 Maximilian Muenchhoff
Maximilian Muenchhoff Andrew J. Prendergast2,3
Andrew J. Prendergast2,3
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 August 2014
Sec. Vaccines and Molecular Therapeutics
Volume 5 - 2014 | https://doi.org/10.3389/fimmu.2014.00391
This article is part of the Research TopicUnderstanding the Ontogeny of the Immune System to Promote Immune-Mediated Health for LifeView all 20 articles
The developing immune system is adapted to the exposure to a plethora of pathogenic and non-pathogenic antigens encountered in utero and after birth, requiring a fine balance between protective immunity and immune tolerance. In early stages of life, this tolerogenic state of the innate and adaptive immune system and the lack of immunological memory render the host more susceptible to infectious pathogens like HIV. HIV pathogenesis is different in children, compared to adults, with more rapid disease progression and a substantial lack of control of viremia compared to adults. Plasma viral load remains high during infancy and only declines gradually over several years in line with immune maturation, even in rare cases where children maintain normal CD4 T-lymphocyte counts for several years without antiretroviral therapy (ART). These pediatric slow progressors also typically show low levels of immune activation despite persistently high viremia, resembling the phenotype of natural hosts of SIV infection. The lack of immunological memory places the fetus and the newborn at higher risk of infections; however, it may also provide an opportunity for unique interventions. Frequencies of central memory CD4+ T-lymphocytes, one of the main cellular reservoirs of HIV, are very low in the newborn child, so immediate ART could prevent the establishment of persistent viral reservoirs and result in “functional cure.” However, as recently demonstrated in the case report of the “Mississippi child” who experienced viral rebound after more than 2 years off ART, additional immunomodulatory strategies might be required for sustained viral suppression after ART cessation. In this review, we discuss the interactions between HIV and the developing immune system in children and the potential implications for therapeutic and prophylactic interventions.
In the dynamic developmental period from fetal life to adolescence, the human organism is more susceptible and vulnerable to chronic viral infections (1). HIV pathogenesis in early life is characterized by faster disease progression and shorter time to AIDS and death, compared to adults. The innate and adaptive arms of the immune system are more tolerogenic soon after birth and fail to control viral replication in infancy. Persistent viremia, reactivation of co-infections, and microbial translocation during chronic infection drive HIV pathogenesis through increased immune activation, resulting in immune dysregulation and functional immune exhaustion that exacerbate disease progression in a positive feedback loop (2–4) as visualized in Figure 1.
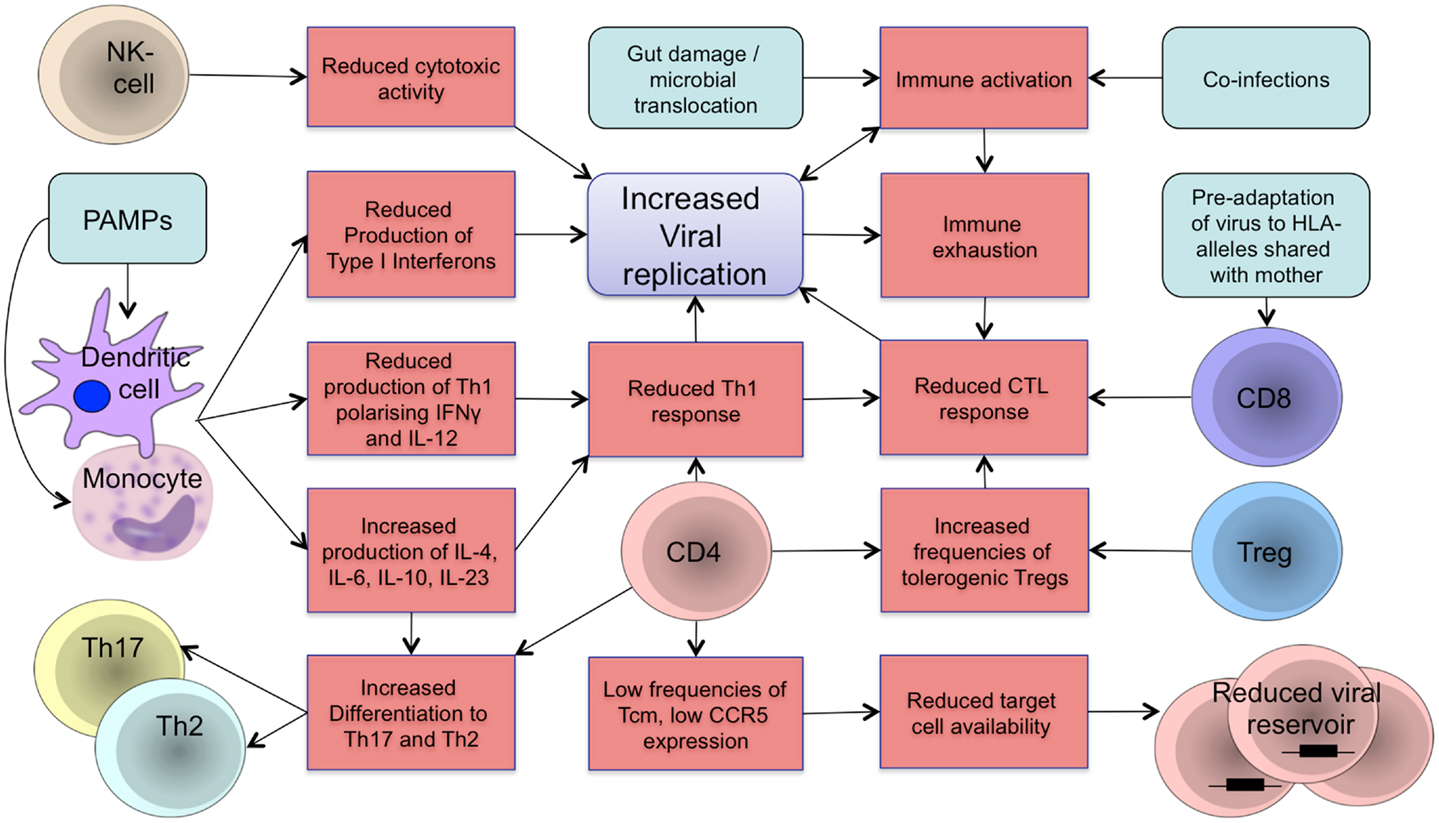
Figure 1. Schematic representation of components of the developing immune system in HIV infection. The developing immune system is characterized by an attenuated pro-inflammatory innate immune response with a shift toward Th2/Th17 polarizing cytokines to protect the organism against extracellular pathogens and reduce the risk of autoimmunity and inflammation. A tolerogenic state of the immune system is supported by increased frequencies of regulatory T-cells that suppress the activity of CD4 and CD8 T-cells. Microbial translocation and viral replication result in immune activation and immune exhaustion that lead to immune dysfunction and loss of immune control resulting in further viral replication in a positive feedback loop. Differences in target cell availability and memory differentiation in early life affect the size and composition of the viral reservoir in children. Main aspects of the developing immune system in regards to vertical HIV infection are summarized. Treg: regulatory T-cells; NK cell: natural killer cell; PAMPs: pathogen-associated molecular patterns; CTL: cytotoxic T-lymphocyte.
However, the recent report of a child who apparently achieved “functional cure” for more than 2 years following early therapeutic intervention (5) has raised major interest in the development of therapeutic strategies that could potentially be applied for cure even beyond the pediatric setting. In this review, we will discuss immunity to HIV in early life and how it affects pathogenesis and disease outcome, but also offers unique opportunities for prophylactic and therapeutic interventions in pediatric HIV infection.
Of the estimated 35 million people living with HIV today, 3.3 million are children (UNAIDS 2013 report). Administration of combination antiretroviral therapy (ART) can reduce risk of transmission to approximately 1–2% (6) and WHO now recommends the use of triple-drug ART regimens in all pregnant and breastfeeding women regardless of CD4 count, either lifelong (formerly referred to as option B+) or only during pregnancy and breastfeeding (formerly referred to as option B) (7). Despite increasing availability of ART, many women are diagnosed with HIV late in pregnancy and prevention of mother-to-child transmission (PMTCT) coverage rates remain suboptimal in many countries, resulting in 260,000 children becoming newly infected with HIV in 2012.
Without PMTCT, the overall risk of HIV transmission is up to 40% in sub-Saharan Africa; infection can occur in utero (5–10%), intrapartum (15%), or post-partum via breastfeeding (15%) (8). Interestingly, MTCT rates are much lower (<7%) in the natural hosts of SIV infection (such as sooty mangabeys), compared to humans (30–40%) or rhesus macaques (25–75%) (9). During breastfeeding, no transmissions have been observed in sooty mangabeys, despite high plasma and breast milk viral loads (10). This has been attributed to paucity of CCR5+ CD4+ target cells for SIV infection (11), since the vast majority of vertically transmitted SIV/HIV-strains are R5-tropic and therefore depend on CCR5 as a co-receptor to infect CD4+ T-cells (12). Indeed, a recent study has shown very low to absent populations of CCR5+ CD4+ T-cells in the peripheral blood compartment and especially in the intestinal tract of sooty mangabeys compared to rhesus macaques (13). In human infants, CCR5+ CD4+ T-cells are virtually absent in cord blood (14). However, CCR5+ CD4+ T-cells are abundant in the intestinal mucosa and these cells are highly susceptible to HIV infection (15). The increased availability of target cells in the intestinal tract, in combination with the effectiveness of the placental barrier prior to labor, are both likely to contribute to the higher risk of intra- and post-partum, compared to in utero, transmission.
Once infection is established, the course of disease progression in vertically infected infants is generally faster than in adults, with a rapid decline of CD4 cells and onset of recurrent infections, failure to thrive, and delayed neurodevelopment. In adult HIV infection, survival times decrease with increasing age at seroconversion, with a median time to AIDS and death of approximately a decade without ART (16). In contrast, mortality among vertically infected children in Africa exceeds 50% by the age of 2 years in the absence of ART (17). However, precise timing of transmission is an important determinant of survival: in a recent meta-analysis, 1-year mortality was 26% among children infected postnatally through breastfeeding, compared to 52% if infected perinatally (18). Another study that distinguished in utero, intrapartum and postnatal infection found median survival times from infection of 208, 380, and >500 days, respectively (19). Furthermore, lower mortality rates have been observed that the later HIV is acquired postnatally via breastfeeding (20). Interestingly, a cohort of children with hemophilia, who acquired HIV infection between the age of 5 and 14 years, had significantly longer AIDS-free survival times than adults (16). Long-term survival rates in vertically infected untreated children are estimated to be higher than initially appreciated with a probability of 20–30% surviving up to 10 years if infected perinatally and 16 years if infected via breastfeeding (20, 21).
Absolute CD4 count and viral load levels, which are well established clinical markers of disease progression in adults, have to be interpreted differently in children. The absolute CD4 count is three- to fourfold higher in the newborn compared to adults and gradually declines with age to reach adult levels after about 6 years or later (22, 23). The percentage of CD4+ T-cells is less variable with age and often used as an indicator of CD4 depletion in young children. HIV-1 plasma RNA levels are generally higher in vertically infected children than in adults and peak viremia (in the region of 5–7 log copies per milliliter) is observed around 3 months of age (24). Peak viremia is higher in children infected intrapartum than in utero (25) and lower in postnatally infected children, suggesting improved viral control by the more mature immune system (26, 27). Whereas in adult infection, a steady state viral set point is reached within several weeks of infection (28, 29), viremia persists at high levels, and only declines slowly until a quasi-set point is reached after several years in children who survive (30). This gradual control of viral replication has been associated with increasing maturation and development of the immune system (1).
In the initial response to infections, the innate immune system performs two broad roles. On the one hand, it directly inactivates or attenuates invading pathogens, and on the other hand, it instructs the adaptive arm of the immune system to generate appropriate effector responses. Innate immune cells, such as macrophages, monocytes, and dendritic cells interact with pathogen-associated molecular patterns (PAMPs) through pattern recognition receptors (PRRs) that have evolved as substrate-specific receptors that can discriminate between self- and non-self and constantly sample the intra- and extracellular milieu [reviewed in Ref. (31)]. PRRs include the Toll-like receptors (TLRs), which are mainly located within the membranes of the cell surface and endosomes, and intracellular sensors such as RIG-1 like receptors (RLRs). HIV is principally detected by the innate immune system through the recognition of viral nucleic acids that are present in the cytosol of productively infected cells during the viral replication cycle and in the endosomes of phagocytosing cells. For example, cell-free virus that is phagocytosed by dendritic cells is recognized in the endosome by TLR-7 and TLR-9, which bind single-stranded RNA and DNA, respectively. Most PAMP/PRR interactions activate signaling pathways that converge in transcriptional activation of pro-inflammatory cytokines and type-1 interferons (IFNs). Type 1 IFNs bind to receptors of infected and neighboring cells and induce expression of IFN-stimulated genes (ISGs) that synergistically inhibit viral replication and spread (32).
The importance of these innate immune mechanisms in viral infections has been demonstrated in children with gene mutations in the TLR3 signaling pathway, who are prone to severe herpes simplex encephalitis (33). Certain polymorphisms in the TLR-9 gene were associated with increased risk of mother-to-child transmission of HIV in one study (34), but further independent studies on innate immune signaling in pediatric HIV infection are required. Genetic polymorphisms of APOBEC3G, a host restriction factor that is induced by type-1 IFNs and directly inhibits replication of HIV, are associated with increased viral loads (35), lower CD4 counts (36), and more rapid disease progression in HIV-infected children (37).
Antigen-presenting innate immune cells such as dendritic cells play a key role not only in inducing pathways that directly inhibit viral replication but also in regulating the host adaptive immune response to HIV infection. Upon TLR stimulation, neonatal dendritic cells and monocytes are less polyfunctional and produce less type-1 IFNs than adult cells (38). However, it has been noted that neonatal innate immune cells do not have a generalized deficit in secretion of pro-inflammatory cytokines, but rather display distinct cytokine profiles to shape differential adaptive immune responses compared to adults. The most consistently reported difference in neonatal innate immune cell cytokine patterns is a relatively low production of Th1-polarizing cytokines, such as Interleukin-12 and IFN-gamma, in favor of Th2- and Th17-inducing Interleukin-10, Interleukin-6, and Interleukin-23 (38, 39) [reviewed in Ref. (40)]. This immunoregulatory strategy has likely evolved for two reasons: first, to dampen pro-inflammatory Th1 responses that could be damaging in utero and after birth when the newborn is exposed to an abundance of newly encountered antigens; and second, to ensure protection against extracellular bacterial and fungal pathogens, which can be rapidly fatal soon after birth (40). However, as a trade-off, the skewed Th1/Th2 ratio leaves the infant more susceptible to intracellular pathogens such as HIV [reviewed in Ref. (1)]. Interestingly, HIV-exposed but uninfected children show enhanced pro-inflammatory cytokine responses upon stimulation of innate immune cells compared to unexposed children, indicating that altered innate immune ontogeny could contribute to the increased vulnerability of this population to morbidity and poor growth (41).
The intrinsically low level of Th1 responses induced by neonatal innate immune cells is reinforced by the dysregulation and depletion of dendritic cells upon HIV infection [reviewed in Ref. (42)]. Most studies have been undertaken in chronically HIV-infected adults, but perinatally infected children also have reduced frequencies and functionality of dendritic cells and other innate immune cell populations including natural killer (NK) cells (43–45).
Natural killer cells are innate lymphocytes that play a critical role in the control of viral infections through interactions with other host immune cells via the secretion of chemokines and cytokines or by direct cellular cytotoxicity against infected target cells [reviewed in Ref. (46)]. NK cells recognize infected cells either through interaction with antibodies bound to the target cell surface in a process termed antibody-dependent cellular cytotoxicity (ADCC) or through interactions of an intricate network of inhibitory and activating cell surface receptors with ligands on infected cells. For example, NK cells are activated by reduced expression levels of MHC-class-I-molecules on the cell surface of HIV-infected cells via signaling of the inhibitory killer cell Ig-like receptors (KIRs), which is an important complementary mechanism to CD8 T-cell responses that depend on MHC-class-I-recognition [reviewed in Ref. (47)].
Frequencies of neonatal NK cells are similar to adults (48), but there are differences in expression patterns of inhibitory and activating cell surface receptors (49) and reduced cytotoxic activity of cord blood NK cells has been reported consistently (50, 51) [reviewed in Ref. (52)]. NK cells isolated from vertically HIV-infected children have decreased cytolytic activity compared to uninfected children (53) and exhibit reduced ADCC compared to adult cells (54), but neonatal NK cells have been shown to be capable of suppressing replication of CCR5-tropic HIV in vitro through a non-cytotoxic chemokine-mediated mechanism (55). Total numbers of NK cells in vertically HIV-infected children decline with age (56) and are only partially reconstituted upon initiation of ART (45). Vertical HIV infection is also associated with increased NK cell activation, differentiation, and loss of perforin expression, which might impair NK cell function and compromise their role in protecting the organism against HIV, co-infections, and cancer (56).
The maternal and fetal immune systems are finely adapted toward increased immunological tolerance to avoid uncontrolled damage to the fetus by allogeneic T-cells from the mother on the one hand and pathological reactions to maternal- and self-antigens by the fetus on the other (1). There is immune adaptation away from Th1 and toward Th2 responses during pregnancy (57), although the concept of pregnancy as a generalized state of immunosuppression is no longer supported (58, 59): for example, pregnant women appear to respond well to vaccines during pregnancy (60–62), and even mount anti-fetal CD8+ T-cell response without pregnancy loss (63). The hyporesponsiveness of the fetal adaptive immune system to antigens was previously interpreted as a functional deficit of effector cells, whereas currently the induction of immune tolerance is thought to be an active process that is largely mediated by regulatory T-cells (Tregs) [reviewed in Ref. (64)]. After depletion of CD4+ CD25+ FoxP3+ Tregs, fetal CD4 and CD8 T-cells have been shown to be highly responsive to stimulation ex vivo (65). In utero exposure to maternal alloantigens induces the development of tolerogenic Tregs that suppress activity of effector T-cells and persist into adulthood (66). The abundant population of Tregs that peaks in the second trimester and gradually declines to adult levels around birth (67) has been shown to originate from fetal hematopoetic stem cells (HSC); these differ from adult HSC based on functional and transcriptional analyses and give rise to a distinct lymphocyte lineage (68).
The role of Tregs in the pathogenesis of HIV infection remains controversial, but several studies have proposed a modulating function in HIV-specific immune responses, chronic immune activation, and inflammation [reviewed in (69)]. Most studies have reported a decrease in absolute numbers of Tregs in chronic HIV infection; however, as the frequency of Tregs is often reported as a percentage of CD4+ T-cells, there appears to be a selective expansion of different Treg subsets within the CD4 population during the CD4 decline that characterizes chronic infection (70). In HIV-infected children, one study found a positive correlation between the proportion of Tregs and HIV viral load, and a negative correlation with CD4 count suggesting a selective expansion of Tregs in pediatric HIV infection (71). Interestingly, in the same study these increased frequencies of Tregs also correlated with the proportion of activated CD8+ T-cells, suggesting the ineffectiveness of these cells to limit immune activation. However, another study in vertically HIV-infected children, showed a depletion of Tregs compared to HIV-negative children when measured as proportion of total T-cells (CD3+ lymphocytes) that was associated with increased levels of immune activation (72). Another study found an association between altered frequencies of Treg subsets and autoantibody production in HIV-positive children highlighting the potential role of Tregs in immunoregulatory pathways that may be disrupted in HIV infection (73). In HIV-exposed but uninfected infants, a significant increase in HIV-specific CD4+ and CD8+ T-cell responses was observed after depletion of Tregs in cord blood mononuclear cells in vitro (73, 74) indicating a suppressive function in pathogen-specific adaptive immunity.
HIV-specific CD8+ T cells are thought to play a key role in control of viral replication based on studies in humans (75, 76) and SIV-infected monkey models (77, 78). In adults, the rapid decline in viral load during acute infection from several million copies/ml to a viral load set point 3 logs lower is temporally associated with the appearance of HIV-specific CD8+ T-cells (75, 76, 79). Cytotoxic CD8+ T cells interact directly with HIV-infected target cells via the T-cell receptor that recognizes viral peptides presented on the cell surface by MHC-class-I molecules. The HLA-class-I type of an individual affects the rate of disease progression: certain HLA alleles such as HLA-B35 are associated with more rapid progression to AIDS, whereas protective alleles such as HLA-B57 and HLA-B27 are associated with viremic control and slow disease progression (80). One of the main mechanisms proposed for the protective effect of these alleles is their peptide binding specificity, allowing CD8+ T cells to target epitopes located particularly in the Gag region, in which an escape mutation that would evade the immune response is associated with a high cost in viral fitness (81, 82). Transmission of viral variants with an accumulation of costly escape mutations that reduce viral replicative capacity facilitates containment of HIV in the new host and reduces viral load and loss of CD4 cells (83, 84).
In pediatric HIV infection, the HLA type of both mother and child is of relevance for disease progression in the child. In mothers with protective HLA alleles, the virus accumulates immune escape mutations that are associated with a loss of viral fitness, thereby attenuating the transmitted virus, resulting in slower disease progression in the child (85). However, as the child inherits 50% of the HLA alleles from the mother, the virus can be partially pre-adapted to the shared alleles so that the recipient child cannot mount an effective immune response against the escaped epitopes and fails to contain viral replication (86). The beneficial effect of protective HLA alleles is therefore greatest if they are not shared between mother and child, and the child can employ them to mount effective immune responses (85). In a study of vertically HIV-infected children and adolescents, the protective allele HLA-B57 was overrepresented in the group of children with slow disease progression (87). In a recent genomic study of over 1000 vertically HIV-infected children, the protective HLA alleles HLA-B57, -B27, -B14, -Cw8, and -DRB1*10 were the SNPs most strongly associated with low baseline HIV-RNA levels (35).
The adaptive immune system starts to develop as early as 7–9 weeks of gestation when T-cell progenitor cells populate the thymus (88). In vertically HIV-infected children, HIV-specific CD8+ T cell responses can be detected from birth (89, 90), but less frequently and at a lower magnitude than in older children or adults (90–92). However, these responses seem to be insufficient to control viremia, as there is typically no rapid decline in viral load in pediatric HIV infection as opposed to the decline in viral load after acute infection in adults [see above and Ref. (25)]. In infants, neither breadth nor magnitude of HIV-specific CD8 T-cell responses at 1 month of age correlate with viremic control or survival at 12 months (93), but Gag-specific CD8 T cell responses are higher in infants who survive to 1 year of age (94). The magnitude and breadth of Gag-specific CD8 T cell responses correlated negatively with viral load in one study (95), but not in another (96). Also, in a study of older children and adolescents who were categorized as progressors and non-progressors, there was no difference in magnitude and breadth of HIV-specific CD8 T cell responses between the two groups (87). However, magnitude and breadth of HIV-specific CD8 responses increase with age (90, 92, 96).
Broad Gag-specific CD8+ T cell responses have consistently been shown to be associated with reduced viral load (97, 98) in HIV-infected adults. But whereas Gag is the most immunogenic HIV-protein in adults, infants preferentially target epitopes in the more variable proteins Env and Nef (92, 95) with an increasing proportion of Gag-specific responses only later in life (95).
In addition to the specificity and magnitude, the effectiveness of a CD8 T cell response to control viral replication is also determined by its quality as measured by the ability to degranulate and produce different effector cytokines simultaneously (99, 100). The frequency of HIV-specific CD8 T cell that exhibit three different effector functions or more (CD107+, IL-2+, IFN-gamma+) is reduced in children younger than 2 years but increases with age (95). In another study, the functional profile of HIV-specific CD8 T cell responses predicted the rate of subsequent disease progression in perinatally HIV-infected infants showing that more polyfunctional responses were associated with slower disease progression (101).
Other functional measures that have been described for effective CD8 T cell responses include the proliferative capacity of a T cell clone upon antigen stimulation in vitro (102). In children, the proliferative capacity of HIV-specific CD8 T cells has thus far not been studied in detail and published data are only available for CD4 T cells at present.
To maintain fully functional CD8+ T cell responses, the help of CD4+ T cells with the same antigen-specificity is required (103, 104). In the mouse model, depletion of CD4 T helper cells during acute infection results in ineffective CD8 T cell memory responses (105, 106). CD4+ T cells are the major target for HIV and massive depletion of CD4+ T cells results during acute infection in adults (107, 108), with ongoing preferential infection of activated and HIV-specific CD4 T cells in chronic infection (109). This depletion of HIV-specific CD4+ T helper cells has been proposed as a main mechanism for failure to control HIV successfully, given their central role in orchestrating diverse cellular and humoral immune functions (110).
In addition to providing help to CD8 T cells (111), HIV-specific CD4 T cells have been shown to have intrinsic direct cytotoxic activity against infected cells (112–114). Increased breadth, especially of Gag-specific CD4 T cell responses, is inversely correlated to viral load in chronically infected adults; elite or viremic controllers show a higher ratio of Gag- vs. Env-specific CD4 responses (115). Another study in a large cohort of ART-naïve HIV-1-clade-C infected adults also found that the magnitude of Gag-specific CD4 T cell responses was associated with higher CD4 counts and lower viral loads (116).
As discussed above in relation to innate immunity, there is a selective impairment of CD4 T cell responses in early childhood with a shift from Th1 toward Th2 type responses (117, 118) and poor generation of persistent Th1 memory responses (119). This has been attributed to low production of Interleukin-12 (120), which is required for Th1-polarization and maintenance of Th1 effector functions (121, 122), but does not reach adult levels until adolescence. The bias toward Th2 responses in early life is reinforced by the increased capacity of neonatal monocytes and dendritic cells to produce Interleukin-10, an immunomodulatory cytokine that induces a shift of T cell induction toward Th2 and Th17 profiles (38).
During fetal development in non-pathologic sterile conditions, the T cell compartment is considered to be mostly naïve and tolerogenic, although a recent study highlighted the existence of CD4 T cell effector memory subsets with Th1, Th2, and Th17 functional profiles in cord blood of healthy newborn infants (123). HIV-specific CD4 T cell responses can be primed in utero (124), but are detected in infants at significantly lower frequencies than in chronically infected adults and increase with age (92, 95). These low levels of HIV-specific Th1 responses in early life coincide with the Th1/Th2 bias and the expanded population of suppressive regulatory T cells as described above. In the macaque model, SIV-specific CD4 T cell responses are suppressed in infants by regulatory T cells that show greater in vitro suppressive activity and are present at higher frequencies than in adults and have been associated with impaired immune control (125).
Gag-specific CD4 T cell responses were detectable at higher frequencies at 3 months of age in vertically HIV-infected children who survived to 12 months, compared to those who died, and Gag-specific responses at 3 and 6 months of age were negatively correlated with VL (94). Another study detected Gag-specific CD4 responses only in a minority of older vertically infected children (median age 9.9 years). Those with detectable responses had lower viral loads than non-responders and the magnitude of the response was inversely correlated with viral load; no association was observed between CD8 T cell responses and viral load (96).
The functional capability of CD4 T cells to produce IL-2 is causally linked to the proliferative capacity of both CD4 and CD8 T cell subsets, a parameter consistently correlated with immune control and slow disease progression [reviewed in Ref. (126)]. HIV-specific proliferative responses of CD4 T cells are detected only in a fraction of viremic chronically infected adults and are inversely correlated with VL if present (127, 128); IL-2 producing HIV-specific CD4 responses are associated with slow disease progression in adults (129, 130). Interleukin-2 is also important for the differentiation, activation, and proliferation of NK cells, Th1 and Th2 CD4 T cells, B cells, Tregs, and memory CD8 T cells, highlighting the key immunoregulatory role of these CD4 T cells in the immune response (131).
The proliferative capacity of HIV-specific CD4 T cells is selectively impaired in vertically infected children with uncontrolled viral replication (124, 132), but, in contrast to adult infection, can be rescued with administration of ART (133). The magnitude of the response increases with age and is strongest in children maintaining viral suppression on ART (133). Interestingly, proliferative and IL-2 producing CD4 responses were also detected in HIV-exposed uninfected infants but were mostly absent in infected children (134). Polyfunctional HIV-specific CD4 T cell responses, especially Interleukin-2 producing cells, were less frequently detected in younger children, but if present, they were associated with low viral loads and remarkably slow disease progression (95). However, it remains unclear whether these responses actually mediate viral control or are simply a proxy for undisrupted immunoregulatory networks.
Chronic HIV infection and persistent antigenic stimulation lead to more terminally differentiated T cell populations with loss of effector functions and proliferative capacity upon antigenic stimulation [reviewed in Ref. (3)]. This state of “Immune Exhaustion” is characterized by the upregulation of inhibitory co-receptors on CD4 and CD8 T cells such as PD-1 (135, 136), Tim-3 (137), CD160 (138), LAG-3 (139), and CTLA-4 (140) that correlate with markers of disease progression in chronic HIV infection (136, 137, 139–141). As immune functions can be partially restored upon antibody-mediated blockade of these receptors (136, 137, 139, 140), they present an attractive target for immunotherapeutic intervention in HIV infection, although immune-related adverse events remain a concern (142, 143).
In pediatric subjects, few studies have been conducted to elucidate the role of immune exhaustion in chronic HIV infection. One study found an expansion of Tim-3+ CD4 and CD8 T cells in vertically HIV-infected adolescents and a correlation of Tim-3 expression on CD8 T cells with viral load (144). In the same study, PD-1 expression only correlated with the frequency of activated CD8 T cells. This is consistent with another report that described a correlation of PD-1 expression on CD8 T cells with immune activation markers and the magnitude of HIV-specific CD8 T cell responses, but not with viral load, suggesting that immune exhaustion is driven more by chronic immune activation than directly by viral replication (96).
Persistent systemic immune activation plays a key role in the pathogenesis of HIV infection and is regarded as the driving force in generalized immune dysregulation, chronic inflammation (145, 146), depletion of CD4 T cells (147, 148), and progression to AIDS (149) [reviewed in (4)]. Recent studies suggest that much of the CD4 depletion is caused by a process called “pyroptosis,” whereby abortive HIV infection of CD4+ T cells causes a highly inflammatory form of cell death, which drives accumulation of further CD4 cells and an ongoing cycle of inflammation and cell death (150, 151). T cell immune activation, as commonly measured by expression of activation markers such as HLA-DR and CD38 on CD4 and CD8 T cells, is a stronger predictor of disease progression than viral load in adults (152). Similarly, in vertically HIV-infected infants, the subsequent rate of disease progression can be predicted by T cell activation levels at 1–2 months of age (153). Both CD4 and CD8 immune activation show a strong inverse correlation with CD4 percentage, but interestingly no correlation is observed between immune activation and viral load in children (72, 154). In vertically HIV-infected children who maintain high CD4 counts without ART (often referred to as pediatric slow progressors), immune activation levels are strikingly low despite high viral loads (Muenchhoff et al., unpublished data). This resembles the phenotype of non-pathogenic infection in the natural hosts of SIV, such as African Green Monkeys and Sooty Mangabeys (155), in whom CD4 counts are maintained despite high levels of viremia, due to attenuated immune activation (156). In acute infection, sooty mangabeys also show high levels of immune activation and a robust innate immune response that is associated with a generalized upregulation of Interferon Stimulated Genes (ISGs, see innate immunity), but this initial response is rapidly resolved, suggesting the involvement of active immunoregulatory mechanisms that dampen detrimental excessive immune activation (157).
After initiation of ART, immune activation levels in children and adults decrease (154) but usually remain elevated compared to HIV-negative controls and this residual immune activation despite ART is associated with incomplete immune reconstitution, increased non-AIDS comorbidities, and mortality (158–160) [reviewed in Ref. (161)]. Persistent immune activation is associated with increased levels of pro-inflammatory cytokines and elevated coagulation markers that have been linked to cardiovascular disease and cancer in adults [reviewed in Ref. (4, 162)], emphasizing a potential role for adjunctive immunomodulatory interventions together with ART to reduce residual immune activation in people living with HIV.
The cause of systemic immune activation in HIV infection has not yet been fully elucidated but appears to arise from a complex interplay between HIV and the host involving several molecular and cellular mechanisms including innate and adaptive immune responses that create a pro-inflammatory milieu, fibrosis of lymphoid tissue, uncontrolled co-infections, and breach of the intestinal barrier resulting in translocation of microbial products into the systemic circulation [reviewed in Ref. (161)].
From early stages of HIV infection, the gastrointestinal tract is a target for HIV-induced pathology, with severe depletion of mucosal CD4 T cells (in particular Th17 cells), dendritic cells, and innate lymphoid cells (107, 163, 164) disrupting mucosal immunity and the intestinal epithelial architecture [reviewed in (2, 165)]. This breach in the mucosal barrier results in translocation of microbial products from the gut into the systemic circulation leading to immune activation (166). A critical difference between non-pathogenic SIV infection (sooty mangabeys, African green monkeys) and pathogenic infection (rhesus macaques, pig-tailed macaques) is in preservation of CD4+ Th17 cells in the sooty mangabeys and African Green monkeys (167). Markers that are used to measure microbial translocation include direct microbial products like lipopolysaccharide (LPS), a component of the Gram-negative bacterial cell wall, and conserved bacterial ribosomal 16sRNA, or indirect markers such as sCD14 as a soluble marker of monocyte activation (168).
The immaturity of the gut in early life, characterized by increased epithelial permeability (169) and the evolution of the gut microbiota following bacterial and viral colonization (170) might influence the degree of microbial translocation in pediatric populations. Plasma levels of LPS in uninfected children initially increase after birth to reach a plateau at about 6 months of age (171) and then decrease again after the age of 2 years (172).
In vertically HIV-infected infants, levels of microbial translocation are elevated compared to uninfected controls and persist at a higher level after initiation of ART (173). Persistent microbial translocation correlates with cellular and soluble markers of immune activation in children on ART suggesting an important role in immune reconstitution (174). In a recent study in untreated HIV-infected children, microbial translocation was strongly correlated with monocyte and T cell activation and PD-1 expression levels on CD4 and CD8 T cells demonstrating the connection between microbial translocation, immune activation, and immune exhaustion (175).
HIV-specific antibodies have been in the focus of intense research for several decades in the quest for a prophylactic HIV vaccine. Recently, hope has been fueled by the results of the RV144 vaccine trial, which showed modest vaccine efficacy (176) and identified antibodies against the V1V2 HIV-envelope region as a correlate of protection in vaccinees (177). Antibodies can mediate antiviral activities by binding either to free virus or to viral particles on infected cells. The Fc-region of the bound antibody can mediate phagocytosis of the virus or infected cells, activation of innate immune cells resulting in ADCC against infected cells (see innate immunity), or activation of the complement system leading to opsonization and lysis of the virus or infected cells [reviewed in Ref. (178)]. An important function of antibodies especially in regard to sterilizing immunity is the ability to neutralize the virus by binding to the viral surface and inhibit cellular infection. The surface of HIV consists of the gp41 trans-membrane protein and the heavily glycosylated gp120 surface protein, which are both highly variable between different viral variants. Therefore, antibodies elicited by an effective protective vaccine would need to be broadly neutralizing across different viral subtypes (179).
These broadly neutralizing antibodies (bNABs) have been shown to occur naturally in a minority (20%) of HIV-infected subjects over the course of several years post-infection (180–182) during a process of intense somatic hypermutation (183). A recent study identified several young children with HIV-specific antibodies that had surprisingly high neutralizing breadth (184). Interestingly, the proportion of children with high neutralization scores was greater than in large-scale cohort studies of chronically HIV-infected adults and high levels of neutralizing breadth were already reached at an age of ~2 years, suggesting accelerated affinity maturation. These findings were especially surprising as it had previously been shown that infants produce lower antibody titers with less diversified somatic mutation upon infection and vaccination shortly after birth [reviewed in Ref. (185)]. As higher set point, viral load levels are associated with increased emergence of bNABs in adults (186) and in these young children (184), it has been hypothesized that the high antigenic load in pediatric HIV infection boosts development of NAB breadth. Also other factors like the shift toward Th2 responses in early life (as discussed above) could promote B-cell function and somatic hypermutation. Differential proportions of IgG-subclasses in children with a preponderance of IgG1 and IgG3 (185) that have higher neutralizing potency (187, 188) could also contribute to this finding. CD4+ T follicular helper (Tfh) cells are thought to be crucial in providing help to B cells in the germinal center to support antibody maturation [reviewed in Ref. (110)], but there have been no studies of Tfh cells in HIV-infected children to date. Further investigation in this field could yield important findings for vaccine development.
The case report of an in utero HIV-infected child who appeared to have been “functionally cured” after early initiation of ART was received with much excitement. The child initiated ART within 30 h of life and stayed on ART until 18 months of age when ART was discontinued by the mother. In subsequent visits to the clinic half a year later and in the successive follow-up period, viral load levels remained undetectable in the absence of ART and no signs of ongoing viral replication or HIV-specific T-cell or antibody responses were detected (5), hence the child was labeled “functionally cured.” However, after more than 2 years of follow up with undetectable HIV-RNA levels after treatment cessation, there was a recent report of rebound viremia in this child.
The main obstacle to HIV eradication is a pool of long-lasting, treatment-resistant viral reservoirs that only decay slowly on ART and are a source for viral rebound after treatment cessation [reviewed in Ref. (189, 190)]. Different cell types and body compartments are thought to contribute to the viral reservoir, but major sources of replication-competent pro-viral DNA during effective ART are resting central and transitional memory CD4+ T cells (191). In this regard, children might be partially protected against the early establishment of viral reservoirs in these cell populations, because neonates have much lower frequencies of CD4 T cells of central memory phenotype than adults (22, 23).
The frequency of latently infected CD4 T cells in children strongly depends on early initiation of ART and the time to achieve virologic suppression (192). A cross-sectional study found low to undetectable levels of integrated pro-viral DNA in CD4 cells of children starting treatment before 6 months of age after more than 3 years of follow-up (193). A recent longitudinal study compared perinatally infected youth initiating treatment before or after 3 months of age (194). In contrast to the late treatment group, all children starting ART before 3 months had undetectable HIV-RNA plasma levels using highly sensitive assays but integrated pro-viral DNA could be detected by PCR in all subjects even after follow-up periods of up to 17 years. In the early-treated group, transitional memory CD4 T cells had a larger contribution to the pool of infected cells than longer lasting central memory CD4 T cells. These findings are consistent with a study of adult post-treatment controllers who initiated ART during acute HIV infection (195), indicating that early treatment can prevent seeding of long-lived cellular reservoirs. It has to be noted that only a fraction of integrated viral DNA codes for replication-competent virus (196) and so the standard assay to measure the size of the functional latent viral reservoir at present is a limiting dilution viral outgrowth assay (197, 198). Using this assay, only one of the early-treated children in this study had inducible replication-competent virus, as opposed to all children receiving ART at a later stage, although limited sample availability in pediatric studies reduces the sensitivity of these assays. Caution also has to be taken as rebound viremia is observed even in patients with extraordinarily low viral burden in the peripheral blood compartment (199) and a large proportion of the viral reservoir is actually located within the gut and other tissues that are not accessed in most studies (200). Reliable biomarkers to predict successful drug-free remission upon treatment cessation are therefore needed.
Apart form timing of ART initiation, the size and constitution of the viral reservoir is affected by host immunological factors including persistent immune activation that fuels replenishment of the reservoir during ART [reviewed in Ref. (161)] and effective immune responses that can reduce the pool of infected memory cells (201, 202). To achieve a cure of HIV in a broader group of patients will therefore most likely require a multifaceted approach that could include induction of potent HIV-specific immunity prior to reversion of HIV-latency in combination with immunomodulatory interventions.
In summary, immunity to HIV in early life differs from that in adulthood not only in quantity but also in quality. This results from the adaptations of the immune system to the abundance of antigenic stimulation and pathogen exposure in utero and after birth. The initial ontogeny of the immune system is characterized by a more tolerogenic state of innate and adaptive immunity that prevents potentially harmful autoimmunity and inflammatory responses, and is geared toward preferential protection against extracellular pathogens. As a consequence, vertically acquired HIV infection is poorly controlled and marked by rapid progression to AIDS and death without effective interventions. Chronic infection results in systemic immune activation that drives immunopathology with functional immune exhaustion, increased susceptibility to co-infections, and inflammatory comorbidities. Coverage rates of PMTCT, early infant diagnosis, and pediatric ART need to be improved worldwide to reduce the global disease burden among children. For those children who become infected despite PMTCT, novel therapeutic strategies need to be developed targeting persistent immune dysregulation and the viral reservoir to potentially achieve drug-free remission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Prendergast AJ, Klenerman P, Goulder PJ. The impact of differential antiviral immunity in children and adults. Nat Rev Immunol (2012) 12(9):636–48. doi: 10.1038/nri3277
2. Brenchley JM. Mucosal immunity in human and simian immunodeficiency lentivirus infections. Mucosal Immunol (2013) 6(4):657–65. doi:10.1038/mi.2013.15
3. Khaitan A, Unutmaz D. Revisiting immune exhaustion during HIV infection. Curr HIV/AIDS Rep (2011) 8(1):4–11. doi:10.1007/s11904-010-0066-0
4. Paiardini M, Muller-Trutwin M. HIV-associated chronic immune activation. Immunol Rev (2013) 254(1):78–101. doi:10.1111/imr.12079
5. Persaud D, Gay H, Ziemniak C, Chen YH, Piatak M Jr., Chun TW, et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N Engl J Med (2013) 369(19):1828–35. doi:10.1056/NEJMoa1302976
6. Siegfried N, van der Merwe L, Brocklehurst P, Sint TT. Antiretrovirals for reducing the risk of mother-to-child transmission of HIV infection. Cochrane Database Syst Rev (2011) (7):CD003510. doi:10.1002/14651858.CD003510.pub3
7. World Health Organisation. Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection. (2013). Available from: http://wwwwhoint/hiv/pub/guidelines/arv2013/en/
8. Working Group on Mother-to-Child Transmission of HIV. Rates of mother-to-child transmission of HIV-1 in Africa, America and Europe: results from 13 perinatal studies. J Acquir Immune Defic Syndr Hum Retrovirol (1995) 8:506–10. doi:10.1097/00042560-199504120-00011
9. Chahroudi A, Meeker T, Lawson B, Ratcliffe S, Else J, Silvestri G. Mother-to-infant transmission of simian immunodeficiency virus is rare in sooty mangabeys and is associated with low viremia. J Virol (2011) 85(12):5757–63. doi:10.1128/JVI.02690-10
10. Amedee AM, Rychert J, Lacour N, Fresh L, Ratterree M. Viral and immunological factors associated with breast milk transmission of SIV in rhesus macaques. Retrovirology (2004) 1:17. doi:10.1186/1742-4690-1-17
11. Pandrea I, Onanga R, Souquiere S, Mouinga-Ondeme A, Bourry O, Makuwa M, et al. Paucity of CD4+ CCR5+ T cells may prevent transmission of simian immunodeficiency virus in natural nonhuman primate hosts by breast-feeding. J Virol (2008) 82(11):5501–9. doi:10.1128/JVI.02555-07
12. van’t Wout AB, Kootstra NA, Mulder-Kampinga GA, Albrecht-van Lent N, Scherpbier HJ, Veenstra J, et al. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J Clin Invest (1994) 94(5):2060–7. doi:10.1172/JCI117560
13. Chahroudi A, Cartwright E, Lee ST, Mavigner M, Carnathan DG, Lawson B, et al. Target cell availability, rather than breast milk factors, dictates mother-to-infant transmission of SIV in sooty mangabeys and rhesus macaques. PLoS Pathog (2014) 10(3):e1003958. doi:10.1371/journal.ppat.1003958
14. Shalekoff S, Gray GE, Tiemessen CT. Age-related changes in expression of CXCR4 and CCR5 on peripheral blood leukocytes from uninfected infants born to human immunodeficiency virus type 1-infected mothers. Clin Diagn Lab Immunol (2004) 11(1):229–34. doi:10.1128/CDLI.11.1.229
15. Bunders MJ, van der Loos CM, Klarenbeek PL, van Hamme JL, Boer K, Wilde JC, et al. Memory CD4(+)CCR5(+) T cells are abundantly present in the gut of newborn infants to facilitate mother-to-child transmission of HIV-1. Blood (2012) 120(22):4383–90. doi:10.1182/blood-2012-06-437566
16. Babiker A, Darby S, De Angelis D, Ewart D, Porter K, Beral V, et al. Time from HIV-1 seroconversion to AIDS and death before widespread use of highly-active antiretroviral therapy: a collaborative re-analysis. Lancet (2000) 355(9210):1131–7. doi:10.1016/S0140-6736(00)02061-4
17. Newell ML, Coovadia H, Cortina-Borja M, Rollins N, Gaillard P, Dabis F, et al. Mortality of infected and uninfected infants born to HIV-infected mothers in Africa: a pooled analysis. Lancet (2004) 364(9441):1236–43. doi:10.1016/S0140-6736(04)17140-7
18. Becquet R, Marston M, Dabis F, Moulton LH, Gray G, Coovadia HM, et al. Children who acquire HIV infection perinatally are at higher risk of early death than those acquiring infection through breastmilk: a meta-analysis. PLoS One (2012) 7(2):e28510. doi:10.1371/journal.pone.0028510
19. Marinda E, Humphrey JH, Iliff PJ, Mutasa K, Nathoo KJ, Piwoz EG, et al. Child mortality according to maternal and infant HIV status in Zimbabwe. Pediatr Infect Dis J (2007) 26(6):519–26. doi:10.1097/01.inf.0000264527.69954.4c
20. Marston M, Becquet R, Zaba B, Moulton LH, Gray G, Coovadia H, et al. Net survival of perinatally and postnatally HIV-infected children: a pooled analysis of individual data from sub-Saharan Africa. Int J Epidemiol (2011) 40(2):385–96. doi:10.1093/ije/dyq255
21. Ferrand RA, Corbett EL, Wood R, Hargrove J, Ndhlovu CE, Cowan FM, et al. AIDS among older children and adolescents in Southern Africa: projecting the time course and magnitude of the epidemic. AIDS (2009) 23(15):2039–46. doi:10.1097/QAD.0b013e32833016ce
22. Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol (2003) 112(5):973–80. doi:10.1016/j.jaci.2003.07.003
23. Schatorje EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Paediatric reference values for the peripheral T cell compartment. Scand J Immunol (2012) 75(4):436–44. doi:10.1111/j.1365-3083.2012.02671.x
24. European Collaborative S. Level and pattern of HIV-1-RNA viral load over age: differences between girls and boys? AIDS (2002) 16(1):97–104. doi:10.1097/00002030-200201040-00012
25. Mphatswe W, Blanckenberg N, Tudor-Williams G, Prendergast A, Thobakgale C, Mkhwanazi N, et al. High frequency of rapid immunological progression in African infants infected in the era of perinatal HIV prophylaxis. AIDS (2007) 21(10):1253–61. doi:10.1097/QAD.0b013e3281a3bec2
26. Obimbo EM, Wamalwa D, Richardson B, Mbori-Ngacha D, Overbaugh J, Emery S, et al. Pediatric HIV-1 in Kenya: pattern and correlates of viral load and association with mortality. J Acquir Immune Defic Syndr (2009) 51(2):209–15. doi:10.1097/QAI.0b013e31819c16d8
27. Biggar RJ, Janes M, Pilon R, Miotti P, Taha TE, Broadhead R, et al. Virus levels in untreated African infants infected with human immunodeficiency virus type 1. J Infect Dis (1999) 180(6):1838–43. doi:10.1086/315122
28. Lyles RH, Munoz A, Yamashita TE, Bazmi H, Detels R, Rinaldo CR, et al. Natural history of human immunodeficiency virus type 1 viremia after seroconversion and proximal to AIDS in a large cohort of homosexual men. Multicenter AIDS Cohort Study. J Infect Dis (2000) 181(3):872–80. doi:10.1086/315339
29. Saathoff E, Pritsch M, Geldmacher C, Hoffmann O, Koehler RN, Maboko L, et al. Viral and host factors associated with the HIV-1 viral load setpoint in adults from Mbeya Region, Tanzania. J Acquir Immune Defic Syndr (2010) 54(3):324–30. doi:10.1097/QAI.0b013e3181cf30ba
30. McIntosh K, Shevitz A, Zaknun D, Kornegay J, Chatis P, Karthas N, et al. Age- and time-related changes in extracellular viral load in children vertically infected by human immunodeficiency virus. Pediatr Infect Dis J (1996) 15(12):1087–91. doi:10.1097/00006454-199612000-00006
31. Rustagi A, Gale M Jr. Innate antiviral immune signaling, viral evasion and modulation by HIV-1. J Mol Biol (2014) 426(6):1161–77. doi:10.1016/j.jmb.2013.12.003
32. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25(3):373–81. doi:10.1016/j.immuni.2006.08.007
33. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science (2007) 317(5844):1522–7. doi:10.1126/science.1139522
34. Ricci E, Malacrida S, Zanchetta M, Mosconi I, Montagna M, Giaquinto C, et al. Toll-like receptor 9 polymorphisms influence mother-to-child transmission of human immunodeficiency virus type 1. J Transl Med (2010) 8:49. doi:10.1186/1479-5876-8-49
35. Qin M, Brummel S, Singh KK, Fenton T, Spector SA. Associations of host genetic variants on CD4+ lymphocyte count and plasma HIV-1 RNA in antiretroviral naive children. Pediatr Infect Dis J (2014). doi:10.1097/INF.0000000000000330
36. Bunupuradah T, Imahashi M, Iampornsin T, Matsuoka K, Iwatani Y, Puthanakit T, et al. Association of APOBEC3G genotypes and CD4 decline in Thai and Cambodian HIV-infected children with moderate immune deficiency. AIDS Res Ther (2012) 9(1):34. doi:10.1186/1742-6405-9-34
37. Singh KK, Wang Y, Gray KP, Farhad M, Brummel S, Fenton T, et al. Genetic variants in the host restriction factor APOBEC3G are associated with HIV-1-related disease progression and central nervous system impairment in children. J Acquir Immune Defic Syndr (2013) 62(2):197–203. doi:10.1097/QAI.0b013e31827ab612
38. Kollmann TR, Crabtree J, Rein-Weston A, Blimkie D, Thommai F, Wang XY, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J Immunol (2009) 183(11):7150–60. doi:10.4049/jimmunol.0901481
39. Belderbos ME, van Bleek GM, Levy O, Blanken MO, Houben ML, Schuijff L, et al. Skewed pattern of Toll-like receptor 4-mediated cytokine production in human neonatal blood: low LPS-induced IL-12p70 and high IL-10 persist throughout the first month of life. Clin Immunol (2009) 133(2):228–37. doi:10.1016/j.clim.2009.07.003
40. Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat Rev Immunol (2007) 7(5):379–90. doi:10.1038/nri2075
41. Reikie BA, Adams RC, Leligdowicz A, Ho K, Naidoo S, Ruck CE, et al. Altered innate immune development in HIV-exposed uninfected infants. J Acquir Immune Defic Syndr (2014) 66(3):245–55. doi:10.1097/QAI.0000000000000161
42. Miller E, Bhardwaj N. Dendritic cell dysregulation during HIV-1 infection. Immunol Rev (2013) 254(1):170–89. doi:10.1111/imr.12082
43. Selvaraj A, Pilakka-Kanthikeel S, Bhavani PK, Hanna LE, Pahwa S, Swaminathan S. Defective dendritic cell response to Toll-like receptor 7/8 agonists in perinatally HIV-infected children. Pathogens Dis (2013) 69(3):184–93. doi:10.1111/2049-632X.12067
44. Usuga X, Montoya CJ, Landay AL, Rugeles MT. Characterization of quantitative and functional innate immune parameters in HIV-1-infected Colombian children receiving stable highly active antiretroviral therapy. J Acquir Immune Defic Syndr (2008) 49(4):348–57. doi:10.1097/QAI.0b013e31818c16ff
45. Azzoni L, Rutstein RM, Chehimi J, Farabaugh MA, Nowmos A, Montaner LJ. Dendritic and natural killer cell subsets associated with stable or declining CD4+ cell counts in treated HIV-1-infected children. J Infect Dis (2005) 191(9):1451–9. doi:10.1086/429300
46. Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol (2013) 31:163–94. doi:10.1146/annurev-immunol-032712-100001
47. Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol (2002) 20:217–51. doi:10.1146/annurev.immunol.20.092501.134942
48. Nakazawa T, Agematsu K, Yabuhara A. Later development of Fas ligand-mediated cytotoxicity as compared with granule-mediated cytotoxicity during the maturation of natural killer cells. Immunology (1997) 92(2):180–7. doi:10.1046/j.1365-2567.1997.00343.x
49. Sundstrom Y, Nilsson C, Lilja G, Karre K, Troye-Blomberg M, Berg L. The expression of human natural killer cell receptors in early life. Scand J Immunol (2007) 66(2–3):335–44. doi:10.1111/j.1365-3083.2007.01980.x
50. Yabuhara A, Kawai H, Komiyama A. Development of natural killer cytotoxicity during childhood: marked increases in number of natural killer cells with adequate cytotoxic abilities during infancy to early childhood. Pediatr Res (1990) 28(4):316–22. doi:10.1203/00006450-199010000-00002
51. Dalle JH, Menezes J, Wagner E, Blagdon M, Champagne J, Champagne MA, et al. Characterization of cord blood natural killer cells: implications for transplantation and neonatal infections. Pediatr Res (2005) 57(5 Pt 1):649–55. doi:10.1203/01.PDR.0000156501.55431.20
52. Guilmot A, Hermann E, Braud VM, Carlier Y, Truyens C. Natural killer cell responses to infections in early life. J Innate Immun (2011) 3(3):280–8. doi:10.1159/000323934
53. Ballan WM, Vu BA, Long BR, Loo CP, Michaelsson J, Barbour JD, et al. Natural killer cells in perinatally HIV-1-infected children exhibit less degranulation compared to HIV-1-exposed uninfected children and their expression of KIR2DL3, NKG2C, and NKp46 correlates with disease severity. J Immunol (2007) 179(5):3362–70. doi:10.4049/jimmunol.179.5.3362
54. Ziegner U, Campbell D, Weinhold K, Frank I, Rutstein R, Starr SE. Deficient antibody-dependent cellular cytotoxicity against human immunodeficiency virus (HIV)-expressing target cells in perinatal HIV infection. Clin Diagn Lab Immunol (1999) 6(5):718–24.
55. Bernstein HB, Kinter AL, Jackson R, Fauci AS. Neonatal natural killer cells produce chemokines and suppress HIV replication in vitro. AIDS Res Hum Retroviruses (2004) 20(11):1189–95. doi:10.1089/0889222042544983
56. Slyker JA, Lohman-Payne B, John-Stewart GC, Dong T, Mbori-Ngacha D, Tapia K, et al. The impact of HIV-1 infection and exposure on natural killer (NK) cell phenotype in Kenyan infants during the first year of life. Front Immunol (2012) 3:399. doi:10.3389/fimmu.2012.00399
57. Wegmann TG, Lin H, Guilbert L, Mosmann TR. Bidirectional cytokine interactions in the maternal-fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol Today (1993) 14(7):353–6. doi:10.1016/0167-5699(93)90235-D
58. Pazos M, Sperling RS, Moran TM, Kraus TA. The influence of pregnancy on systemic immunity. Immunol Res (2012) 54(1–3):254–61. doi:10.1007/s12026-012-8303-9
59. Kourtis AP, Read JS, Jamieson DJ. Pregnancy and infection. N Engl J Med (2014) 370(23):2211–8. doi:10.1056/NEJMra1213566
60. Sperling RS, Engel SM, Wallenstein S, Kraus TA, Garrido J, Singh T, et al. Immunogenicity of trivalent inactivated influenza vaccination received during pregnancy or postpartum. Obstet Gynecol (2012) 119(3):631–9. doi:10.1097/AOG.0b013e318244ed20
61. Ohfuji S, Fukushima W, Deguchi M, Kawabata K, Yoshida H, Hatayama H, et al. Immunogenicity of a monovalent 2009 influenza A (H1N1) vaccine among pregnant women: lowered antibody response by prior seasonal vaccination. J Infect Dis (2011) 203(9):1301–8. doi:10.1093/infdis/jir026
62. Healy CM. Vaccines in pregnant women and research initiatives. Clin Obstet Gynecol (2012) 55(2):474–86. doi:10.1097/GRF.0b013e31824f3acb
63. Lissauer D, Piper K, Goodyear O, Kilby MD, Moss PA. Fetal-specific CD8+ cytotoxic T cell responses develop during normal human pregnancy and exhibit broad functional capacity. J Immunol (2012) 189(2):1072–80. doi:10.4049/jimmunol.1200544
64. Mold JE, McCune JM. At the crossroads between tolerance and aggression: revisiting the “layered immune system” hypothesis. Chimerism (2011) 2(2):35–41. doi:10.4161/chim.2.2.16329
65. Michaelsson J, Mold JE, McCune JM, Nixon DF. Regulation of T cell responses in the developing human fetus. J Immunol (2006) 176(10):5741–8. doi:10.4049/jimmunol.176.10.5741
66. Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science (2008) 322(5907):1562–5. doi:10.1126/science.1164511
67. Takahata Y, Nomura A, Takada H, Ohga S, Furuno K, Hikino S, et al. CD25+CD4+ T cells in human cord blood: an immunoregulatory subset with naive phenotype and specific expression of forkhead box p3 (Foxp3) gene. Exp Hematol (2004) 32(7):622–9. doi:10.1016/j.exphem.2004.03.012
68. Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science (2010) 330(6011):1695–9. doi:10.1126/science.1196509
69. Simonetta F, Bourgeois C. CD4+FOXP3+ regulatory T-cell subsets in human immunodeficiency virus infection. Front Immunol (2013) 4:215. doi:10.3389/fimmu.2013.00215
70. Chevalier MF, Weiss L. The split personality of regulatory T cells in HIV infection. Blood (2013) 121(1):29–37. doi:10.1182/blood-2012-07-409755
71. Freguja R, Gianesin K, Mosconi I, Zanchetta M, Carmona F, Rampon O, et al. Regulatory T cells and chronic immune activation in human immunodeficiency virus 1 (HIV-1)-infected children. Clin Exp Immunol (2011) 164(3):373–80. doi:10.1111/j.1365-2249.2011.04383.x
72. Prendergast A, O’Callaghan M, Menson E, Hamadache D, Walters S, Klein N, et al. Factors influencing T cell activation and programmed death 1 expression in HIV-infected children. AIDS Res Hum Retroviruses (2012) 28(5):465–8. doi:10.1089/AID.2011.0113
73. Arguello RJ, Balbaryski J, Barboni G, Candi M, Gaddi E, Laucella S. Altered frequency and phenotype of CD4+ forkhead box protein 3+ T cells and its association with autoantibody production in human immunodeficiency virus-infected paediatric patients. Clin Exp Immunol (2012) 168(2):224–33. doi:10.1111/j.1365-2249.2012.04569.x
74. Legrand FA, Nixon DF, Loo CP, Ono E, Chapman JM, Miyamoto M, et al. Strong HIV-1-specific T cell responses in HIV-1-exposed uninfected infants and neonates revealed after regulatory T cell removal. PLoS One (2006) 1:e102. doi:10.1371/journal.pone.0000102
75. Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol (1994) 68(9):6103–10.
76. Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol (1994) 68(7):4650–5.
77. Friedrich TC, Valentine LE, Yant LJ, Rakasz EG, Piaskowski SM, Furlott JR, et al. Subdominant CD8+ T-cell responses are involved in durable control of AIDS virus replication. J Virol (2007) 81(7):3465–76. doi:10.1128/JVI.02392-06
78. Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science (1999) 283(5403):857–60. doi:10.1126/science.283.5403.857
79. Rodriguez B, Sethi AK, Cheruvu VK, Mackay W, Bosch RJ, Kitahata M, et al. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA (2006) 296(12):1498–506. doi:10.1001/jama.296.12.1498
80. Goulder PJ, Walker BD. HIV and HLA class I: an evolving relationship. Immunity (2012) 37(3):426–40. doi:10.1016/j.immuni.2012.09.005
81. Crawford H, Lumm W, Leslie A, Schaefer M, Boeras D, Prado JG, et al. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J Exp Med (2009) 206(4):909–21. doi:10.1084/jem.20081984
82. Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, et al. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J Virol (2007) 81(22):12382–93. doi:10.1128/JVI.01543-07
83. Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, et al. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog (2012) 8(11):e1003041. doi:10.1371/journal.ppat.1003041
84. Goepfert PA, Lumm W, Farmer P, Matthews P, Prendergast A, Carlson JM, et al. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J Exp Med (2008) 205(5):1009–17. doi:10.1084/jem.20072457
85. Thobakgale CF, Prendergast A, Crawford H, Mkhwanazi N, Ramduth D, Reddy S, et al. Impact of HLA in mother and child on disease progression of pediatric human immunodeficiency virus type 1 infection. J Virol (2009) 83(19):10234–44. doi:10.1128/JVI.00921-09
86. Goulder PJ, Brander C, Tang Y, Tremblay C, Colbert RA, Addo MM, et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature (2001) 412(6844):334–8. doi:10.1038/35085576
87. Sharp ER, Willberg CB, Kuebler PJ, Abadi J, Fennelly GJ, Dobroszycki J, et al. Immunodominance of HIV-1 specific CD8+ T-cell responses is related to disease progression rate in vertically infected adolescents. PLoS One (2011) 6(7):e21135. doi:10.1371/journal.pone.0021135
88. Haynes BF, Heinly CS. Early human T cell development: analysis of the human thymus at the time of initial entry of hematopoietic stem cells into the fetal thymic microenvironment. J Exp Med (1995) 181(4):1445–58. doi:10.1084/jem.181.4.1445
89. Luzuriaga K, Holmes D, Hereema A, Wong J, Panicali DL, Sullivan JL. HIV-1-specific cytotoxic T lymphocyte responses in the first year of life. J Immunol (1995) 154(1):433–43.
90. Thobakgale CF, Ramduth D, Reddy S, Mkhwanazi N, de Pierres C, Moodley E, et al. Human immunodeficiency virus-specific CD8+ T-cell activity is detectable from birth in the majority of in utero-infected infants. J Virol (2007) 81(23):12775–84. doi:10.1128/JVI.00624-07
91. Scott ZA, Chadwick EG, Gibson LL, Catalina MD, McManus MM, Yogev R, et al. Infrequent detection of HIV-1-specific, but not cytomegalovirus-specific, CD8(+) T cell responses in young HIV-1-infected infants. J Immunol (2001) 167(12):7134–40. doi:10.4049/jimmunol.167.12.7134
92. Shalekoff S, Meddows-Taylor S, Gray GE, Sherman GG, Coovadia AH, Kuhn L, et al. Identification of human immunodeficiency virus-1 specific CD8+ and CD4+ T cell responses in perinatally-infected infants and their mothers. AIDS (2009) 23(7):789–98. doi:10.1097/QAD.0b013e328329c784
93. Lohman BL, Slyker JA, Richardson BA, Farquhar C, Mabuka JM, Crudder C, et al. Longitudinal assessment of human immunodeficiency virus type 1 (HIV-1)-specific gamma interferon responses during the first year of life in HIV-1-infected infants. J Virol (2005) 79(13):8121–30. doi:10.1128/JVI.79.13.8121-8130.2005
94. Nqoko B, Day CL, Mansoor N, De Kock M, Hughes EJ, Hawkridge T, et al. HIV-specific gag responses in early infancy correlate with clinical outcome and inversely with viral load. AIDS Res Hum Retroviruses (2011) 27(12):1311–6. doi:10.1089/aid.2011.0081
95. Huang S, Dunkley-Thompson J, Tang Y, Macklin EA, Steel-Duncan J, Singh-Minott I, et al. Deficiency of HIV-Gag-specific T cells in early childhood correlates with poor viral containment. J Immunol (2008) 181(11):8103–11. doi:10.4049/jimmunol.181.11.8103
96. Prendergast A, Goodliffe H, Clapson M, Cross R, Tudor-Williams G, Riddell A, et al. Gag-specific CD4+ T-cell responses are associated with virological control of paediatric HIV-1 infection. AIDS (2011) 25(10):1329–31. doi:10.1097/QAD.0b013e3283478575
97. Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med (2007) 13(1):46–53. doi:10.1038/nm1520
98. Geldmacher C, Currier JR, Herrmann E, Haule A, Kuta E, McCutchan F, et al. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J Virol (2007) 81(5):2440–8. doi:10.1128/JVI.01847-06
99. Akinsiku OT, Bansal A, Sabbaj S, Heath SL, Goepfert PA. Interleukin-2 production by polyfunctional HIV-1-specific CD8 T cells is associated with enhanced viral suppression. J Acquir Immune Defic Syndr (2011) 58(2):132–40. doi:10.1097/QAI.0b013e318224d2e9
100. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood (2006) 107(12):4781–9. doi:10.1182/blood-2005-12-4818
101. Thobakgale CF, Streeck H, Mkhwanazi N, Mncube Z, Maphumulo L, Chonco F, et al. Short communication: CD8(+) T cell polyfunctionality profiles in progressive and nonprogressive pediatric HIV type 1 infection. AIDS Res Hum Retroviruses (2011) 27(9):1005–12. doi:10.1089/AID.2010.0227
102. Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol (2002) 3(11):1061–8. doi:10.1038/ni845
103. Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, Ghrayeb J, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science (2003) 302(5645):659–62. doi:10.1126/science.1088774
104. Lichterfeld M, Kaufmann DE, Yu XG, Mui SK, Addo MM, Johnston MN, et al. Loss of HIV-1-specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1-specific CD4+ T cells. J Exp Med (2004) 200(6):701–12. doi:10.1084/jem.20041270
105. Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science (2003) 300(5617):337–9. doi:10.1126/science.1082305
106. Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science (2003) 300(5617):339–42. doi:10.1126/science.1083317
107. Guadalupe M, Reay E, Sankaran S, Prindiville T, Flamm J, McNeil A, et al. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J Virol (2003) 77(21):11708–17. doi:10.1128/JVI.77.21.11708-11717.2003
108. Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature (2005) 434(7037):1093–7. doi:10.1038/nature03501
109. Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature (2002) 417(6884):95–8. doi:10.1038/417095a
110. Streeck H, D’Souza MP, Littman DR, Crotty S. Harnessing CD4(+) T cell responses in HIV vaccine development. Nat Med (2013) 19(2):143–9. doi:10.1038/nm.3054
111. Chevalier MF, Julg B, Pyo A, Flanders M, Ranasinghe S, Soghoian DZ, et al. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J Virol (2011) 85(2):733–41. doi:10.1128/JVI.02030-10
112. Norris PJ, Moffett HF, Yang OO, Kaufmann DE, Clark MJ, Addo MM, et al. Beyond help: direct effector functions of human immunodeficiency virus type 1-specific CD4(+) T cells. J Virol (2004) 78(16):8844–51. doi:10.1128/JVI.78.16.8844-8851.2004
113. Soghoian DZ, Streeck H. Cytolytic CD4(+) T cells in viral immunity. Expert Rev Vaccines (2010) 9(12):1453–63. doi:10.1586/erv.10.132
114. Soghoian DZ, Jessen H, Flanders M, Sierra-Davidson K, Cutler S, Pertel T, et al. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci Transl Med (2012) 4(123):123ra25. doi:10.1126/scitranslmed.3003165
115. Ranasinghe S, Flanders M, Cutler S, Soghoian DZ, Ghebremichael M, Davis I, et al. HIV-specific CD4 T cell responses to different viral proteins have discordant associations with viral load and clinical outcome. J Virol (2012) 86(1):277–83. doi:10.1128/JVI.05577-11
116. Ramduth D, Day CL, Thobakgale CF, Mkhwanazi NP, de Pierres C, Reddy S, et al. Immunodominant HIV-1 Cd4+ T cell epitopes in chronic untreated clade C HIV-1 infection. PLoS One (2009) 4(4):e5013. doi:10.1371/journal.pone.0005013
117. Barrios C, Brandt C, Berney M, Lambert PH, Siegrist CA. Partial correction of the TH2/TH1 imbalance in neonatal murine responses to vaccine antigens through selective adjuvant effects. Eur J Immunol (1996) 26(11):2666–70. doi:10.1002/eji.1830261118
118. Prescott SL, Macaubas C, Holt BJ, Smallacombe TB, Loh R, Sly PD, et al. Transplacental priming of the human immune system to environmental allergens: universal skewing of initial T cell responses toward the Th2 cytokine profile. J Immunol (1998) 160(10):4730–7.
119. Adkins B, Bu Y, Guevara P. The generation of Th memory in neonates versus adults: prolonged primary Th2 effector function and impaired development of Th1 memory effector function in murine neonates. J Immunol (2001) 166(2):918–25. doi:10.4049/jimmunol.166.2.918
120. Upham JW, Lee PT, Holt BJ, Heaton T, Prescott SL, Sharp MJ, et al. Development of interleukin-12-producing capacity throughout childhood. Infect Immun (2002) 70(12):6583–8. doi:10.1128/IAI.70.12.6583-6588.2002
121. Cleary AM, Tu W, Enright A, Giffon T, Dewaal-Malefyt R, Gutierrez K, et al. Impaired accumulation and function of memory CD4 T cells in human IL-12 receptor beta 1 deficiency. J Immunol (2003) 170(1):597–603. doi:10.4049/jimmunol.170.1.597
122. Yap G, Pesin M, Sher A. Cutting edge: IL-12 is required for the maintenance of IFN-gamma production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J Immunol (2000) 165(2):628–31. doi:10.4049/jimmunol.165.2.628
123. Zhang X, Mozeleski B, Lemoine S, Deriaud E, Lim A, Zhivaki D, et al. CD4 T cells with effector memory phenotype and function develop in the sterile environment of the fetus. Sci Transl Med (2014) 6(238):238ra72. doi:10.1126/scitranslmed.3008748
124. Ramduth D, Thobakgale CF, Mkhwanazi NP, De Pierres C, Reddy S, van der Stok M, et al. Detection of HIV type 1 gag-specific CD4(+) T cell responses in acutely infected infants. AIDS Res Hum Retroviruses (2008) 24(2):265–70. doi:10.1089/aid.2007.0096
125. Hartigan-O’Connor DJ, Abel K, McCune JM. Suppression of SIV-specific CD4+ T cells by infant but not adult macaque regulatory T cells: implications for SIV disease progression. J Exp Med (2007) 204(11):2679–92. doi:10.1084/jem.20071068
126. Imami N, Westrop SJ, Grageda N, Herasimtschuk AA. Long-term non-progression and broad HIV-1-specific proliferative T-cell responses. Front Immunol (2013) 4:58. doi:10.3389/fimmu.2013.00058
127. Rosenberg ES, Billingsley JM, Caliendo AM, Boswell SL, Sax PE, Kalams SA, et al. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science (1997) 278(5342):1447–50. doi:10.1126/science.278.5342.1447
128. McNeil AC, Shupert WL, Iyasere CA, Hallahan CW, Mican JA, Davey RT Jr., et al. High-level HIV-1 viremia suppresses viral antigen-specific CD4(+) T cell proliferation. Proc Natl Acad Sci USA (2001) 98(24):13878–83. doi:10.1073/pnas.251539598
129. Boaz MJ, Waters A, Murad S, Easterbrook PJ, Vyakarnam A. Presence of HIV-1 Gag-specific IFN-gamma+IL-2+ and CD28+IL-2+ CD4 T cell responses is associated with nonprogression in HIV-1 infection. J Immunol (2002) 169(11):6376–85. doi:10.4049/jimmunol.169.11.6376
130. Emu B, Sinclair E, Favre D, Moretto WJ, Hsue P, Hoh R, et al. Phenotypic, functional, and kinetic parameters associated with apparent T-cell control of human immunodeficiency virus replication in individuals with and without antiretroviral treatment. J Virol (2005) 79(22):14169–78. doi:10.1128/JVI.79.22.14169-14178.2005
131. Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity (2013) 38(1):13–25. doi:10.1016/j.immuni.2013.01.004
132. Scott ZA, Beaumier CM, Sharkey M, Stevenson M, Luzuriaga K. HIV-1 replication increases HIV-specific CD4+ T cell frequencies but limits proliferative capacity in chronically infected children. J Immunol (2003) 170(11):5786–92. doi:10.4049/jimmunol.170.11.5786
133. Feeney ME, Draenert R, Roosevelt KA, Pelton SI, McIntosh K, Burchett SK, et al. Reconstitution of virus-specific CD4 proliferative responses in pediatric HIV-1 infection. J Immunol (2003) 171(12):6968–75. doi:10.4049/jimmunol.171.12.6968
134. Kuhn L, Meddows-Taylor S, Gray G, Tiemessen C. Human immunodeficiency virus (HIV)-specific cellular immune responses in newborns exposed to HIV in utero. Clin Infect Dis (2002) 34(2):267–76. doi:10.1086/338153
135. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439(7077):682–7. doi:10.1038/nature04444
136. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443(7109):350–4. doi:10.1038/nature05115
137. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med (2008) 205(12):2763–79. doi:10.1084/jem.20081398
138. Peretz Y, He Z, Shi Y, Yassine-Diab B, Goulet JP, Bordi R, et al. CD160 and PD-1 co-expression on HIV-specific CD8 T cells defines a subset with advanced dysfunction. PLoS Pathog (2012) 8(8):e1002840. doi:10.1371/journal.ppat.1002840
139. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol (2009) 10(1):29–37. doi:10.1038/ni.1679
140. Kaufmann DE, Kavanagh DG, Pereyra F, Zaunders JJ, Mackey EW, Miura T, et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat Immunol (2007) 8(11):1246–54. doi:10.1038/ni1515
141. Zhang JY, Zhang Z, Wang X, Fu JL, Yao J, Jiao Y, et al. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood (2007) 109(11):4671–8. doi:10.1182/blood-2006-09-044826
142. Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Dis (2013) 12(2):130–46. doi:10.1038/nrd3877
143. Porichis F, Kaufmann DE. HIV-specific CD4 T cells and immune control of viral replication. Curr Opin HIV AIDS (2011) 6(3):174–80. doi:10.1097/COH.0b013e3283454058
144. Tandon R, Giret MT, Sengupta D, York VA, Wiznia AA, Rosenberg MG, et al. Age-related expansion of Tim-3 expressing T cells in vertically HIV-1 infected children. PLoS One (2012) 7(9):e45733. doi:10.1371/journal.pone.0045733
145. Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med (2008) 5(10):e203. doi:10.1371/journal.pmed.0050203
146. Rodger AJ, Fox Z, Lundgren JD, Kuller LH, Boesecke C, Gey D, et al. Activation and coagulation biomarkers are independent predictors of the development of opportunistic disease in patients with HIV infection. J Infect Dis (2009) 200(6):973–83. doi:10.1086/605447
147. Serrano-Villar S, Gutierrez C, Vallejo A, Hernandez-Novoa B, Diaz L, Abad Fernandez M, et al. The CD4/CD8 ratio in HIV-infected subjects is independently associated with T-cell activation despite long-term viral suppression. J Infect (2013) 66(1):57–66. doi:10.1016/j.jinf.2012.09.013
148. Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood (2004) 104(4):942–7. doi:10.1182/blood-2003-09-3333
149. Hazenberg MD, Otto SA, van Benthem BH, Roos MT, Coutinho RA, Lange JM, et al. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS (2003) 17(13):1881–8. doi:10.1097/00002030-200309050-00006
150. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2014) 505(7484):509–14. doi:10.1038/nature12940
151. Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science (2014) 343(6169):428–32. doi:10.1126/science.1243640
152. Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis (1999) 179(4):859–70. doi:10.1086/314660
153. Paul ME, Mao C, Charurat M, Serchuck L, Foca M, Hayani K, et al. Predictors of immunologic long-term nonprogression in HIV-infected children: implications for initiating therapy. J Allergy Clin Immunol (2005) 115(4):848–55. doi:10.1016/j.jaci.2004.11.054
154. Ssewanyana I, Baker CA, Ruel T, Bousheri S, Kamya M, Dorsey G, et al. The distribution and immune profile of T cell subsets in HIV-infected children from Uganda. AIDS Res Hum Retroviruses (2009) 25(1):65–71. doi:10.1089/aid.2008.0138
155. Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV hosts: showing AIDS the door. Science (2012) 335(6073):1188–93. doi:10.1126/science.1217550
156. Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, McClure HM, et al. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity (2003) 18(3):441–52. doi:10.1016/S1074-7613(03)00060-8
157. Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest (2009) 119(12):3556–72. doi:10.1172/JCI40115
158. Hunt PW, Martin JN, Sinclair E, Epling L, Teague J, Jacobson MA, et al. Valganciclovir reduces T cell activation in HIV-infected individuals with incomplete CD4+ T cell recovery on antiretroviral therapy. J Infect Dis (2011) 203(10):1474–83. doi:10.1093/infdis/jir060
159. Sauce D, Larsen M, Fastenackels S, Pauchard M, Ait-Mohand H, Schneider L, et al. HIV disease progression despite suppression of viral replication is associated with exhaustion of lymphopoiesis. Blood (2011) 117(19):5142–51. doi:10.1182/blood-2011-01-331306
160. Lederman MM, Calabrese L, Funderburg NT, Clagett B, Medvik K, Bonilla H, et al. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J Infect Dis (2011) 204(8):1217–26. doi:10.1093/infdis/jir507
161. Klatt NR, Chomont N, Douek DC, Deeks SG. Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev (2013) 254(1):326–42. doi:10.1111/imr.12065
162. Phillips AN, Neaton J, Lundgren JD. The role of HIV in serious diseases other than AIDS. AIDS (2008) 22(18):2409–18. doi:10.1097/QAD.0b013e3283174636
163. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med (2004) 200(6):749–59. doi:10.1084/jem.20040874
164. Xu H, Wang X, Liu DX, Moroney-Rasmussen T, Lackner AA, Veazey RS. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol (2012) 5(6):658–69. doi:10.1038/mi.2012.39
165. Klatt NR, Funderburg NT, Brenchley JM. Microbial translocation, immune activation, and HIV disease. Trends Microbiol (2013) 21(1):6–13. doi:10.1016/j.tim.2012.09.001
166. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med (2006) 12(12):1365–71. doi:10.1038/nm1511
167. Paiardini M. Th17 cells in natural SIV hosts. Curr Opin HIV AIDS (2010) 5(2):166–72. doi:10.1097/COH.0b013e328335c161
168. Abad-Fernandez M, Vallejo A, Hernandez-Novoa B, Diaz L, Gutierrez C, Madrid N, et al. Correlation between different methods to measure microbial translocation and its association with immune activation in long-term suppressed HIV-1-infected individuals. J Acquir Immune Defic Syndr (2013) 64(2):149–53. doi:10.1097/QAI.0b013e31829a2f12
169. Sherman MP. New concepts of microbial translocation in the neonatal intestine: mechanisms and prevention. Clin Perinatol (2010) 37(3):565–79. doi:10.1016/j.clp.2010.05.006
170. Fanaro S, Chierici R, Guerrini P, Vigi V. Intestinal microflora in early infancy: composition and development. Acta Paediatr (2003) 91(441):48–55. doi:10.1111/j.1651-2227.2003.tb00646.x
171. Kourtis AP, Ibegbu CC, Wiener J, King CC, Tegha G, Kamwendo D, et al. Role of intestinal mucosal integrity in HIV transmission to infants through breast-feeding: the BAN study. J Infect Dis (2013) 208(4):653–61. doi:10.1093/infdis/jit221
172. Wallet MA, Rodriguez CA, Yin L, Saporta S, Chinratanapisit S, Hou W, et al. Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T-cell activation following therapy. AIDS (2010) 24(9):1281–90. doi:10.1097/QAD.0b013e328339e228
173. Papasavvas E, Azzoni L, Foulkes A, Violari A, Cotton MF, Pistilli M, et al. Increased microbial translocation in </= 180 days old perinatally human immunodeficiency virus-positive infants as compared with human immunodeficiency virus-exposed uninfected infants of similar age. Pediatr Infect Dis J (2011) 30(10):877–82. doi:10.1097/INF.0b013e31821d141e
174. Pilakka-Kanthikeel S, Huang S, Fenton T, Borkowsky W, Cunningham CK, Pahwa S. Increased gut microbial translocation in HIV-infected children persists in virologic responders and virologic failures after antiretroviral therapy. Pediatr Infect Dis J (2012) 31(6):583–91. doi:10.1097/INF.0b013e31824da0f5
175. Pilakka-Kanthikeel S, Kris A, Selvaraj A, Swaminathan S, Pahwa S. Immune activation is associated with increased gut microbial translocation in treatment-naive, HIV-infected children in a resource-limited setting. J Acquir Immune Defic Syndr (2014) 66(1):16–24. doi:10.1097/QAI.0000000000000096
176. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med (2009) 361(23):2209–20. doi:10.1056/NEJMoa0908492
177. Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med (2012) 366(14):1275–86. doi:10.1056/NEJMoa1113425
178. Burton DR. Antibodies, viruses and vaccines. Nat Rev Immunol (2002) 2(9):706–13. doi:10.1038/nri891
179. Walker BD, Burton DR. Toward an AIDS vaccine. Science (2008) 320(5877):760–4. doi:10.1126/science.1152622
180. Sather DN, Armann J, Ching LK, Mavrantoni A, Sellhorn G, Caldwell Z, et al. Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J Virol (2009) 83(2):757–69. doi:10.1128/JVI.02036-08
181. Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature (2011) 477(7365):466–70. doi:10.1038/nature10373
182. Gray ES, Madiga MC, Hermanus T, Moore PL, Wibmer CK, Tumba NL, et al. The neutralization breadth of HIV-1 develops incrementally over four years and is associated with CD4+ T cell decline and high viral load during acute infection. J Virol (2011) 85(10):4828–40. doi:10.1128/JVI.00198-11
183. Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science (2010) 329(5993):856–61. doi:10.1126/science.1187659
184. Goo L, Chohan V, Nduati R, Overbaugh J. Early development of broadly neutralizing antibodies in HIV-1-infected infants. Nat Med (2014) 20(6):655–8. doi:10.1038/nm.3565
185. Siegrist CA. Neonatal and early life vaccinology. Vaccine (2001) 19(25–26):3331–46. doi:10.1016/S0264-410X(01)00028-7
186. Piantadosi A, Panteleeff D, Blish CA, Baeten JM, Jaoko W, McClelland RS, et al. Breadth of neutralizing antibody response to human immunodeficiency virus type 1 is affected by factors early in infection but does not influence disease progression. J Virol (2009) 83(19):10269–74. doi:10.1128/JVI.01149-09
187. Scharf O, Golding H, King LR, Eller N, Frazier D, Golding B, et al. Immunoglobulin G3 from polyclonal human immunodeficiency virus (HIV) immune globulin is more potent than other subclasses in neutralizing HIV type 1. J Virol (2001) 75(14):6558–65. doi:10.1128/JVI.75.14.6558-6565.2001
188. Baba TW, Liska V, Hofmann-Lehmann R, Vlasak J, Xu W, Ayehunie S, et al. Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nat Med (2000) 6(2):200–6. doi:10.1038/72309
189. Lafeuillade A. Eliminating the HIV reservoir. Curr HIV/AIDS Rep (2012) 9(2):121–31. doi:10.1007/s11904-012-0115-y
190. Pace MJ, Agosto L, Graf EH, O’Doherty U. HIV reservoirs and latency models. Virology (2011) 411(2):344–54. doi:10.1016/j.virol.2010.12.041
191. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med (2009) 15(8):893–900. doi:10.1038/nm.1972
192. Persaud D, Palumbo PE, Ziemniak C, Hughes MD, Alvero CG, Luzuriaga K, et al. Dynamics of the resting CD4(+) T-cell latent HIV reservoir in infants initiating HAART less than 6 months of age. AIDS (2012) 26(12):1483–90. doi:10.1097/QAD.0b013e3283553638
193. Ananworanich J, Puthanakit T, Suntarattiwong P, Chokephaibulkit K, Kerr SJ, Fromentin R, et al. Reduced markers of HIV persistence and restricted HIV-specific immune responses after early antiretroviral therapy in children. AIDS (2014) 28(7):1015–20. doi:10.1097/QAD.0000000000000178
194. Luzuriaga K, Tabak B, Garber M, Chen YH, Ziemniak C, McManus MM, et al. Reduced HIV reservoirs after early treatment HIV-1 proviral reservoirs decay continuously under sustained virologic control in early-treated HIV-1-infected children. J Infect Dis (2014). doi:10.1093/infdis/jiu297
195. Saez-Cirion A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog (2013) 9(3):e1003211. doi:10.1371/journal.ppat.1003211
196. Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell (2013) 155(3):540–51. doi:10.1016/j.cell.2013.09.020
197. Siliciano JD, Siliciano RF. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol Biol (2005) 304:3–15. doi:10.1385/1-59259-907-9:003
198. Laird GM, Eisele EE, Rabi SA, Lai J, Chioma S, Blankson JN, et al. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS Pathog (2013) 9(5):e1003398. doi:10.1371/journal.ppat.1003398
199. Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis (2008) 197(5):714–20. doi:10.1086/527324
200. Yukl SA, Gianella S, Sinclair E, Epling L, Li Q, Duan L, et al. Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J Infect Dis (2010) 202(10):1553–61. doi:10.1086/656722
201. Descours B, Avettand-Fenoel V, Blanc C, Samri A, Melard A, Supervie V, et al. Immune responses driven by protective human leukocyte antigen alleles from long-term nonprogressors are associated with low HIV reservoir in central memory CD4 T cells. Clin Infect Dis (2012) 54(10):1495–503. doi:10.1093/cid/cis188
Keywords: HIV, pediatric, innate immunity, adaptive immunity, immune responses, immune activation, immune exhaustion, viral reservoir
Citation: Muenchhoff M, Prendergast AJ and Goulder PJR (2014) Immunity to HIV in early life. Front. Immunol. 5:391. doi: 10.3389/fimmu.2014.00391
Received: 23 June 2014; Paper pending published: 07 July 2014;
Accepted: 30 July 2014; Published online: 12 August 2014.
Edited by:
Arnaud Marchant, Université Libre de Bruxelles, BelgiumReviewed by:
Nicolas Chomont, Vaccine and Gene Therapy Institute-Florida, USACopyright: © 2014 Muenchhoff, Prendergast and Goulder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maximilian Muenchhoff, Peter Medawar Building, South Parks Road, OX1 3SY Oxford, UK e-mail:bWF4aW1pbGlhbi5tdWVuY2hob2ZmQHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.