 Natalia Castaño-Rodríguez
Natalia Castaño-Rodríguez Nadeem O. Kaakoush
Nadeem O. Kaakoush Hazel M. Mitchell
Hazel M. Mitchell- School of Biotechnology and Biomolecular Sciences, The University of New South Wales, Sydney, NSW, Australia
Chronic inflammation has been associated with an increased risk of several human malignancies, a classic example being gastric adenocarcinoma (GC). Development of GC is known to result from infection of the gastric mucosa by Helicobacter pylori, which initially induces acute inflammation and, in a subset of patients, progresses over time to chronic inflammation, gastric atrophy, intestinal metaplasia, dysplasia, and finally intestinal-type GC. Germ-line encoded receptors known as pattern-recognition receptors (PRRs) are critical for generating mature pro-inflammatory cytokines that are crucial for both Th1 and Th2 responses. Given that H. pylori is initially targeted by PRRs, it is conceivable that dysfunction within genes of this arm of the immune system could modulate the host response against H. pylori infection, and subsequently influence the emergence of GC. Current evidence suggests that Toll-like receptors (TLRs) (TLR2, TLR3, TLR4, TLR5, and TLR9), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) (NOD1, NOD2, and NLRP3), a C-type lectin receptor (DC-SIGN), and retinoic acid-inducible gene (RIG)-I-like receptors (RIG-I and MDA-5), are involved in both the recognition of H. pylori and gastric carcinogenesis. In addition, polymorphisms in genes involved in the TLR (TLR1, TLR2, TLR4, TLR5, TLR9, and CD14) and NLR (NOD1, NOD2, NLRP3, NLRP12, NLRX1, CASP1, ASC, and CARD8) signaling pathways have been shown to modulate the risk of H. pylori infection, gastric precancerous lesions, and/or GC. Further, the modulation of PRRs has been suggested to suppress H. pylori-induced inflammation and enhance GC cell apoptosis, highlighting their potential relevance in GC therapeutics. In this review, we present current advances in our understanding of the role of the TLR and NLR signaling pathways in the pathogenesis of GC, address the involvement of other recently identified PRRs in GC, and discuss the potential implications of PRRs in GC immunotherapy.
Introduction
Of the three main types of stomach cancer, gastric adenocarcinoma (GC), non-Hodgkin’s lymphoma, and gastrointestinal stromal tumors, approximately 95% are GC, which remains one of the most commonly diagnosed cancers in the world (1). In 2012, stomach cancer was the fifth most common cancer worldwide, with 952,000 new cases diagnosed, accounting for 6.8% of the total cancer cases (1). Furthermore, it is the third leading cause of cancer-related deaths worldwide, accounting for 8.8% of total deaths from cancer, with 5-year relative survival rates lower than 30%, except in Japan where mass screening has been undertaken for several years (2).
Gastric cancer is a heterogeneous pathology with respect to anatomical location and histological subtypes (Figure 1A). In relation to location, GC may occur in the cardia or non-cardia region of the stomach. Cardia GC has been associated with gastro-esophageal reflux, Helicobacter pylori infection, and atrophic gastritis, male gender, smoking, and diet (3). Epidemiological studies assessing the worldwide incidence of GC by anatomical location have shown an increase in the incidence of cardia GC, however, in high GC risk areas, non-cardia GC remains the most frequent pathology (4). Further, even though cardia and non-cardia GC have been considered etiologically different phenomena, it has been demonstrated that cancer of the cardia among individuals from areas with a high risk of GC represents a subset of cardia GC that is associated with H. pylori-related atrophic gastritis and resembles non-cardia GC pathogenesis (5, 6).
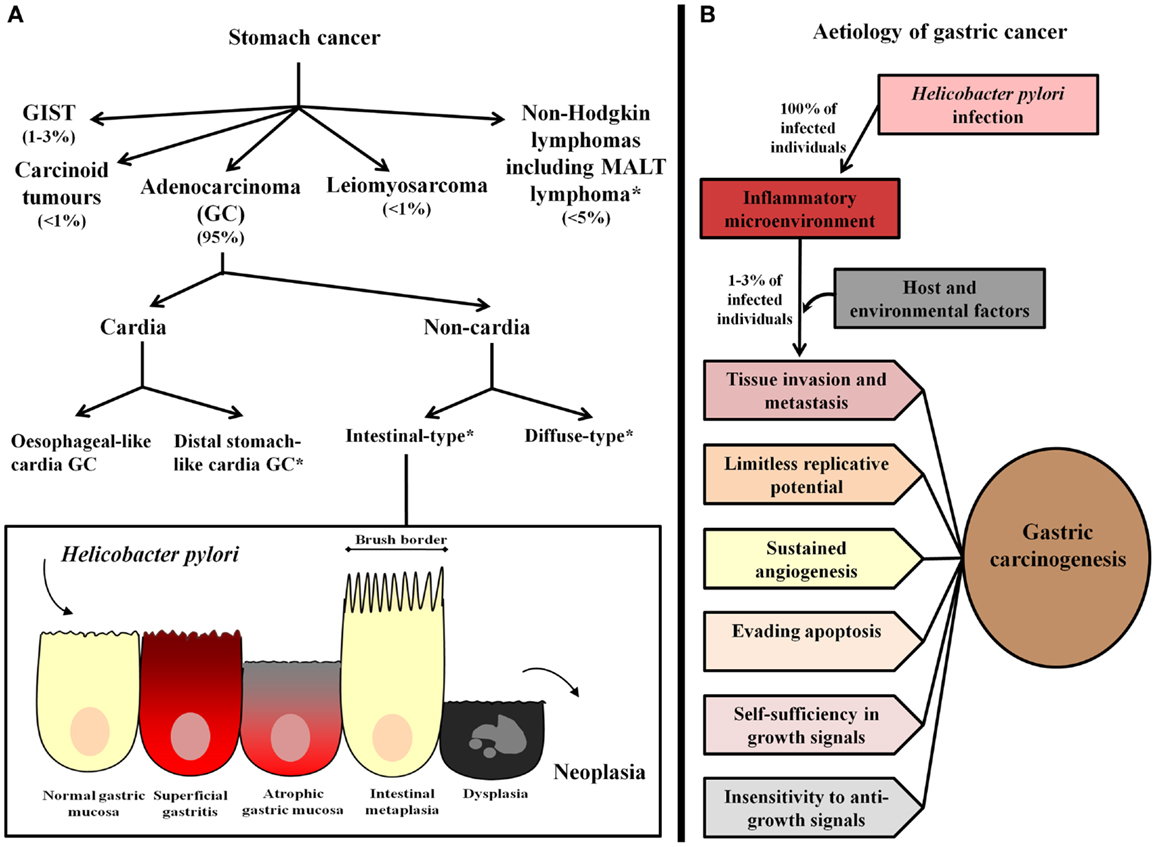
Figure 1. Gastric cancer classification and etiology. (A) Stomach cancer comprises gastric adenocarcinoma (GC), non-Hodgkin lymphomas, including mucosa-associated lymphoid tissue (MALT) lymphoma, and the rare gastrointestinal stromal tumors (GIST), leiomyosarcoma, and carcinoid tumors. The most common type, GC, has been classified as cardia and non-cardia GC according to anatomical location. Cardia GC is divided into two different etiological entities, esophageal-like cardia GC, which is associated with gastro-esophageal reflux, smoking, and diet, and is frequent in areas with a low risk of GC and distal stomach-like cardia GC, which is associated with the presence of H. pylori and gastric atrophy, and is the most frequent cardia GC variant in areas with a high risk of GC. Non-cardia GC is further subdivided into two histological variants called intestinal-type and diffuse-type GC. Intestinal-type GC, according to the widely accepted Correa’s cascade (7), is a biological continuum that commences as chronic gastritis and progresses to atrophic gastritis, intestinal metaplasia, dysplasia, and finally, GC. *Stomach cancer subtypes that have been associated with Helicobacter pylori infection. (B) H. pylori infection causes chronic inflammation of the gastric mucosa of all infected individuals, and in combination with host and environmental factor, leads to the development of GC in a subset of infected individuals (1–3%). In these subjects, inflammation represents the seventh hallmark of cancer and an enabling characteristic that facilitates the acquisition of the other established hallmarks that collectively dictate malignant growth (tissue invasion/metastasis, limitless replicative potential, sustained angiogenesis, evasion of programed-cell death (apoptosis), self-sufficiency in growth signals, and insensitivity to growth-inhibitory signals) (8, 9).
According to the Lauren Classification, non-cardia GC has been further subdivided into the two histological variants intestinal-type and diffuse-type. Intestinal-type GC is characterized by the formation of gland-like structures, distal stomach localization, and a predilection for older individuals. It is also more frequent in males (2:1 ratio) and in subjects of lower socioeconomic status (10). This type of GC is often preceded by a precancerous phase that starts with the transition of normal mucosa into multifocal atrophic gastritis. This initial histological alteration is followed by intestinal metaplasia, dysplasia, and finally adenocarcinoma (11). On the other hand, diffuse-type GC is poorly differentiated, affects younger individuals, and has been highly associated with genetic susceptibility (the variant hereditary diffuse GC, which is associated with germ-line mutations in CDH1, a gene encoding E-cadherin) (12, 13). Additionally, it is not associated with the formation of precancerous lesions and has been found to affect the entire surface of the stomach. This type of GC is present equally between the two sexes and is associated with a worse prognosis in comparison to intestinal-type GC (10, 12).
Most GC cases are sporadic and arise due to the combination of a permissive environment interacting with a susceptible host. Several factors that contribute to the development of GC have been identified; these include bacterial (H. pylori), host, and environmental factors (12).
Helicobacter pylori is a Gram-negative bacterium that infects nearly 50% of the human population (14). In the gastric mucosa, the majority of Helicobacter pylori are found within the mucus layer but they can also be attached to epithelial cells leading to the maintenance, spread, and severity of the infection (15). H. pylori infection has been associated with the development of a range of diseases, including peptic ulcer disease (10%), non-cardia GC (1–3%), and gastric mucosa-associated lymphoid tissue (MALT) lymphoma (<0.1%) (14, 16–18). Furthermore, this bacterium has been associated with three distinct phenotypes in the infected host: (1) a corpus-predominant gastritis, which has the potential to lead to atrophic gastritis, hypochlorhydria, and to the development of GC; (2) a duodenal ulcer phenotype in which an antrum-predominant gastritis leads to increased gastric acid secretion; and (3) a benign phenotype in which the bacterial infection causes a mild mixed gastritis that has a minor effect on gastric acid production (19).
Helicobacter pylori infection is transmitted by direct human-to-human transmission, via either the oral–oral route, fecal–oral route, or both (14). H. pylori is acquired early in life, the majority of individuals being infected before the age of 10 years with close family members being a common source of infection (20–22). It has been postulated that early acquisition of infection might be associated with the broad pathological spectrum associated with H. pylori infection and the highly persistent GC incidence rates in genetically susceptible populations who have migrated to developed countries. In the absence of antibiotic therapy, H. pylori infection generally persists for life (23).
Natural colonization by H. pylori is restricted to humans, primates, and domestic animals such as cats (23–25). H. pylori is considered to be the dominant microorganism in the human stomach as the majority of bacteria cannot survive in the low gastric pH (26). Several other factors make the human stomach an unfavorable environment for bacterial colonization including peristalsis, poor nutrient availability, and host innate and adaptive immunity (23). The ability of H. pylori to survive and colonize the stomach relates to a number of mechanisms. Most importantly H. pylori, unlike other bacteria, produces large amounts of the enzyme urease, which hydrolyzes urea to ammonia, which subsequently interacts with hydrogen ions in the stomach to form ammonium (27, 28). In addition, H. pylori is able to regulate gene expression in response to changes in pH (29). Further, H. pylori expresses multiple paralogous outer membrane proteins, including the blood-group antigen-binding adhesin (BabA), the sialic-acid binding adhesin (SabA), and the outer inflammatory protein (OipA), which appear to bind to receptors on the surface of gastric epithelial cells, which reduces the rate of bacterial elimination as a result of peristalsis (30, 31). H. pylori counteracts the lack of nutrients by inducing tissue inflammation and using specific systems that facilitate the transport and uptake of nutritional resources (23). In addition, H. pylori has been reported to produce antibacterial peptides that might decrease competition from other microorganisms (32).
Further, a number of other factors have been shown to help H. pylori evade the host immune system. For example, the vacuolating cytotoxin (VacA) produced by some strains of H. pylori has been shown to inhibit T-cell proliferation as well as antigen presentation by B cells and to alter the normal functions of CD8+ T cells, mast cells, and macrophages (33–36). In addition, gamma-glutamyl transpeptidase, another immunosuppressive factor of H. pylori, has been associated with inhibition of T-cell proliferation by induction of a cell cycle arrest in the G1 phase (37). Furthermore, H. pylori has been shown to use arginase to down-regulate the production of inducible nitric oxide synthase by macrophages (38).
The fact that more than one H. pylori strain can colonize the gastric mucosa provides the opportunity for H. pylori to acquire new genetic sequences and to undergo recombination events (23). One of the most remarkable differences among H. pylori strains is the presence or absence of a 40-kb DNA insertion element known as the cytotoxin-associated gene pathogenicity island (cag PAI) (39). This region contains between 27 and 31 genes flanked by 31-bp repeats and encodes the most widely investigated H. pylori virulence factor, the cytotoxin-associated antigen A (CagA) (40, 41). H. pylori strains expressing CagA represent 60–70% of Western strains and approximately 100% of East Asian strains (39, 42). CagA is a 120- to 140-kDa protein that is translocated into host cells through a type IV secretion system following attachment to gastric epithelial cells (43). Following translocation, CagA is tyrosine phosphorylated at the EPIYA (glutamate–proline–isoleucine–tyrosine–alanine) motifs by members of the host cell kinase families known as proto-oncogene proteins Abl and Src (18). In Western populations strains, EPIYA-A, EPIYA-B, and varying numbers of EPIYA-C motifs have been reported, while in H. pylori strains from East Asian populations, EPIYA-A and EPIYA-B with EPIYA-D motifs, are found (44). Both phosphorylated and non-phosphorylated CagA result in alterations in the gastric epithelium including: (1) the activation of the protein tyrosine phosphatase, non-receptor type 11 (SHP-2), (2) alterations in cell scattering and proliferation, (3) alterations in cell structure and cell motility, (4) perturbation of epithelial cell differentiation and polarity, (5) alteration of tight junctions, and (6) aberrant activation of β-catenin (45–47). Furthermore, numerous studies have shown that cag PAI-positive H. pylori strains are associated with an increased risk of gastric diseases including peptic ulcer disease, premalignant gastric lesions and GC (48–51). Further details of the interplay between H. pylori virulence factors and gastric epithelial cells and GC, can be found in an excellent review by Posselt et al. (44).
In the last two decades, a large number of epidemiological studies have established the association between H. pylori and the subsequent risk of developing both intestinal-type and diffuse-type GC (52–57). This finding has been consistent among different populations. For example, in the study by Parsonnet et al. (57), conducted in Caucasian, African-American, and Asian individuals, subjects infected with H. pylori who had antibodies against CagA were shown to be more likely than uninfected subjects to develop both intestinal-type and diffuse-type GC (OR: 5.1, 95% CI: 2.1–12.2 and OR: 10.1, 95% CI: 2.2–47.4, respectively). Consistently, a further study conducted in a Japanese population showed that, although the association was stronger in cases with intestinal-type GC (OR: 3.2, 95% CI: 1.8–5.8), there was also a positive association between H. pylori infection and diffuse-type GC (OR: 3.0, 95% CI: 1.0–8.8) (53). Further, a study conducted in a Spanish population showed no differences in H. pylori infection between the two GC histological subtypes (58). Similarly, a recent study in German individuals showed that H. pylori prevalence was comparable in patients with intestinal-type (82.1%) and diffuse-type (77.9%) GC (59).
Interestingly, more recent studies, assessing H. pylori infection through Western blot (CagA) for the detection of past infection, have shown an unprecedented association between H. pylori and GC that can be explained by a reduction of the misclassification that might take place when samples are analyzed with the enzyme-linked immunosorbent assay (ELISA) alone (60, 61). For example, Ekstrome et al. (60) conducted a population-based study, comprising 298 GC patients and 244 controls, in which the OR for H. pylori infection among non-cardia GC was 21.0 (95% CI: 8.3–53.4). Further, Siman et al. (61) showed that H. pylori significantly increased the risk of non-cardia GC showing an OR of 17.8 (95% CI: 4.2–74.8).
While H. pylori infection has been established as the most important risk factor for GC and was classified as a class 1 carcinogen by the World Health Organization in 1994, the etiology of GC also involves host and environmental factors. This is evidenced by the fact that only 1–3% of H. pylori-infected patients develop GC, and that progression to GC in some subjects occurs even after eradication of the bacterium (18).
Given that H. pylori is initially targeted by germ-line encoded receptors known as pattern-recognition receptors (PRRs), it is conceivable that dysfunction within genes of this arm of the immune system would affect the magnitude and direction of the host inflammatory response against the infection, resulting in an increased risk of GC development. Recent studies clearly show that PRRs are critical for generating mature pro-inflammatory cytokines that are crucial for both Th1 and Th2 responses during H. pylori infection, and these immune responses have been directly associated with gastric immunopathology. In this review, we present current advances in the understanding of the role of PRRs, mainly the Toll-like receptor (TLR) and nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) signaling pathways, in the pathogenesis of GC, and discuss future directions for continued research in this area. In the first section, we highlight the relevance of inflammation in GC. In subsequent sections, we address new developments in the TLR and NLR signaling pathways in GC, the role of other PRRs in GC, and the new frontier of therapeutic application of these concepts.
Inflammation in Gastric Cancer
It is well established that most cancer cell genotypes are the manifestation of six essential alterations in cell physiology that collectively dictate malignant growth: (1) self-sufficiency in growth signals, (2) insensitivity to growth-inhibitory signals, (3) evasion of programed-cell death (apoptosis), (4) limitless replicative potential, (5) sustained angiogenesis, and (6) tissue invasion/metastasis (8). Recently, inflammation has been considered the seventh hallmark of cancer and an enabling characteristic that facilitates the acquisition of the other hallmarks (Figure 1B). Inflammation initiated by innate immune cells, mainly macrophage subtypes, mast cells, myeloid progenitors, and neutrophils (62–65), designed to fight infections and heal wounds, can instead result in unintentional support of multiple cancer hallmark functions, thereby manifesting the widely accepted tumor-promoting consequences of inflammatory responses (9). In addition, active evasion by cancer cells from attack and elimination by immune cells, mainly CD8+ cytotoxic T lymphocytes, CD4+ Type 1 helper T cells, and natural killer (NK) cells, highlights the dual role of an immune system that both antagonizes and promotes cancer development and progression (9).
In the context of tumor enhancement, it has been proposed that once inflammation is initiated, tissue integrity is compromised leading to the multistage process of carcinogenesis by altering targets and pathways that are pivotal for normal tissue homeostasis (66). The mechanisms that are connected to these alterations include production of mutagenic reactive oxygen and nitrogen species as well as synthesis of cytokines and growth factors that favor tumor cell growth (67). In addition, inflammation provides a source of other bioactive molecules to the tumor microenvironment, including survival factors that limit cell death, pro-angiogenic factors, extracellular matrix-modifying enzymes that facilitate angiogenesis, invasion, and metastasis, and inductive signals that lead to activation of the epithelial–mesenchymal transition (a developmental regulatory program that enables epithelial cells to invade, resist apoptosis, and disseminate) (9). Interestingly, inflammation can be considered a “perigenetic alteration” of cancer cells because it may promote growth, expansion, and invasion of tumors even without the involvement of further genetic mutations or epigenetic alterations (68).
In 1988, Correa proposed a human model of intestinal-type gastric carcinogenesis (7). The model hypothesized a sequence of events progressing from acute inflammation to chronic inflammation, to atrophy, to intestinal metaplasia, to dysplasia, to carcinoma in situ, and finally to invasive GC. A subsequent study by Correa evaluated the gastric precancerous process in a Colombian population (7). The results of this cross-sectional study led to the widely accepted conclusion that the severity of atrophy correlates with the prevalence of metaplasia and that the severity of metaplasia correlates with the prevalence of dysplasia, suggesting that the process is indeed a biological continuum (69).
Given that inflammation is a hallmark of gastric carcinogenesis, polymorphisms in genes encoding pro-inflammatory cytokines/chemokines have been the focus of much research in recent years. To date, polymorphisms in the interleukin (IL)-1 family genes have been the most widely studied, including polymorphisms in IL1A, IL1B, and IL1RN that encode IL-1α, IL-1β, and their endogenous receptor antagonist IL-1RA, respectively. In particular, IL-1β, a potent endogenous pyrogen and an important component in the development of Th2-mediated immunity (70, 71), has been associated with lipid peroxidation, DNA damage, inhibition of gastric acid secretion, increased H. pylori colonization, and induction of gastric atrophy and dysplasia in the presence or absence of H. pylori (72). Global meta-analyses have shown that the IL1B-511 T allele is significantly associated with an increased risk of developing GC in Caucasians but not Asians or Mestizos (73, 74). Furthermore, IL-1 receptor signaling is known to induce the production of genes that not only stimulate tumor growth but are also involved in angiogenesis and metastasis such as matrix metalloproteinases, basic fibroblast growth factor, vascular endothelial growth factor, vascular cell adhesion molecule 1, intercellular adhesion molecule 1, monocytic chemotactic protein 1, and CXCL-2 (75). To date, only one study has addressed the role of IL1R1 (also known as CD121A) in GC and H. pylori infection. The study, conducted in a Caucasian population, showed an increased risk of H. pylori infection in those harboring the IL1R1 Hinfl A allele (OR: 2.01, P-value: 0.009) but failed to show an association with GC (76). In addition, a recent meta-analysis on the endogenous receptor antagonist IL-1RA has shown the IL1RN*22 genotype to increase the risk of gastric precancerous lesions, supporting a role for this polymorphism in the early stages of gastric carcinogenesis (OR: 2.27, 95% CI: 1.40–3.70) (77). A further meta-analysis that included 39 case–control studies, showed statistically significant associations between the IL1RN*22 genotype and both intestinal-type and diffuse-type GC, showing ORs of 1.83 and 1.72, respectively (78). Further examples of polymorphisms in pro-inflammatory cytokines/chemokines that play an essential role promoting inflammation in the context of gastrointestinal carcinogenesis are IL-4 (IL4-590C/T and -168T/C) (79), IL-6 (IL6-174 G/C) (80–82), IL-8 (IL8-251 A/T, +396 T/G, and +781 C/T) (79, 83), IL-10 (IL10-1082 A/G, −819 C/T, and −592 C/A) (84–86), IL-12 (IL12A-701 C/A, −798 T/A, +277 G/A, and −504 T/G) (87), IL-17 (IL17-197 G/A and +7488 T/C) (79), IL-18 (IL18-137 G/C) (88), and TNF-α (TNFA −238 G/A, −308 G/A, and −857 C/T) (89). In addition to this, a recent comprehensive review on this topic recommended the investigation of other polymorphisms in IL1B (3954 C/T and −1473G/C), IL4 (–168T/C), IL6 (572 G/C and 597 G/A), and IL17 (+7488A/G and −197G/A), given their potential relevance in GC (79).
While extensive evidence supports the important role of pro-inflammatory cytokines/chemokines in gastric carcinogenesis, given that PRRs, mainly TLRs and NLRs, are important modulators of intestinal epithelial barrier function, epithelial repair, and immune homeostasis in the gastrointestinal tract (90), and that signal transduction from these receptors converges upon a common set of signaling molecules, including the activation of the transcription factors nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and the activator protein 1 (AP-1) that lead to the production of pro-inflammatory cytokines/chemokines (e.g., IL-1α, IL-1β, IL-6, IL-8, IL-10, and TNF-α) as well as members of the interferon (IFN) regulatory transcription factor family that mediate type I IFN-dependent responses, defects in PRRs function could be even more important than defects in pro-inflammatory cytokines/chemokines per se in the instauration of an inflammation-related disorder such as GC.
Pattern-Recognition Receptors in Gastric Cancer
Innate immunity refers to responses that do not require previous exposure to an immune stimulus and represents the first line of host defense in the response to pathogens. PRRs are part of the innate immune system and are pivotal for the detection of invariant microbial motifs. PRRs have been divided into five distinct genetic and functional clades: TLRs, NLRs, C-type lectin receptors (CLRs), retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), and absent in melanoma 2 (AIM2)-like receptors (ALRs) (91, 92). PRRs are commonly expressed by cells of the innate immune system such as monocytes, macrophages, dendritic cells (DCs), neutrophils, and epithelial cells, as well as cells of the adaptive immune system (93).
Toll-like receptors and CLRs scan the extracellular milieu and endosomal compartments for pathogen-associated molecular patterns (PAMPs), which are highly conserved microbial structures that are essential for microbial survival (94), while intracellular PRRs, including NLRs, RLRs, and ALRs, cooperate to provide cytosolic surveillance (92, 93).
In H. pylori infection, the first physical–chemical barriers for the pathogen are the mucus layer, gastric epithelial cells, autophagy, and PRRs (TLRs, NLRs, CLRs, and RLRs) (Figure 2).
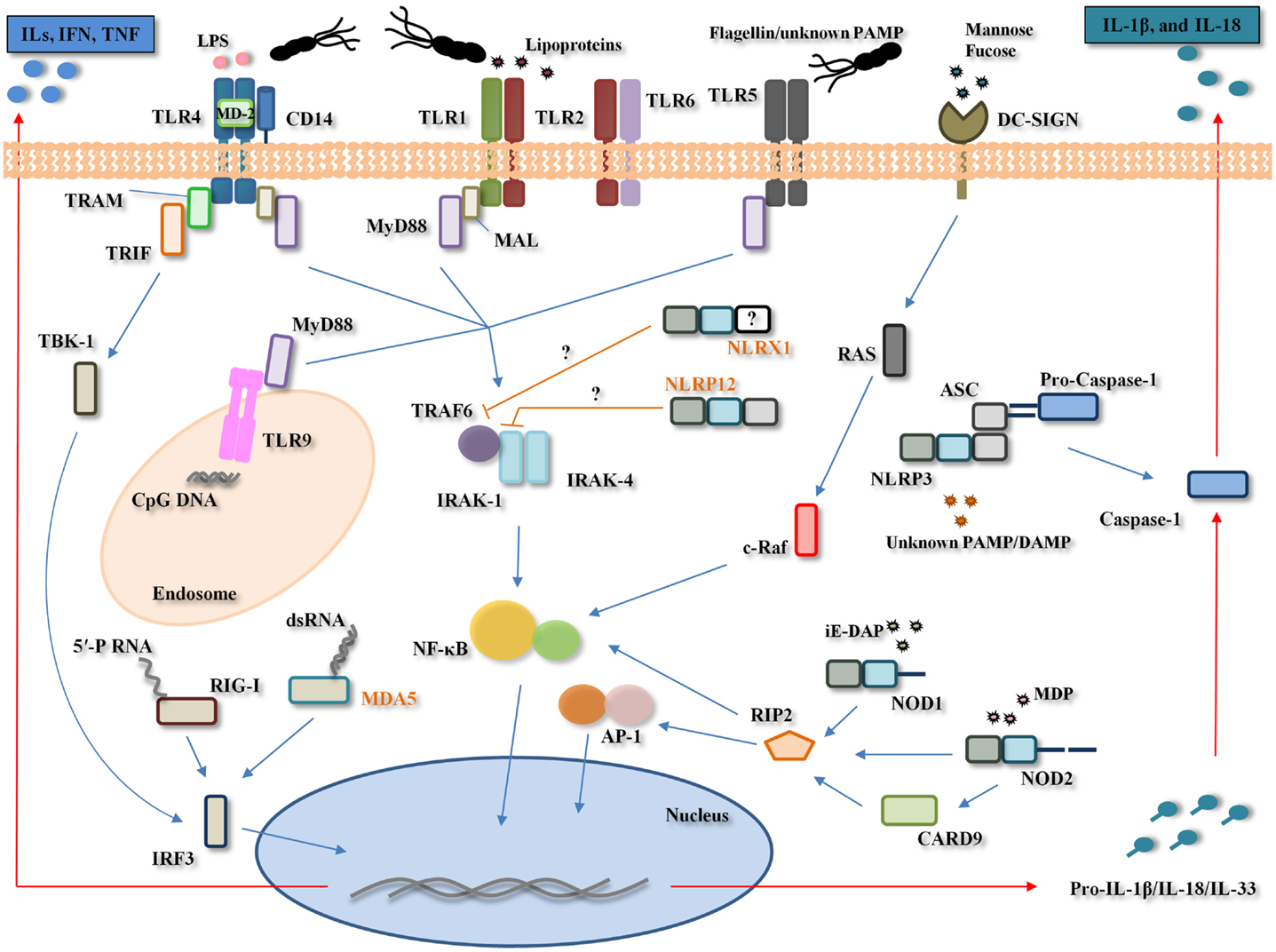
Figure 2. Pattern-recognition receptors involvement in Helicobacter pylori infection. H. pylori is recognized by the Toll-like receptors (TLRs) (TLR2, TLR4, TLR5, and TLR9), NOD-like receptors (NLRs) (NOD1, NOD2, NLRP3, and possibly, NLRP12 and NLRX1), RIG-I like receptors (RLRs) (RIG-I and possibly, MDA-5), and C-type lectin receptors (CLRs) (DC-SIGN). TLR4 poorly recognizes H. pylori lipopolysaccharide (LPS) to generate pro-inflammatory cytokines (e.g., IL-1α, IL-1β, IL-6, IL-8, IL-10, and TNF-α) and interferons (IFNs) through the myeloid differentiation primary response gene 88 (MyD88)-dependent and -independent pathways, respectively. TLR2 recognizes H. pylori LPS/peptigoglycan/unknown pathogen-associated molecular pattern (PAMP) while TLR5 poorly recognizes H. pylori flagella and TLR9 recognizes H. pylori DNA (unmethylated CpG motifs). H. pylori recognition by these three TLRs leads to nuclear factor-κB (NF-κB) activation. NOD1 and NOD2 recognize H. pylori peptidoglycan-derived peptides [γ-d-glutamyl-meso-diaminopimelic acid (iE-DAP) and muramyl dipeptide (MDP)], leading to the activation of both transcription factors NF-κB and activator protein (AP)-1. The NLRP3 inflammasome, comprising NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1, recognizes a yet unknown H. pylori PAMP and/or damage-associated molecular pattern (DAMP), and through caspase-1 cleavage, leads to the maturation and secretion of interleukin (IL)-1β and IL-18. NLRX1 and NLRP12, two known negative regulators of NF-κB, appear to be significantly down-regulated during H. pylori infection in vitro, however, their exact role during H. pylori infection remains unclear. RIG-I recognizes H. pylori 5′-triphosphorylated RNA (5′-PRNA) while MDA-5 possibly recognizes H. pylori dsRNA. The dendritic cell-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN) recognizes H. pylori fucosylated ligands and this interaction appears to counteract the pro-inflammatory immune response to H. pylori. Only one generic cell type depicting all TLRs, NLRs, RLRs, and CLRs involved in H. pylori recognition is shown here for simplicity. MAL, MyD88 adaptor-like protein, also named TIRAP; TRAM, translocating chain-associating membrane protein; TRIF, TIR domain containing adaptor inducing interferon-beta protein; TBK-1, TANK-binding kinase 1; IRF3, IFN-regulatory factor 3; TRAF6, TNF receptor-associated factor 6; IRAK, interleukin 1 receptor-associated kinase; RAS, proto-oncogene ras; c-RAF, proto-oncogene protein ras; RIP2, receptor-interacting serine/threonine-protein kinase 2, also known as RICK; CARD9, caspase activation and recruitment domain; MD-2, myeloid differentiation protein-2; ILs: interleukins. Names in orange correspond to molecules with a probable but not established role in the host response to H. pylori.
Toll-Like Receptors and Helicobacter pylori-Related Gastric Cancer
Toll-Like Receptors Recognition of Helicobacter pylori
The involvement of the TLR signaling pathway in infectious, autoimmune, and inflammatory diseases is well accepted (95). During H. pylori infection, TLRs on gastric epithelial and immune cells recognize diverse PAMPs such as flagellin/unknown PAMP (TLR5), unmethylated CpG motifs (TLR9), and lipopolysaccharide (LPS) (TLR4 and TLR2).
TLR4 was initially identified as the potential signaling receptor for H. pylori LPS on gastric epithelial cells (96–99). After forming a complex with the LPS-binding protein (LBP), LPS interacts with the monocyte differentiation antigen CD14 (CD14), and subsequently with the myeloid differentiation protein-2 (MD-2) (100). Together with TLR4, this complex induces the TLR4-mediated MyD88-dependent signal transduction pathway, which leads to the rapid activation of transcription factors, mainly NF-κB, and cytokines such as TNF-α, IL-1β, IL-6, and IL-12 (95). On the other hand, stimulation of TLR4 by LPS also facilitates the activation of a MyD88-independent pathway that activates IFN-regulatory factor (IRF) 3 and involves the late phase of NF-κB activation, both of which lead to the production of IFN-β and the expression of IFN-inducible genes (101, 102). In addition to LPS, the H. pylori secretory protein HP0175, through its ability to bind to TLR4, was shown to transactivate the epidermal growth factor receptor (EGFR) and stimulate the EGFR-dependent vascular endothelial growth factor production in the GC cell line AGS, which have been linked to H. pylori-associated gastroduodenal diseases, ulcerogenesis, and carcinogenesis (103).
Although early studies concluded that TLR4 is the first innate immune response against H. pylori (104, 105), later studies suggested that TLR4 had a limited role, given that H. pylori LPS appeared to bind poorly to LBP, resulting in it being inefficiently transferred to CD14 (106). Consequently, recent studies addressing the role of other TLRs during H. pylori infection, have found TLR2 to be the initial barrier against H. pylori infection (107–112). A potential explanation for these inter-study differences in relation to the TLRs response to H. pylori might be attributed to cell type (i.e., epithelial versus immune cells), origin of the cell studied (i.e., peritoneal versus bone marrow derived macrophages), and the type of inflammatory response measured (i.e., type of cytokines), and thus, currently any conclusions regarding the role of TLR4 must be treated with caution.
In contrast, there is strong evidence supporting an important role for TLR2 in H. pylori infection, with both animal and cell culture experiments suggesting that TLR2 ligands (LPS or other) exist in H. pylori and related Helicobacter species (112–114), and that TLR2 may be involved in the innate immune sensing of these bacteria by epithelial cells (113). Furthermore, an interesting publication by Smith et al. (115) showed that H. pylori LPS functions as a classic TLR2 ligand and induces a discrete pattern of chemokine expression in epithelial cells, which involves modulation of the expression of the signaling protein tribbles 3 (TRIB3), a molecule implicated in the regulation of NF-κB.
Yet, the most likely scenario is that both TLR4 and TLR2 are involved in the early immune response against H. pylori as has been demonstrated by a number of investigators (116–118). For example, Obonyo et al. (116) showed that both TLR2 and TLR4 were crucial signaling receptors for H. pylori activation of the host immune response leading to the secretion of cytokines. Further, Yokota et al. (118) not only showed that H. pylori LPS was initially targeted by TLR2 as described by others, but, for the first time, showed that this TLR2 activation leads to cell proliferation and TLR4 expression via the MEK1/2-ERK1/2 pathway. The final outcome of this signaling pathway is increased proliferation of gastric epithelial cells and the instauration of a strong inflammatory reaction. Once this response is instaurated, H. pylori could then enhance inflammatory reactions mediated by TLR4 agonists such as other bacterial LPS, which would also contribute to gastric inflammation and subsequent carcinogenesis (118). Further, the heat-shock protein 60, an immune-potent antigen of H. pylori, has been shown to activate NF-κB and induce IL-8 production through TLR2 and TLR4 pathways in gastric epithelial cells, a phenomenon that is likely to contribute to the development of gastric inflammation caused by H. pylori infection (117).
In addition, TLR9 appears to play an important role in H. pylori recognition. Interestingly, Rad et al. (112) identified TLR9-mediated recognition of H. pylori DNA as a main H. pylori-induced intracellular TLR signaling pathway in DCs. Further, a study using a murine model of H. pylori infection has suggested that TLR9 signaling is involved in the suppression of H. pylori-induced gastritis in the early phase of infection via down-regulation of Th1-type cytokines modulated by IFN-α (119). In addition, a recent study has shown that the gastric epithelia of children respond to H. pylori infection by increasing the expression of TLR2, TLR4, TLR5, and TLR9, as well as the cytokines IL-8, IL-10, and TNF-α (120).
Although TLR5 interaction with H. pylori induces only weak receptor activation (121), TLR5 has been involved in the inflammatory response to H. pylori. An interesting publication by Smith et al. (107), using HEK293 cells transfected with specific TLR expression constructs and MKN45 cells expressing dominant negative versions of TLR2, TLR4, and TLR5, which block the activity of wild-type forms of these receptors, has demonstrated that live H. pylori induces NF-κB activation and chemokine gene expression due to ligation of TLR2 and TLR5. A further study that aimed to explore the involvement of TLR2 and TLR5 in THP-1 cells and HEK293 cell lines (stably transfected with TLR2 or TLR5) during H. pylori infection, has indicated that H. pylori-induced expression of TLR2 and TLR5 can qualitatively shift cag PAI-dependent to cag PAI-independent pro-inflammatory signaling pathways with possible impact on the outcome of H. pylori-associated diseases (122). Given the established TLR5 evasion of α and ε Proteobacteria including H. pylori (123), the TLR5-mediated inflammatory responses during H. pylori infection described by Smith et al. (107) and Kumar Pachathundikandi et al. (122) are likely to be flagellin-independent, and therefore, a still unknown H. pylori factor might be responsible for this.
The importance of TLRs recognition during H. pylori infection and GC development is further supported by the acquired characteristics that enable H. pylori to survive in the human stomach and cause chronic inflammation. For example, H. pylori LPS is characterized by a modification of the lipid A component of LPS that makes it less pro-inflammatory (124) and has been reported to exhibit a 1000-fold reduction in bioactivity as compared to Escherichia coli LPS (125). Also, the flagellin of this bacterium has been shown to be poorly recognized due to modifications in the TLR5 recognition site of the N-terminal D1 domain of flagellin (123).
Toll-Like Receptors and Gastric Carcinogenesis
While TLR2, TLR4, TLR5, and TLR9 appear to be important for H. pylori recognition, their role in the evolution of gastritis to more advanced lesions remains unclear. Interestingly, Schmausser et al. (126) showed that TLR9 was not detectable in intestinal metaplasia or dysplasia and was only focally detected in 6 out of 22 gastric carcinomas, while TLR4 and TLR5 were strongly expressed by gastric carcinomas. Consistently, a study by Pimentel-Nunes et al. (127) showed a statistically significant trend for a progressive increase of TLR2, TLR4, and TLR5 expression from normal mucosa to gastric dysplasia (mean expression in normal mucosa: 0.1, gastritis: 1.0, metaplasia: 2.2, and dysplasia: 2.8, P-value <0.01), with dysplasia presenting more than 90% positive epithelial cells showing strong expression (2.8, 95% CI: 2.7–3). In addition, these authors showed a significant trend for decrease in TOLLIP and PPARγ, two TLR signaling pathway inhibitors, which was associated with increasing levels of CDX-2, a marker for adenocarcinoma, from normal mucosa to carcinoma (P-value <0.05) (128). Fernandez-Garcia et al. (129) have also reported increased expression of TLR3, TLR4, and TLR9 in GC, and furthermore, these authors noted that TLR3 expression by cancer cells was significantly associated with a poor overall survival in patients with resectable tumors, which lead them to suggest that TLR3 might be an indicator of tumor aggressiveness. Similarly, Yakut et al. (130) investigating the association between serum IL-1β, TLR4 levels, pepsinogen I and II, gastrin 17, vascular endothelial growth factor, and H. pylori CagA status in patients with a range of gastric precancerous lesions, concluded that serum TLR4 levels could be used as a biomarker to differentiate individuals presenting with dysplasia from those with other gastric precancerous lesions, the mean TLR4 level in patients with dysplasia (0.56 ± 0.098 ng/mL) being significantly higher than in patients with H. pylori positive chronic non-atrophic gastritis (0.10 ± 0.15 ng/mL), chronic atrophic gastritis (0.06 ± 0.07 ng/mL), and intestinal metaplasia (0.12 ± 0.18 ng/mL). Furthermore, while TLRs have been shown to be expressed at the apical and basolateral pole of both normal gastric epithelial cells and in H. pylori gastritis, in metaplasia, dysplastic, and neoplastic epithelial cells all TLRs are expressed diffusely and homogeneously throughout the cytoplasm, with no apparent polarization, which may suggest an increased activation of these diffusely over-expressed receptors during gastric carcinogenesis (126, 128).
In recent years, TLRs have been associated with tumor development and progression processes including cell proliferation, epithelial–mesenchymal transition, angiogenesis, metastasis, and immunosuppression. Interestingly, Chochi et al. (104) not only showed that H. pylori augmented the growth of GC via the LPS-TLR4 pathway but also found that this bacterium attenuated the antitumor activity and IFN-γ-mediated cellular immunity of human mononuclear cells. In addition, Song et al. (131) have suggested that flagellin-activated TLR5 enhances the proliferation of GC cells through an ERK-dependent pathway. Furthermore, Tye et al. (132) have proposed a novel role for TLR2 in promoting gastric tumorigenesis independent of inflammation, whereby up-regulation of TLR2 within epithelial tumor cells, rather than infiltrating inflammatory cells, by the uncontrolled activation of the oncogenic transcription factor STAT3, promoted gastric tumor cell proliferation, and survival via up-regulation of anti-apoptotic genes [e.g., BCL2-related protein A1 (BCL2A1), baculoviral IAP repeat containing 3 (BIRC3), and B-cell CLL/lymphoma 3 (BCL3)]. Further, two processes that facilitate carcinogenesis and involve TLRs have recently been described by Li et al. (133). Using LPS-treated CD14-knockdown GC cells, these authors showed that CD14, an important co-receptor in the TLR4 complex, promotes tumor cell epithelial–mesenchymal transition and invasion through TNF-α (133).
In addition, the expression of tumor-associated molecules known to be important in gastric carcinogenesis has been linked to the activation of the TLR signaling pathway. For example, prostaglandin-endoperoxide synthase 2 (PTGS2), which is also termed cyclooxygenase 2 (COX2), a key enzyme that catalyzes the conversion of arachidonic acid to prostaglandins, has been shown to play a pivotal role in gastric inflammation and carcinogenesis (134). For example, a study by Chang et al. (108), using clinical H. pylori isolates, has shown that H. pylori acts through TLR2/TLR9 to activate both the PI-PLCγ/PKCα/c-Src/IKKα/β and NIK/IKKα/β pathways, resulting in the phosphorylation and degradation of IκBα, which in turn leads to the stimulation of NF-κB and the expression of PTGS2.
Further, as compared with normal cells, cancer cells are more metabolically active and generate more reactive oxygen species (ROS), which affects cell survival. Several studies have suggested that ROS can act as secondary messengers and control a range of signaling cascades, leading to sustained proliferation of cancer cells (135, 136). In the context of gastric carcinogenesis, H. pylori-infected gastric epithelial cells have been shown to generate ROS (137). Interestingly, Yuan et al. (138) recently suggested that TLR4 expression in GC correlated with tumor stage and that activation of TLR4 contributed to GC cell proliferation via mitochondrial ROS production through up-regulation of phosphorylated Akt and NF-κB p65 activation and nuclear translocation.
However, the involvement of TLRs in GC might be more complex than initially suspected as TLRs not only recognize antigenic determinants of viruses, bacteria, protozoa, and fungi, but are also involved in the detection of damage-associated molecular patterns (DAMPs) (e.g., extracellular adenosine triphosphate, hyaluran, extracellular glucose, monosodium urate crystals) (139). Release of DAMPs, which are especially targeted by TLR2 and TLR4 (140–145) during cancer progression may cause chronic inflammation leading to down-regulation of the ζ chain of the T-cell and NK cell activating receptors [for comprehensive information on this topic see the review by Baniyash et al. (146)], which entails T-cell and NK cell dysfunction, a phenomenon observed in some malignancies such as GC (147, 148), colon (149), prostate (150), cervical (151), and pancreatic cancer (152). In addition to immunosuppression, DAMPS appear to facilitate other processes during gastric carcinogenesis. For example, Wu et al. (153) have recently showed that hyaluronan, derived from malignant cells, induced long-lived tumor-associated neutrophils and subsequent malignant cell migration in gastric carcinomas via a TLR4/PI3K interaction.
Collectively, TLRs might be involved in both gastric carcinogenesis mediated by H. pylori infection (a tumor-promoting consequence of inflammatory responses) and in GC perpetuation associated with immunosuppression (active evasion by cancer cells from attack and elimination by immune cells) and increased metastasis.
Genetic Polymorphisms Involved in the Toll-Like Receptor Signaling Pathway and Gastric Cancer
In recent years, a number of investigations have attempted to establish the relationship between polymorphisms in molecules of the TLR signaling pathway and risk of GC. Recent studies, conducted in several populations, have shown associations between the polymorphisms TLR1 rs5743618 (Ile602Ser) (154), TLR2 −196 to −174del (155–158), TLR2 rs3804099 (157), TLR4 rs4986790 (Asp299Gly) (155, 157, 159), TLR4 rs4986791 (Thr399Ile) (160), TLR4 rs10116253 (161), TLR4 rs10983755 (162), TLR4 rs11536889 (+3725G/C) (155), TLR4 rs1927911 (161), TLR5 rs5744174 (158), TLR9 rs187084 (−1486 T/C) (163), and CD14 rs2569190 (−260 C/T) (155, 164–167), and risk of GC development in an ethnic-specific manner (Table 1). In addition, three polymorphisms located in the TLR4 mRNA promoter region (sites −2081, −2026, and −1601) and TLR4 Thr135Ala at the leucine-rich repeat (LRR), have been associated with poorly differentiated GC (168, 169).
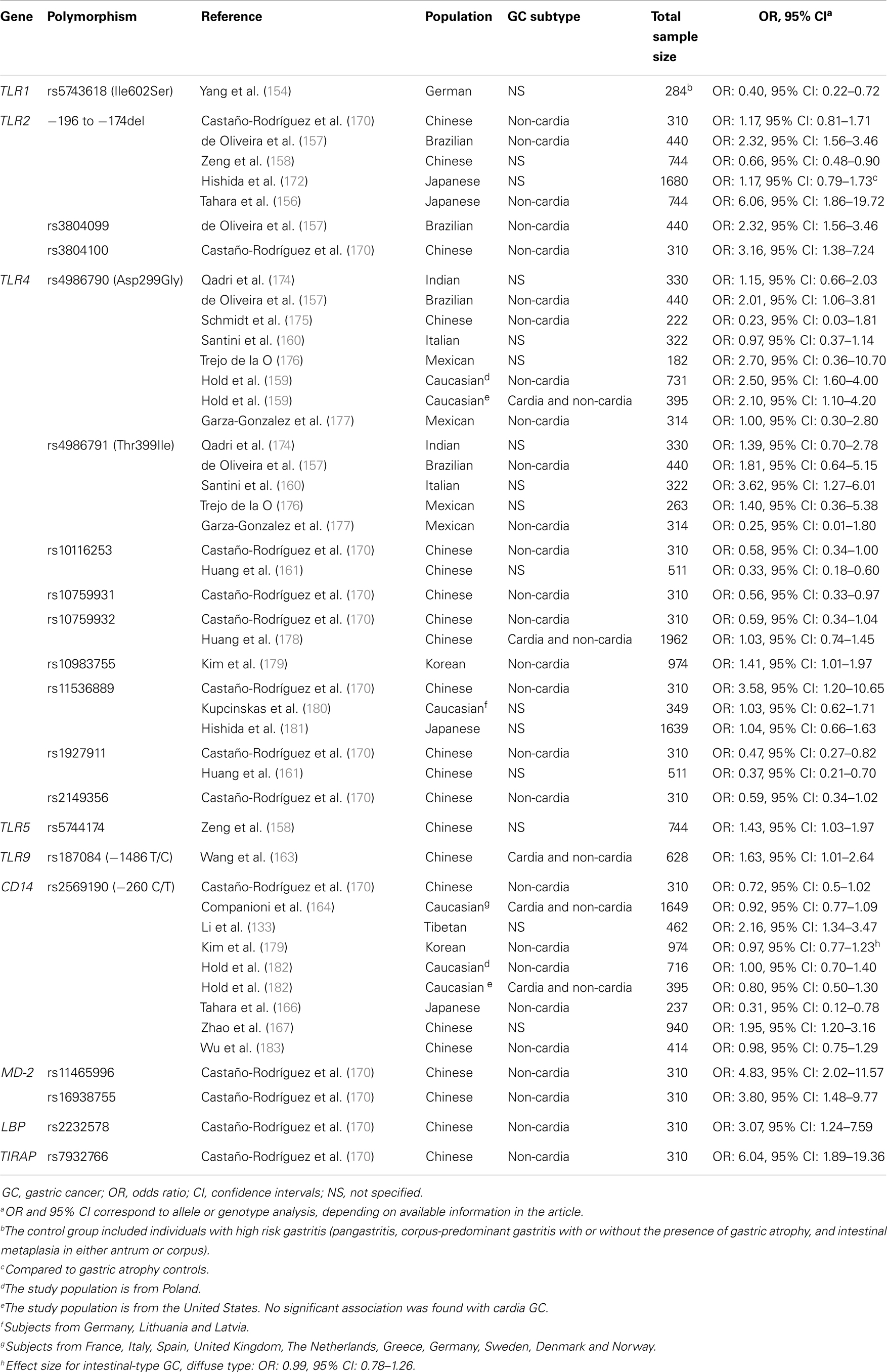
Table 1. Genetic polymorphisms in the Toll-like receptor signalling pathway that have been studied in relation to gastric cancer (170).
Interestingly, some of these polymorphisms including TLR4 Asp299Gly (159, 184), TLR4 Thr399Ile (184, 185), TLR4 rs10759932 (186), CD14-260 C/T (187), and TLR2 −196 to −174del (157), appear to be involved in the biological continuum that results in intestinal-type GC as they have also been associated with gastric precancerous lesions (Table 2).
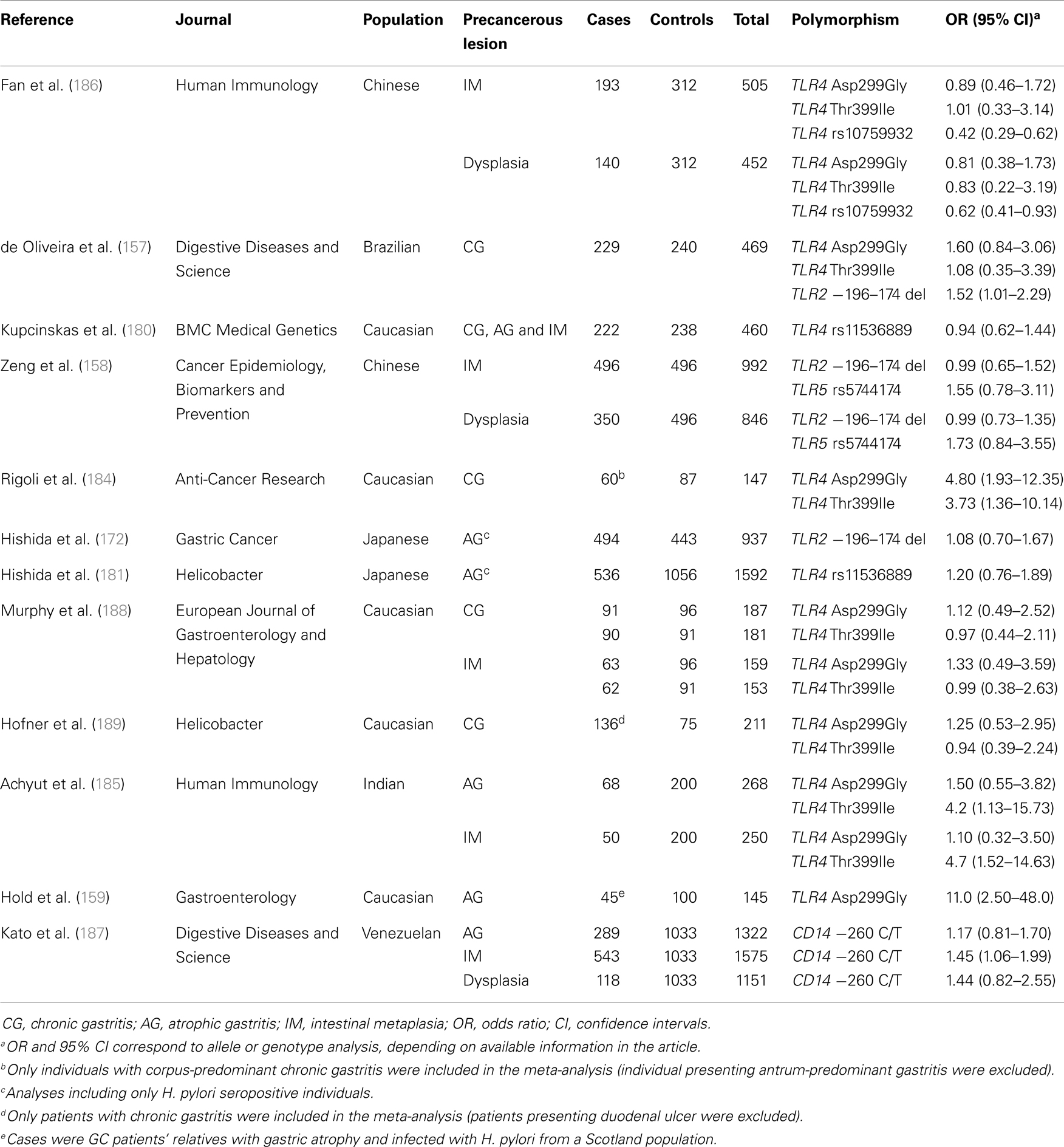
Table 2. Genetic polymorphisms in the Toll-like receptor signalling pathway that have been studied in relation to gastric precancerous lesions.
Given that some authors have failed to show specific associations between polymorphisms in the TLR signaling pathway, especially in TLR2, TLR4, and CD14, and gastric precancerous lesions/GC (157, 160, 162, 164, 172, 174–178, 180–183, 185, 188, 189), we performed the first global meta-analysis to assess the role of TLR2, TLR4, and CD14 polymorphisms in gastric carcinogenesis (155), in an attempt to clarify the limited and current conflicting evidence, and to establish the true impact of the TLR signaling pathway in GC. Our meta-analysis, which included 18 case–control studies conducted in Caucasian, Asian, and Latin American populations, showed that TLR4 Asp299Gly was a definitive risk factor for GC in Western populations (pooled OR: 1.87, 95% CI: 1.31–2.65). In addition, there was a potential association between TLR2 −196 to −174 and GC in Japanese (pooled OR: 1.18, 95% CI: 0.96–1.45) (155). Interestingly, a recent meta-analysis on TLR2 −196 to −174 and the risk of GC, conducted by Cheng et al. (190), failed to reproduce the findings in our meta-analysis, however, their stratification by ethnicity analyses included subjects from both Japan and China, which might explain the different outcomes. A further meta-analysis conducted by Chen et al. (191) that included 21 case–control studies showed an overall increased risk of GC in individuals harboring TLR4 Asp299Gly (Allele analysis, OR: 1.84, 95% CI: 1.41–2.39) and TLR4 Thr399Ile (Allele analysis, OR: 1.97, 95% CI: 1.22–3.18). Consistently, in stratified analyses by ethnicity, these authors only found an association between TLR4 Asp299Gly (Allele analysis, OR: 1.90, 95% CI: 1.43–2.51) and TLR4 Thr399Ile (Allele analysis, OR: 2.84, 95% CI: 1.56–5.15) in Caucasian individuals (191). Further, Zhao et al. (192) in an updated version of a meta-analysis that was initially conducted by Zhang et al. (193), on the risk of TLR4 polymorphisms and risk of cancer in general, found a significant association with GC after stratifying by cancer type (OR: 2.00, 95% CI: 1.53–2.62). In addition, Zou et al. (194), through a meta-analysis that included 10 case–control studies, not only found that TLR4 Asp299Gly was associated with GC (OR: 1.87, 95% CI: 1.44–2.44), especially non-cardia GC (OR: 2.03, 95% CI: 1.51–2.72), but also gastric precancerous lesions (OR: 2.47, 95% CI: 1.57–3.88), especially in H. pylori-infected individuals (OR: 3.43, 95% CI: 1.92–6.13).
Given limited evidence regarding the association between polymorphisms in other molecules of the TLR signaling pathway and the risk of GC, and the fact that 42% of cases of GC worldwide occur in the Chinese population, we conducted a case–control study comprising 310 ethnic Chinese individuals (87 non-cardia GC cases and 223 controls with functional dyspepsia), in which 25 polymorphisms involved in the TLR signaling pathway were investigated (170). Seven polymorphisms showed significant associations with GC (TLR4 rs11536889, TLR4 rs10759931, TLR4 rs1927911, TLR4 rs10116253, TLR4 rs10759932, TLR4 rs2149356, and CD14 −260 C/T). In multivariate analyses, TLR4 rs11536889 remained a risk factor for GC even after adjustment (OR: 3.58, 95% CI: 1.20–10.65). Further, TLR4 rs10759932 decreased the risk of H. pylori infection (OR: 0.59, 95% CI: 0.41–0.86) (170). Strikingly, statistical analyses assessing the joint effect of H. pylori and the selected polymorphisms revealed that H. pylori-infected individuals harboring TLR2 rs3804100, TLR2 −196 to −174del, TLR4 rs11536889, MD-2 rs11465996, MD-2 rs16938755, LBP rs2232578, and TIRAP rs7932766 were at most risk of developing GC (Table 1) (170).
The functional relevance of a number of these polymorphisms has already been established. For example, two polymorphisms in TLR4, Asp299Gly, and Thr399Ile, have been shown to disrupt the normal structure of the extracellular domain of TLR4, and thus, as a result, may reduce responsiveness to H. pylori by diminishing the binding affinity of the bacterial ligands (195). In addition, the TLR4 rs11536889 polymorphism, which is located in the center of the 2818-bp TLR4 3′ untranslated region (UTR), has recently been shown by Sato et al. (196) to contribute to the translational regulation of TLR4, possibly by binding to microRNAs. Further, these authors elegantly demonstrated that subjects harboring TLR4 rs11536889 exhibited higher levels of TLR4 receptors on monocytes and secreted higher levels of IL-8 in response to LPS (196). In addition, TLR4 rs10759932 has been shown to decrease the expression of forkhead box protein P3 (FOXP3), the most specific marker for natural regulatory T (Treg) cells (197). FOXP3+ Treg cells, which suppress the immune response of antigen-specific T cells, have been demonstrated to play a key role in immunologic tolerance (198). Notably, recent studies have not only shown that in vivo depletion of FOXP3+ Treg cells in H. pylori-infected mice leads to increased gastric inflammation and reduced bacterial colonization (199), but also recruitment of FOXP3+ Treg cells is increased in H. pylori-related human disorders including gastritis (200, 201), duodenal ulcer (202), and GC (200, 203, 204), suggesting that FOXP3+ Treg cells might contribute to lifelong persistence of H. pylori infection. Also, TLR1 rs5743618 appears to impair the surface expression of TLR1 of NK cells and NK cells-derived IFN-γ production (154). Further, TLR2 −196 to −174 has been associated with decreased transcriptional activity of TLR2 (205, 206). Similarly, it has been demonstrated that TLR9 rs187084 down-regulates TLR9 expression (207).
Further, CD14 has been shown to activate macrophages/monocytes to release Th1-type cytokines including IL-12, thus, establishing the chronic inflammation stimulated by H. pylori infection (208–210). A Th1 predominant response has been extensively associated with the pathogenesis of H. pylori-related gastric disease (211–213). Currently, however, controversy exists regarding the influence of CD14 −260 on expression of soluble CD14 (sCD14). According to a number of studies, the CD14 −260 T allele is believed to increase sCD14 production and therefore, serum sCD14 levels (214–217). In contrast, it has been reported that elevated sCD14 levels are associated with H. pylori infection, especially in subjects with the CD14 −260 CC genotype (167). Alternatively, others have argued that this polymorphism has no effect on transcription (218). Since the evidence to date is conflicting, more functional studies are required to clarify this issue.
Overall, it is clear that genetic variability in genes of the TLR signaling pathway plays an important role in GC pathogenesis. Investigations of polymorphisms in different molecules of this pathway among different populations could provide novel insights into targeted treatment in genetically susceptible individuals, and thus, improve primary and secondary prevention of H. pylori-related GC in high risk populations.
NOD-Like Receptors and Helicobacter pylori-Related Gastric Cancer
NOD-Like Receptors Recognition of Helicobacter pylori
The NLR family not only recognizes PAMPs but also DAMPs in the cytoplasm (93). The NLRs characteristic structure includes a central nucleotide-binding and oligomerization (NACHT) domain that is present in all NLR family members, a C-terminal LRRs and an N-terminal caspase recruitment (CARD) or pyrin (PYD) domain.
Based on phylogenetic analysis of NACHT domains, the NLR family has been shown to comprise three subfamilies: (1) the NOD family which includes NOD1-2, NOD 3 (NLRC3), NOD4 (NLRC5), NOD5 (NLRX1), and CIITA, (2) the NLRPs including NLRP1-14 (also known as NALPs), and (3) the IPAF subfamily, which consists of IPAF (NLRC4) and NAIP (93).
The NACHT domain belongs to a family of P-loop NTPases known as the signal transduction ATPases with numerous domains (STAND) (219). This domain permits activation of the signaling complex via adenosine ATP-dependent oligomerization (94). NACHT domain oligomerization is essential for the activation of NLRs, forming high molecular weight complexes, probably hexamers or heptamers that characterize inflammasomes (molecular complexes involved in the activation of inflammatory caspases for the maturation and secretion of IL-1β, IL-18, and possibly IL-33) and NOD signalosomes (complexes that are assembled upon oligomerization of NOD1 or NOD2 and lead to NF-κB activation through the receptor-interacting protein-2) (94). CARD and PYD are death domains that mediate homotypic protein–protein interactions for down-stream signaling (93, 94). These domains are characterized by six α helices that form trimers or dimers with other members of the same subfamily (94). The third domain, the LRR region, has been implicated in ligand sensing and autoregulation of not only NLRs but TLRs (93, 94). The LRR is formed by tandem repeats of a structural unit consisting of a β strand and an α helix and is composed of 20–30 amino acids that form a horse-shoe shaped structure rich in the hydrophobic amino acid leucine (220). The NLRPs LRR gene is made up of tandem repeats of exons of exactly 171 nucleotides, which encode one central LRR and two halves of the neighboring LRRs (221). This particular modular organization possibly allows extensive alternative splicing of the LRR region leading to maximum variability in the ligand-sensing unit (94). However, a recent publication by Tenthorey et al. (222) analyzing a panel of 43 chimeric NAIPs, showed that LRR was unnecessary for NAIP/NLRC4 inflammasome ligand specificity, leading them to propose a model in which NAIP activation is instead triggered by ligand binding to NACHT-associated helical domains. This recent evidence suggests that the ligand-sensing function of the LRR domain in NLRs, which has been supported primarily by analogy to the well-established ligand-sensing function of the LRR region in TLRs, needs to be re-examined.
The most widely studied NLRs during H. pylori infection are NOD1 and NOD2, which are expressed in epithelial and antigen-presenting cells, and are known to specifically recognize peptidoglycan-derived peptides (γ-d-glutamyl-meso-diaminopimelic acid and muramyl dipeptide, respectively). An early study, attempting to determine the mechanism whereby H. pylori delivers peptidoglycan to cytosolic host NOD1, demonstrated that H. pylori peptidoglycan is delivered to the host cell via a type IV secretion system (223). More recently, Hutton et al. (224) showed, for the first time, that cholesterol-rich microdomains called lipid rafts, were important for the type IV secretion system-dependent peptidoglycan delivery and subsequent NF-κB activation and IL-8 production, mediated by NOD1. Interestingly, Kaparakis et al. (225) reported a novel mechanism in Gram-negative bacteria, including H. pylori, for the delivery of peptidoglycan to cytosolic NOD1 in host cells that involves outer membrane vesicles that enter epithelial cells through lipid rafts. In addition, Necchi et al. (226) demonstrated the formation of a particle-rich cytoplasmic structure (PaCS) in H. pylori-infected human gastric epithelium having metaplastic or dysplastic foci, where VacA, CagA, urease, outer membrane proteins, NOD1 receptor, ubiquitin-activating enzyme E1, polyubiquitinated proteins, proteasome components, and potentially oncogenic proteins like SHP-2 and ERKs colocalized, inferring that this structure is likely to modulate inflammatory and proliferative responses during H. pylori infection.
The recent finding that NF-κB and AP-1 complexes can be physically translocated to the nucleus in response to NOD1 activation has led to the view that NOD1 is likely to be essential for the induction of both NF-κB and AP-1 activation during H. pylori infection (227). A number of studies have shown up-regulation of NOD1 expression in diverse human cell lines challenged with H. pylori in a cag PAI-dependent manner (228–230). Further, H. pylori cag PAI-positive strains have recently been shown to activate the NOD1 pathway through two components of the IFN-γ signaling pathway, STAT1 and IRF1 (228). Similarly, expression of NOD2 was shown to significantly sensitize HEK293 cells to H. pylori-induced NF-κB activation in a cag PAI-dependent manner (231). Further, NOD2, but not NOD1, seems to be required for induction of pro-IL-1β and NLRP3 in H. pylori-infected DCs (232).
A limited number of studies have assessed the interaction between NLRPs and other inflammasome-associated molecules, and H. pylori. NLRPs represent the largest NLR subfamily (14 genes have been identified in humans) and are believed to be the scaffolding proteins of inflammasomes (221, 233). NLRPs interact and recruit the adaptor apoptosis-associated speck-like protein (ASC) via PYD-PYD interaction (94). ASC (also known as PYCARD), a key component required for inflammasome formation, is formed by an N-terminal PYD and a C-terminal CARD (234, 235). This interaction leads to the recruitment of caspase-1, an intracellular aspartate specific cysteine protease, which subsequently leads to the maturation and release of pro-inflammatory cytokines (236).
An early study by Tomita et al. (237) demonstrated that in H. pylori positive patients antral IL-18 mRNA expression was increased as compared with H. pylori negative patients, however, mature IL-18 protein and active caspase-1 were found to be present in both infected and non-infected gastric mucosa. Interestingly, in the following year, Potthoff et al. (238) reported activation of caspase-3, -8, and -9, but not caspase-1, in AGS cells challenged with H. pylori. However, this finding is in contrast with subsequent studies, which have demonstrated an important role for NLRPs and inflammasome-related molecules in H. pylori infection. For example, Basak et al. (96) demonstrated that H. pylori LPS could activate caspase-1 through Rac1/PAK1 signaling, and that activated caspase-1 played a role in LPS-induced IL-1β maturation (96). Further, ASC-deficient mice challenged with H. pylori have been shown to exhibit higher bacterial loads and significantly lower levels of gastritis, when compared with wild-type mice, and were incapable of producing IL-1β or IL-18 and produced less INF-γ in response to H. pylori infection (239). Later, Hitzler et al. (240) showed in both cultured DCs and in vivo that H. pylori infection activates caspase-1, leading to IL-1β/IL-18 processing and secretion. Consistently, three studies, using human GC cell lines, gastric tissue, and murine models, confirmed increased expression of caspase-1, IL-1β, and IL-18 in H. pylori-infected cells (171, 241, 242). Further, Jiang et al. (243), also using a murine model, have reported the expression of NLRP3 inflammasome-related molecules as well as serum IL-1β, IL-18, and IL-33 levels to be significantly increased in H. pylori-infected mice. More recently, a study by Kim et al. (232) has shown that secretion of IL-1β by DCs infected with H. pylori requires TLR2, NOD2, and the NLRP3 inflammasome.
Given that little is known about the role of NLRPs, inflammasomes, or other molecules involved in the NLR signaling pathways in response to H. pylori infection, we recently assessed the gene expression of 84 different molecules involved in the NLR signaling pathways, through quantitative real-time PCR, using THP-1-derived macrophages infected with two strains of H. pylori, GC026 (GC) and 26695 (gastritis) (173). Our gene expression analyses showed five genes encoding NLRs to be significantly regulated in H. pylori-challenged cells (NLRC4, NLRC5, NLRP9, NLRP12, and NLRX1) (173). Interestingly, NLRP12 and NLRX1, two known NF-κB negative regulators, were markedly down-regulated, while NFKB1 and several NF-κB target genes encoding pro-inflammatory cytokines (IFNB1, IL12A, IL-12B, IL6, and TNF), chemokines (CXCL1, CXCL2, and CCL5) and molecules involved in carcinogenesis (PTGS2 and BIRC3) were markedly up-regulated, in THP-1 cells infected with a highly virulent H. pylori strain isolated from a GC patient. These findings highlight the relevance of the NLR signaling pathways in gastric carcinogenesis and its close interaction with NF-κB (173).
Overall, current evidence clearly shows that, in response to H. pylori, members of the NOD and NLRP subfamilies are critical for generating mature pro-inflammatory cytokines/chemokines that are crucial for Th1 responses and lead to H. pylori-related gastric disorders.
NOD-Like Receptors and Gastric Carcinogenesis
The role of the NLR signaling pathways in the biological continuum that characterizes GC remains relatively unexplored as a very limited number of studies have addressed this issue. For example, Allison et al. (228) have shown that NOD1 expression was significantly increased in human gastric biopsies displaying severe gastritis, when compared with those without gastritis, as well as in gastric tumor tissues, as compared with paired non-tumor tissues. In contrast, Jee et al. (244), who analyzed human GC tissues and GC cell lines, showed that a significant decrease in the expression of caspase-1 was associated with poor survival and was inversely correlated with p53 expression.
Given the reported interaction of H. pylori with NLRs and the importance of this in the development of gastric inflammation and subsequent carcinogenesis, as well as the production of DAMPs during tumor formation (245), further comprehensive studies of the functional relevance of NLRs activation during chronic gastritis, atrophic gastritis, intestinal metaplasia, dysplasia, and GC are clearly warranted.
Genetic Polymorphisms Involved in the NOD-Like Receptor Signaling Pathway and Gastric Cancer
The majority of studies examining the association between polymorphisms involved in the NLR signaling pathways and the risk of GC have focused on NOD1 and NOD2 polymorphisms. Studies, conducted in a number of populations, have investigated the association between the polymorphisms NOD1 rs2907749 (246), NOD1 rs7789045 (246), NOD1 rs2075820 (E266K) (179, 247), NOD1 rs5743336 (180), NOD2 rs7205423 (246), NOD2 rs7202124 (164), NOD2 rs2111235 (164), NOD2 rs5743289 (164), NOD2 rs2066842 (P268S) (248, 249), NOD2 rs2066844 (R702W) (250), NOD2 rs2066845 (G908R) (184), and NOD2 rs2066847 (L1007insC) (184, 250), and risk of gastric precancerous lesions and GC (Table 3). Further, a recent meta-analysis by Liu et al. (251) that included six case–control studies has shown consistent associations between NOD2 R702W, G908R, and L1007insC, and risk of GC.
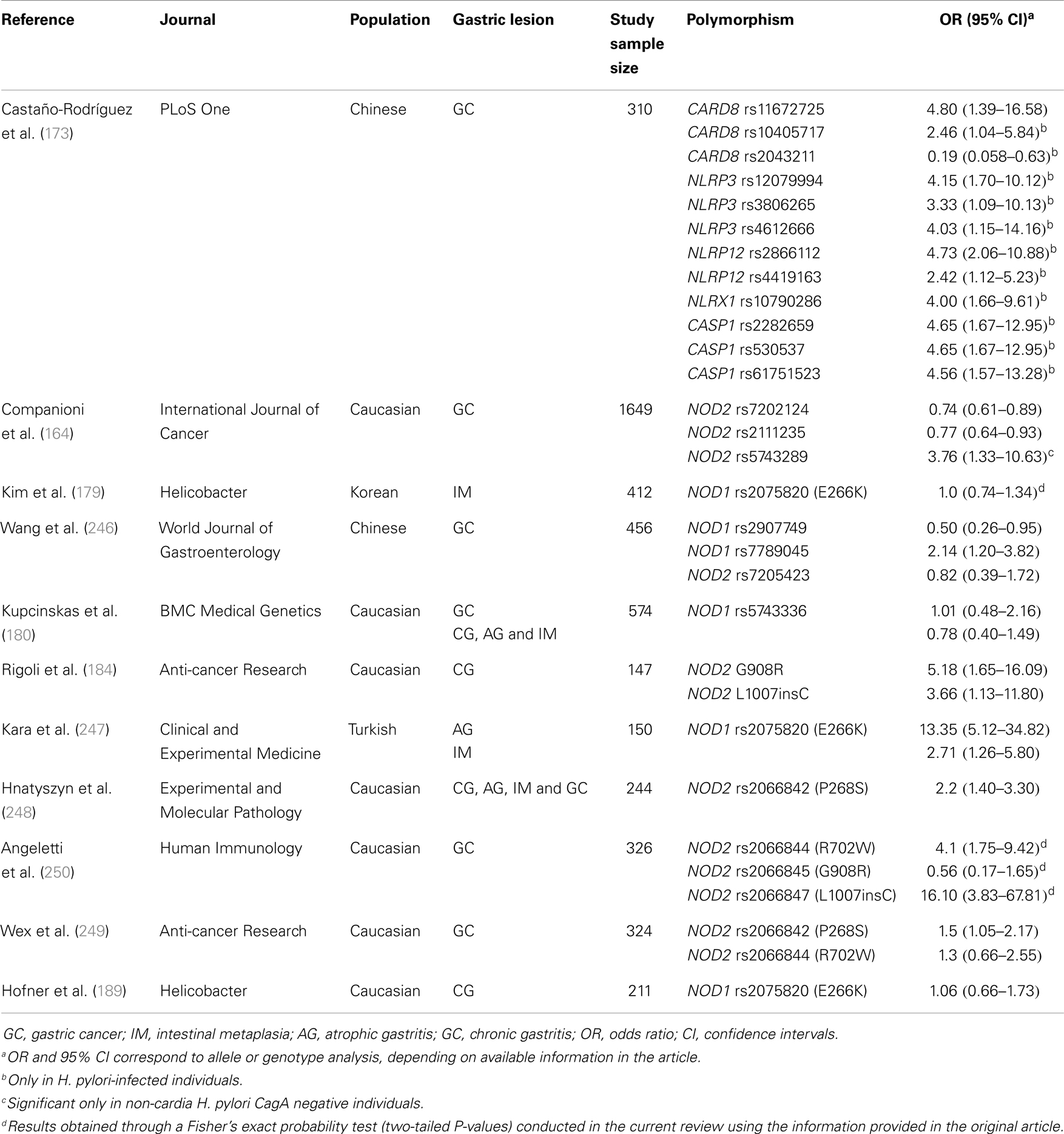
Table 3. Genetic polymorphisms in the NOD-like receptor signalling pathway that have been studied in relation to gastric precancerous lesions and gastric cancer.
Given the documented relevance of other NLRs in H. pylori infection and related GC, and that polymorphisms in genes such as NLRP3 (252–255) and CARD8 (255, 256) have been associated with inflammatory gastrointestinal disorders, we addressed, for the first time, the association between 51 polymorphisms in six genes (NLRP3, NLRP12, NLRX1, CASP1, ASC, and CARD8) involved in the NLR signaling pathways and risk of GC in a high risk Chinese population (173). In this study, we found novel associations between CARD8 rs11672725 and the risk of GC, and NLRP12 rs2866112 and the risk of H. pylori infection (Table 3). Further, we showed that the concomitant presence of polymorphisms involved in the NLR signaling pathways (CARD8, NLRP3, CASP1, and NLRP12) and H. pylori infection dramatically increased the risk of GC in Chinese (Table 3) (173).
The functional relevance of a number of these polymorphisms has been examined. For example, the introduction of NOD2 R702W, a polymorphism located in the LRR of NOD2, into the HEK293 cell line, resulted in abrogation of H. pylori-induced activation of NF-κB signaling (231). Further, Maeda et al. (257) observed increased NF-κB activation in response to muramyl dipeptide in mice harboring a NOD2 mutation that is homologous to NOD2 rs5743293 (3020insC) in humans. However, it is worth noting that the conclusions described by Maeda et al. (257) must be interpreted with care given that the authors subsequently found a duplication of the 3’ end of the wild-type Nod2 locus, including exon 11, which was targeted by the mutation, and therefore, they are currently working to recreate a mutant strain without such a duplication.
Given that investigation of the role of polymorphisms involved in the NLR signaling pathways in GC is a relatively recent field of research, further studies are required to assess the association between these polymorphisms and GC in a range of human populations, especially those at high risk of GC.
Other Pattern-Recognition Receptors and Helicobacter pylori-Related Gastric Cancer
A further two PRR subfamilies, RLRs and CLRs, have been studied in relation to H. pylori infection and gastric carcinogenesis. It is well known that RLRs (RIG-I, MDA-5, and LGP2) induce type I IFN in response to different RNA viruses, however, investigation on the role of RIG-I-like receptors in the recognition of RNA derived from intracellular bacteria is very limited. Interestingly, a study by Rad et al. (112), which used mice lacking simultaneously up to four different TLRs, apart from identifying TLR2 and TLR9 to be important H. pylori recognizing PRRs, also showed that H. pylori 5′-triphosphorylated RNA can be sensed by RIG-I and can contribute to the TLR-independent type I IFN response to this bacteria in DCs. Further, Tatsuta et al. (258) have recently shown that MDA-5 expression was significantly increased in the human gastric antral mucosa of H. pylori-infected individuals. In addition, these authors showed that increased MDA-5 levels correlated with atrophy and intestinal metaplasia in the corpus of these individuals (258).
C-type lectin receptors bind to carbohydrates (mannose- or fucose-containing glycans) present on pathogens to tailor immune responses to viruses, bacteria, and fungi. DC-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) is a CLR expressed on the surface of both macrophages and DCs. Interestingly, it has been shown that H. pylori harbors fucosylated ligands that can be recognized by DC-SIGN (259). Further, H. pylori DC-SIGN ligands appear to actively dissociate the signaling complex down-stream of DC-SIGN (KSR1–CNK–Raf-1) to suppress pro-inflammatory cytokine production (259). In addition, H. pylori LPS Lewis blood-group antigens can bind to DC-SIGN in a fucose or galactose-dependent manner (260, 261) and this interaction appears to inhibit a Th1 response in DCs (262). It has also been demonstrated that H. pylori-induced IL-10 production in monocyte-derived DCs is significantly suppressed by the addition of anti-DC-SIGN, TLR2, or TLR4 antibodies, either alone or in combination, before H. pylori stimulation (263). Further, in vitro and in vivo experiments have shown that the expression of DC-SIGN is significantly higher in H. pylori-infected individuals as compared with that in their uninfected counterparts (264, 265).
To date, no studies have been conducted to determine the association between genetic polymorphisms involved in the RLR and CLR signaling pathways and GC, however, Kutikhin and Yuzhalin (266) have comprehensively analyzed the oncogenic potential of both RLRs and CLRs, suggesting that future oncogenomic investigations should focus on polymorphisms in MRC1 (rs1926736, rs2478577, rs2437257, and rs691005), CD209 (rs2287886, rs735239, rs4804803, and rs735240), CLEC7A (rs16910526), and RIG-I (rs36055726, rs11795404, and rs10813831).
Given the limited but consistent current evidence suggesting a role of RLRs and CLRs in H. pylori infection, and the documented interaction between these signaling pathways and other important PRRs in GC such as TLRs (267, 268) and NLRs (269, 270), further studies assessing the implications of RLRs and CLRs in H. pylori-related inflammation and subsequent carcinogenesis need to be conducted.
Pattern-Recognition Receptors as Therapeutics Targets in Gastric Cancer
Pattern-recognition receptors are increasingly recognized as important players in immunotherapy as PRRs-specific agonists elicit a potent immune response to cancers, allergic diseases, and chronic viral infections, while reducing the risk of an uncontrolled and detrimental systemic inflammatory response (for comprehensive information on this topic refer to the reviews by Hedayat et al. (271) and Paul-Clark et al. (272).
In the context of gastric carcinogenesis, Tye et al. (132), using a GC murine model (gp130F/F) displaying elevated gastric TLR2 expression levels, have elegantly shown that genetic and antibody-mediated therapeutic targeting of TLR2 leads to a substantial reduction in stomach size and overall tumor burden, including the number of gastric tumors. A further example is presented in the study by Gradisar et al. (273), which suggested that MD-2 is one of the important targets of curcumin (diferuloylmethane), the main component of the spice turmeric (Curcuma longa) that is widely used for gastric disorders in the Indian subcontinent, in its suppression of the innate immune response to bacterial infection. Furthermore, curcumin was recently shown to polarize myeloid-derived suppressor cells, extracted from a human GC xenograft mouse model, toward a M1-like phenotype with an increased expression of CCR7 and decreased expression of the CLR dectin 1, being both observed in vivo (tumor tissue) and in vitro (splenic myeloid-derived suppressor cells from tumor-bearing mice) (274). In addition, a study by Yang et al. (171) demonstrated that the combination of catechins and sialic acid is effective in suppressing the inflammatory responses mediated by the inflammasome/caspase-1 signaling pathway in gastric epithelial cells during H. pylori infection. Also, poly(I:C), an agonist of TLR3 and RLRs, has been shown to have a pro-apoptotic effect in vitro, and has significantly inhibited xenograft growth of human GC in a mouse model, through up-regulation of RLRs (RIG-I, MDA-5, and LGP2) as well as an increased expression of Bcl-2 family members, suggesting that it may be a promising chemotherapeutic agent against GC (275).
Given that modulation of PRRs has been proven to be relevant in gastric carcinogenesis through diverse mechanisms, including suppression of H. pylori-induced inflammation and enhancement of cancer cell apoptosis, this approach should be considered a new and promising angle of immunotherapy in GC.
Conclusion
In conclusion, abundant evidence supports the pivotal role of PRRs in gastric carcinogenesis as these receptors of the innate immune system, including TLRs, NLRs, CLRs, and RLRs, have been shown to recognize diverse components of H. pylori, the major risk factor of GC. In addition, PRRs are also involved in gastric carcinogenesis per se as these receptors are known to exert tumor-promoting functions (cell proliferation, epithelial–mesenchymal transition, angiogenesis, and metastasis) as well as immunosuppression during cancer. Given that host genetic variability in the TLR and NLR signaling pathways are known to be associated with an increased risk of H. pylori infection, the development of gastric precancerous lesions and GC, this knowledge has the potential to allow better prevention of GC through selective treatment and surveillance of individuals harboring high risk genetic profiles. Finally, given that PRRs are increasingly being used as a target for immunotherapy against both cancer and infectious diseases, the established relevance of PRRs in H. pylori infection and GC, could suggest that PRR agonists and/or antagonists may potentially improve the outcome of GC. Based on the extensive evidence presented in the current review, we propose a synergistic interaction between PRRs and H. pylori, which over time, could facilitate the sequence of events that characterizes GC development including inflammation, atrophy, intestinal metaplasia, dysplasia, and finally, GC (Figure 3).
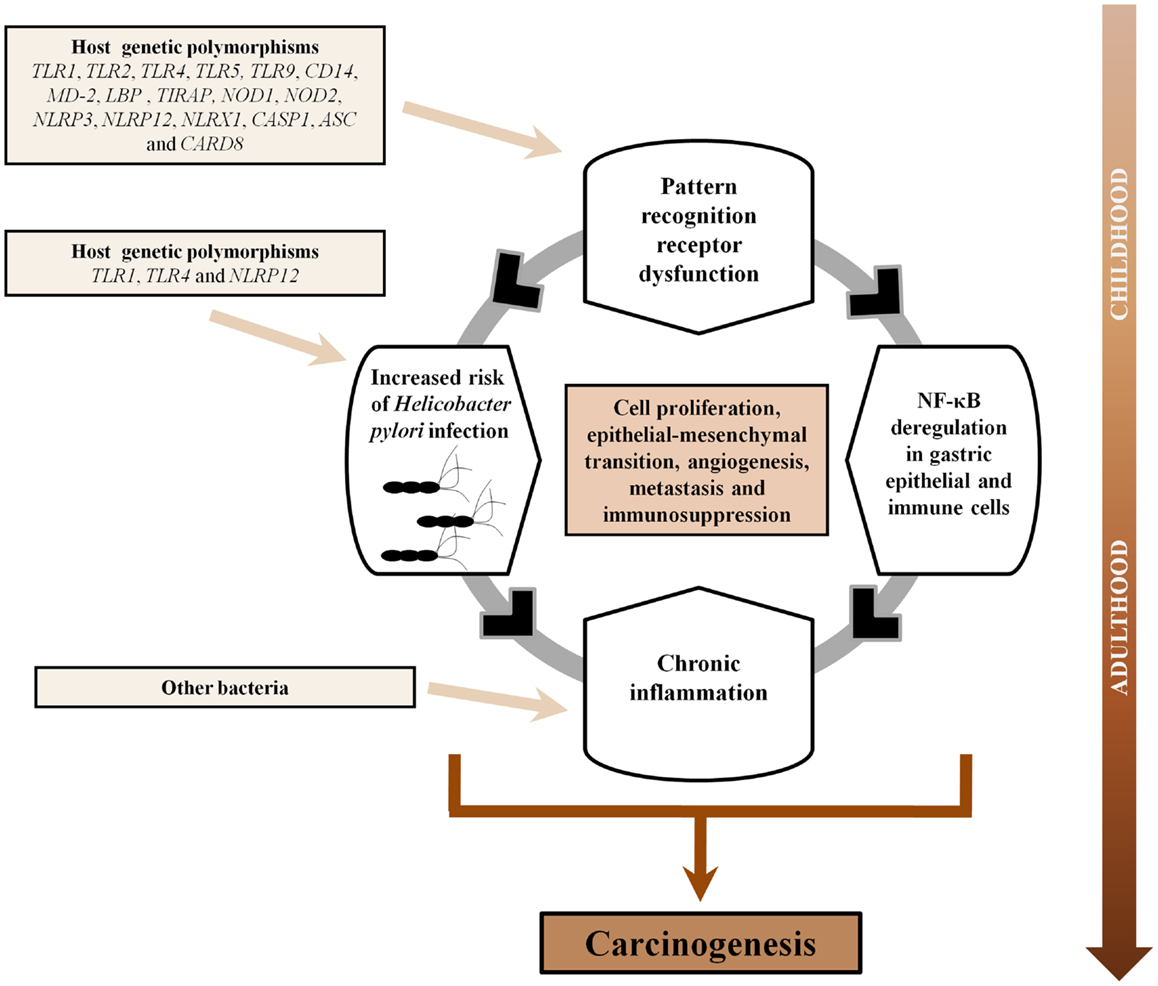
Figure 3. Pattern-recognition receptors and gastric carcinogenesis. Based on this comprehensive literature review, we propose a synergistic interaction between pattern-recognition receptors (PRRs) and Helicobacter pylori in gastric carcinogenesis. The association between PRRs and risk of GC might be a continuum commencing in childhood. Individuals harboring polymorphisms in PRRs could not only be more susceptible to acquisition of H. pylori in childhood but also would present deregulation of NF-κB in gastric epithelial and immune cells, and subsequent uncontrolled production of cytokines/chemokines, due to dysfunctional PRRs. This in turn would impact upon the direction and magnitude of the chronic inflammatory response to H. pylori. As H. pylori, the dominant bacterium in the stomach, gradually disappears upon the development of gastric atrophy, it is plausible that other microbial species might bloom in its absence and perpetuate local inflammation through further PRRs activation. Over time, the combination of these events would facilitate a number of features that promote gastric cancer development including cell proliferation, epithelial–mesenchymal transition, angiogenesis, metastasis, and immunosuppression.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Nadeem O. Kaakoush is supported by an Early Career fellowship from the National Health and Medical Research Council, Australia.
References
1. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.0, cancer incidence and mortality worldwide. IARC CancerBase No. 11 [Internet]. Lyon: International Agency for Research on Cancer (2013).
3. Mccoll KE. Cancer of the gastric cardia. Best Pract Res Clin Gastroenterol (2006) 20:687–96. doi: 10.1016/j.bpg.2006.03.005
4. Ferro A, Peleteiro B, Malvezzi M, Bosetti C, Bertuccio P, Levi F, et al. Worldwide trends in gastric cancer mortality (1980–2011), with predictions to 2015, and incidence by subtype. Eur J Cancer (2014) 50:1330–44. doi:10.1016/j.ejca.2014.01.029
5. Cavaleiro-Pinto M, Peleteiro B, Lunet N, Barros H. Helicobacter pylori infection and gastric cardia cancer: systematic review and meta-analysis. Cancer Causes Control (2011) 22:375–87. doi:10.1007/s10552-010-9707-2
6. Hansen S, Vollset SE, Derakhshan MH, Fyfe V, Melby KK, Aase S, et al. Two distinct aetiologies of cardia cancer; evidence from premorbid serological markers of gastric atrophy and Helicobacter pylori status. Gut (2007) 56:918–25. doi:10.1136/gut.2006.114504
8. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi:10.1016/S0092-8674(00)81683-9
9. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
10. Fuchs CS, Mayer RJ. Gastric carcinoma. N Engl J Med (1995) 333:32–41. doi:10.1056/NEJM199507063330107
11. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process – First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res (1992) 52:6735–40.
12. Li H, Stoicov C, Cai X, Wang TC, Houghton J. Helicobacter and gastric cancer disease mechanisms: host response and disease susceptibility. Curr Gastroenterol Rep (2003) 5:459–67. doi:10.1007/s11894-003-0034-6
13. Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. E-cadherin alterations in hereditary disorders with emphasis on hereditary diffuse gastric cancer. Prog Mol Biol Transl Sci (2013) 116:337–59. doi:10.1016/B978-0-12-394311-8.00015-7
14. Kusters JG, Van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev (2006) 19:449–90. doi:10.1128/CMR.00054-05
15. Costa NR, Mendes N, Marcos NT, Reis CA, Caffrey T, Hollingsworth MA, et al. Relevance of MUC1 mucin variable number of tandem repeats polymorphism in H. pylori adhesion to gastric epithelial cells. World J Gastroenterol (2008) 14:1411–4. doi:10.3748/wjg.14.1411
16. Atherton JC. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu Rev Pathol (2006) 1:63–96. doi:10.1146/annurev.pathol.1.110304.100125
17. Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med (2002) 347:1175–86. doi:10.1056/NEJMra020542
18. Wroblewski LE, Peek RM Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev (2010) 23:713–39. doi:10.1128/CMR.00011-10
19. El-Omar EM, Penman ID, Ardill JE, Chittajallu RS, Howie C, Mccoll KE. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology (1995) 109:681–91. doi:10.1016/0016-5085(95)90374-7
20. Kivi M, Tindberg Y, Sorberg M, Casswall TH, Befrits R, Hellstrom PM, et al. Concordance of Helicobacter pylori strains within families. J Clin Microbiol (2003) 41:5604–8. doi:10.1128/JCM.41.12.5604-5608.2003
21. Konno M, Fujii N, Yokota S, Sato K, Takahashi M, Mino E, et al. Five-year follow-up study of mother-to-child transmission of Helicobacter pylori infection detected by a random amplified polymorphic DNA fingerprinting method. J Clin Microbiol (2005) 43:2246–50. doi:10.1128/JCM.43.5.2246-2250.2005
22. Rowland M, Daly L, Vaughan M, Higgins A, Bourke B, Drumm B. Age-specific incidence of Helicobacter pylori. Gastroenterology (2006) 130:65–72;quiz211. doi:10.1053/j.gastro.2005.11.004
23. Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology (2009) 136:1863–73. doi:10.1053/j.gastro.2009.01.073
24. Neiger R, Simpson KW. Helicobacter infection in dogs and cats: facts and fiction. J Vet Intern Med (2000) 14:125–33. doi:10.1111/j.1939-1676.2000.tb02225.x
25. Simpson KW, Strauss-Ayali D, Straubinger RK, Scanziani E, Mcdonough PL, Straubinger AF, et al. Helicobacter pylori infection in the cat: evaluation of gastric colonization, inflammation and function. Helicobacter (2001) 6:1–14. doi:10.1046/j.1523-5378.2001.00010.x
26. Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A (2006) 103:732–7. doi:10.1073/pnas.0506655103
27. Bauerfeind P, Garner R, Dunn BE, Mobley HL. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut (1997) 40:25–30.
28. Marshall BJ, Barrett LJ, Prakash C, Mccallum RW, Guerrant RL. Urea protects Helicobacter (Campylobacter) pylori from the bactericidal effect of acid. Gastroenterology (1990) 99:697–702.
29. Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect Immun (2003) 71:3529–39. doi:10.1128/IAI.71.6.3529-3539.2003
30. Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science (1998) 279:373–7. doi:10.1126/science.279.5349.373
31. Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science (2002) 297:573–8. doi:10.1126/science.1069076
32. Putsep K, Branden CI, Boman HG, Normark S. Antibacterial peptide from H. pylori. Nature (1999) 398:671–2. doi:10.1038/19439
33. Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science (2003) 301:1099–102. doi:10.1126/science.1086871
34. Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci U S A (2004) 101:7727–32. doi:10.1073/pnas.0401528101
35. Supajatura V, Ushio H, Wada A, Yahiro K, Okumura K, Ogawa H, et al. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J Immunol (2002) 168:2603–7. doi:10.4049/jimmunol.168.6.2603
36. Torres VJ, Vancompernolle SE, Sundrud MS, Unutmaz D, Cover TL. Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J Immunol (2007) 179:5433–40. doi:10.4049/jimmunol.179.8.5433
37. Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, et al. Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology (2007) 132:1820–33. doi:10.1053/j.gastro.2007.02.031
38. Gobert AP, Mcgee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, et al. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A (2001) 98:13844–9. doi:10.1073/pnas.241443798
39. Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A (1996) 93:14648–53. doi:10.1073/pnas.93.25.14648
40. Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, et al. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol (1998) 28:37–53. doi:10.1046/j.1365-2958.1998.00770.x
41. Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci U S A (1993) 90:5791–5. doi:10.1073/pnas.90.12.5791
42. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature (1997) 388:539–47. doi:10.1038/41483
43. Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science (2000) 287:1497–500. doi:10.1126/science.287.5457.1497
44. Posselt G, Backert S, Wessler S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal (2013) 11:77. doi:10.1186/1478-811X-11-77
45. Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science (2002) 295:683–6. doi:10.1126/science.1067147
46. Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene (2007) 26:4617–26. doi:10.1038/sj.onc.1210251
47. Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci U S A (1999) 96:14559–64. doi:10.1073/pnas.96.25.14559
48. Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res (1995) 55:2111–5.
49. Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst (2002) 94:1680–7. doi:10.1093/jnci/94.22.1680
50. Kuipers EJ, Uyterlinde AM, Pena AS, Roosendaal R, Pals G, Nelis GF, et al. Long-term sequelae of Helicobacter pylori gastritis. Lancet (1995) 345:1525–8. doi:10.1016/S0140-6736(95)91084-0
51. Plummer M, Van Doorn LJ, Franceschi S, Kleter B, Canzian F, Vivas J, et al. Helicobacter pylori cytotoxin-associated genotype and gastric precancerous lesions. J Natl Cancer Inst (2007) 99:1328–34. doi:10.1093/jnci/djm120
52. Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med (1991) 325:1132–6. doi:10.1056/NEJM199110173251604
53. Nomura AM, Lee J, Stemmermann GN, Nomura RY, Perez-Perez GI, Blaser MJ. Helicobacter pylori CagA seropositivity and gastric carcinoma risk in a Japanese American population. J Infect Dis (2002) 186:1138–44. doi:10.1086/343808
54. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med (1991) 325:1127–31. doi:10.1056/NEJM199110173251603
55. Pinto-Santini D, Salama NR. The biology of Helicobacter pylori infection, a major risk factor for gastric adenocarcinoma. Cancer Epidemiol Biomarkers Prev (2005) 14:1853–8. doi:10.1158/1055-9965.EPI-04-0784
56. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med (2001) 345:784–9. doi:10.1056/NEJMoa001999
57. Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut (1997) 40:297–301.
58. Seoane A, Bessa X, Balleste B, O’Callaghan E, Panades A, Alameda F, et al. [Helicobacter pylori and gastric cancer: relationship with histological subtype and tumor location]. Gastroenterol Hepatol (2005) 28:60–4. doi:10.1157/13070701
59. Bornschein J, Selgrad M, Warnecke M, Kuester D, Wex T, Malfertheiner P. H. pylori infection is a key risk factor for proximal gastric cancer. Dig Dis Sci (2010) 55:3124–31. doi:10.1007/s10620-010-1351-x
60. Ekstrom AM, Held M, Hansson LE, Engstrand L, Nyren O. Helicobacter pylori in gastric cancer established by CagA immunoblot as a marker of past infection. Gastroenterology (2001) 121:784–91. doi:10.1053/gast.2001.27999
61. Siman JH, Engstrand L, Berglund G, Forsgren A, Floren CH. Helicobacter pylori and CagA seropositivity and its association with gastric and oesophageal carcinoma. Scand J Gastroenterol (2007) 42:933–40. doi:10.1080/00365520601173863
62. Coffelt SB, Lewis CE, Naldini L, Brown JM, Ferrara N, De Palma M. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am J Pathol (2010) 176:1564–76. doi:10.2353/ajpath.2010.090786
63. De Palma M, Murdoch C, Venneri MA, Naldini L, Lewis CE. Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol (2007) 28:519–24. doi:10.1016/j.it.2007.09.004
64. Denardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev (2010) 29:309–16. doi:10.1007/s10555-010-9223-6
65. Johansson M, Denardo DG, Coussens LM. Polarized immune responses differentially regulate cancer development. Immunol Rev (2008) 222:145–54. doi:10.1111/j.1600-065X.2008.00600.x
66. Wu MS, Chen CJ, Lin JT. Host-environment interactions: their impact on progression from gastric inflammation to carcinogenesis and on development of new approaches to prevent and treat gastric cancer. Cancer Epidemiol Biomarkers Prev (2005) 14:1878–82. doi:10.1158/1055-9965.EPI-04-0792
68. Tsuji S, Kawai N, Tsujii M, Kawano S, Hori M. Review article: inflammation-related promotion of gastrointestinal carcinogenesis – a perigenetic pathway. Aliment Pharmacol Ther (2003) 18(Suppl 1):82–9. doi:10.1046/j.1365-2036.18.s1.22.x
69. Correa P, Haenszel W, Cuello C, Zavala D, Fontham E, Zarama G, et al. Gastric precancerous process in a high risk population: cross-sectional studies. Cancer Res (1990) 50:4731–6.
70. Dinarello CA. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J Endotoxin Res (2004) 10:201–22. doi:10.1179/096805104225006129
71. Helmby H, Grencis RK. Interleukin 1 plays a major role in the development of Th2-mediated immunity. Eur J Immunol (2004) 34:3674–81. doi:10.1002/eji.200425452
72. Hwang IR, Kodama T, Kikuchi S, Sakai K, Peterson LE, Graham DY, et al. Effect of interleukin 1 polymorphisms on gastric mucosal interleukin 1beta production in Helicobacter pylori infection. Gastroenterology (2002) 123:1793–803. doi:10.1053/gast.2002.37043
73. Camargo MC, Mera R, Correa P, Peek RM Jr, Fontham ET, Goodman KJ, et al. Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms and gastric cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev (2006) 15:1674–87. doi:10.1158/1055-9965.EPI-06-0189
74. Xue H, Lin B, Ni P, Xu H, Huang G. Interleukin-1B and interleukin-1 RN polymorphisms and gastric carcinoma risk: a meta-analysis. J Gastroenterol Hepatol (2010) 25:1604–17. doi:10.1111/j.1440-1746.2010.06428.x
75. Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol (2011) 166:1–15. doi:10.1111/j.1365-2249.2011.04440.x
76. Hartland S, Newton JL, Griffin SM, Donaldson PT. A functional polymorphism in the interleukin-1 receptor-1 gene is associated with increased risk of Helicobacter pylori infection but not with gastric cancer. Dig Dis Sci (2004) 49:1545–50. doi:10.1023/B:DDAS.0000042262.14969.2d
77. Peleteiro B, Lunet N, Carrilho C, Duraes C, Machado JC, La Vecchia C, et al. Association between cytokine gene polymorphisms and gastric precancerous lesions: systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev (2010) 19:762–76. doi:10.1158/1055-9965.EPI-09-0917
78. Persson C, Canedo P, Machado JC, El-Omar EM, Forman D. Polymorphisms in inflammatory response genes and their association with gastric cancer: a HuGE systematic review and meta-analyses. Am J Epidemiol (2011) 173:259–70. doi:10.1093/aje/kwq370
79. Yuzhalin A. The role of interleukin DNA polymorphisms in gastric cancer. Hum Immunol (2011) 72:1128–36. doi:10.1016/j.humimm.2011.08.003
80. Wang J, He W, Liu J, Nong L, Wei Y, Yang F. Association of IL-6 polymorphisms with gastric cancer risk: evidences from a meta-analysis. Cytokine (2012) 59:176–83. doi:10.1016/j.cyto.2012.03.032
81. Yin YW, Hu AM, Sun QQ, Liu HL, Wang Q, Zeng YH, et al. Association between interleukin-6 gene -174 G/C polymorphism and the risk of coronary heart disease: a meta-analysis of 20 studies including 9619 cases and 10,919 controls. Gene (2012) 503:25–30. doi:10.1016/j.gene.2012.04.075
82. Da Costa DM, Neves-Filho EH, Alves MK, Rabenhorst SH. Interleukin polymorphisms and differential methylation status in gastric cancer: an association with Helicobacter pylori infection. Epigenomics (2013) 5:167–75. doi:10.2217/epi.13.7
83. Xue H, Liu J, Lin B, Wang Z, Sun J, Huang G. A meta-analysis of interleukin-8 -251 promoter polymorphism associated with gastric cancer risk. PLoS One (2012) 7:e28083. doi:10.1371/journal.pone.0028083
84. Ni P, Xu H, Xue H, Lin B, Lu Y. A meta-analysis of interleukin-10-1082 promoter polymorphism associated with gastric cancer risk. DNA Cell Biol (2012) 31:582–91. doi:10.1089/dna.2011.1440
85. Yu Z, Liu Q, Huang C, Wu M, Li G. The interleukin 10 −819C/T polymorphism and cancer risk: a HuGE review and meta-analysis of 73 studies including 15,942 cases and 22,336 controls. OMICS (2013) 17:200–14. doi:10.1089/omi.2012.0089
86. Zhuang W, Wu XT, Zhou Y, Liu L, Liu GJ, Wu TX, et al. Interleukin10 −592 promoter polymorphism associated with gastric cancer among Asians: a meta-analysis of epidemiologic studies. Dig Dis Sci (2010) 55:1525–32. doi:10.1007/s10620-009-0922-1
87. Yuzhalin AE, Kutikhin AG. Interleukin-12: clinical usage and molecular markers of cancer susceptibility. Growth Factors (2012) 30:176–91. doi:10.3109/08977194.2012.678843
88. Haghshenas MR, Hosseini SV, Mahmoudi M, Saberi-Firozi M, Farjadian S, Ghaderi A. IL-18 serum level and IL-18 promoter gene polymorphism in Iranian patients with gastrointestinal cancers. J Gastroenterol Hepatol (2009) 24:1119–22. doi:10.1111/j.1440-1746.2009.05791.x
89. Gorouhi F, Islami F, Bahrami H, Kamangar F. Tumour-necrosis factor-A polymorphisms and gastric cancer risk: a meta-analysis. Br J Cancer (2008) 98:1443–51. doi:10.1038/sj.bjc.6604277
90. Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol (2011) 11:9–20. doi:10.1038/nri2891
91. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol (2011) 29:707–35. doi:10.1146/annurev-immunol-031210-101405
92. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11:997–1004. doi:10.1038/ni.1932
93. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi:10.1016/j.cell.2010.01.040
94. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi:10.1146/annurev.immunol.021908.132715
95. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol (2003) 21:335–76. doi:10.1146/annurev.immunol.21.120601.141126
96. Basak C, Pathak SK, Bhattacharyya A, Mandal D, Pathak S, Kundu M. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and PAK1-mediated caspase-1 activation play essential roles in interleukin-1beta release from Helicobacter pylori lipopolysaccharide-stimulated macrophages. J Biol Chem (2005) 280:4279–88. doi:10.1074/jbc.M412820200
97. Kawahara T, Teshima S, Oka A, Sugiyama T, Kishi K, Rokutan K. Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect Immun (2001) 69:4382–9. doi:10.1128/IAI.69.7.4382-4389.2001
98. Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, et al. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem (2001) 276:44856–64. doi:10.1074/jbc.M105381200
99. Su B, Ceponis PJ, Lebel S, Huynh H, Sherman PM. Helicobacter pylori activates Toll-like receptor 4 expression in gastrointestinal epithelial cells. Infect Immun (2003) 71:3496–502. doi:10.1128/IAI.71.6.3496-3502.2003
100. Thomas CJ, Kapoor M, Sharma S, Bausinger H, Zyilan U, Lipsker D, et al. Evidence of a trimolecular complex involving LPS, LPS binding protein and soluble CD14 as an effector of LPS response. FEBS Lett (2002) 531:184–8. doi:10.1016/S0014-5793(02)03499-3
101. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4:499–511. doi:10.1038/nri1391
102. Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol (2001) 167:5887–94. doi:10.4049/jimmunol.167.10.5887
103. Basu S, Pathak SK, Chatterjee G, Pathak S, Basu J, Kundu M. Helicobacter pylori protein HP0175 transactivates epidermal growth factor receptor through TLR4 in gastric epithelial cells. J Biol Chem (2008) 283:32369–76. doi:10.1074/jbc.M805053200
104. Chochi K, Ichikura T, Kinoshita M, Majima T, Shinomiya N, Tsujimoto H, et al. Helicobacter pylori augments growth of gastric cancers via the lipopolysaccharide-toll-like receptor 4 pathway whereas its lipopolysaccharide attenuates antitumor activities of human mononuclear cells. Clin Cancer Res (2008) 14:2909–17. doi:10.1158/1078-0432.CCR-07-4467
105. Ishihara S, Rumi MA, Kadowaki Y, Ortega-Cava CF, Yuki T, Yoshino N, et al. Essential role of MD-2 in TLR4-dependent signaling during Helicobacter pylori-associated gastritis. J Immunol (2004) 173:1406–16. doi:10.4049/jimmunol.173.2.1406
106. Cunningham MD, Seachord C, Ratcliffe K, Bainbridge B, Aruffo A, Darveau RP. Helicobacter pylori and Porphyromonas gingivalis lipopolysaccharides are poorly transferred to recombinant soluble CD14. Infect Immun (1996) 64:3601–8.
107. Smith MF Jr, Mitchell A, Li G, Ding S, Fitzmaurice AM, Ryan K, et al. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem (2003) 278:32552–60. doi:10.1074/jbc.M305536200
108. Chang YJ, Wu MS, Lin JT, Sheu BS, Muta T, Inoue H, et al. Induction of cyclooxygenase-2 overexpression in human gastric epithelial cells by Helicobacter pylori involves TLR2/TLR9 and c-Src-dependent nuclear factor-kappaB activation. Mol Pharmacol (2004) 66:1465–77. doi:10.1124/mol.104.005199
109. Gobert AP, Bambou JC, Werts C, Balloy V, Chignard M, Moran AP, et al. Helicobacter pylori heat shock protein 60 mediates interleukin-6 production by macrophages via a toll-like receptor (TLR)-2-, TLR-4-, and myeloid differentiation factor 88-independent mechanism. J Biol Chem (2004) 279:245–50. doi:10.1074/jbc.M307858200
110. Lepper PM, Triantafilou M, Schumann C, Schneider EM, Triantafilou K. Lipopolysaccharides from Helicobacter pylori can act as antagonists for Toll-like receptor 4. Cell Microbiol (2005) 7:519–28. doi:10.1111/j.1462-5822.2005.00482.x
111. Mandell L, Moran AP, Cocchiarella A, Houghton J, Taylor N, Fox JG, et al. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun (2004) 72:6446–54. doi:10.1128/IAI.72.11.6446-6454.2004
112. Rad R, Ballhorn W, Voland P, Eisenacher K, Mages J, Rad L, et al. Extracellular and intracellular pattern recognition receptors cooperate in the recognition of Helicobacter pylori. Gastroenterology (2009) 136:2247–57. doi:10.1053/j.gastro.2009.02.066
113. Chaouche-Drider N, Kaparakis M, Karrar A, Fernandez MI, Carneiro LA, Viala J, et al. A commensal Helicobacter sp. of the rodent intestinal flora activates TLR2 and NOD1 responses in epithelial cells. PLoS One (2009) 4:e5396. doi:10.1371/journal.pone.0005396
114. Sayi A, Kohler E, Toller IM, Flavell RA, Muller W, Roers A, et al. TLR-2-activated B cells suppress Helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J Immunol (2011) 186:878–90. doi:10.4049/jimmunol.1002269
115. Smith SM, Moran AP, Duggan SP, Ahmed SE, Mohamed AS, Windle HJ, et al. Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J Immunol (2011) 186:2462–71. doi:10.4049/jimmunol.1000864
116. Obonyo M, Sabet M, Cole SP, Ebmeyer J, Uematsu S, Akira S, et al. Deficiencies of myeloid differentiation factor 88, Toll-like receptor 2 (TLR2), or TLR4 produce specific defects in macrophage cytokine secretion induced by Helicobacter pylori. Infect Immun (2007) 75:2408–14. doi:10.1128/IAI.01794-06
117. Takenaka R, Yokota K, Ayada K, Mizuno M, Zhao Y, Fujinami Y, et al. Helicobacter pylori heat-shock protein 60 induces inflammatory responses through the Toll-like receptor-triggered pathway in cultured human gastric epithelial cells. Microbiology (2004) 150:3913–22. doi:10.1099/mic.0.27527-0
118. Yokota S, Okabayashi T, Rehli M, Fujii N, Amano K. Helicobacter pylori lipopolysaccharides upregulate toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect Immun (2010) 78:468–76. doi:10.1128/IAI.00903-09
119. Otani K, Tanigawa T, Watanabe T, Nadatani Y, Sogawa M, Yamagami H, et al. Toll-like receptor 9 signaling has anti-inflammatory effects on the early phase of Helicobacter pylori-induced gastritis. Biochem Biophys Res Commun (2012) 426:342–9. doi:10.1016/j.bbrc.2012.08.080
120. Lagunes-Servin H, Torres J, Maldonado-Bernal C, Perez-Rodriguez M, Huerta-Yepez S, Madrazo De La Garza A, et al. Toll-like receptors and cytokines are upregulated during Helicobacter pylori infection in children. Helicobacter (2013) 18:423–32. doi:10.1111/hel.12067
121. Gewirtz AT, Yu Y, Krishna US, Israel DA, Lyons SL, Peek RM Jr. Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J Infect Dis (2004) 189:1914–20. doi:10.1086/386289
122. Kumar Pachathundikandi S, Brandt S, Madassery J, Backert S. Induction of TLR-2 and TLR-5 expression by Helicobacter pylori switches cagPAI-dependent signalling leading to the secretion of IL-8 and TNF-alpha. PLoS One (2011) 6:e19614. doi:10.1371/journal.pone.0019614
123. Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, Logan SM, et al. Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A (2005) 102:9247–52. doi:10.1073/pnas.0502040102
124. Muotiala A, Helander IM, Pyhala L, Kosunen TU, Moran AP. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect Immun (1992) 60:1714–6.
125. Moran AP, Lindner B, Walsh EJ. Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J Bacteriol (1997) 179:6453–63.
126. Schmausser B, Andrulis M, Endrich S, Muller-Hermelink HK, Eck M. Toll-like receptors TLR4, TLR5 and TLR9 on gastric carcinoma cells: an implication for interaction with Helicobacter pylori. Int J Med Microbiol (2005) 295:179–85. doi:10.1016/j.ijmm.2005.02.009
127. Pimentel-Nunes P, Afonso L, Lopes P, Roncon-Albuquerque R Jr, Goncalves N, Henrique R, et al. Increased expression of toll-like receptors (TLR) 2, 4 and 5 in gastric dysplasia. Pathol Oncol Res (2011) 17:677–83. doi:10.1007/s12253-011-9368-9
128. Pimentel-Nunes P, Goncalves N, Boal-Carvalho I, Afonso L, Lopes P, Roncon-Albuquerque R Jr, et al. Helicobacter pylori induces increased expression of Toll-like receptors and decreased Toll-interacting protein in gastric mucosa that persists throughout gastric carcinogenesis. Helicobacter (2013) 18:22–32. doi:10.1111/hel.12008
129. Fernandez-Garcia B, Eiro N, Gonzalez-Reyes S, Gonzalez L, Aguirre A, Gonzalez LO, et al. Clinical significance of toll-like receptor 3, 4, and 9 in gastric cancer. J Immunother (2014) 37:77–83. doi:10.1097/CJI.0000000000000016
130. Yakut M, Ormeci N, Erdal H, Keskin O, Karayel Z, Tutkak H, et al. The association between precancerous gastric lesions and serum pepsinogens, serum gastrin, vascular endothelial growth factor, serum interleukin-1 Beta, serum toll-like receptor-4 levels and Helicobacter pylori Cag A status. Clin Res Hepatol Gastroenterol (2013) 37:302–11. doi:10.1016/j.clinre.2012.09.013
131. Song EJ, Kang MJ, Kim YS, Kim SM, Lee SE, Kim CH, et al. Flagellin promotes the proliferation of gastric cancer cells via the Toll-like receptor 5. Int J Mol Med (2011) 28:115–9. doi:10.3892/ijmm.2011.656
132. Tye H, Kennedy CL, Najdovska M, Mcleod L, Mccormack W, Hughes N, et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell (2012) 22:466–78. doi:10.1016/j.ccr.2012.08.010
133. Li K, Dan Z, Hu X, Gesang L, Ze Y, Bianba Z. CD14 regulates gastric cancer cell epithelial mesenchymal transition and invasion in vitro. Oncol Rep (2013) 30:2725–32. doi:10.3892/or.2013.2733
134. Wu WK, Sung JJ, Lee CW, Yu J, Cho CH. Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Lett (2010) 295:7–16. doi:10.1016/j.canlet.2010.03.015
135. Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell (2006) 10:175–6. doi:10.1016/j.ccr.2006.08.015
136. Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res (2007) 67:10823–30. doi:10.1158/0008-5472.CAN-07-0783
137. Park JH, Kim TY, Jong HS, Chun YS, Park JW, Lee CT, et al. Gastric epithelial reactive oxygen species prevent normoxic degradation of hypoxia-inducible factor-1alpha in gastric cancer cells. Clin Cancer Res (2003) 9:433–40.
138. Yuan X, Zhou Y, Wang W, Li J, Xie G, Zhao Y, et al. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis (2013) 4:e794. doi:10.1038/cddis.2013.334
139. Kutikhin AG. Impact of Toll-like receptor 4 polymorphisms on risk of cancer. Hum Immunol (2011) 72:193–206. doi:10.1016/j.humimm.2010.11.003
140. Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem (2002) 277:15028–34. doi:10.1074/jbc.M200497200
141. Guillot L, Balloy V, Mccormack FX, Golenbock DT, Chignard M, Si-Tahar M. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol (2002) 168:5989–92. doi:10.4049/jimmunol.168.12.5989
142. Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol (2002) 168:5233–9. doi:10.4049/jimmunol.168.10.5233
143. Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol (2001) 167:2887–94. doi:10.4049/jimmunol.167.5.2887
144. Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med (2002) 195:99–111. doi:10.1084/jem.20001858
145. Vabulas RM, Ahmad-Nejad P, Da Costa C, Miethke T, Kirschning CJ, Hacker H, et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem (2001) 276:31332–9. doi:10.1074/jbc.M103217200
146. Baniyash M. TCR zeta-chain downregulation: curtailing an excessive inflammatory immune response. Nat Rev Immunol (2004) 4:675–87. doi:10.1038/nri1434
147. Kim CW, Choi SH, Chung EJ, Lee MJ, Byun EK, Ryu MH, et al. Alteration of signal-transducing molecules and phenotypical characteristics in peripheral blood lymphocytes from gastric carcinoma patients. Pathobiology (1999) 67:123–8. doi:10.1159/000028061
148. Takahashi A, Kono K, Amemiya H, Iizuka H, Fujii H, Matsumoto Y. Elevated caspase-3 activity in peripheral blood T cells coexists with increased degree of T-cell apoptosis and down-regulation of TCR zeta molecules in patients with gastric cancer. Clin Cancer Res (2001) 7:74–80.
149. Matsuda M, Petersson M, Lenkei R, Taupin JL, Magnusson I, Mellstedt H, et al. Alterations in the signal-transducing molecules of T cells and NK cells in colorectal tumor-infiltrating, gut mucosal and peripheral lymphocytes: correlation with the stage of the disease. Int J Cancer (1995) 61:765–72. doi:10.1002/ijc.2910610605
150. Healy CG, Simons JW, Carducci MA, Deweese TL, Bartkowski M, Tong KP, et al. Impaired expression and function of signal-transducing zeta chains in peripheral T cells and natural killer cells in patients with prostate cancer. Cytometry (1998) 32: doi:10.1002/(SICI)1097-0320(19980601)32:2<109::AID-CYTO6>3.0.CO;2-G
151. Kono K, Ressing ME, Brandt RM, Melief CJ, Potkul RK, Andersson B, et al. Decreased expression of signal-transducing zeta chain in peripheral T cells and natural killer cells in patients with cervical cancer. Clin Cancer Res (1996) 2:1825–8.
152. Schmielau J, Nalesnik MA, Finn OJ. Suppressed T-cell receptor zeta chain expression and cytokine production in pancreatic cancer patients. Clin Cancer Res (2001) 7:933s–9s.
153. Wu Y, Zhao Q, Peng C, Sun L, Li XF, Kuang DM. Neutrophils promote motility of cancer cells via a hyaluronan-mediated TLR4/PI3K activation loop. J Pathol (2011) 225:438–47. doi:10.1002/path.2947
154. Yang CA, Scheibenbogen C, Bauer S, Kleinle C, Wex T, Bornschein J, et al. A frequent Toll-like receptor 1 gene polymorphism affects NK- and T-cell IFN-gamma production and is associated with Helicobacter pylori-induced gastric disease. Helicobacter (2013) 18:13–21. doi:10.1111/hel.12001
155. Castaño-Rodríguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM. The role of TLR2, TLR4 and CD14 genetic polymorphisms in gastric carcinogenesis: a case-control study and meta-analysis. PLoS One (2013) 8:e60327. doi:10.1371/journal.pone.0060327
156. Tahara T, Arisawa T, Wang F, Shibata T, Nakamura M, Sakata M, et al. Toll-like receptor 2 −196 to 174del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci (2007) 98:1790–4. doi:10.1111/j.1349-7006.2007.00590.x
157. de Oliveira JG, Rossi AF, Nizato DM, Miyasaki K, Silva AE. Profiles of gene polymorphisms in cytokines and Toll-like receptors with higher risk for gastric cancer. Dig Dis Sci (2013) 58:978–88. doi:10.1007/s10620-012-2460-5
158. Zeng HM, Pan KF, Zhang Y, Zhang L, Ma JL, Zhou T, et al. Genetic variants of toll-like receptor 2 and 5, helicobacter pylori infection, and risk of gastric cancer and its precursors in a chinese population. Cancer Epidemiol Biomarkers Prev (2011) 20:2594–602. doi:10.1158/1055-9965.EPI-11-0702
159. Hold GL, Rabkin CS, Chow WH, Smith MG, Gammon MD, Risch HA, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology (2007) 132:905–12. doi:10.1053/j.gastro.2006.12.026
160. Santini D, Angeletti S, Ruzzo A, Dicuonzo G, Galluzzo S, Vincenzi B, et al. Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms in gastric cancer of intestinal and diffuse histotypes. Clin Exp Immunol (2008) 154:360–4. doi:10.1111/j.1365-2249.2008.03776.x
161. Huang L, Yuan K, Liu J, Ren X, Dong X, Tian W, et al. Polymorphisms of the TLR4 gene and risk of gastric cancer. Gene (2014) 537:46–50. doi:10.1016/j.gene.2013.12.030
162. Kim J, Cho YA, Choi IJ, Lee YS, Kim SY, Hwang JA, et al. Effects of polymorphisms of innate immunity genes and environmental factors on the risk of noncardia gastric cancer. Cancer Res Treat (2013) 45:313–24. doi:10.4143/crt.2013.45.4.313
163. Wang X, Xue L, Yang Y, Xu L, Zhang G. TLR9 promoter polymorphism is associated with both an increased susceptibility to gastric carcinoma and poor prognosis. PLoS One (2013) 8:e65731. doi:10.1371/journal.pone.0065731
164. Companioni O, Bonet C, Munoz X, Weiderpass E, Panico S, Tumino R, et al. Polymorphisms of Helicobacter pylori signaling pathway genes and gastric cancer risk in the European Prospective Investigation into Cancer-Eurgast cohort. Int J Cancer (2014) 134:92–101. doi:10.1002/ijc.28357
165. Li K, Dan Z, Hu XJ, Gesang LB, Ze YG, Bianba ZX, et al. Association of CD14/-260 polymorphism with gastric cancer risk in Highland Tibetans. World J Gastroenterol (2014) 20:2688–94. doi:10.3748/wjg.v20.i10.2688
166. Tahara T, Arisawa T, Shibata T, Hirata I, Nakano H. Association of polymorphism of TLR4 and CD14 genes with gastroduodenal diseases in Japan. Inflammopharmacology (2007) 15:124–8. doi:10.1007/s10787-006-1567-8
167. Zhao D, Sun T, Zhang X, Guo Y, Yu D, Yang M, et al. Role of CD14 promoter polymorphisms in Helicobacter pylori infection – related gastric carcinoma. Clin Cancer Res (2007) 13:2362–8. doi:10.1158/1078-0432.CCR-06-2612
168. Ohara T, Kanoh Y, Tani N, Ohdaira H, Suzuki Y, Kameyama J, et al. Single nucleotide polymorphism typing of the human toll-like receptor 4 gene at the 2-kb upstream region of the 5′ untranslated region: new enclosure strategy for the risk grouping of poorly-differentiated gastric adenocarcinoma patients. Mol Med Rep (2009) 2:17–21. doi:10.3892/mmr_00000055
169. Ohara T, Morishita T, Suzuki H, Hibi T. Heterozygous Thr 135 Ala polymorphism at leucine-rich repeat (LRR) in genomic DNA of toll-like receptor 4 in patients with poorly-differentiated gastric adenocarcinomas. Int J Mol Med (2006) 18:59–63.
170. Castaño-Rodríguez N, Kaakoush NO, Pardo AL, Goh KL, Fock KM, Mitchell HM. Genetic polymorphisms in the Toll-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer. Hum Immunol (2014). doi:10.1016/j.humimm.2014.06.001
171. Yang JC, Yang HC, Shun CT, Wang TH, Chien CT, Kao JY. Catechins and sialic acid attenuate Helicobacter pylori-triggered epithelial caspase-1 activity and eradicate Helicobacter pylori infection. Evid Based Complement Alternat Med (2013) 2013:248585. doi:10.1155/2013/248585
172. Hishida A, Matsuo K, Goto Y, Naito M, Wakai K, Tajima K, et al. No associations of Toll-like receptor 2 (TLR2) −196 to −174del polymorphism with the risk of Helicobacter pylori seropositivity, gastric atrophy, and gastric cancer in Japanese. Gastric Cancer (2010) 13:251–7. doi:10.1007/s10120-010-0567-y
173. Castaño-Rodríguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLoS One (2014) 9:e98899. doi:10.1371/journal.pone.0098899
174. Qadri Q, Rasool R, Afroze D, Naqash S, Gulzar GM, Yousuf A, et al. Study of TLR4 and IL-8 gene polymorphisms in H. pylori-induced inflammation in gastric cancer in an ethnic Kashmiri population. Immunol Invest (2013) 43:324–36. doi:10.3109/08820139.2013.854378
175. Schmidt HM, Ha DM, Taylor EF, Kovach Z, Goh KL, Fock KM, et al. Variation in human genetic polymorphisms, their association with Helicobacter pylori acquisition and gastric cancer in a multi-ethnic country. J Gastroenterol Hepatol (2011) 26:1725–32. doi:10.1111/j.1440-1746.2011.06799.x
176. Trejo-de la O A, Torres J, Perez-Rodriguez M, Camorlinga-Ponce M, Luna LF, Abdo-Francis JM, et al. TLR4 single-nucleotide polymorphisms alter mucosal cytokine and chemokine patterns in Mexican patients with Helicobacter pylori-associated gastroduodenal diseases. Clin Immunol (2008) 129:333–40. doi:10.1016/j.clim.2008.07.009
177. Garza-Gonzalez E, Bosques-Padilla FJ, Mendoza-Ibarra SI, Flores-Gutierrez JP, Maldonado-Garza HJ, Perez-Perez GI. Assessment of the toll-like receptor 4 Asp299Gly, Thr399Ile and interleukin-8 −251 polymorphisms in the risk for the development of distal gastric cancer. BMC Cancer (2007) 7:70. doi:10.1186/1471-2407-7-70
178. Huang H, Wu J, Jin G, Zhang H, Ding Y, Hua Z, et al. A 5′-flanking region polymorphism in toll-like receptor 4 is associated with gastric cancer in a Chinese population. J Biomed Res (2010) 24:100–6. doi:10.1016/S1674-8301(10)60017-6
179. Kim EJ, Lee JR, Chung WC, Jung SH, Sung HJ, Lee YW, et al. Association between genetic polymorphisms of NOD 1 and Helicobacter pylori-induced gastric mucosal inflammation in healthy Korean population. Helicobacter (2013) 18:143–50. doi:10.1111/hel.12020
180. Kupcinskas J, Wex T, Bornschein J, Selgrad M, Leja M, Juozaityte E, et al. Lack of association between gene polymorphisms of Angiotensin converting enzyme, Nod-like receptor 1, Toll-like receptor 4, FAS/FASL and the presence of Helicobacter pylori-induced premalignant gastric lesions and gastric cancer in Caucasians. BMC Med Genet (2011) 12:112. doi:10.1186/1471-2350-12-112
181. Hishida A, Matsuo K, Goto Y, Mitsuda Y, Hiraki A, Naito M, et al. Toll-like receptor 4 +3725 G/C polymorphism, Helicobacter pylori seropositivity, and the risk of gastric atrophy and gastric cancer in Japanese. Helicobacter (2009) 14:47–53. doi:10.1111/j.1523-5378.2009.00659.x
182. Hold GL, Rabkin CS, Gammon MD, Berry SH, Smith MG, Lissowska J, et al. CD14-159C/T and TLR9-1237T/C polymorphisms are not associated with gastric cancer risk in Caucasian populations. Eur J Cancer Prev (2009) 18:117–9. doi:10.1097/CEJ.0b013e3283101292
183. Wu MS, Cheng TY, Shun CT, Lin MT, Chen LC, Lin JT. Functional polymorphisms of CD14 and toll-like receptor 4 in Taiwanese Chinese with Helicobacter pylori-related gastric malignancies. Hepatogastroenterology (2006) 53:807–10.
184. Rigoli L, Di Bella C, Fedele F, Procopio V, Amorini M, Lo Giudice G, et al. TLR4 and NOD2/CARD15 genetic polymorphisms and their possible role in gastric carcinogenesis. Anticancer Res (2010) 30:513–7.
185. Achyut BR, Ghoshal UC, Moorchung N, Mittal B. Association of Toll-like receptor-4 (Asp299Gly and Thr399Ileu) gene polymorphisms with gastritis and precancerous lesions. Hum Immunol (2007) 68:901–7. doi:10.1016/j.humimm.2007.10.006
186. Fan YF, Wu YM, Liu H, Yu Y, Jiang YY, Xue YZ, et al. TLR4 polymorphisms associated with developing gastric pre-cancer lesions in a Chinese Han population. Hum Immunol (2014) 75:176–81. doi:10.1016/j.humimm.2013.11.002
187. Kato I, Canzian F, Plummer M, Franceschi S, Van Doorn LJ, Vivas J, et al. Polymorphisms in genes related to bacterial lipopolysaccharide/peptidoglycan signaling and gastric precancerous lesions in a population at high risk for gastric cancer. Dig Dis Sci (2007) 52:254–61. doi:10.1007/s10620-006-9303-1
188. Murphy G, Thornton J, Mcmanus R, Swan N, Ryan B, Hughes DJ, et al. Association of gastric disease with polymorphisms in the inflammatory-related genes IL-1B, IL-1RN, IL-10, TNF and TLR4. Eur J Gastroenterol Hepatol (2009) 21:630–5. doi:10.1097/MEG.0b013e3283140eea
189. Hofner P, Gyulai Z, Kiss ZF, Tiszai A, Tiszlavicz L, Toth G, et al. Genetic polymorphisms of NOD1 and IL-8, but not polymorphisms of TLR4 genes, are associated with Helicobacter pylori-induced duodenal ulcer and gastritis. Helicobacter (2007) 12:124–31. doi:10.1111/j.1523-5378.2007.00481.x
190. Cheng C, Lingyan W, Yi H, Cheng Z, Huadan Y, Xuting X, et al. Association between TLR2, MTR, MTRR, XPC, TP73, TP53 genetic polymorphisms and gastric cancer: a meta-analysis. Clin Res Hepatol Gastroenterol (2014). doi:10.1016/j.clinre.2013.12.009
191. Chen J, Hu S, Liang S, Chen Q, Yang Q, Zheng W, et al. Associations between the four toll-like receptor polymorphisms and the risk of gastric cancer: a meta-analysis. Cancer Biother Radiopharm (2013) 28:674–81. doi:10.1089/cbr.2012.1395
192. Zhao X, Liu L, Kang S, Zhang D. An updated meta-analysis about the association of Asp299Gly in Toll-like receptor 4 gene with risk of cancer. Eur J Cancer (2013) 49:2068–70. doi:10.1016/j.ejca.2013.01.031
193. Zhang K, Zhou B, Wang Y, Rao L, Zhang L. The TLR4 gene polymorphisms and susceptibility to cancer: a systematic review and meta-analysis. Eur J Cancer (2013) 49:946–54. doi:10.1016/j.ejca.2012.09.022
194. Zou TH, Wang ZH, Fang JY. Positive association between Toll-like receptor 4 gene +896A/G polymorphism and susceptibility to gastric carcinogenesis: a meta-analysis. Tumour Biol (2013) 34:2441–50. doi:10.1007/s13277-013-0795-y
195. El-Omar EM, Ng MT, Hold GL. Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene (2008) 27:244–52. doi:10.1038/sj.onc.1210912
196. Sato K, Yoshimura A, Kaneko T, Ukai T, Ozaki Y, Nakamura H, et al. A single nucleotide polymorphism in 3′-untranslated region contributes to the regulation of Toll-like receptor 4 translation. J Biol Chem (2012) 287:25163–72. doi:10.1074/jbc.M111.338426
197. Liu J, Radler D, Illi S, Klucker E, Turan E, Von Mutius E, et al. TLR2 polymorphisms influence neonatal regulatory T cells depending on maternal atopy. Allergy (2011) 66:1020–9. doi:10.1111/j.1398-9995.2011.02573.x
198. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol (2005) 6:345–52. doi:10.1038/ni1178
199. Rad R, Brenner L, Bauer S, Schwendy S, Layland L, Da Costa CP, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology (2006) 131:525–37. doi:10.1053/j.gastro.2006.05.001
200. Jang TJ. The number of Foxp3-positive regulatory T cells is increased in Helicobacter pylori gastritis and gastric cancer. Pathol Res Pract (2010) 206:34–8. doi:10.1016/j.prp.2009.07.019
201. Kandulski A, Wex T, Kuester D, Peitz U, Gebert I, Roessner A, et al. Naturally occurring regulatory T cells (CD4+, CD25high, FOXP3+) in the antrum and cardia are associated with higher H. pylori colonization and increased gene expression of TGF-beta1. Helicobacter (2008) 13:295–303. doi:10.1111/j.1523-5378.2008.00612.x
202. Kindlund B, Sjoling A, Hansson M, Edebo A, Hansson LE, Sjovall H, et al. FOXP3-expressing CD4(+) T-cell numbers increase in areas of duodenal gastric metaplasia and are associated to CD4(+) T-cell aggregates in the duodenum of Helicobacter pylori-infected duodenal ulcer patients. Helicobacter (2009) 14:192–201. doi:10.1111/j.1523-5378.2009.00673.x
203. Enarsson K, Lundgren A, Kindlund B, Hermansson M, Roncador G, Banham AH, et al. Function and recruitment of mucosal regulatory T cells in human chronic Helicobacter pylori infection and gastric adenocarcinoma. Clin Immunol (2006) 121:358–68. doi:10.1016/j.clim.2006.07.002
204. Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, Edebo A, et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun (2005) 73:523–31. doi:10.1128/IAI.73.1.523-531.2005
205. Junpee A, Tencomnao T, Sanprasert V, Nuchprayoon S. Association between Toll-like receptor 2 (TLR2) polymorphisms and asymptomatic bancroftian filariasis. Parasitol Res (2010) 107:807–16. doi:10.1007/s00436-010-1932-9
206. Noguchi E, Nishimura F, Fukai H, Kim J, Ichikawa K, Shibasaki M, et al. An association study of asthma and total serum immunoglobin E levels for Toll-like receptor polymorphisms in a Japanese population. Clin Exp Allergy (2004) 34:177–83. doi:10.1111/j.1365-2222.2004.01839.x
207. Tao K, Fujii M, Tsukumo S, Maekawa Y, Kishihara K, Kimoto Y, et al. Genetic variations of Toll-like receptor 9 predispose to systemic lupus erythematosus in Japanese population. Ann Rheum Dis (2007) 66:905–9. doi:10.1136/ard.2006.065961
208. Guiney DG, Hasegawa P, Cole SP. Helicobacter pylori preferentially induces interleukin 12 (IL-12) rather than IL-6 or IL-10 in human dendritic cells. Infect Immun (2003) 71:4163–6. doi:10.1128/IAI.71.7.4163-4166.2003
209. Mohammadi M, Nedrud J, Redline R, Lycke N, Czinn SJ. Murine CD4 T-cell response to Helicobacter infection: TH1 cells enhance gastritis and TH2 cells reduce bacterial load. Gastroenterology (1997) 113:1848–57. doi:10.1016/S0016-5085(97)70004-0
210. Smythies LE, Waites KB, Lindsey JR, Harris PR, Ghiara P, Smith PD. Helicobacter pylori-induced mucosal inflammation is Th1 mediated and exacerbated in IL-4, but not IFN-gamma, gene-deficient mice. J Immunol (2000) 165:1022–9. doi:10.4049/jimmunol.165.2.1022
211. Bamford KB, Fan X, Crowe SE, Leary JF, Gourley WK, Luthra GK, et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology (1998) 114:482–92. doi:10.1016/S0016-5085(98)70531-1
212. Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol (2001) 166:7456–61. doi:10.4049/jimmunol.166.12.7456
213. Hafsi N, Voland P, Schwendy S, Rad R, Reindl W, Gerhard M, et al. Human dendritic cells respond to Helicobacter pylori, promoting NK cell and Th1-effector responses in vitro. J Immunol (2004) 173:1249–57. doi:10.4049/jimmunol.173.2.1249
214. Baldini M, Lohman IC, Halonen M, Erickson RP, Holt PG, Martinez FDA. Polymorphism* in the 5′ flanking region of the CD14 gene is associated with circulating soluble CD14 levels and with total serum immunoglobulin E. Am J Respir Cell Mol Biol (1999) 20:976–83. doi:10.1165/ajrcmb.20.5.3494
215. Hubacek JA, Rothe G, Pit’ha J, Skodova Z, Stanek V, Poledne R, et al. C(−260) →T polymorphism in the promoter of the CD14 monocyte receptor gene as a risk factor for myocardial infarction. Circulation (1999) 99:3218–20. doi:10.1161/01.CIR.99.25.3218
216. Karhukorpi J, Yan Y, Niemela S, Valtonen J, Koistinen P, Joensuu T, et al. Effect of CD14 promoter polymorphism and H. pylori infection and its clinical outcomes on circulating CD14. Clin Exp Immunol (2002) 128:326–32. doi:10.1046/j.1365-2249.2002.01837.x
217. Levan TD, Bloom JW, Bailey TJ, Karp CL, Halonen M, Martinez FD, et al. A common single nucleotide polymorphism in the CD14 promoter decreases the affinity of Sp protein binding and enhances transcriptional activity. J Immunol (2001) 167:5838–44. doi:10.4049/jimmunol.167.10.5838
218. Liang XH, Cheung W, Heng CK, Liu JJ, Li CW, Lim B, et al. CD14 promoter polymorphisms have no functional significance and are not associated with atopic phenotypes. Pharmacogenet Genomics (2006) 16:229–36. doi:10.1097/01.fpc.0000197466.14340.0f
219. Leipe DD, Koonin EV, Aravind L. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J Mol Biol (2004) 343:1–28. doi:10.1016/j.jmb.2004.08.023
220. Bella J, Hindle KL, Mcewan PA, Lovell SC. The leucine-rich repeat structure. Cell Mol Life Sci (2008) 65:2307–33. doi:10.1007/s00018-008-8019-0
221. Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ (2007) 14:10–22. doi:10.1038/sj.cdd.4402038
222. Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE. Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol Cell (2014) 54:17–29. doi:10.1016/j.molcel.2014.02.018
223. Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol (2004) 5:1166–74. doi:10.1038/ni1131
224. Hutton ML, Kaparakis-Liaskos M, Turner L, Cardona A, Kwok T, Ferrero RL. Helicobacter pylori exploits cholesterol-rich microdomains for induction of NF-kappaB-dependent responses and peptidoglycan delivery in epithelial cells. Infect Immun (2010) 78:4523–31. doi:10.1128/IAI.00439-10
225. Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC, et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol (2010) 12:372–85. doi:10.1111/j.1462-5822.2009.01404.x
226. Necchi V, Sommi P, Ricci V, Solcia E. In vivo accumulation of Helicobacter pylori products, NOD1, ubiquitinated proteins and proteasome in a novel cytoplasmic structure. PLoS One (2010) 5:e9716. doi:10.1371/journal.pone.0009716
227. Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol (2009) 183:8099–109. doi:10.4049/jimmunol.0900664
228. Allison CC, Ferrand J, Mcleod L, Hassan M, Kaparakis-Liaskos M, Grubman A, et al. Nucleotide oligomerization domain 1 enhances IFN-gamma signaling in gastric epithelial cells during Helicobacter pylori infection and exacerbates disease severity. J Immunol (2013) 190:3706–15. doi:10.4049/jimmunol.1200591
229. Boonyanugomol W, Chomvarin C, Hahnvajanawong C, Sripa B, Kaparakis-Liaskos M, Ferrero RL. Helicobacter pylori cag pathogenicity island (cagPAI) involved in bacterial internalization and IL-8 induced responses via NOD1- and MyD88-dependent mechanisms in human biliary epithelial cells. PLoS One (2013) 8:e77358. doi:10.1371/journal.pone.0077358
230. Grubman A, Kaparakis M, Viala J, Allison C, Badea L, Karrar A, et al. The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides. Cell Microbiol (2010) 12:626–39. doi:10.1111/j.1462-5822.2009.01421.x
231. Rosenstiel P, Hellmig S, Hampe J, Ott S, Till A, Fischbach W, et al. Influence of polymorphisms in the NOD1/CARD4 and NOD2/CARD15 genes on the clinical outcome of Helicobacter pylori infection. Cell Microbiol (2006) 8:1188–98. doi:10.1111/j.1462-5822.2006.00701.x
232. Kim DJ, Park JH, Franchi L, Backert S, Nunez G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1beta production in Helicobacter pylori infected dendritic cells. Eur J Immunol (2013) 43:2650–8. doi:10.1002/eji.201243281
233. Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol (2003) 4:95–104. doi:10.1038/nrm1019
234. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430:213–8. doi:10.1038/nature02664
235. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi:10.1016/S1097-2765(02)00599-3
236. Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell (2004) 117:561–74. doi:10.1016/j.cell.2004.05.004
237. Tomita T, Jackson AM, Hida N, Hayat M, Dixon MF, Shimoyama T, et al. Expression of Interleukin-18, a Th1 cytokine, in human gastric mucosa is increased in Helicobacter pylori infection. J Infect Dis (2001) 183:620–7. doi:10.1086/318541
238. Potthoff A, Ledig S, Martin J, Jandl O, Cornberg M, Obst B, et al. Significance of the caspase family in Helicobacter pylori induced gastric epithelial apoptosis. Helicobacter (2002) 7:367–77. doi:10.1046/j.1523-5378.2002.00112.x
239. Benoit BN, Kobayashi M, Kawakubo M, Takeoka M, Sano K, Zou J, et al. Role of ASC in the mouse model of Helicobacter pylori infection. J Histochem Cytochem (2009) 57:327–38. doi:10.1369/jhc.2008.952366
240. Hitzler I, Sayi A, Kohler E, Engler DB, Koch KN, Hardt WD, et al. Caspase-1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL-1beta and IL-18. J Immunol (2012) 188:3594–602. doi:10.4049/jimmunol.1103212
241. Shimada M, Ando T, Peek RM, Watanabe O, Ishiguro K, Maeda O, et al. Helicobacter pylori infection upregulates interleukin-18 production from gastric epithelial cells. Eur J Gastroenterol Hepatol (2008) 20:1144–50. doi:10.1097/MEG.0b013e32830edb15
242. Chen Y, Wang Y, Xu W, Zhang Z. Analysis on the mechanism of Helicobacter pylori-induced apoptosis in gastric cancer cell line BGC-823. Int J Mol Med (2005) 16:741–5. doi:10.3892/ijmm.16.4.741
243. Jiang J, Liu S, Luo J, Li X, Tang S, Yu M, et al. [The expressions of NLRP3 inflammasome and its downstream molecules in the mouse model of Helicobacter pylori infection]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2013) 29:785–8.
244. Jee CD, Lee HS, Bae SI, Yang HK, Lee YM, Rho MS, et al. Loss of caspase-1 gene expression in human gastric carcinomas and cell lines. Int J Oncol (2005) 26:1265–71. doi:10.3892/ijo.26.5.1265
245. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12:860–75. doi:10.1038/nrc3380
246. Wang P, Zhang L, Jiang JM, Ma D, Tao HX, Yuan SL, et al. Association of NOD1 and NOD2 genes polymorphisms with Helicobacter pylori related gastric cancer in a Chinese population. World J Gastroenterol (2012) 18:2112–20. doi:10.3748/wjg.v18.i17.2112
247. Kara B, Akkiz H, Doran F, Bayram S, Erken E, Gumurdullu Y, et al. The significance of E266K polymorphism in the NOD1 gene on Helicobacter pylori infection: an effective force on pathogenesis? Clin Exp Med (2010) 10:107–12. doi:10.1007/s10238-009-0077-6
248. Hnatyszyn A, Szalata M, Stanczyk J, Cichy W, Slomski R. Association of c.802C >T polymorphism of NOD2/CARD15 gene with the chronic gastritis and predisposition to cancer in H. pylori infected patients. Exp Mol Pathol (2010) 88:388–93. doi:10.1016/j.yexmp.2010.03.003
249. Wex T, Ebert MP, Kropf S, Dierkes J, Schuttler K, Rocken C, et al. Gene polymorphisms of the NOD-2/CARD-15 gene and the risk of gastric cancer in Germany. Anticancer Res (2008) 28:757–62.
250. Angeletti S, Galluzzo S, Santini D, Ruzzo A, Vincenzi B, Ferraro E, et al. NOD2/CARD15 polymorphisms impair innate immunity and increase susceptibility to gastric cancer in an Italian population. Hum Immunol (2009) 70:729–32. doi:10.1016/j.humimm.2009.04.026
251. Liu J, He C, Xu Q, Xing C, Yuan Y. NOD2 polymorphisms associated with cancer risk: a meta-analysis. PLoS One (2014) 9:e89340. doi:10.1371/journal.pone.0089340
252. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet (2009) 41:71–6. doi:10.1038/ng.285
253. Pontillo A, Vendramin A, Catamo E, Fabris A, Crovella S. The missense variation Q705K in CIAS1/NALP3/NLRP3 gene and an NLRP1 haplotype are associated with celiac disease. Am J Gastroenterol (2011) 106:539–44. doi:10.1038/ajg.2010.474
254. Pontillo A, Brandao L, Guimaraes R, Segat L, Araujo J, Crovella S. Two SNPs in NLRP3 gene are involved in the predisposition to type-1 diabetes and celiac disease in a pediatric population from northeast Brazil. Autoimmunity (2010) 43:583–9. doi:10.3109/08916930903540432
255. Roberts RL, Topless RK, Phipps-Green AJ, Gearry RB, Barclay ML, Merriman TR. Evidence of interaction of CARD8 rs2043211 with NALP3 rs35829419 in Crohn’s disease. Genes Immun (2010) 11:351–6. doi:10.1038/gene.2010.11
256. Yang SK, Kim H, Hong M, Lim J, Choi E, Ye BD, et al. Association of CARD8 with inflammatory bowel disease in Koreans. J Hum Genet (2011) 56:217–23. doi:10.1038/jhg.2010.170
257. Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science (2005) 307:734–8. doi:10.1126/science.1103685
258. Tatsuta T, Imaizumi T, Shimoyama T, Sawaya M, Kunikazu T, Matsumiya T, et al. Expression of melanoma differentiation associated gene 5 is increased in human gastric mucosa infected with Helicobacter pylori. J Clin Pathol (2012) 65:839–43. doi:10.1136/jclinpath-2011-200590
259. Gringhuis SI, Den Dunnen J, Litjens M, Van Der Vlist M, Geijtenbeek TB. Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat Immunol (2009) 10:1081–8. doi:10.1038/ni.1778
260. Miszczyk E, Rudnicka K, Moran AP, Fol M, Kowalewicz-Kulbat M, Druszczynska M, et al. Interaction of Helicobacter pylori with C-type lectin dendritic cell-specific ICAM grabbing nonintegrin. J Biomed Biotechnol (2012) 2012:206463. doi:10.1155/2012/206463
261. Van Die I, Van Vliet SJ, Nyame AK, Cummings RD, Bank CM, Appelmelk B, et al. The dendritic cell-specific C-type lectin DC-SIGN is a receptor for Schistosoma mansoni egg antigens and recognizes the glycan antigen Lewis x. Glycobiology (2003) 13:471–8. doi:10.1093/glycob/cwg052
262. Bergman MP, Engering A, Smits HH, Van Vliet SJ, Van Bodegraven AA, Wirth HP, et al. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J Exp Med (2004) 200:979–90. doi:10.1084/jem.20041061
263. Chang LL, Wang SW, Wu IC, Yu FJ, Su YC, Chen YP, et al. Impaired dendritic cell maturation and IL-10 production following H. pylori stimulation in gastric cancer patients. Appl Microbiol Biotechnol (2012) 96:211–20. doi:10.1007/s00253-012-4034-z
264. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest (2012) 122:1082–96. doi:10.1172/JCI61029
265. Wu J, Lin K, Zeng J, Liu W, Yang F, Wang X, et al. Role of DC-SIGN in Helicobacter pylori infection of gastrointestinal cells. Front Biosci (Landmark Ed) (2014) 19:825–34. doi:10.2741/4250
266. Kutikhin AG, Yuzhalin AE. C-type lectin receptors and RIG-I-like receptors: new points on the oncogenomics map. Cancer Manag Res (2012) 4:39–53. doi:10.2147/CMAR.S28983
267. Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med (2003) 197:1107–17. doi:10.1084/jem.20021787
268. Gringhuis SI, Den Dunnen J, Litjens M, Van Het Hof B, Van Kooyk Y, Geijtenbeek TB. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity (2007) 26:605–16. doi:10.1016/j.immuni.2007.03.012
269. Kankkunen P, Teirila L, Rintahaka J, Alenius H, Wolff H, Matikainen S. (1,3)-beta-glucans activate both dectin-1 and NLRP3 inflammasome in human macrophages. J Immunol (2010) 184:6335–42. doi:10.4049/jimmunol.0903019
270. Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, Von Messling V, et al. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog (2013) 9:e1003256. doi:10.1371/journal.ppat.1003256
271. Hedayat M, Takeda K, Rezaei N. Prophylactic and therapeutic implications of toll-like receptor ligands. Med Res Rev (2012) 32:294–325. doi:10.1002/med.20214
272. Paul-Clark MJ, George PM, Gatheral T, Parzych K, Wright WR, Crawford D, et al. Pharmacology and therapeutic potential of pattern recognition receptors. Pharmacol Ther (2012) 135:200–15. doi:10.1016/j.pharmthera.2012.05.007
273. Gradisar H, Keber MM, Pristovsek P, Jerala R. MD-2 as the target of curcumin in the inhibition of response to LPS. J Leukoc Biol (2007) 82:968–74. doi:10.1189/jlb.1206727
274. Tu SP, Jin H, Shi JD, Zhu LM, Suo Y, Lu G, et al. Curcumin induces the differentiation of myeloid-derived suppressor cells and inhibits their interaction with cancer cells and related tumor growth. Cancer Prev Res (Phila) (2012) 5:205–15. doi:10.1158/1940-6207.CAPR-11-0247
Keywords: stomach neoplasm, Helicobacter pylori, inflammation, pattern-recognition receptors, Toll-like receptors, NOD-like receptors, genetic polymorphism, therapeutics
Citation: Castaño-Rodríguez N, Kaakoush NO and Mitchell HM (2014) Pattern-recognition receptors and gastric cancer. Front. Immunol. 5:336. doi: 10.3389/fimmu.2014.00336
Received: 15 April 2014; Paper pending published: 25 May 2014;
Accepted: 03 July 2014; Published online: 22 July 2014.
Edited by:
Anton G. Kutikhin, Russian Academy of Medical Sciences, RussiaReviewed by:
Masaaki Murakami, Hokkaido University, JapanArseniy E. Yuzhalin, University of Oxford, UK
Si Ming Man, St. Jude Children’s Research Hospital, USA
Copyright: © 2014 Castaño-Rodríguez, Kaakoush and Mitchell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hazel M. Mitchell, School of Biotechnology and Biomolecular Sciences, Laboratory 301A, Biological Sciences Building, The University of New South Wales, Sydney, NSW 2052, Australia e-mail: h.mitchell@unsw.edu.au