshold of 5 g/l for IgG has been set for adults as this is the cut-off used in the French DEFI study (5). Patients must be older than 4 years of age and must not have a secondary cause for their hypogammaglobulinemia. If patients do not meet category A criteria, other criteria do not apply and a diagnosis of CVID cannot be entertained (Figure 1).
The most important feature of these criteria is clinical evidence of ISF (category B). Patients must have predisposition to infections and/or autoimmune disease, as a direct result of their immune defect. We have not been prescriptive about the numbers of upper or lower respiratory tract infections, as these will be influenced by the patient’s socio-demographic circumstances. While there may be debate about the specificity of each criterion in category B, the intention is to distinguish symptomatic patients from those who are well. These symptomatic patients must have laboratory evidence of immune dysfunction (category C) or the characteristic histological findings (category D) associated with CVID (Table 2).
None of the category C criteria are specific but in combination support the diagnosis of CVID. The majority of CVID patients will have a reduction of IgA and/or IgM (1, 4). Most will have impaired vaccine responses (49). It is, however, important to compare antibody responses to those of normal controls, as many CVID patients who have received their primary immunization series are likely to have developed memory B cells and are able to generate protective antibody levels. Using protective antibody levels as eligibility criteria for IVIG/SCIG is unacceptable in symptomatic patients as it will exclude a significant proportion of patients who will benefit from IVIG/SCIG treatment (48, 49).
Vaccine responses in normal controls have been published (50, 52, 53). Patients receiving a single booster dose of tetanus toxoid should achieve an antibody response of at least 1 IU/ml (50). Although a response of 1 IU/ml has been suggested following diphtheria immunization, our experience and original immunization studies show that this vaccine is much less immunogenic than the tetanus vaccine (50). Even otherwise healthy individuals may fail to reach this level, particularly persons over 50 years of age. The inclusion of diphtheria vaccine responses is thus problematic.
Adults and children receiving the Haemophilus influenzae type B (HIB) vaccine should reach antibody levels of at least 1.0 μg/ml rather than the protective level of 0.15 μg/ml (52, 53). Adults receiving the Pneumovax® should achieve a protective level of 1.3 μg/ml for at least 70% of serotypes while children should reach 50% (64, 65). Adequate but transient vaccine responses are also included in category C criteria (21, 29). This may reflect in vivo failure of B cell memory in some patients.
We have included absent isohemagglutinins in these criteria providing the patient is not blood group AB (30). Although absent isohemagglutinins are part of the PAGID/ESID (1999) criteria, in our experience it is rare for a diagnosis of CVID to be made or for IVIG/SCIG to be prescribed, purely on the basis of absent isohemagglutinins in a patient with reduced IgG levels.
Most patients with CVID have impaired memory B cells and these constitute a diagnostic criterion in category C (9). It is, however, important for memory B cell subsets to be measured on at least two occasions, as we have shown the numbers can vary on repeat testing (73). Genetic predisposition (mutations of TACI, BAFF receptor, MSH5, etc.) to CVID is included in category C diagnostic criteria (42). This criterion is primarily intended for patients who may have been enrolled in clinical research studies. In the interim, we strongly discourage routine sequencing of these genes for diagnostic purposes until their utility is established.
Reduction or deficiency of IgG3 has been included in the new diagnostic criteria. There is considerable peer-reviewed literature suggesting IgG3 deficiency should be considered an important biomarker for a humoral immune defect (25, 26). We have included serological manifestations of significant autoimmunity in category C. This would include positive tests for lupus anticoagulant, Coombs test, etc. In most large series of CVID patients there are approximately 15% of patients who have severe autoimmunity and some have minimal infections in spite of their profound hypogammaglobulinemia. These patients will qualify as having CVID according to the Ameratunga et al. (2013) criteria, providing they have not been treated with rituximab.
Finally, some CVID patients have characteristic histological findings, which have been included in the Ameratunga et al. (2013) criteria (category D). These require biopsy for confirmation. In the context of primary (category A), symptomatic (category B) hypogammaglobulinemia, the presence of these relatively specific histological markers obviates the need to undertake vaccine responses, measure memory B cells, etc. Most of the histological criteria described in category D can occur with other disorders. Because category D will only apply if patients have already met category A and B criteria (Figure 1), this will confer specificity for CVID. Category C and D criteria may serve as an useful check list when assessing patients with hypogammaglobulinemia (21). We have shown that careful review of category C and D criteria may help to distinguish CVID from secondary causes of hypogammaglobulinemia (46, 74, 75).
As in the past, a diagnostic rectal biopsy could be undertaken to confirm the absence of plasma cells (19). Although a minority of CVID patients have plasma cells, the absence of these cells in a gut biopsy is a characteristic feature of CVID (21). Incorporating histological features in the diagnostic criteria may be useful where patients have already commenced IVIG/SCIG as there are risks in stopping treatment to assess vaccine responses.
We have designated a category of possible CVID for those patients meeting category A criteria (or AC or AD) but not category B criteria (Figure 1). Some asymptomatic patients with profound hypogammaglobulinemia may need to be treated with IVIG/SCIG as they may be at risk of severe viral infections or bacterial sepsis (28).
There are other patients who have mild hypogammaglobulinemia (IgG > 5 g/l) who are otherwise well or have only mild symptoms. We have termed these patients hypogammaglobulinemia of uncertain significance (HGUS) (21). In the future, it may be useful to sub classify HGUS patients depending on whether (sHGUS) or not (aHGUS) they have symptoms attributable to ISF. Prospective studies will indicate if the prognosis for these two sub groups is different.
The Ameratunga et al. (2013) criteria are intended primarily for clinical use and we therefore felt it was important to link them closely to treatment (Figure 1) (21). As with the Jones criteria for acute rheumatic fever, CVID no longer needs to be a diagnosis of exclusion.
The revised ESID registry (2014) criteria for probable CVID have been recently released (http://esid.org/Working-Parties/Registry/Diagnosis-criteria) and are shown in Table 3. These are structured in a similar way to the Ameratunga et al. (2013) criteria but have not been given named categories. In contrast to the previous ESID/PAGID (1999) criteria, patients are required to have symptoms of their immune deficiency or a family history of antibody deficiency to be eligible for a diagnosis of CVID. Increased susceptibility to infection along with autoimmunity, unexplained polyclonal lymphoproliferation, or granulomatous disease qualifies patients for further consideration of CVID. This is similar to the Ameratunga et al. (2013) category B criteria, where a diagnosis of CVID cannot be made in the absence of symptoms (21).
Symptomatic patients are required to have a marked decrease of IgG as well as IgA and/or IgM. Again immunoglobulin levels 2 SD below mean is required for the relevant population. Immunoglobulin levels need to be repeated to confirm persistent reduction. This would exclude transient reductions in immunoglobulins that can sometimes be seen following viral infections or use of medications (44). Although a large number of patients with mild hypogammaglobulinemia will qualify for further investigation, the subsequent criteria will increase the specificity of the diagnosis.
Patients with hypogammaglobulinemia must then have either impaired antibody responses to vaccines and/or absent isohemagglutinins or reduced numbers of switched memory B cells for further consideration of CVID. Unlike the previous ESID/PAGID (1999) criteria, protective antibody levels are deemed to be required.
Like the Ameratunga et al. (2013) criteria, the revised ESID registry (2014) criteria do not specify which vaccines should be used. It is likely vaccine protocols will evolve with the availability and experience with new vaccines. As stated in Table 4, the increasing use of Prevnar13® in routine childhood vaccine schedules will make assessing pneumococcal polysaccharide responses increasingly problematic. In contrast to the revised ESID registry (2014) criteria, the Ameratunga et al. (2013) criteria require vaccine responses to be compared to the normal population, as a significant proportion of presumed CVID patients have protective antibody responses to tetanus and Pneumovax® (48, 49).
In contrast to the previous ESID/PAGID (1999) criteria, vaccine responses do not play such a pivotal role in the revised ESID registry (2014) criteria for CVID. Like the Ameratunga et al. (2013) criteria, it is therefore possible for patients to qualify as having CVID with normal vaccine challenge responses if they have either absent isohemagglutinins or reduced switched memory B cell numbers (Table 3). The requirement for reduced memory B cells in the revised ESID registry (2014) criteria is not mandatory and patients with absent B cells may still qualify for the diagnosis if they have impaired vaccine responses and/or absent isohemagglutinins.
As with all criteria described here, secondary causes of hypogammaglobulinemia must be excluded. The previous ESID/PAGID (1999) criteria specified 2 years as the eligible age for diagnosis, while the Ameratunga et al. (2013) and revised ESID registry criteria specify 4 years. The older age for diagnosis will help exclude monogenic defects as well as many cases of transient hypogammaglobulinemia of infancy. Unlike the ESID/PAGID (1999) and Ameratunga et al. (2013) criteria, the revised ESID registry (2014) criteria excludes severe T-cell defects from the spectrum of CVID, since these patients are deemed to have a combined immune deficiency. It is likely the genetic defect will differ from those with a largely humoral deficiency.
Granulomatous disease and lymphoproliferation are included in the initial set of clinical criteria in the revised ESID registry (2014) criteria (Table 3). This will require histological confirmation. Although not explicitly stated, our interpretation is that lymphoproliferation will include nodular lymphoid hyperplasia of the gut, nodular regenerative hyperplasia of the liver and lymphoid interstitial pneumonitis. These characteristic histological features and are included in category D of the Ameratunga et al. (2013) criteria. We assume an increase in CD21 low B cells may be included in the lymphoproliferation criterion of the revised ESID (2014) criteria. Like the Ameratunga et al. (2013) criteria, secondary causes for the histological lesions will be excluded by subsequent criteria. Nodular regenerative hyperplasia of the liver for example can occur with azathioprine treatment.
Like the Ameratunga et al. (2013) criteria, the revised ESID registry (2014) criteria may allow the diagnosis of CVID in patients who have already commenced IVIG/SCIG treatment. The presence of lymphoproliferation or reduced switched memory B cells will allow the diagnosis as long as other criteria are satisfied. This may obviate the need to stop IVIG/SCIG treatment to assess vaccine responses. Neither the Ameratunga et al. (2013) criteria nor the revised ESID (2014) registry criteria attempt to address the complex situation of hypogammaglobulinemia/CVID associated with malignancy. It can be very difficult to determine if hypogammaglobulinemia/CVID is the cause or the effect of malignancy.
Unlike the Ameratunga et al. (2013) criteria, the absence of plasma cells is not included in the revised ESID registry (2014) criteria. As indicated above, this is a useful feature in patients who have already commenced IVIG/SCIG replacement and may be identified by a diagnostic rectal biopsy. This is perhaps the most useful histological marker of CVID. The Ameratunga et al. (2013) criteria may be less useful if there are no characteristic histological lesions in patients who have already commenced IVIG/SCIG treatment. The emphasis on reduced switched memory B cells in the revised ESID registry (2014) criteria may allow the diagnosis in patients who have already commenced IVIG/SCIG. The revised ESID registry (2014) criteria require switched memory B cells to be below 70% of the normal population. Reference intervals will therefore have to be established for each laboratory. Given the variability in these cells, the Ameratunga et al. (2013) criteria require the assay to be repeated (73).
In comparison with the Ameratunga et al. (2013) criteria, transient vaccine responses are not included in the ESID registry (2014) criteria or breakthrough infections in spite of prophylactic antibiotics. Failure of vaccines to prevent infections, e.g., human papillomavirus is also not included in the revised criteria. The latter two may, however, be covered in the initial criterion of increased susceptibility to infection in the revised ESID registry (2013) criteria. Sequence variations in genes (TACI, BAFF receptor, MSH5, etc.) predisposing to CVID and IgG3 deficiency are not included in the revised ESID registry criteria (2014). Prospective studies will determine the value of these criteria (21).
The previous ESID/PAGID (1999) criteria had a category of possible CVID for patients not meeting the complete criteria for CVID (Table 1). The revised ESID registry (2014) criteria have a category of unclassified hypogammaglobulinemia for patients who do not meet all the criteria for CVID. Studies of cohorts of hypogammaglobulinemia patients will indicate if these categories are equivalent to patients with HGUS in the Ameratunga et al. (2013) criteria.
Like the previous ESID/PAGID (1999) criteria, diagnosis has not been linked to eligibility to IVIG/SCIG treatment in the revised ESID registry (2014) criteria, as these are intended for clinical research rather than therapy. However, it is inevitable these criteria will be used by clinicians to determine which patients will qualify for immunoglobulin treatment in the absence of specific national guidelines. Cohorts such as the NZ CVID/hypogammaglobulinemia study will be important in determining if the revised ESID registry (2014) criteria can be used to determine eligibility for treatment. It will be important to undertake prospective head to head comparisons of these criteria in hypogammaglobulinemic patients as we have done for memory B cells in CVID (73).
The latter two (Ameratunga et al. 2013 and the revised ESID registry 2014) criteria are based on the framework established by the ESID/PAGID 1999 diagnostic criteria. Most patients diagnosed with CVID will have substantially reduced IgG, reduction in other isotypes, impaired memory B cells, and impaired vaccine responses. The majority will thus qualify as having CVID by all three criteria. It should be noted that none of these criteria address partial antibody deficiency syndromes. This is an area that will need to be addressed in future studies.
It will be important to determine how these criteria perform in real-life clinical situations in ethnically diverse populations across the globe. We expect most if not all patients deemed to have idiopathic primary hypogammaglobulinemia, will qualify as having CVID, which will secure their eligibility for treatment with IVIG/SCIG (45, 68). Equally, it is hoped fewer asymptomatic patients with mild hypogammaglobulinemia will be treated with IVIG/SCIG.
Regardless of which criteria are used, it is essential that sound clinical judgment is exercised when diagnosing and treating these patients. It will be important to offer IVIG/SCIG for patients with bronchiectasis with higher levels of IgG for example (sHGUS) (76). Similarly, when assessing the cause of recurrent infections, there may be other contributing factors, which could be potentially treated in patients with hypogammaglobulinemia. The immune defect may not be the dominant predisposing factor for infections in some such individuals. Correcting functional or anatomical predisposing factors such as chronic sinus disease may reduce the number of infections and the patient may not need to be treated with IVIG/SCIG in spite of the hypogammaglobulinemia. This is why it is critical for these patients to be under the care of experienced clinical immunologists.
Rohan Ameratunga conceived the idea of the new diagnostic and treatment criteria for CVID. Maia Brewerton, Charlotte Slade, Anthony Jordan, David Gillis, Richard Steele, Wikke Koopmans, and See-Tarn Woon contributed references and made editorial changes necessary to support the manuscript.
Rohan Ameratunga has received an unrestricted educational grant from Octapharma. The other co-authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank our patients for participating in these studies for the benefit of others. In turn, we hope our analysis of various CVID diagnostic and treatment criteria will assist our patients, colleagues, and funders of IVIG/SCIG. We thank the Auckland Medical Research Foundation and the Paykel trust for grant support. We thank colleagues from around the world for their valuable comments.
1. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol (1999) 92(1):34–48. doi:10.1006/clim.1999.4725
2. Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q J Med (1993) 86(1):31–42.
3. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol (2007) 27(3):308–16. doi:10.1007/s10875-007-9075-1
4. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood (2008) 112(2):277–86. doi:10.1182/blood-2007-11-124545
5. Oksenhendler E, Gérard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis (2008) 46(10):1547–54. doi:10.1086/587669
6. Park JH, Resnick ES, Cunningham-Rundles C. Perspectives on common variable immune deficiency. Ann N Y Acad Sci (2011) 1246:41–9. doi:10.1111/j.1749-6632.2011.06338.x
7. Gathmann B, Mahlaoui N; CEREDIH, Gérard L, Oksenhendler E, Warnatz K, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 134(1):116–26.e. doi:10.1016/j.jaci.2013.12.1077
8. Ballow M. Vaccines in the assessment of patients for immune deficiency. J Allergy Clin Immunol (2012) 130(1):283–4.e. doi:10.1016/j.jaci.2012.04.028
9. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood (2008) 111(1):77–85. doi:10.1182/blood-2007-06-091744
10. Malphettes M, Gérard L, Carmagnat M, Mouillot G, Vince N, Boutboul D, et al. Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin Infect Dis (2009) 49(9):1329–38. doi:10.1086/606059
11. Popa V. Lymphocytic interstitial pneumonia of common variable immunodeficiency. Ann Allergy (1988) 60(3):203–6.
12. Ameratunga R, Becroft DM, Hunter W. The simultaneous presentation of sarcoidosis and common variable immune deficiency. Pathology (2000) 32(4):280–2. doi:10.1080/713688933
13. Fasano MB, Sullivan KE, Sarpong SB, Wood RA, Jones SM, Johns CJ, et al. Sarcoidosis and common variable immunodeficiency. Report of 8 cases and review of the literature. Medicine (Baltimore) (1996) 75(5):251–61. doi:10.1097/00005792-199609000-00002
14. Fuss IJ, Friend J, Yang Z, He JP, Hooda L, Boyer J, et al. Nodular regenerative hyperplasia in common variable immunodeficiency. J Clin Immunol (2013) 33(4):748–58. doi:10.1007/s10875-013-9873-6
15. Malamut G, Ziol M, Suarez F, Beaugrand M, Viallard JF, Lascaux AS, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol (2008) 48(1):74–82. doi:10.1016/j.jhep.2007.08.011
16. Luzi G, Zullo A, Iebba F, Rinaldi V, Sanchez Mete L, Muscaritoli M, et al. Duodenal pathology and clinical-immunological implications in common variable immunodeficiency patients. Am J Gastroenterol (2003) 98(1):118–21. doi:10.1111/j.1572-0241.2003.07159.x
17. Malamut G, Verkarre V, Suarez F, Viallard JF, Lascaux AS, Cosnes J, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterol (2010) 105(10):2262–75. doi:10.1038/ajg.2010.214
18. Agarwal S, Smereka P, Harpaz N, Cunningham-Rundles C, Mayer L. Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflamm Bowel Dis (2011) 17(1):251–9. doi:10.1002/ibd.21376
19. Daniels JA, Lederman HM, Maitra A, Montgomery EA. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): a clinicopathologic study and review. Am J Surg Pathol (2007) 31(12):1800–12. doi:10.1097/PAS.0b013e3180cab60c
20. Quinti I, Soresina A, Guerra A, Rondelli R, Spadaro G, Agostini C, et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol (2011) 31(3):315–22. doi:10.1007/s10875-011-9511-0
21. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol (2013) 174(2):203–11. doi:10.1111/cei.12178
22. Agarwal S, Cunningham-Rundles C. Assessment and clinical interpretation of reduced IgG values. Ann Allergy Asthma Immunol (2007) 99(3):281–3. doi:10.1016/S1081-1206(10)60665-5
23. Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol (2009) 145(6):709–27. doi:10.1111/j.1365-2141.2009.07669.x
24. Knight AK, Cunningham-Rundles C. Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun Rev (2006) 5(2):156–9. doi:10.1016/j.autrev.2005.10.002
25. Abrahamian F, Agrawal S, Gupta S. Immunological and clinical profile of adult patients with selective immunoglobulin subclass deficiency: response to intravenous immunoglobulin therapy. Clin Exp Immunol (2010) 159(3):344–50. doi:10.1111/j.1365-2249.2009.04062.x
26. Olinder-Nielsen AM, Granert C, Forsberg P, Friman V, Vietorisz A, Björkander J. Immunoglobulin prophylaxis in 350 adults with IgG subclass deficiency and recurrent respiratory tract infections: a long-term follow-up. Scand J Infect Dis (2007) 39(1):44–50. doi:10.1080/00365540600951192
27. Musher DM, Manof SB, Liss C, McFetridge RD, Marchese RD, Bushnell B, et al. Safety and antibody response, including antibody persistence for 5 years, after primary vaccination or revaccination with pneumococcal polysaccharide vaccine in middle-aged and older adults. J Infect Dis (2010) 201(4):516–24. doi:10.1086/649839
28. Koopmans W, Woon ST, Brooks AE, Dunbar PR, Browett P, Ameratunga R. Clinical variability of family members with the C104R mutation in transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI). J Clin Immunol (2013) 33(1):68–73. doi:10.1007/s10875-012-9793-x
29. Grabenstein JD, Manoff SB. Pneumococcal polysaccharide 23-valent vaccine: long-term persistence of circulating antibody and immunogenicity and safety after revaccination in adults. Vaccine (2012) 30(30):4435–44. doi:10.1016/j.vaccine.2012.04.052
30. Tiller TL Jr, Buckley RH. Transient hypogammaglobulinemia of infancy: review of the literature, clinical and immunologic features of 11 new cases, and long-term follow-up. J Pediatr (1978) 92(3):347–53. doi:10.1016/S0022-3476(78)80417-X
31. Pan-Hammarstrom Q, Salzer U, Du L, Björkander J, Cunningham-Rundles C, Nelson DL, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet (2007) 39(4):429–30. doi:10.1038/ng0407-429
32. Salzer U, Bacchelli C, Buckridge S, Pan-Hammarström Q, Jennings S, Lougaris V, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood (2009) 113(9):1967–76. doi:10.1182/blood-2008-02-141937
33. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (pan-American group for immunodeficiency) and ESID (European society for immunodeficiencies). Clin Immunol (1999) 93(3):190–7. doi:10.1006/clim.1999.4799
34. Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O, Ledue TB, Craig WY. Reference distributions for immunoglobulins A, G, and M: a practical, simple, and clinically relevant approach in a large cohort. J Clin Lab Anal (1998) 12(6):363–70. doi:10.1002/(SICI)1098-2825(1998)12:6<371::AID-JCLA7>3.0.CO;2-T
35. van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJ, van Tol MJ, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med (2006) 354(18):1901–12. doi:10.1056/NEJMoa051568
36. van Zelm MC, Smet J, Adams B, Mascart F, Schandené L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest (2010) 120(4):1265–74. doi:10.1172/JCI39748
37. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol (2003) 4(3):261–8. doi:10.1038/ni902
38. Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest (2010) 120(1):214–22. doi:10.1172/JCI40231
39. Ameratunga R, Woon ST, Brewerton M, Koopmans W, Jordan A, Brothers S, et al. Primary immune deficiency disorders in the South Pacific: the clinical utility of a customized genetic testing program in New Zealand. Ann N Y Acad Sci (2011) 1238:53–64. doi:10.1111/j.1749-6632.2011.06238.x
40. Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2011) 2:54. doi:10.3389/fimmu.2011.00054
41. Ameratunga R, Woon ST. Customised molecular diagnosis of primary immune deficiency disorders in New Zealand: an efficient strategy for a small developed country. N Z Med J (2009) 122(1304):46–53.
42. Orange JS, Glessner JT, Resnick E, Sullivan KE, Lucas M, Ferry B, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol (2011) 127(6):1360–7.e. doi:10.1016/j.jaci.2011.02.039
43. Sekine H, Ferreira RC, Pan-Hammarström Q, Graham RR, Ziemba B, de Vries SS, et al. Role for Msh5 in the regulation of Ig class switch recombination. Proc Natl Acad Sci U S A (2007) 104(17):7193–8. doi:10.1073/pnas.0700815104
44. Smith J, Fernando T, McGrath N, Ameratunga R. Lamotrigine-induced common variable immune deficiency. Neurology (2004) 62(5):833–4. doi:10.1212/01.WNL.0000113754.29225.5D
45. Duraisingham SS, Buckland MS, Grigoriadou S, Longhurst HJ. Secondary antibody deficiency. Expert Rev Clin Immunol (2014) 10(5):583–91. doi:10.1586/1744666X.2014.902314
46. Empson M, Sinclair J, O’Donnell J, Ameratunga R, Fitzharris P, Steele R, et al. The assessment and management of primary antibody deficiency. N Z Med J (2004) 117(1195):U914.
47. de Silva NR, Gunawardena S, Rathnayake D, Wickramasingha GD. Spectrum of primary immunodeficiency disorders in Sri Lanka. Allergy Asthma Clin Immunol (2013) 9(1):50. doi:10.1186/1710-1492-9-50
48. Agarwal S, Cunningham-Rundles C. Treatment of hypogammaglobulinemia in adults: a scoring system to guide decisions on immunoglobulin replacement. J Allergy Clin Immunol (2013) 131(6):1699–701. doi:10.1016/j.jaci.2013.01.036
49. Goldacker S, Draeger R, Warnatz K, Huzly D, Salzer U, Thiel J, et al. Active vaccination in patients with common variable immunodeficiency (CVID). Clin Immunol (2007) 124(3):294–303. doi:10.1016/j.clim.2007.04.011
50. Thierry-Carstensen B, Jordan K, Uhlving HH, Dalby T, Sørensen C, Jensen AM, et al. A randomised, double-blind, non-inferiority clinical trial on the safety and immunogenicity of a tetanus, diphtheria and monocomponent acellular pertussis (TdaP) vaccine in comparison to a tetanus and diphtheria (Td) vaccine when given as booster vaccinations to healthy adults. Vaccine (2012) 30(37):5464–71. doi:10.1016/j.vaccine.2012.06.073
51. Ladhani S, Ramsay M, Flood J, Campbell H, Slack M, Pebody R, et al. Haemophilus influenzae serotype B (Hib) seroprevalence in England and Wales in 2009. Euro Surveill (2012) 17(46):20313.
52. Hawdon N, Nix EB, Tsang RS, Ferroni G, McCready WG, Ulanova M. Immune response to Haemophilus influenzae type b vaccination in patients with chronic renal failure. Clin Vaccine Immunol (2012) 19(6):967–9. doi:10.1128/CVI.00101-12
53. Dentinger CM, Hennessy TW, Bulkow LR, Reasonover AL, Romero-Steiner S, Holder PF, et al. Immunogenicity and reactogenicity to Haemophilus influenzae type B (Hib) conjugate vaccine among rural Alaska adults. Hum Vaccin (2006) 2(1):24–8. doi:10.4161/hv.2.1.2445
54. Orange JS, Ballow M, Stiehm ER, Ballas ZK, Chinen J, De La Morena M, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the basic and clinical immunology interest section of the American academy of allergy, asthma & immunology. J Allergy Clin Immunol (2012) 130(3 Suppl):S1–24. doi:10.1016/j.jaci.2012.07.002
55. Lee H, Nahm MH, Kim KH. The effect of age on the response to the pneumococcal polysaccharide vaccine. BMC Infect Dis (2010) 10:60. doi:10.1186/1471-2334-10-60
56. O’Brien KL, Hochman M, Goldblatt D. Combined schedules of pneumococcal conjugate and polysaccharide vaccines: is hyporesponsiveness an issue? Lancet Infect Dis (2007) 7(9):597–606. doi:10.1016/S1473-3099(07)70210-4
57. Henckaerts I, Goldblatt D, Ashton L, Poolman J. Critical differences between pneumococcal polysaccharide enzyme-linked immunosorbent assays with and without 22F inhibition at low antibody concentrations in pediatric sera. Clin Vaccine Immunol (2006) 13(3):356–60. doi:10.1128/CVI.13.3.356-360.2006
58. Concepcion NF, Frasch CE. Pneumococcal type 22f polysaccharide absorption improves the specificity of a pneumococcal-polysaccharide enzyme-linked immunosorbent assay. Clin Diagn Lab Immunol (2001) 8(2):266–72.
59. Balloch A, Licciardi PV, Tang ML. Serotype-specific anti-pneumococcal IgG and immune competence: critical differences in interpretation criteria when different methods are used. J Clin Immunol (2013) 33(2):335–41. doi:10.1007/s10875-012-9806-9
60. Jódar L, Butler J, Carlone G, Dagan R, Goldblatt D, Käyhty H, et al. Serological criteria for evaluation and licensure of new pneumococcal conjugate vaccine formulations for use in infants. Vaccine (2003) 21(23):3265–72. doi:10.1016/S0264-410X(03)00230-5
61. Lee LH, Frasch CE, Falk LA, Klein DL, Deal CD. Correlates of immunity for pneumococcal conjugate vaccines. Vaccine (2003) 21(17–18):2190–6. doi:10.1016/S0264-410X(03)00025-2
62. Ameratunga R, Woon ST, Neas K, Love DR. The clinical utility of molecular diagnostic testing for primary immune deficiency disorders: a case based review. Allergy Asthma Clin Immunol (2010) 6(1):12. doi:10.1186/1710-1492-6-12
63. Jokinen JT, Ahman H, Kilpi TM, Mäkelä PH, Käyhty MH. Concentration of antipneumococcal antibodies as a serological correlate of protection: an application to acute otitis media. J Infect Dis (2004) 190(3):545–50. doi:10.1086/422531
64. Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol (2005) 94(5 Suppl 1):S1–63. doi:10.1016/S1081-1206(10)61142-8
65. Paris K, Sorensen RU. Assessment and clinical interpretation of polysaccharide antibody responses. Ann Allergy Asthma Immunol (2007) 99(5):462–4. doi:10.1016/S1081-1206(10)60572-8
66. Rezaei N, Siadat SD, Aghamohammadi A, Moin M, Pourpak Z, Norouzian D, et al. Serum bactericidal antibody response 1 year after meningococcal polysaccharide vaccination of patients with common variable immunodeficiency. Clin Vaccine Immunol (2010) 17(4):524–8. doi:10.1128/CVI.00389-09
67. Ochs HD, Davis SD, Wedgwood RJ. Immunologic responses to bacteriophage phi-X 174 in immunodeficiency diseases. J Clin Invest (1971) 50(12):2559–68. doi:10.1172/JCI106756
68. Driessen GJ, Dalm VA, van Hagen PM, Grashoff HA, Hartwig NG, van Rossum AM, et al. Common variable immunodeficiency and idiopathic primary hypogammaglobulinemia: two different conditions within the same disease spectrum. Haematologica (2013) 98(10):1617–23. doi:10.3324/haematol.2013.085076
69. Chapel H, Lucas M, Patel S, Lee M, Cunningham-Rundles C, Resnick E, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol (2012) 130(5):1197–8.e. doi:10.1016/j.jaci.2012.05.046
70. Seppanen M, Aghamohammadi A, Rezaei N. Is there a need to redefine the diagnostic criteria for common variable immunodeficiency? Expert Rev Clin Immunol (2013) 10(1):1–5. doi:10.1586/1744666X.2014.870478
71. Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol (2011) 111:47–107. doi:10.1016/B978-0-12-385991-4.00002-7
72. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for CVID. Expert Rev Clin Immunol (2014) 10(2):183–6. doi:10.1586/1744666X.2014.875274
73. Koopmans W, Woon ST, Zeng IS, Jordan A, Brothers S, Browett P, et al. Variability of memory B cell markers in a cohort of common variable immune deficiency patients over six months. Scand J Immunol (2013) 77(6):470–5. doi:10.1111/sji.12028
74. Ameratunga RV, Casey P, Parry S, Kendi C. Hypogammaglobulinemia factitia – Munchausen syndrome presenting as common variable immune deficiency. Allergy Asthma Clin Immunol (2013) 9(1):36. doi:10.1186/1710-1492-9-36
75. Ameratunga R, Lindsay K, Woon S-T, Jordan A, Anderson NE, Koopmans W. New diagnostic criteria could distinguish common variable immunodeficiency disorder from anticonvulsant-induced hypogammaglobulinemia. Clin Exp Neuroimmunology (in press). doi:10.1111/cen3.12135
Keywords: common variable immunodeficiency, diagnostic criteria, HGUS
Citation: Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, Koopmans W and Woon S-T (2014) Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front. Immunol. 5:415. doi: 10.3389/fimmu.2014.00415
Received: 30 June 2014; Accepted: 17 August 2014;
Published online: 15 September 2014.
Edited by:
Luigi Daniele Notarangelo, Harvard Medical School, USAReviewed by:
Antonio Condino-Neto, University of São Paulo, BrazilCopyright: © 2014 Ameratunga, Brewerton, Slade, Jordan, Gillis, Steele, Koopmans and Woon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rohan Ameratunga, Auckland Healthcare Services, Park Road, Grafton, Auckland 1010, New Zealand e-mail:cm9oYW5hQGFkaGIuZ292dC5ueg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.