 Leticia Huergo-Zapico1
Leticia Huergo-Zapico1 Andrea Acebes-Huerta1
Andrea Acebes-Huerta1 Alejandro López-Soto1
Alejandro López-Soto1 Mónica Villa-Álvarez1
Mónica Villa-Álvarez1 Ana Pilar Gonzalez-Rodriguez2
Ana Pilar Gonzalez-Rodriguez2 Segundo Gonzalez1*
Segundo Gonzalez1*- 1Department of Functional Biology, University Institute of Oncology (IUOPA), University of Oviedo, Oviedo, Spain
- 2Department of Hematology, Hospital Universitario Central de Asturias, Oviedo, Spain
NKG2D is an activating receptor expressed by NK and T cells primarily involved in the elimination of transformed and infected cells. NKG2D ligands are self-proteins restrictedly expressed in healthy tissues, but induced in response to signaling pathways commonly associated with transformation. Proliferative, tumor suppressor, and stress signaling pathways linked to the tumorigenic process induce the expression of NKG2D ligands, initiating an immune response against the incipient tumor. Nevertheless, the activity of NKG2D ligands is counter-regulated in vivo by the immunoediting of cancer cells, resulting in the expression of multiple mechanisms of immune evasion in advanced tumors. The redundancy of NKG2D ligands, besides increasing the complexity of their regulation, may impair the generation of these immune evasion mechanisms. In this review, we attempt to integrate the mechanisms and pathways involved in the regulation of NKG2D ligand expression in cancer.
Human NKG2D and Its Ligands
NKG2D is a type-II transmembrane-anchored glycoprotein constitutively expressed on the surface of NK, CD8 T, and γδ T cells (1). At the cell membrane, human NKG2D associates with DAP10, while mouse NKG2D may associate with both DAP10 and DAP12, which activate downstream signaling pathways resulting in the activation of NK cells and co-stimulation of CD8 T cells (2). Cross-linking of NKG2D receptor alone on freshly isolated NK cells does not seem to trigger a significant cell-mediated cytotoxic response; and simultaneous cross-linking of other activating receptors or stimulation of NK cells with IL-15 or high dose of IL-2 is rather necessary to kill tumor target cells (3).
NKG2D is a receptor for multiple ligands (NKG2DLs), which are distant members of the MHC class I family. In humans, NKG2D is a receptor for MICA and MICB (MICA/B) and ULBP1–6 molecules (4–8) (Figure 1). NKG2DLs are restrictedly expressed in healthy cells avoiding autoimmunity, but they are frequently over-expressed in infected and transformed cells, acting as a danger signal that favors the perforin-mediated elimination of transformed cells, thus acting as an extrinsic mechanism of cancer surveillance that complements the intrinsic processes that control growth and proliferation of tumor cells.
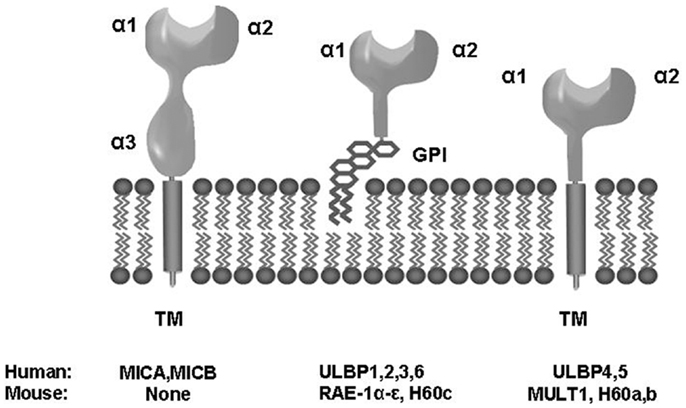
Figure 1. Biochemical and structural properties of human and mouse NKG2D ligands. TM, transmembrane; GPI, glycosylphosphatidylinositol.
NKG2D is Involved in the Immune Surveillance of Cancers
NKG2D-deficient mice are more prone to develop cancer (9), and neutralization of NKG2D increases the sensitivity of mice to carcinogen-induced sarcomas (10). Conversely, murine NK cells may efficiently eliminate cancer cells expressing NKG2DLs in vivo, highlighting the relevant role of this receptor in the cancer immune surveillance in mice (11–13). In humans, NKG2DLs are restrictedly expressed in healthy cells, but a broad expression of these proteins is observed in transformed cells, rendering them more susceptible to NK cell-mediated killing (1, 14–16). Moreover, cell-associated NKG2DL expression correlates with the outcome in colorectal (16, 17), breast, (18) and pancreatic cancers (19) and hepatocellular carcinoma (20). However, it is noteworthy that high expression of ULBP2 and ULBP4 has been paradoxically associated with poor prognosis in ovarian cancer, and it does not appear to be related to an increase of the shedding of these molecules (21, 22). Further, the development of immune evasion mechanisms by cancer cells has been associated with poor prognosis in different types of cancer (see below). Thus, the serum level of soluble MICA (sMICA) is an independent prognostic factor for multiple myeloma (23) and advanced hepatocellular carcinoma (24). sMICA and soluble MICB (sMICB) serum levels were correlated with disease stage and survival rates in oral squamous cell carcinoma patients (25), whereas soluble ULBP2 (sULBP2) was associated with poor prognosis in melanoma (26), chronic lymphocytic leukemia (27), and lung cancer (28).
Albeit these data clearly indicate an important role for the NKG2D response in the immune surveillance of human cancers, the molecular bases underlying the regulation of NKG2DL expression have not been fully elucidated. Nevertheless, increasing evidence indicates that the activation of oncogenic pathways commonly associated with the tumorigenic process may be responsible for the induction of NKG2DL expression and the activation of the NKG2D response (29) (Table 1).
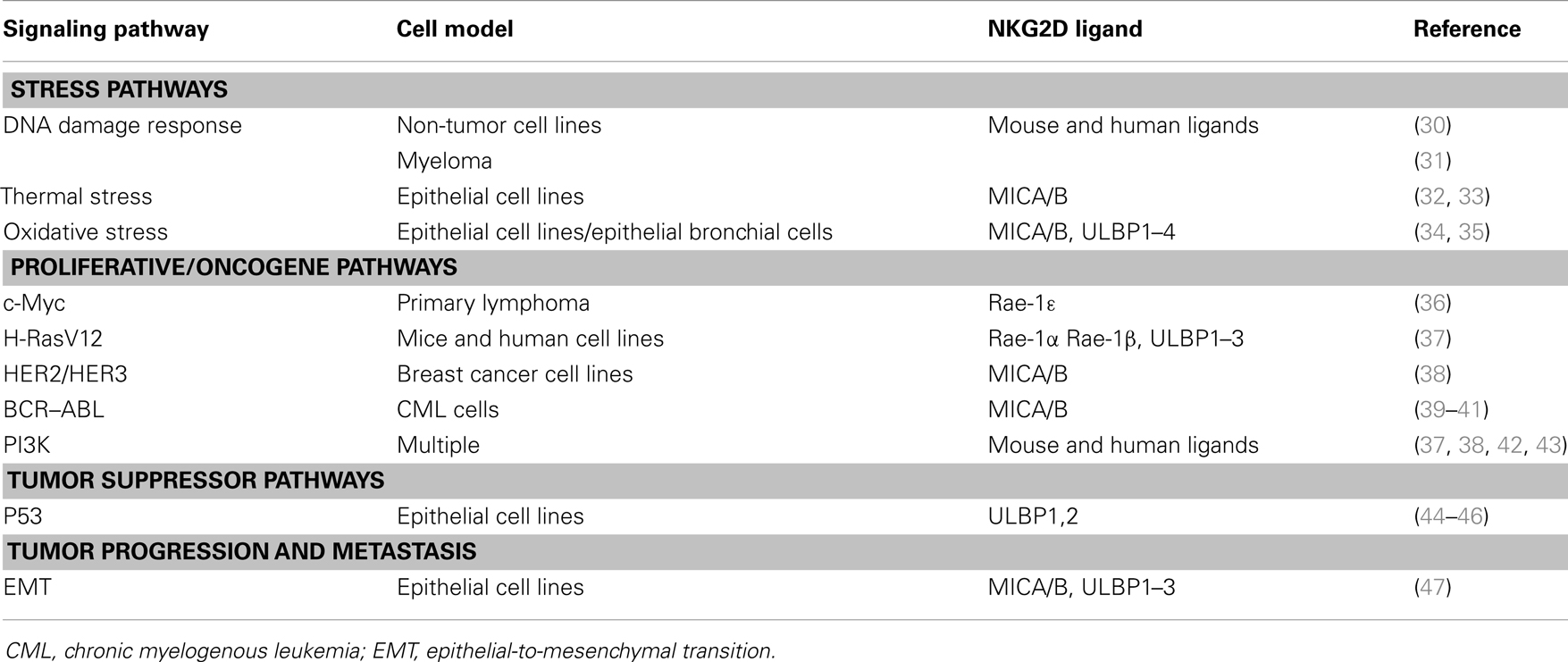
Table 1. Main signaling pathways that regulate NKG2D ligand expression.
DNA Damage and NKG2D Response
DNA damage potentially represents an initiating event for carcinogenesis as it is frequently observed in precancerous lesions. As a result of the DNA damage response (DDR), cell cycle checkpoints are activated, stopping the cell cycle to give the cell time to repair the damage, and, if such damage is too great, to trigger apoptosis. DNA damage checkpoint activation is controlled by two kinases: ATM (ataxia telangiectasia, mutated), which responds to DNA double-strand breaks and disruptions in chromatin structure; and ATR (ATM- and Rad3-related), which primarily responds to stalled replication forks. Activated ATM or ATR initiates a protein kinase cascade that includes the activation of the checkpoint kinases CHK1 and CHK2, and p53. In a seminal paper, it was shown that DNA damage also activates the immune response as an additional extrinsic control mechanism that favors the elimination of damaged and cancer cells through the induction of NKG2DL expression (30). It is worth-mentioning that the up-regulation of NKG2DLs occurs, at least in mice, in a p53-independent manner, which is particularly relevant given that a p53 expression is lost in a majority of clinical aggressive and invasive cancers. However, the downstream regulators of the DDR involved in NKG2DL expression remain ill-defined.
DNA damage response is activated in response to ionizing radiation and chemotherapy. Consequently, the immune response may contribute to the efficacy of these conventional cancer treatments. Many viruses with distinct replication strategies, such as DNA viruses and retroviruses, may also activate the DDR, which may potentially activate the NKG2D response. The replication of viral DNA in the nucleus has the potential to trigger the DDR if the ends of viral genomes are exposed and recognized as double stranded breaks. Retroviral DNA integration creates a discontinuity in the host cell chromatin, also activating the DDR. Likewise, induction of NKG2DLs by two retroviral infections, caused by Abelson murine leukemia virus and human immunodeficiency virus (HIV), supports a role of DDR in the NKG2D-mediated anti-viral response (48, 49). A more universal role of this pathway in the elimination of virus-infected cells remains to be established.
NKG2DL expression may also be induced as a result of DNA replication stress and activation of ATM/ATR in human T cells that proliferate in response to mitogens, antigens, and superantigens (50). This has been proposed to limit undesired immune responses; however, the underlying mechanisms and the biological significance of NKG2DL expression in T cells and other healthy cells are poorly understood.
Other Types of Cellular Stress Regulate NKG2D Response
Different forms of cellular stress frequently associated with the tumorigenic process are involved in the regulation of NKG2DL expression. Early reports showed that heat shock was able to induce MICA and MICB expression (32). This induction of MICA/B transcription was lately described as the result of the binding of heat shock factor 1 (HSF1) to the promoters of MICA and MICB genes (33). Several studies showed that oxidative stress also induced MICA/B gene and protein expression in several tumor cell lines (34, 51). In normal human bronchial epithelial cells, oxidative stress induced MICA and ULBPs expression through the activation of ERK (35). Hypoxia has also been shown to down-regulate the surface expression of MICA in osteosarcoma cell lines through the induction of hypoxia-inducible factor 1α (HIF-1α), but independently of nitric oxide (NO) production (52).
Proliferative Signals Induce NKG2D Response
Early reports showed that MICA/B mRNA and protein expression are mostly limited to proliferating epithelial cells, but they are scarcely expressed in quiescent cells (32, 33, 50). Recently, proliferation has been associated with the induction of the expression of murine RAE-1 family of NKG2DLs in mouse fibroblasts, and of MICA/B and ULBP2 in HCT116 colorectal cancer cells (42). The induction of NKG2DL expression was independent of stress pathways. Instead, Raet1 genes, but not MULT1 and H60b, were found to be direct targets of E2F family of transcription factors, which plays a major role during the G1/S transition and cell cycle reentry. This provides a mechanistic link between proliferation and NKG2D response. Moreover, murine RAE-1ε induction in primary fibroblast cultures was blocked by inhibiting proliferative pathways such as cyclin-dependent kinases, PI3K–mTOR, and MAPK pathways (42).
Unrestrained proliferation is a common characteristic of cancer cells. Albeit it has not always been mechanistically linked to cell proliferation, it has been reported that inappropriate over-expression of several oncogenes induced NKG2DL expression independently of DDR. However, the expression of certain oncogenes (K-ras and c-myc or Akt and c-myc) was not sufficient to induce NKG2DL expression in primary ovarian epithelial cells (30). This probably reflects that transformation is a multistep process, and changes in many pathways, such as tumor suppressor pathways, are required for both transforming a normal cell into a cancer cell and for activating the NKG2D response. In fact, it has been reported that several oncogenic pathways up-regulate NKG2DLs. For instance, NKG2DLs are induced on spontaneously arising tumors in an Eμ-myc murine model of lymphoma (36). The induction of NKG2DLs involves the activity of c-Myc, which is a master regulator of proliferation. Similarly, the oncogenic H-RasV12 protein up-regulates the expression of Rae-1α and Rae-1β in mouse and of ULBP1–3 in human cells (37). H-RasV12 mainly regulated the expression of Rae-1 by post-transcriptional mechanisms via Raf–MAPK/MEK and PI3K pathways. Ras signals also contribute to the induction of NKG2DL expression in primary fibroblast cultures in response to the serum, which may be blocked by epidermal growth factor receptor (EGFR) inhibitors (37). EGFR comprises a family of four closely related transmembrane tyrosine kinase receptors (also known as HER1–4). Amplification or over-expression of the HER2/HER3 dimer is involved in the progression of different cancers, particularly in certain types of aggressive breast cancer. It has been shown that over-expression of HER2/HER3 activates the expression of MICA/B, mainly through the PI3K/AKT pathway (38). Philadelphia chromosome is a specific chromosomal translocation associated with chronic myelogenous leukemia (CML). It results in BCR–ABL fusion protein, with a continuously activated tyrosine kinase activity, thus promoting the unrestrained cell division of leukemia cells. BCR–ABL kinase activity was also linked to NKG2DL expression and the activity of NK cells (39, 40). Thus, BCR/ABL induces the MICA surface expression on CML leukemia cells, whereas it was absent on healthy hematopoietic cells (41).
Induction of host cell proliferation is a common viral strategy that favors viral survival and replication. However, it may also induce NKG2DL expression, resulting in an NKG2D-dependent elimination of the infected cell. Thus, the activation of PI3K pathway by the murine cytomegalovirus results in the expression of RAE-1 family of NKG2DLs (43). Similarly, adenovirus serotype 5 (Ad5) E1A oncogene up-regulates RAE-1 expression, but not murine ULBP-like transcript 1 (53).
Overall, these data suggest that proliferative signals generated by oncogene activation and virus infection and, possibly by other physiological situations, may alert the immune surveillance response through the induction of the NKG2D response.
Tumor Suppression and Senescence may Induce NKG2DL Expression
The deregulation of the activity of oncogene proteins is counter-regulated by tumor suppressor proteins. These proteins repress cellular proliferation and the loss of their function is an additional characteristic associated with tumorigenesis. For instance, in the Eμ-myc murine model of lymphoma, sustained over-expression of Myc signaling induces p19Arf expression, resulting in apoptosis through the inhibition of Mdm2 and stabilization of the p53 tumor suppressor protein. Thus, a combination of deregulated activity of Myc and the loss of a tumor suppressor protein (either p19Arf or p53) is involved in both tumor progression and in the induction of Rae-1 in lymphoma cells (36). Thus, despite the existence of few experimental data, the current knowledge about tumor biology suggests that the combination of oncogenes and tumor suppressors may be involved in the regulation of NKG2DL expression.
Accordingly, the tumor suppressor p53 has been associated with the regulation of NKG2D response. Over-expression of wild-type p53 in cancer cell lines strongly up-regulated the expression of ULBP1 and ULBP2, but not of other NKG2DLs, upon binding of p53 to response elements located in the intronic regions of these genes (44, 45). Moreover, the small molecule inhibitor Nutlin-3a decreased ULBP2 levels in a p53-dependent manner, through the increase in cellular levels of the suppressive miR-34 (46).
Additionally to growth arrest and apoptosis, p53 may induce senescence in response to diverse forms of cellular stress, including telomere shortening, DNA damage, oncogene activation, oxidative stress, and chemotherapeutic drug administration. The senescence program activates p53 and p16INK4a–pRb tumor suppressor pathways, which establish an irreversible form of cell cycle arrest. The most fully described function of senescence in vivo is the ability to establish a potent barrier to tumorigenesis in response to oncogene activation or DNA damage. A typical characteristic of senescent cells, known as “senescence-associated secretory phenotype,” is the secretion of inflammatory cytokines and chemokines that may recruit and activate distinct adaptive and innate immune cells subsets, including NK and T cells.
It has been shown that chemotherapeutic drug treatment of myeloma cells induces DDRs and activates the expression of NKG2DLs, mainly in senescent cells, resulting in their elimination by NK cells through the recognition by NKG2D and DNAM-1 receptors (31). NKG2DLs are also up-regulated on activated senescent hepatic stellate cells (54). Further, a recent study in a mouse model of senescence generated by induction of p53 expression also showed that NK cells participate in the elimination of senescent cells in an NKG2D-dependent manner (55). Nevertheless, induction of p53 did not increase NKG2DL expression, but, instead, induced senescent cell to secrete chemokines that recruit NK cells.
NKG2DL Expression during Tumor Progression
It has been well established that NKG2D may protect the host from tumor initiation (9–13); however, less information exists regarding the role of NKG2D in tumor progression and metastasis. Early reports showed the loss of MICA/B expression in metastatic melanoma suggesting the immune selection of MICA/B negative tumors (56). Similarly, we have recently reported that epithelial-to-mesenchymal transition (EMT) is an immunological checkpoint that controls tumor progression through NKG2D-mediated immune responses (47, 57). EMT is one of the first steps of the metastatic cascade, which results in the loss of epithelial characteristics and the gain of mesenchymal properties by malignant cells, such as migration and invasiveness. EMT induction by Snail1 over-expression, GSK-3 inhibition, or TGF-β stimulation up-regulated NKG2DL expression in epithelial cells, rendering EMT cells more susceptible to NK cell-mediated killing. Sp1 and Sp3 transcription factors are key regulators of the basal transcription of MICA/B and ULBP1–3 (33, 58, 59); and induction of Sp1 activity in colorectal tumor cells was involved in the up-regulation of NKG2DL expression during the EMT process. In vivo, MICA/B and ULBP1 proteins are expressed in healthy colon mucosa cells, displaying a polarized apical distribution; and no interaction with NKG2D-bearing immune cells was observed (47, 57). The polarized expression of MICA and ULBP1 was also observed in malignant cells retaining epithelial characteristics, whereas a loss of the polarization of NKG2DL expression was observed in malignant cells that suffer EMT. Moreover, a dramatic increase in the number of NKG2D-bearing tumor-infiltrating T lymphocytes was observed in specimens lacking MICA expression, suggesting that the loss of the epithelial integrity and polarity, characteristics of the EMT process, may allow the diffusion of MICA/B proteins along the membrane of mesenchymal cells, resulting in their elimination by NKG2D-bearing immune cells. Overall, these data suggest that tumor progression and metastasis, a key characteristic of malignant cancers, may also be under the control of the NKG2D response.
Tumor Evasion of NKG2D Response
NKG2D-deficient mice expressed higher amounts of NKG2DLs in cancer cells than similar tumors in wild-type mice, which suggests that NKG2DL expression in a host may be counter-regulated by the immunoediting of cancer cells (9). This is in agreement with the description of a plethora of immune evasion mechanisms that impair the NKG2D-mediated response. Repression of NKG2DL expression is frequently observed in advanced tumors and metastases compared with primary tumors, and this may be regulated by epigenetic mechanisms. In advanced tumors, histone deacetylases (HDACs) may interact with the promoter region of NKG2DL genes, regulating the chromatin structure and impairing the access of transcription factors to their promoters. Thus, HDAC1 may inhibit the transcription of MICA/B on leukemia cells (60), whereas HDAC3 may repress the transcription of ULBP1–3 in epithelial tumors (59). Consequently, HDAC inhibitors stimulate the expression of NKG2DLs on tumor cells (60–63). Other mechanisms, such as the over-expression of suppressive microRNAs in tumors, may also account for the repression of NKG2DL expression (46, 64–66). Contrarily, sustained NKG2DL expression and prolonged interaction with NKG2D receptor also leads to a strong down-regulation of NK cell cytotoxic activity (67, 68).
NKG2DLs may be also shed as soluble proteins (69, 70), which may cause the endocytosis and degradation of NKG2D receptor in immune cells (2). Interaction of MICA on the surface of tumor cells with the chaperon molecule ERp5 plays a relevant role in sMICA shedding (71). ERp5 may form a transitory disulfide bond with MICA, which induces a conformational change, allowing the proteolytic cleavage of MICA by proteases. Likewise, membrane ERp5 was functionally associated with sMICA shedding in chronic lymphocytic leukemia patients (72); and sMICA serum levels have been associated with ERp5 expression in multiple myeloma (73) and Hodgkin lymphoma (74). sULBP2 may also be released from tumor cells by proteolytic cleavage (75). However, engagement of sULBP2 not necessarily down-regulates NKG2D receptor. sULBP4 may be shed from tumor cells by alternative splicing mechanisms (76). Additionally, cancer cells may release MICA, MICB, and ULBPs into exosomes (77, 78). This event appears to be ligand-dependent, since ULBP2 is mainly shed by metalloproteinases, whereas ULBP3 is abundantly released as part of exosomes. Moreover, in the case of MICA, it may be allele-dependent (79). Exosomes containing NKG2DLs selectively down-regulate NKG2D expression on CD8 T and NK cells.
Concluding Remarks
As discussed above, relevant biological pathways associated with tumorigenesis, including proliferative, tumor suppressor, and stress pathways, are key signals involved in the induction of the NKG2D response in cancer. Many of these pathways are frequently activated in viral infections and in other pathological and physiological situations, providing a common link in the regulation of NKG2DLs between these pathologies and cancer. Nevertheless, little information regarding to the regulation of NKG2DLs in non-cancer-conditions is available, and future research in this field is needed.
However, the activity of NKG2DLs is counter-regulated in vivo by the immunoediting of cancer cells and for the development of immune evasion mechanisms in virus. Likewise, advanced tumors develop multiple mechanisms of immune evasion that frequently modify NKG2DL expression, thus making difficult to differentiate the intrinsic mechanisms of NKG2DL regulation from the consequences of the immune selection. As described above, distinct NKG2DLs are differentially regulated by specific oncogenic pathways. Thus, the redundancy of NKG2DLs presumably increases the range of different pathological situations in which NKG2D response may be activated and may also impair the generation of immune evasion mechanisms.
Author Contributions
All authors participated in the preparation, discussion, and writing of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Fondo de Investigaciones Sanitarias Spanish Grant FIS PI12/01280.
References
1. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285(5428):727–9. doi:10.1126/science.285.5428.727
2. Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science (1999) 285(5428):730–2. doi:10.1126/science.285.5428.730
3. Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood (2006) 107(1):159–66. doi:10.1182/blood-2005-04-1351
4. Bahram S, Bresnahan M, Geraghty DE, Spies T. A second lineage of mammalian major histocompatibility complex class I genes. Proc Natl Acad Sci U S A (1994) 91(14):6259–63. doi:10.1073/pnas.91.14.6259
5. Cosman D, Müllberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity (2001) 14(2):123–33. doi:10.1016/S1074-7613(01)00095-4
6. Radosavljevic M, Cuillerier B, Wilson MJ, Clément O, Wicker S, Gilfillan S, et al. A cluster of ten novel MHC class I related genes on human chromosome 6q24.2-q25.3. Genomics (2002) 79(1):114–23. doi:10.1006/geno.2001.6673
7. Chalupny NJ, Sutherland CL, Lawrence WA, Rein-Weston A, Cosman D. ULBP4 is a novel ligand for human NKG2D. Biochem Biophys Res Commun (2003) 305(1):129–35. doi:10.1016/S0006-291X(03)00714-9
8. Bacon L, Eagle RA, Meyer M, Easom N, Young NT, Trowsdale J. Two human ULBP/RAET1 molecules with transmembrane regions are ligands for NKG2D. J Immunol (2004) 173(2):1078–84.
9. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity (2008) 28(4):571–80. doi:10.1016/j.immuni.2008.02.016
10. Smyth MJ, Swann J, Cretney E, Zerafa N, Yokoyama WM, Hayakawa Y. NKG2D function protects the host from tumor initiation. J Exp Med (2005) 202(5):583–8. doi:10.1084/jem.20050994
11. Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A (2001) 98(20):11521–6. doi:10.1073/pnas.201238598
12. Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature (2001) 413(6852):165–71. doi:10.1038/35093109
13. Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, et al. Regulation of cutaneous malignancy by gammadelta T cells. Science (2001) 294(5542):605–9. doi:10.1126/science.1063916
14. Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A (1999) 96(12):6879–84. doi:10.1073/pnas.96.12.6879
15. Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG, et al. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood (2003) 102(4):1389–96. doi:10.1182/blood-2003-01-0019
16. Watson NF, Spendlove I, Madjd Z, McGilvray R, Green AR, Ellis IO, et al. Expression of the stress-related MHC class I chain-related protein MICA is an indicator of good prognosis in colorectal cancer patients. Int J Cancer (2006) 118(6):1445–52. doi:10.1002/ijc.21510
17. McGilvray RW, Eagle RA, Watson NF, Al-Attar A, Ball G, Jafferji I, et al. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res (2009) 15(22):6993–7002. doi:10.1158/1078-0432.CCR-09-0991
18. de Kruijf EM, Sajet A, van Nes JG, Putter H, Smit VT, Eagle RA, et al. NKG2D ligand tumor expression and association with clinical outcome in early breast cancer patients: an observational study. BMC Cancer (2012) 12:24. doi:10.1186/1471-2407-12-24
19. Duan X, Deng L, Chen X, Lu Y, Zhang Q, Zhang K, et al. Clinical significance of the immunostimulatory MHC class I chain-related molecule A and NKG2D receptor on NK cells in pancreatic cancer. Med Oncol (2011) 28(2):466–74. doi:10.1007/s12032-010-9480-9
20. Kamimura H, Yamagiwa S, Tsuchiya A, Takamura M, Matsuda Y, Ohkoshi S, et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. J Hepatol (2012) 56(2):381–8. doi:10.1016/j.jhep.2011.06.017
21. Li K, Mandai M, Hamanishi J, Matsumura N, Suzuki A, Yagi H, et al. Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: high expression of ULBP2 is an indicator of poor prognosis. Cancer Immunol Immunother (2009) 58(5):641–52. doi:10.1007/s00262-008-0585-3
22. McGilvray RW, Eagle RA, Rolland P, Jafferji I, Trowsdale J, Durrant LG. ULBP2 and RAET1E NKG2D ligands are independent predictors of poor prognosis in ovarian cancer patients. Int J Cancer (2010) 127(6):1412–20. doi:10.1002/ijc.25156
23. Rebmann V, Schütt P, Brandhorst D, Opalka B, Moritz T, Nowrousian MR, et al. Soluble MICA as an independent prognostic factor for the overall survival and progression-free survival of multiple myeloma patients. Clin Immunol (2007) 123(1):114–20. doi:10.1016/j.clim.2006.11.007
24. Li JJ, Pan K, Gu MF, Chen MS, Zhao JJ, Wang H, et al. Prognostic value of soluble MICA levels in the serum of patients with advanced hepatocellular carcinoma. Chin J Cancer (2013) 32(3):141–8. doi:10.5732/cjc.012.10025
25. Tamaki S, Kawakami M, Ishitani A, Kawashima W, Kasuda S, Yamanaka Y, et al. Soluble MICB serum levels correlate with disease stage and survival rate in patients with oral squamous cell carcinoma. Anticancer Res (2010) 30(10):4097–101.
26. Paschen A, Sucker A, Hill B, Moll I, Zapatka M, Nguyen XD, et al. Differential clinical significance of individual NKG2D ligands in melanoma: soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin Cancer Res (2009) 15(16):5208–15. doi:10.1158/1078-0432.CCR-09-0886
27. Nückel H, Switala M, Sellmann L, Horn PA, Dürig J, Dührsen U, et al. The prognostic significance of soluble NKG2D ligands in B-cell chronic lymphocytic leukemia. Leukemia (2010) 24(6):1152–9. doi:10.1038/leu.2010.74
28. Yamaguchi K, Chikumi H, Shimizu A, Takata M, Kinoshita N, Hashimoto K, et al. Diagnostic and prognostic impact of serum-soluble UL16-binding protein 2 in lung cancer patients. Cancer Sci (2012) 103(8):1405–13. doi:10.1111/j.1349-7006.2012.02330.x
29. López-Soto A, Huergo-Zapico L, Acebes-Huerta A, Villa-Alvarez M, Gonzalez S. NKG2D signaling in cancer immunosurveillance. Int J Cancer (2014). doi:10.1002/ijc.28775 (in press).
30. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature (2005) 436(7054):1186–90. doi:10.1038/nature03884
31. Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood (2009) 113(15):3503–11. doi:10.1182/blood-2008-08-173914
32. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A (1996) 93(22):12445–50. doi:10.1073/pnas.93.22.12445
33. Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol (2007) 178(2):961–9.
34. Yamamoto K, Fujiyama Y, Andoh A, Bamba T, Okabe H. Oxidative stress increases MICA and MICB gene expression in a human colon carcinoma cell line (CaCo-2). Biochim Biophys Acta (2001) 1526:10–2. doi:10.1016/S0304-4165(01)00099-X
35. Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol (2006) 291(2):L222–31. doi:10.1152/ajplung.00327.2005
36. Unni AM, Bondar T, Medzhitov R. Intrinsic sensor of oncogenic transformation induces a signal for innate immunosurveillance. Proc Natl Acad Sci U S A (2008) 105(5):1686–91. doi:10.1073/pnas.0701675105
37. Liu XV, Ho SS, Tan JJ, Kamran N, Gasser S. Ras activation induces expression of Raet1 family NK receptor ligands. J Immunol (2012) 189(4):1826–34. doi:10.4049/jimmunol.1200965
38. Okita R, Mougiakakos D, Ando T, Mao Y, Sarhan D, Wennerberg E, et al. HER2/HER3 signaling regulates NK cell-mediated cytotoxicity via MHC class I chain-related molecule A and B expression in human breast cancer cell lines. J Immunol (2012) 188(5):2136–45. doi:10.4049/jimmunol.1102237
39. Terme M, Borg C, Guilhot F, Masurier C, Flament C, Wagner EF, et al. BCR/ABL promotes dendritic cell-mediated natural killer cell activation. Cancer Res (2005) 65(14):6409–17. doi:10.1158/0008-5472.CAN-04-2675
40. Cebo C, Da RochaS, Wittnebel S, Turhan AG, Abdelali J, Caillat-Zucman S, et al. The decreased susceptibility of BCR/ABL targets to NK cell-mediated lysis in response to imatinib mesylate involves modulation of NKG2D ligands, GM1 expression, and synapse formation. J Immunol (2006) 176(2):864–72.
41. Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela JM, et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol (2006) 176(8):5108–16.
42. Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med (2012) 209(13):2409–22. doi:10.1084/jem.20120565
43. Tokuyama M, Lorin C, Delebecque F, Jung H, Raulet DH, Coscoy L. Expression of the RAE-1 family of stimulatory NK-cell ligands requires activation of the PI3K pathway during viral infection and transformation. PLoS Pathog (2011) 7(9):e1002265. doi:10.1371/journal.ppat.1002265
44. Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res (2011) 71(18):5998–6009. doi:10.1158/0008-5472.CAN-10-3211
45. Li H, Lakshmikanth T, Garofalo C, Enge M, Spinnler C, Anichini A, et al. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle (2011) 10(19):3346–58. doi:10.4161/cc.10.19.17630
46. Heinemann A, Zhao F, Pechlivanis S, Eberle J, Steinle A, Diederichs S, et al. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res (2012) 72(2):460–71. doi:10.1158/0008-5472.CAN-11-1977
47. López-Soto A, Huergo-Zapico L, Galván JA, Rodrigo L, de Herreros AG, Astudillo A, et al. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J Immunol (2013) 190(8):4408–19. doi:10.4049/jimmunol.1202950
48. Gourzi P, Leonova T, Papavasiliou FN. A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity (2006) 24(6):779–86. doi:10.1016/j.immuni.2006.03.021
49. Ward J, Davis Z, DeHart J, Zimmerman E, Bosque A, Brunetta E, et al. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog (2009) 5(10):e1000613. doi:10.1371/journal.ppat.1000613
50. Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood (2007) 110(2):606–15. doi:10.1182/blood-2006-10-052720
51. Peraldi MN, Berrou J, Dulphy N, Seidowsky A, Haas P, Boissel N, et al. Oxidative stress mediates a reduced expression of the activating receptor NKG2D in NK cells from end-stage renal disease patients. J Immunol (2009) 182(3):1696–705.
52. Yamada N, Yamanegi K, Ohyama H, Hata M, Nakasho K, Futani H, et al. Hypoxia downregulates the expression of cell surface MICA without increasing soluble MICA in osteosarcoma cells in a HIF-1α-dependent manner. Int J Oncol (2012) 41(6):2005–12. doi:10.3892/ijo.2012.1630
53. Routes JM, Ryan S, Morris K, Takaki R, Cerwenka A, Lanier LL. Adenovirus serotype 5 E1A sensitizes tumor cells to NKG2D-dependent NK cell lysis and tumor rejection. J Exp Med (2005) 202(11):1477–82. doi:10.1084/jem.20050240
54. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell (2008) 134(4):657–67. doi:10.1016/j.cell.2008.06.049
55. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med (2013) 210(10):2057–69. doi:10.1084/jem.20130783
56. Vetter CS, Lieb W, Bröcker EB, Becker JC. Loss of nonclassical MHC molecules MIC-A/B expression during progression of uveal melanoma. Br J Cancer (2004) 91(8):1495–9. doi:10.1038/sj.bjc.6602123
57. López-Soto A, Zapico LH, Acebes-Huerta A, Rodrigo L, Gonzalez S. Regulation of NKG2D signaling during the epithelial-to-mesenchymal transition. Oncoimmunology (2013) 2(9):e25820. doi:10.4161/onci.25820
58. López-Soto A, Quiñones-Lombraña A, López-Arbesú R, López-Larrea C, González S. Transcriptional regulation of ULBP1, a human ligand of the NKG2D receptor. J Biol Chem (2006) 281(41):30419–30. doi:10.1074/jbc.M604868200
59. López-Soto A, Folgueras AR, Seto E, Gonzalez S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: potential implications for the immunosurveillance of cancer. Oncogene (2009) 28(25):2370–82. doi:10.1038/onc.2009.117
60. Kato N, Tanaka J, Sugita J, Toubai T, Miura Y, Ibata M, et al. Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia (2007) 21(10):2103–8. doi:10.1038/sj.leu.2404862
61. Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood (2008) 111(3):1428–36. doi:10.1182/blood-2007-07-101311
62. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res (2005) 65(14):6321–9. doi:10.1158/0008-5472.CAN-04-4252
63. Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res (2005) 65(23):11136–45. doi:10.1158/0008-5472.CAN-05-0599
64. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol (2008) 9(9):1065–73. doi:10.1038/ni.1642
65. Espinoza JL, Takami A, Yoshioka K, Nakata K, Sato T, Kasahara Y, et al. Human microRNA-1245 downregulates the NKG2D receptor in NK cells and impairs NKG2D-mediated functions. Haematologica (2012) 97(9):1295–303. doi:10.3324/haematol.2011.058529
66. Yadav D, Ngolab J, Lim RS, Krishnamurthy S, Bui JD. Cutting edge: down-regulation of MHC class I-related chain A on tumor cells by IFN-gamma-induced microRNA. J Immunol (2009) 182(1):39–43.
67. Coudert JD, Zimmer J, Tomasello E, Cebecauer M, Colonna M, Vivier E, et al. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood (2005) 106(5):1711–7. doi:10.1182/blood-2005-03-0918
68. Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood (2008) 111(7):3571–8. doi:10.1182/blood-2007-07-100057
69. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature (2002) 419(6908):734–8. doi:10.1038/nature01112
70. Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol (2002) 169(8):4098–102.
71. Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature (2007) 447(7143):482–6. doi:10.1038/nature05768
72. Huergo-Zapico L, Gonzalez-Rodriguez AP, Contesti J, Gonzalez E, López-Soto A, Fernandez-Guizan A, et al. Expression of ERp5 and GRP78 on the membrane of chronic lymphocytic leukemia cells: association with soluble MICA shedding. Cancer Immunol Immunother (2012) 61(8):1201–10. doi:10.1007/s00262-011-1195-z
73. Jinushi M, Vanneman M, Munshi NC, Tai YT, Prabhala RH, Ritz J, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci U S A (2008) 105(4):1285–90. doi:10.1073/pnas.0711293105
74. Zocchi MR, Catellani S, Canevali P, Tavella S, Garuti A, Villaggio B, et al. High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D ligands recognition in Hodgkin lymphomas. Blood (2012) 119(6):1479–89. doi:10.1182/blood-2011-07-370841
75. Waldhauer I, Steinle A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res (2006) 66(5):2520–6. doi:10.1158/0008-5472.CAN-05-2520
76. Cao W, Xi X, Hao Z, Li W, Kong Y, Cui L, et al. RAET1E2, a soluble isoform of the UL16-binding protein RAET1E produced by tumor cells, inhibits NKG2D-mediated NK cytotoxicity. J Biol Chem (2007) 282(26):18922–8. doi:10.1074/jbc.M702504200
77. Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol (2008) 180:7249–58.
78. Fernández-Messina L, Ashiru O, Boutet P, Agüera-González S, Skepper JN, Reyburn HT, et al. Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J Biol Chem (2010) 285(12):8543–51. doi:10.1074/jbc.M109.045906
Keywords: NKG2D, MICA, MICB, ULBP, NK cell, T cells, signaling pathways, regulation
Citation: Huergo-Zapico L, Acebes-Huerta A, López-Soto A, Villa-Álvarez M, Gonzalez-Rodriguez AP and Gonzalez S (2014) Molecular bases for the regulation of NKG2D ligands in cancer. Front. Immunol. 5:106. doi: 10.3389/fimmu.2014.00106
Received: 20 December 2013; Paper pending published: 10 February 2014;
Accepted: 03 March 2014; Published online: 21 March 2014.
Edited by:
Massimo Vitale, IRCCS A.O.U. S.Martino-IST, ItalyReviewed by:
Angela Santoni, Sapienza University of Rome, ItalyJacques Zimmer, Centre de Recherche Public de la Santé, Luxembourg
Copyright: © 2014 Huergo-Zapico, Acebes-Huerta, López-Soto, Villa-Álvarez, Gonzalez-Rodriguez and Gonzalez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Segundo Gonzalez, Facultad de Medicina, University of Oviedo, IUOPA, 4a Planta, Despacho D02, Julián Clavería sn, Oviedo 33006, Spain e-mail:c2VndW5kb2dAdW5pb3ZpLmVz