 Heather M. Wilson
Heather M. Wilson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Immunol. , 28 July 2014
Sec. Molecular Innate Immunity
Volume 5 - 2014 | https://doi.org/10.3389/fimmu.2014.00357
This article is part of the Research Topic M1/M2 Macrophages: The Arginine Fork in the Road to Health and Disease View all 31 articles
Macrophages were initially described as “big eaters” due to their phagocytic nature. It is now clear that macrophages have many diverse functions not only in innate immunity and tissue homeostasis but also in metabolism, development, and regeneration. Macrophage functions are driven largely by tissue-derived and pathogenic microenvironmental stimuli that help them adapt to changing conditions within tissues and tailor an appropriate response. The heterogeneity of macrophages has resulted in their classification into subtypes based on their phenotype and function (1). One major classification, based on function, is M1 and M2 macrophages, with destructive and healing properties, respectively (2, 3). As imbalances between M1 and M2 states have been observed in a number of diseases, an understanding of the molecular mechanisms, signaling pathways, and transcription factors controlling their polarization has obvious therapeutic implications. Recent studies have established strong potential for suppressor of cytokine signaling (SOCS) proteins to regulate M1 and M2 macrophage polarization (4–7). Here, the focus will be on the evidence for this, and the consequences of altered SOCS expressions on macrophage function in health and disease. Overall it is proposed that a high SOCS1 to SOCS3 ratio could be a potential marker for M2 macrophages while high SOCS3 expression is associated with M1 cells.
Suppressor of cytokine signaling proteins are a family of intracellular cytokine-inducible proteins, consisting of eight members (CIS and SOCS1–SOCS7) (8, 9). SOCS1 and SOCS3 are most widely characterized regarding their roles in shaping M1 and M2 macrophage polarization (4–6). They show low expression in resting macrophages, but are rapidly induced on activation. All SOCS family proteins contain an Src homology 2 (SH2) domain, a variable length amino-terminal domain and a conserved carboxy-terminal SOCS box motif that interacts with ubiquitin–ligase machinery (8, 9). SOCS are induced by a variety of stimuli that cause M1 and M2 activation, including cytokines, toll-like receptor (TLR) ligands, angiotensin II, immune complexes, and high glucose (9). The most studied signaling pathway regulated by SOCS is JAK/STAT activation. SOCS negatively regulate JAK/STAT signaling through association with key phosphorylated tyrosine residues on JAK proteins and/or cytokine receptors, and by degradation of signaling molecules mediated via the ubiquitin–proteasome pathway (8, 9). SOCS1 and SOCS3 contain a kinase inhibitory region (KIR) that directly suppresses JAK tyrosine kinase activity. SOCS proteins also influence ERK (10), PI3K (11), Notch (12), MAPK (13), and NF-κB (14) signaling cascades that directs M1 and M2 functions.
SOCS1 regulates M1-macrophage activation by inhibiting the interferon gamma-induced JAK2/STAT1 pathway and TLR/NF-κB signaling (9, 15) (Figure 1). To suppress the latter pathway, SOCS1 binds to the p65 subunit of NF-κB and the TLR adaptor molecule Mal/TIRAP as well as IRAK, facilitating its ubiquitin-mediated proteolysis via ubiquitin ligases recruited by the SOCS box (8, 14–17). SOCS1 indirectly inhibits TLR4 signaling through secondary mechanisms targeting IRF3 and IFN-β induced JAK/STAT pathways (18, 19). Thus, SOCS1 mediates a negative feedback mechanism during TLR4 signaling, via control of both MyD88-dependent and MyD88-independent signaling. SOCS1-deficient mice succumb to severe systemic autoimmune and inflammatory disease (14, 16) and their M1-macrophages display an increased capacity to kill intracellular bacterial pathogens, presumably due to unrestrained IFN-γ/STAT1 and p65 signaling. In line with this, SOCS1 knockout or knockdown M1-activated macrophages show enhanced levels of IL-6, IL-12, MHC class II, and nitric oxide suggesting SOCS1 sustains the properties of M1 macrophages at a less destructive level to prevent overshooting inflammatory responses (4, 18). This explains why SOCS1 promoter hypermethylation, which results in loss of SOCS1 expression leads to enhanced secretion of lipopolysaccharide (LPS)-induced pro-inflammatory cytokines (20). Micro RNA-155 (miR-155) is a critical regulator of innate immunity and TLR signaling (21–23); miR-155 targets and degrades SOCS1 in M1-activated macrophages (21), thus miR-155 induction during activation serves to maximize and extend the inflammatory process.
SOCS1 also regulates M2 macrophage polarization. Expression of macrophage SOCS1, but not SOCS3, is strongly upregulated in an M2 polarizing environment in vitro and in vivo, where it has an important role in acquisition of M2 functional characteristics, such as a high arginase I/low inducible nitric oxide synthase (iNOS) expression ratio (4). Strikingly, this contrasts with macrophages infiltrating an in vivo inflamed M1-activating environment, where macrophages with enhanced SOCS3 but not SOCS1 expression are prominent (5). This suggests that exclusive upregulation of SOCS1, or indeed, a high SOCS1/SOCS3 expression ratio, has potential as a useful and additional in vivo biomarker for M2 (see later). Arginase I expression, as an M2 macrophage marker, can be mediated via activation of either STAT6 (24) or PI3K (25). SOCS1 is important in controlling PI3K activity, supporting a mechanism for regulating arginase I expression in M2 cells; SOCS1 also regulates STAT6 phosphorylation (26). Following activation, SOCS1 knockdown or SOCS1-deficient macrophages show a reciprocal upregulation of SOCS3 expression. SOCS3 inhibits PI3K activation (27), and so the expression of high SOCS1 and low SOCS3 in M2 macrophages could result in greater PI3K activity and more arginase I induction in these cells. An elevated expression of SOCS1 is important for the arginase I-induced suppressive nature of M2 macrophages that attenuate lymphocyte proliferation (28). Moreover, siRNA-mediated knockdown of SOCS1 results in the induction of iNOS in IL-4-pretreated cells stimulated with IFN/LPS (4). Thus, SOCS1 regulates the iNOS/arginase I expression ratio in both M1 and M2 macrophages and helps fine-tune key signaling pathways to mount an appropriate response to changes within the microenvironment.
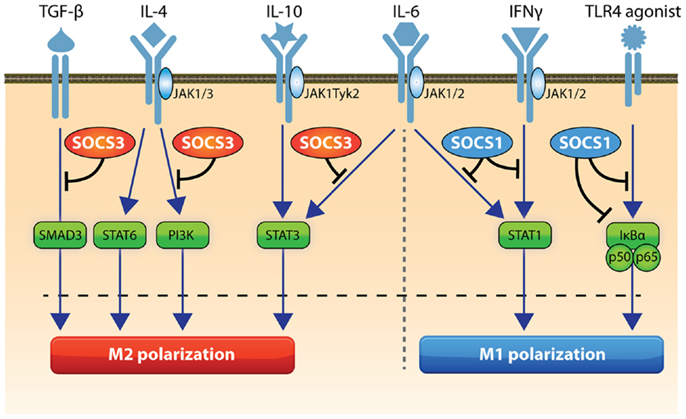
Figure 1. Role of SOCS1 and SOCS3 in macrophage activation. STAT1 and NFκB drive M1 polarization and SOCS1 can inhibit these pathways. SOCS3 can regulate TLR signaling and inhibits IL-6-induced STAT3 activation and SMAD3 and PI3K activity to action an appropriate destructive effect. STAT3, STAT6, and PI3K can drive M2 activation and SOCS3 inhibits STAT3 and PI3K. Pathways that trigger SOCS1 in macrophages include STAT1 and NFκB, while SOCS3 expression can be induced by STAT3, NFκB, NOTCH1, PI3K, and MAP kinase activation.
An important role for SOCS2 in driving M2 polarization and limiting M1 polarization has been shown, with IL-4 activation of macrophages, resulting in enhanced SOCS2 expression (27). Macrophages from SOCS2-/- mice display increased secretion of IFN-γ, IL-1β, and TNF-α in response to LPS in parallel to an increased pro-inflammatory cytokine mRNA expression (29). These BMDMs have higher basal levels of p65–NF-κB compared with macrophages from wild-type mice (29). In another study, SOCS2-deficient macrophages were hyper responsive to IFN-γ, produced more NO and dealt with infection more efficiently (30). SOCS2 has also been described as a feedback inhibitor of TLR-induced activation in dendritic cells (31).
In contrast with SOCS2, a key role for SOCS3 in M1 polarization is proposed (Figure 1). The majority of macrophages activated within an in vivo pro-inflammatory conditioning environment show strong upregulation of SOCS3 expression and this cell population co-express the M1 marker, iNOS (5, 6). Without SOCS3, both human and rodent macrophages have a reduced ability to develop pro-inflammatory features but instead display immunoregulatory characteristics (5, 6). Notably, mice with a targeted deletion of SOCS3 in macrophages and neutrophils demonstrate a reduced IL-12 response and succumb to toxoplasmosis (32). SOCS3 binds to and inhibits gp130-related cytokine receptors and consequently this abrogates IL-6-induced STAT1 gene expression and IL-6-induced STAT3 anti-inflammatory effects (33–35). Therefore, in SOCS3-deficient macrophages, IL-6 signals in a similar manner to the immunosuppressive cytokine IL-10, through prolonged STAT3 activation and dampening of LPS signaling (33). As a result, mice deficient in SOCS3 in myeloid cells are resistant to endotoxic shock (35) with reduced production of pro-inflammatory cytokines. However, one report in the same mice suggests SOCS3 deficiency promotes M1 macrophage activation in spite of enhanced STAT3 activation (7). The reasons for this discrepancy in findings are unclear but could relate to differences in dose and purity of the LPS used in the different studies, as well as and the genes and time-points analyzed after macrophage activation (7, 35). Moreover, the conflicting results for the role of SOCS3 in M1 polarization in isolated macrophages in vitro (5–7) could result from the different technologies and species used (siRNA-mediated knockdown in rat and human macrophages, which avoids the risk of compensatory effects of other SOCS genes (5, 6) versus cells from macrophage-specific SOCS3 knockout mice) (7). Resolving these issues should establish the importance of SOCS3 in modulating macrophage function in vivo.
Studies of SOCS3-deficient macrophages confirm that SOCS3 positively regulates TLR4 signaling and M1 activation by inhibition of IL-6R-mediated STAT3 activation, as well as TGF-β-mediated SMAD3 activation, which is critical for the negative regulation of TLR-induced TNF-α and IL-6 production (5, 6, 33, 36). Since SOCS3 blocks PI3K that feeds and inhibits TLR responses, this could be an alternative mechanism by which SOCS3 augments TLR signaling in M1 macrophages (6). Forced activation of Notch signaling enhances both M1 polarization and anti-tumor activity via SOCS3 induction (12). In line with this, macrophage-specific SOCS3 knockout animals are resistant to tumor transplantation due to reduced secretion of tumor-promoting TNF-α and IL-6, together with elevated MCP2/CCL8 that is anti-tumorigenic (37).
Regulation of SOCS3 in innate cells influences downstream T cell fates. The presence of SOCS3 in macrophages is important in fine-tuning downstream T effector cell priming due to both influences in expression of presenting molecules and altered secretion of T cell polarizing cytokines (6, 7). Mouse SOCS3-deficient dendritic cells display an analogous reduced potential to drive T effector cell responses and a tolerogenic phenotype as a result of enhanced TGFβ production and expansion of Foxp3-positive regulatory T cells (38). These dendritic cells reduce the severity of experimental autoimmune disease. Therefore, regulation of intracellular signaling pathways by SOCS3 in innate cells is critical for the decision of adaptive responses such as T cell fates. The depletion of macrophage SOCS3 in a clinical situation would thus be predicted to dampen both pro-inflammatory innate and adaptive immune responses.
The above studies suggest that macrophage SOCS3 is associated with M1 macrophages and pro-inflammatory responses and is a potential therapeutic target in inflammatory diseases. However, a word of caution should be introduced as this may not be the case in all inflammatory conditions. In diseases, where STAT3 activation exerts a profound inflammatory and pathogenic response (39, 40) then the effects of SOCS3 targeting may not be beneficial. For example, in an IL-1/STAT3 model of chronic arthritis where SOCS3 was deleted in hematopoietic and endothelial cells, animals exhibited more severe disease. Thus, the pathology needs first to be assessed before SOCS3 manipulation as a therapy is considered (37).
The heightened expression of macrophage SOCS1 and SOCS3 proteins have been demonstrated in many pathologies in vivo where this has been proposed, through the molecular mechanisms described above, to enhance or inhibit pathogenesis.
Macrophages are an important feature in glomerulonephritis pathology. Macrophages infiltrating inflamed glomeruli in experimental models are rapidly polarized to express either SOCS1 or SOCS3, but rarely both, with most exclusively expressing SOCS3 (5, 6). The proportion of these SOCS3-expressing macrophages correlates strongly with the severity of immune-mediated injury. Local delivery of IL-4 to inflamed glomeruli has a major effect on reducing the number of SOCS3-expressing glomerular macrophages, and this is reflected by a decrease in the severity of nephritis, supporting a role for SOCS3 in driving M1-mediated injury (5).
Human atherosclerotic plaques exhibit a high expression of macrophage SOCS1 and SOCS3 in unstable inflammatory shoulder regions as compared to stable fibrous area (41). SOCS1 and SOCS3 expression is increased in aortic lesion macrophages from apoE(−/−) mice (42). In human tissue, the percentages of SOCS1-positive, M2 macrophages are decreased in morphologically stable atherosclerotic plaques, whereas percentages of SOCS3-positive, iNOS positive, macrophages are increased in unstable, rupture-prone plaques, suggesting targeting macrophage SOCS3 would be beneficial to dampen inflammation and plaque vulnerability (43). The differing expression ratio of SOCS1:SOCS3 in atherosclerotic plaques again suggests that the ratio could be an indicator of the inflammatory status of human macrophages in vivo. SOCS1 was atheroprotective in mouse models (44) while the absence of macrophage SOCS3 of apoE(−/−) mice attenuates disease, confirming a causal link between macrophage SOCS3 and atherosclerosis (45).
Beneath the gut epithelia, lamina propria macrophages phagocytose bacteria and maintain an M2 phenotype in the steady state. Approximately 10% of these macrophages express SOCS3 in healthy individuals, whereas in inflammatory bowel disease (IBD) patients this increases to 40%, again suggesting SOCS3 expression relates to M1-activated macrophages (46). Peroxisome proliferator-activated receptor-γ (PPARγ) agonists demonstrate efficacy in ameliorating intestinal inflammation associated with IBD. PPARγ expression is upregulated in M2 but not M1 macrophages. In macrophages lacking PPARγ, a significant upregulation of SOCS3 was noted and this could be important if treating IBD with PPARγ agonists (47).
In human tumors, SOCS3 expression identifies macrophages with enhanced tumor killing, whereas SOCS1 expressing macrophages (M2) favor tumor survival (48). Macrophage-specific deletion of SOCS1 leads to reduced susceptibility to melanoma growth and colon carcinogenesis through increased anti-tumor responses (49) and a switch to M1 polarization of tumor-associated macrophage. In contrast, mice with a macrophage-specific deletion of SOCS3, subcutaneously implanted with melanoma cells, did not show a difference in tumor size, although the number of metastasis increased in these mice (37). These SOCS3-deficient macrophages produce less IL-6 and TNF-α upon stimulation with tumor lysates due to aberrant STAT3 activity, again showing a positive link of SOCS and macrophage polarization (37).
SOCS3 restrains macrophage responses to IL-6 and leptin that are systemically upregulated in obesity (50). SOCS1 inhibits insulin signaling and macrophage cytokine secretion, resulting in insulin sensitivity in spite of an obese state (17). Moreover, an increase in SOCS1 expression in mouse macrophages inhibits LPS- and palmitate-induced TLR4 signaling and in so doing prevents systemic inflammation and hepatic insulin resistance (17).
Given the broad role of SOCS in regulating macrophage functions in health and disease, the modulation of macrophage-specific SOCS1 and SOCS3 expression provides new opportunities for therapeutic manipulation of immune and inflammatory responses. However, it is not only macrophages that are affected by SOCS proteins. Other cell types upregulate and react to SOCS proteins to shape cellular functions. Targeting SOCS specifically in macrophages is therefore important as an efficient means of changing the inflammatory response.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol (2011) 11:723–37. doi: 10.1038/nri3073
2. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2000) 164:6166–73. doi:10.4049/jimmunol.164.12.6166
3. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest (2012) 122:787–95. doi:10.1172/JCI59643
4. Whyte CS, Bishop ET, Rückerl D, Gaspar-Pereira S, Barker RN, Allen JE, et al. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol (2011) 90:845–54. doi:10.1189/jlb.1110644
5. Liu Y, Stewart KN, Bishop ET, Marek CM, Kluth DC, Rees AJ, et al. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J Immunol (2008) 180:6270–8. doi:10.4049/jimmunol.180.9.6270
6. Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology (2014) 141:96–110. doi:10.1111/imm.12173
7. Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol (2012) 189:3439–48. doi:10.4049/jimmunol.1201168
8. Strebovsky J, Walker P, Dalpke AH. Suppressor of cytokine signaling proteins as regulators of innate immune signaling. Front Biosci (2012) 17:1627–39.
9. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signaling, and immune regulation. Nat Rev Immunol (2007) 7:454–65. doi:10.1038/nri2093
10. Madonna S, Scarponi C, De Pità O, Albanesi C. Suppressor of cytokine signaling 1 inhibits IFN-gamma inflammatory signaling in human keratinocytes by sustaining ERK1/2 activation. FASEB J (2008) 22:3287–97. doi:10.1096/fj.08-106831
11. Lu Y, Fukuyama S, Yoshida R, Kobayashi T, Saeki K, Shiraishi H, et al. Loss of SOCS3 gene expression converts STAT3 function from anti-apoptotic to pro-apoptotic. J Biol Chem (2006) 281:36683–90. doi:10.1074/jbc.M607374200
12. Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res (2010) 70:4840–9. doi:10.1158/0008-5472.CAN-10-0269
13. Puhr M, Santer FR, Neuwirt H, Marcias G, Hobisch A, Culig Z. SOCS-3 antagonises the proliferative and migratory effects of fibroblast growth factor-2 in prostate cancer by inhibition of p44/p42 MAPK signalling. Endocr Relat Cancer (2010) 17:525–38. doi:10.1677/ERC-10-0007
14. Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity (2002) 17:677–87. doi:10.1016/S1074-7613(02)00449-1
15. Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell (2003) 12:1413–26. doi:10.1016/S1097-2765(03)00490-8
16. Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity (2002) 17:583–91. doi:10.1016/S1074-7613(02)00446-6
17. Sachithanandan N, Graham KL, Galic S, Honeyman JE, Fynch SL, Hewitt KA, et al. Macrophage deletion of SOCS1 increases sensitivity to LPS and palmitic acid and results in systemic inflammation and hepatic insulin resistance. Diabetes (2011) 60:2023–31. doi:10.2337/db11-0259
18. Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem (2004) 279:54708–15. doi:10.1074/jbc.M410992200
19. Gingras S, Parganas E, de Pauw A, Ihle JN, Murray PJ. Re-examination of the role of suppressor of cytokine signaling 1 (SOCS1) in the regulation of toll-like receptor signaling. J Biol Chem (2004) 279:54702–7. doi:10.1074/jbc.M411043200
20. Cheng C, Huang C, Ma TT, Bian EB, He Y, Zhang L, et al. SOCS1 hypermethylation mediated by DNMT1 is associated with lipopolysaccharide-induced inflammatory cytokines in macrophages. Toxicol Lett (2014) 225:488–97. doi:10.1016/j.toxlet.2013.12.023
21. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A (2007) 104:1604–9. doi:10.1073/pnas.0610731104
22. Ruggiero T, Trabucchi M, De Santa F, Zupo S, Harfe BD, McManus MT, et al. LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. FASEB J (2009) 23:2898–908. doi:10.1096/fj.09-131342
23. Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity (2009) 31:220–31. doi:10.1016/j.immuni.2009.06.024
24. Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol (2004) 172:7565–73. doi:10.4049/jimmunol.172.12.7565
25. Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity (2004) 21:227–39. doi:10.1016/j.immuni.2004.07.010
26. Dickensheets HN, Vazquez N, Sheikh F, Gingras S, Murray PJ, Ryan JJ, et al. Suppressor of cytokine signaling-1 is an IL-4-inducible gene in macrophages and feedback inhibits IL-4 signaling. Genes Immun (2007) 8:21–7. doi:10.1038/sj.gene.6364352
27. O’Connor JC, Sherry CL, Guest CB, Freund GG. Type 2 diabetes impairs insulin receptor substrate-2-mediated phosphatidylinositol 3-kinase activity in primary macrophages to induce a state of cytokine resistance to IL-4 in association with overexpression of suppressor of cytokine signaling-3. J Immunol (2007) 178:6886–93. doi:10.4049/jimmunol.178.11.6886
28. Pesce JT, Ramalingam TR, Mentink-Kane MM, Wilson MS, El Kasmi KC, Smith AM, et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog (2009) 5:e1000371. doi:10.1371/journal.ppat.1000371
29. Esper L, Roman-Campos D, Lara A, Brant F, Castro LL, Barroso A, et al. Role of SOCS2 in modulating heart damage and function in a murine model of acute Chagas disease. Am J Pathol (2012) 181:130–40. doi:10.1016/j.ajpath.2012.03.042
30. Zadjali F, Santana-Farre R, Vesterlund M, Carow B, Mirecki-Garrido M, Hernandez-Hernandez I, et al. In SOCS2 KO mice, there was a reduction in both parasitemia and the expression of interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), IL-6, IL-10, SOCS1, and SOCS3 in the spleen. FASEB J (2012) 26:3282–91. doi:10.1096/fj.12-205583
31. Posselt G, Schwarz H, Duschl A, Horejs-Hoeck J. Suppressor of cytokine signaling 2 is a feedback inhibitor of TLR-induced activation in human monocyte-derived dendritic cells. J Immunol (2011) 187:2875–84. doi:10.4049/jimmunol.1003348
32. Whitmarsh RJ, Gray CM, Gregg B, Christian DA, May MJ, Murray PJ, et al. A critical role for SOCS3 in innate resistance to Toxoplasma gondii. Cell Host Microbe (2011) 10:224–36. doi:10.1016/j.chom.2011.07.009
33. Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol (2003) 4:546–50. doi:10.1038/ni932
34. Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. Negatively regulates IL-6 signaling in vivo. Nat Immunol (2003) 4:540–5. doi:10.1038/ni931
35. Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol (2003) 4:551–6. doi:10.1038/ni938
36. Liu X, Zhang Y, Yu Y, Yang X, Cao X. SOCS3 promotes TLR4 response in macrophages by feedback inhibiting TGF-beta1/Smad3 signaling. Mol Immunol (2008) 45:1405–13. doi:10.1016/j.molimm.2007.08.018
37. Hiwatashi K, Tamiya T, Hasegawa E, Fukaya T, Hashimoto M, Kakoi K, et al. Suppression of SOCS3 in macrophages prevents cancer metastasis by modifying macrophage phase and MCP2/CCL8 induction. Cancer Lett (2011) 308:172–80. doi:10.1016/j.canlet.2011.04.024
38. Matsumura Y, Kobayashi T, Ichiyama K, Yoshida R, Hashimoto M, Takimoto T, et al. Selective expansion of foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J Immunol (2007) 179:2170–9. doi:10.4049/jimmunol.179.4.2170
39. Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A (2012) 109:5004–9. doi:10.1073/pnas.1117218109
40. Wong PK, Egan PJ, Croker BA, O’Donnell K, Sims NA, Drake S, et al. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest (2006) 116:1571–81. doi:10.1172/JCI25660
41. Ortiz-Munoz G, Martin-Ventura JL, Hernandez-Vargas P, Mallavia B, Lopez-Parra V, Lopes-Franco O, et al. Suppressors of cytokine signalling modulate JAK/STAT-mediated cell responses during atherosclerosis. Arterioscler Thromb Vasc Biol (2009) 29:525–31. doi:10.1161/ATVBAHA.108.173781
42. Tang J, Kozaki K, Farr AG, Martin PJ, Lindahl P, Betsholtz C, et al. The absence of platelet-derived growth factor-B in circulating cells promotes immune and inflammatory responses in atherosclerosis-prone ApoE-/- mice. Am J Pathol (2005) 167:901–12. doi:10.1016/S0002-9440(10)62061-5
43. Shaikh S, Brittenden J, Lahiri R, Brown PA, Thies F, Wilson HM. Macrophage subtypes in symptomatic carotid artery and femoral artery plaques. Eur J Vasc Endovasc Surg (2012) 44:491–7. doi:10.1016/j.ejvs.2012.08.005
44. Grothusen C, Schuett H, Hillmer A, Lumpe S, Grote K, Ballmaier M. Role of suppressor of cytokine signaling-1 in murine atherosclerosis. PLoS One (2012) 7:e51608. doi:10.1371/journal.pone.0051608
45. Yamamoto TYH, Koga M. Lacking the SOCS3 Gene in macrophages prevents the development of atherosclerosis in ApoE deficient mice. Circ J (2007) 71:170.
46. Li Y, de Haar C, Peppelenbosch MP, van der Woude CJ. SOCS3 in immune regulation of inflammatory bowel disease and inflammatory bowel disease-related cancer. Cytokine Growth Factor Rev (2012) 23:127–38. doi:10.1016/j.cytogfr.2012.04.005
47. Hontecillas R, Horne WT, Climent M, Guri AJ, Evans C, Zhang Y. Immunoregulatory mechanisms of macrophage PPAR-γ in mice with experimental inflammatory bowel disease. Mucosal Immunol (2011) 4:304–13. doi:10.1038/mi.2010.75
48. Heys SD, Stewart KN, McKenzie EJ, Miller ID, Wong SY, Sellar G, et al. Characterisation of tumour-infiltrating macrophages: impact on response and survival in patients receiving primary chemotherapy for breast cancer. Breast Cancer Res Treat (2009) 135:539–48. doi:10.1007/s10549-012-2190-6
49. Hashimoto M, Ayada T, Kinjyo I, Hiwatashi K, Yoshida H, Okada Y, et al. Silencing of SOCS1 in macrophages suppresses tumor development by enhancing antitumor inflammation. Cancer Sci (2009) 100:730–6. doi:10.1111/j.1349-7006.2009.01098.x
Keywords: suppressor of cytokine signaling proteins, macrophage, M1, M2, inflammation
Citation: Wilson HM (2014) SOCS proteins in macrophage polarization and function. Front. Immunol. 5:357. doi: 10.3389/fimmu.2014.00357
Received: 04 June 2014; Paper pending published: 26 June 2014;
Accepted: 12 July 2014; Published online: 28 July 2014.
Edited by:
Laurel L. Lenz, National Jewish Health, USAReviewed by:
Masato Kubo, Tokyo University of Science, JapanCopyright: © 2014 Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:aC5tLndpbHNvbkBhYmRuLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.