 Simone Lipinski
Simone Lipinski Philip Rosenstiel
Philip Rosenstiel- Institute of Clinical Molecular Biology, Christian-Albrechts-University of Kiel, Kiel, Germany
The host’s ability to discriminate friend and foe and to establish a precise homeostasis with its associated microbiota is crucial for its survival and fitness. Among the mediators of intestinal host-microbe interactions, NOD-like receptor (NLR) proteins take center stage. They are present in the epithelial lining and innate immune cells that constantly monitor microbial activities at the intestinal barrier. Dysfunctional NLRs predispose to intestinal inflammation as well as sensitization to extra-intestinal immune-mediated diseases and are linked to the alteration of microbial communities. Here, we review advances in our understanding of their reciprocal relationship in the regulation of intestinal homeostasis and implications for intestinal health.
Introduction
The intestinal epithelium is the largest barrier organ of the human body and the colon harbors the majority of the individual’s microbiota (1). It is estimated that more than 1000 different bacterial species colonize the human gut, outnumbering eukaryotic cells at least by an order of magnitude (2). As many of the bacteria represent facultative pathogens (pathobionts), the integrity of the intestinal barrier must be highly secured. This is accomplished by physical and immunological mechanisms formed by cellular (i.e., epithelial- and mesoderm-derived immune cells) and non-cellular components (e.g., antimicrobial peptides, cytokines, and mucus). On the other hand, an extensive crosstalk between host and microbiota contributes to the normal development and maturation of the intestinal epithelium and immune system (3, 4). The recognition of this complex bacterial community is mediated by phylogenetically ancient innate immune receptors, e.g., Toll-like receptors (TLRs) and NOD-like receptors (NLRs). NLR proteins have co-evolved with intestinal microbial communities and are expressed by intestinal epithelial and immune cells. They are characterized by a central nucleotide-binding and oligomerization domain (NOD or NACHT) and C-terminal leucine-rich repeats (LRRs) (5). Upon activation, NLRs initiate assembly of the inflammasome or signaling cascades [e.g., NF-κB, reactive oxygen species (ROS)] leading to a transient pro-inflammatory environment and, ultimately, aim at resolution of inflammation. Dysfunctional NLR signaling is linked to intestinal inflammation and in fact, polymorphisms in NLR genes are associated with complex chronic inflammatory barrier diseases, such as inflammatory bowel disease (IBD) (6). The two major forms of IBD, Crohn’s disease (CD) and ulcerative colitis (UC) are chronic relapsing-remittent or progressive inflammatory conditions that affect the gastrointestinal tract.
It has become clear that NLRs play a crucial role for the maintenance of structural and functional composition of the intestinal microbiota. Several lines of evidence have been presented that link dysfunctional NLR signaling to an impaired host-microbiota homeostasis that may predispose to subsequent altered inflammatory responses in animal models. Here, we summarize multiple levels of host-microbe crosstalk in the intestine and review the recent findings and consequences of NLRs in physiological and pathological intestinal host-microbe interactions.
The Role of NLRs in the Multiple Levels of Intestinal Host-Microbe Crosstalk – The NOD2 Example
The importance of NOD2 for intestinal homeostasis is emphasized by the finding that genetic variants in NOD2 contribute to dysregulated intestinal inflammatory responses and to manifestation of CD in humans. The three most common single nucleotide polymorphisms (SNPs) are located within the LRR of NOD2 causing either a frameshift mutation (L1007fsinsC), which leads to a truncated LRR or amino acid changes (R702W and G908R) (7–9). Cells that express these variants fail to activate NF-κB upon stimulation with the NOD2 ligand muramyl-dipeptide (MDP) (10, 11). In mouse models of intestinal inflammation, NOD2 has been assigned a protective role, since lack of NOD2 conferred increased susceptibility to DSS and TNBS-induced colitis (12). It must be emphasized that the effects are modest and under regular animal housing conditions no spontaneous inflammatory phenotype has been observed. Although it is still unclear how exactly a loss of NOD2 function predisposes to CD, several mechanisms related to altered host-microbe interactions and consequently increased susceptibility to intestinal inflammation, are currently discussed.
Tolerance, Polarity, and Control of Protective Cellular Programs in IECs
An imprinting function of NOD2 on microbial composition and/or active antibacterial responses against pathogens may be explained by its ability to modulate cellular programs in IECs (summarized in Figure 1). Furthermore, it was demonstrated that the LRR domain of NOD2 already confers antibacterial properties per se. The purified NOD2 LRR domain directly interacted with bacteria leading to bacterial killing, whereas the LRR domains bearing the CD-associated mutation L1007fsinsC lacked antibacterial activity (13). Moreover, NOD2 exhibits additional antibacterial effects by interacting with various proteins, which have been implicated in bacterial clearance. Of these, ATG16L1, a protein involved in antibacterial autophagy (“xenophagy”), was shown to interact with NOD2 and to cooperatively mediate pathogen defense in intestinal epithelial cells (14–16). This is of interest since variants in ATG16L1 are associated with CD (17) and combination of disease-associated alleles of ATG16L1 and NOD2 are assumed to synergistically increase susceptibility for CD (18, 19). Moreover, NOD2 was shown to interact with both components and catalytic proteins of ROS-producing enzymes. ROS production is an integral part of the innate host defense system, and inflammatory responses at mucosal surfaces include moderate (activation of signaling cascades) to excessive (bacterial killing due to oxidative burst) formation of ROS. Intestinal epithelial cells express members of the ROS-generating NADPH-oxidase complex (20) and MDP induces ROS formation (21, 22). NOD2 was shown to interact with the structural NADPH-oxidase component Rac1 (23, 24) and with the DUOX family member DUOX2 (22). Another important facet in the regulation of NOD2 signaling is the specific localization within the intestinal epithelial cell. Despite its intracellular localization, NOD2 can shuttle to the basolateral plasma membrane upon activation (25–27). Moreover, NOD2-mediated cytokine release and defensin production are specifically induced from a membrane complex including Erbin and FRMPD2 from the basolateral side (28).
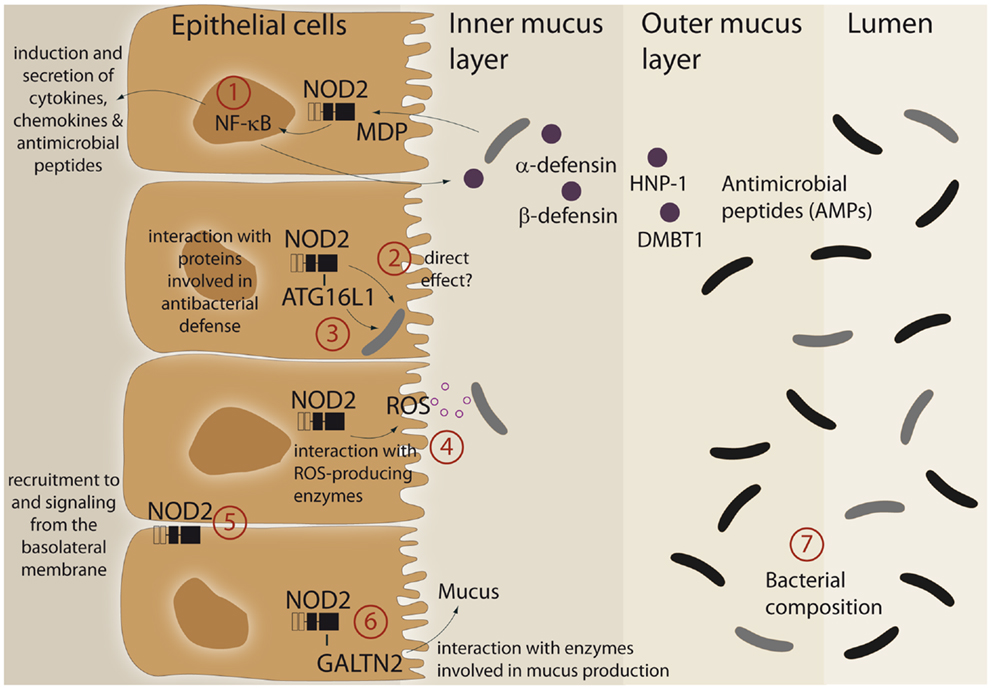
Figure 1. Schematic representation of different functional aspects of the NLR family member NOD2 at the epithelial barrier. (1) Recognition of MDP leads to a complex protective gene expression program including the induction of antimicrobial peptides and cytokines. (2, 3) NOD2 has been shown to interact with the autophagic pathway and may direct xenophagy in a direct manner. A direct antibacterial effect of NOD2 itself has been postulated. (4) There is interaction between NOD2 activation and ROS-generating enzyme complexes that may have an effect on intestinal bacteria. (5) A complex machinery regulates the presence of NOD2 at the plasma membrane (e.g., Erbin and FRMPD2) which may modulate the ability to recognize and act against invasive bacteria. (6) A recent study has shown that NOD2 is involved in mucus generation via modulation of GALNT2. For further details see main text. (7) All aforementioned factors may causally contribute to the reported differences in microbiome composition. For further details see text.
A link between NOD2 and intestinal mucus production has been established with the discovery that NOD2 interacts with GALNT2 (polypeptide N-acetylgalactosaminyltransferase 2), a regulator of mucin biosynthesis. A defect in GALNT2 function due to impaired NOD2 interaction might therefore alter mucin production and hence contribute to CD susceptibility (29).
NOD2 signaling leads to activation of NF-κB and subsequent induction of diverse antimicrobial peptides and proteins like HNP-1 (30), β-defensin-2 (28, 31), and DMBT1 (32), a Scavenger Receptor Cysteine-Rich (SRCR) domain-containing protein, which interacts with and agglutinates several Gram-negative and Gram-positive bacteria [reviewed in Ref. (33)]. Since patients with ileal CD exhibit reduced levels of Paneth cell derived α-defensins HD-5 and -6 (34, 35) and NOD2 is constitutively expressed by Paneth cells (36), several studies investigated an underlying causal role for NOD2. However, contradictory results exist. Whereas patients carrying NOD2 polymorphism had greater reduction of α-defensins (37, 38), no genotype-dependent correlation was found in another study (39). Similarly, conflicting data exists from transgenic mouse models. Nod2-deficient mice displayed reduced mRNA expression of α-defensins compared to wild-type mice (40). These results were challenged by the recent finding that NOD2 knockout mice that were co-housed with their wild-type littermate had equivalent α-defensin profiles and identical antimicrobial activity against commensal and pathogenic bacterial strains (41). Moreover, NOD2-deficient mice were not impaired in Paneth cell numbers compared to wild-type animals (42). Thus, further work needs to clarify the role of NOD2 in regulating mouse α-defensin status (42).
Effects on Microbial Composition
It has been shown that NOD2 is involved in recognition and defense against various intestinal pathogens, including Helicobacter pylori (43), Helicobacter hepaticus (44), Citrobacter (45), Salmonella typhimurium (46), E. coli (47), and Listeria monocytogenes (40, 48). In CD-affected humans, the link between NOD2 status and intestinal dysbiosis has been confirmed in disease patients homozygous for the NOD2 L1007fsinsC mutation. Tissue-attached microbiota from ileal biopsies exhibited higher loads of Bacteroidetes, Firmicutes, and Bacteroides compared to healthy controls. In fecal samples, a similar pattern was observed, however differences were not statistically significant (49). Another study that incorporated CD patients mutated in one of the three major risk alleles (R702W, G908R, and L1007fsinsC) confirmed that genotype and disease phenotype are associated with shifts in their intestinal microbial compositions (50). Nevertheless, NOD2-deficient mice do not develop spontaneous colitis when kept under specific pathogen free (SPF) conditions. With the advent of next-generation sequencing, it has become possible to take an in-depth snapshot of the intestinal bacterial ecosystem and to delineate microbial community structures and composition at the species level. However, considerable differences between published studies exist concerning animal housing and breeding (e.g., hygiene status of animal facility, genetic background, caging effects, use of F2 littermates, or separated WT/knockout strains) study design (age, sex, intestinal sampling location) and sequencing methods (DNA extraction, sequencing, and data analysis). Despite these differences, several independent groups reported that NOD2 status is associated with alterations in the intestinal microbial composition and density (summarized in Figure 2) (47, 49, 51, 52). Increased abundance of members of the phylum Bacteroidetes was detected in weaning mice and persisted throughout development (49). In line with this, RIPK2-deficient mice displayed increased levels of Bacteroides and Firmicutes arguing for a RIK2-dependency (47). Greater fecal abundances within the Alistipes and Bacteroides but an underrepresentation of Prevotellacea along with a decreased diversity and richness in the microbiota was found in NOD2−/− compared to WT mice (51). Recently, another aspect of the complex host genotype-microbe interaction was highlighted. Wild-type mice that received disease-predisposing bacterial communities from NOD2 or RIPK2-deficient mice via co-housing or cross-fostering experiments suffered from increased susceptibility to DSS-induced colitis and colitis-associated carcinogenesis. Reciprocal microbiota transplantation from wild-type donors reduced disease risk in NOD2-deficient mice (53). However, two recent studies reported only minimal differences in gut microbial composition of co-housed, littermate controlled NOD2-deficient, and wild-type mice (41, 54). The latter one showed that shifts in bacterial communities were independent of genotype and correlated with housing conditions (54). In light of the findings from recent co-housing experiments with NOD2 and other NLRs [e.g., Ref. (53, 55)] this might be partly explained by the restoration of disturbed microbiota due to animal co-housing, however, more studies are needed to fully understand the interference of NOD2 with host-microbe interactions.
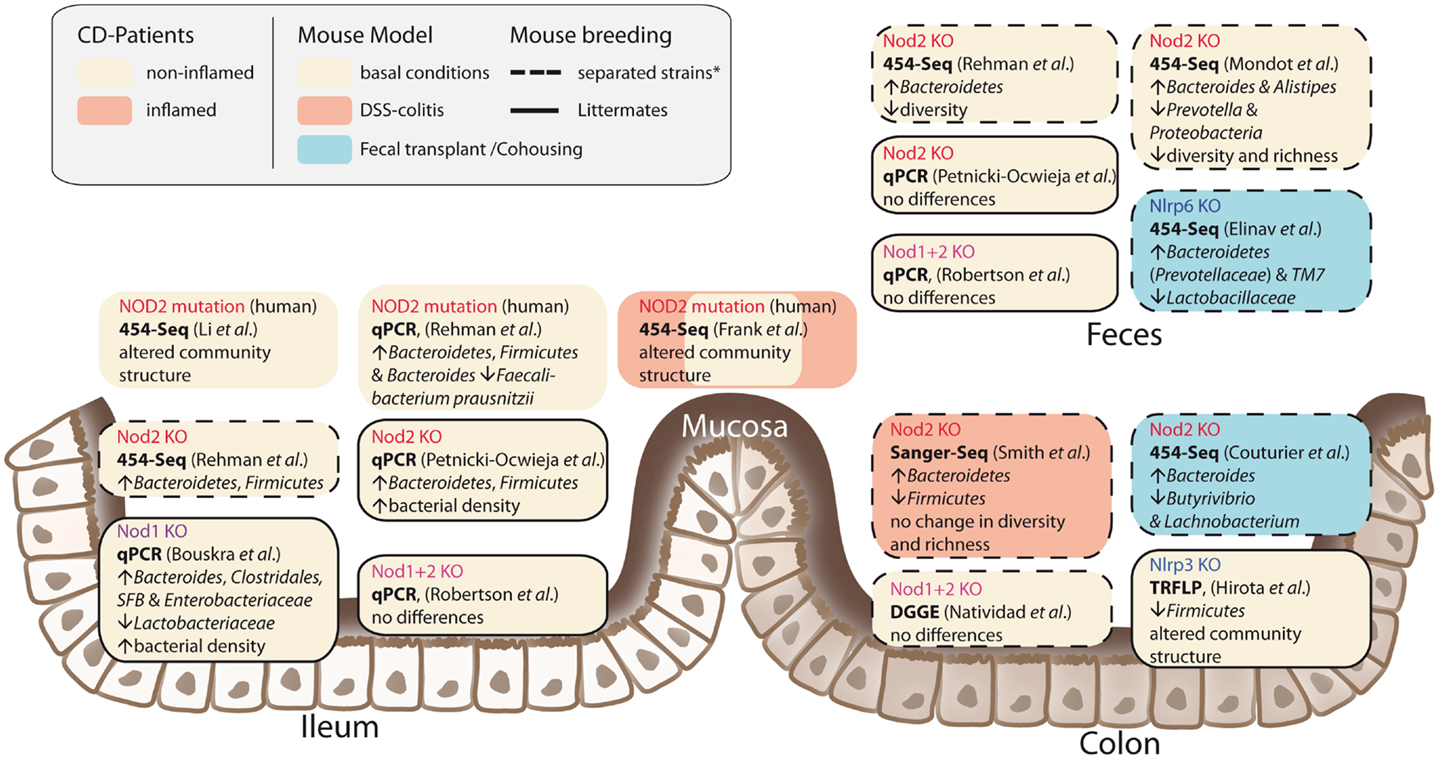
Figure 2. Influences of NLRs on intestinal microbial community structures. The figure summarizes recent studies in humans and mice and depicts different approaches and animal breeding schemes. The asterisk denotes the fact that behind the term separated breeding a variety of strategies is conjoined. For further discussion see main text.
NOD all NODs are Created Equal – Lessons from NOD1
NOD1 and 2 share similar structural composition, detection of peptidoglycan moieties (iE-DAP/NOD1, MDP/NOD2), and downstream signaling pathways, including RIPK2 and NF-κB activation. In contrast to NOD2, the association between genetic variants in the NOD1 gene and susceptibility to IBD is less evident. While some studies identified NOD1 as a risk factor for IBD in some studies (56, 57) this has not been widely replicated (58–60) including a recent meta-analysis (61).
Nevertheless, there is evidence that NOD1-mediated innate immune responses are critically involved in maintaining intestinal homeostasis. Depletion of intestinal microbiota was associated with impaired neutrophil function, which was reversed by administration of NOD1 ligand in the drinking water of mice (62). Moreover, NOD1-deficiency leads to increased susceptibility to H. pylori infection (63), impaired clearance of Clostridium difficile in the intestine, increased bacterial translocation (64), and enhanced colitis-associated colon tumor formation (65). The NOD1-mediated recognition of peptidoglycan was necessary to induce genesis of isolated lymphoid follicles (ILFs) in the intestine, which in turn influenced the composition of the intestinal bacterial community. In NOD1-deficient mice, the total bacterial population was expanded 100-fold, which was largely due to the groups of Clostridiales, Bacteroides, and Enterobacteriaceae (66). Furthermore, lack of NOD1 led to deficiencies in intestinal barrier integrity reflected by lower expression levels of NOD2, Muc2, α- and β-defensin, and keratinocyte-derived chemokine (KC) as compared to their F2 littermates (54). In line with this, the combined knockout of NOD1 and 2 led to increased paracellular permeability, decreased levels of E-cadherin, and lower colonic antimicrobial RegIII-γ expression in comparison to littermate control mice (67). Nevertheless, both studies did not find genotype-specific differences in the relative abundance of intestinal bacteria (54, 67).Thus, as previously pointed out for NOD2, the impact of breeding strategies and housing conditions may strongly interfere with study results and yet it is still too early to draw final conclusions about the role of NOD1 in physiological and pathological host-microbe interactions in the intestine.
IL-1β – the Role of Inflammasome-Type NLRs in the Intestine
Several NLRs form multimeric complexes termed “inflammasomes” that serve as molecular platforms for caspase-1 activation and processing of pro-IL-1-like cytokines into their active forms (68). Until now, this group comprises the NLRPs (NLRs with PYRIN domain) NLRP3, NLRP6, NLRP1, NLRP12, NLRP7, and NLRC4 (69, 70). Although no variants in inflammasome forming NLR genes are among the 163 IBD susceptibility loci (71), their relevance for intestinal health has been shown by various inflammasome-deficient mice in models of intestinal inflammation, as reviewed in Ref. (72). However, in comparison to mice deficient for Nlrp3, Nlrp10, Nlrp12, and Nlrc4, Nlrp6 showed the largest potential to alter microbiota and colitis susceptibility of co-housed mice. In the following paragraph we will therefore focus on the role of Nlrp6 in intestinal host-microbe interactions.
Components of the Nlrp6 inflammasome are expressed in intestinal epithelial cells (73) and throughout the intestinal tract (55), and several studies have demonstrated a protective role of Nlrp6 against colitis and colitis-associated tumor formation (55, 73–75) [reviewed in Ref. (76)]. Importantly, Nlrp6-deficiency was demonstrated to significantly alter intestinal microbiota composition (55). On the genus level, Prevotellaceae (belonging to the Bacteroidetes phylum) were strongly increased, whereas Lactobacilli (Firmicutes phylum) were decreased. In addition, members of the phylum of TM7, which were highly abundant in Nlrp6-deficient mice, have been found to be overrepresented in CD patients (77). Likewise, Prevotellaceae were more prominent in the mucosa tissues of patients with UC compared to healthy individuals (78). The distinct bacterial composition of Nlrp6-knockout mice was transmissible to co-housed adult mice and cross-fostered litters and resulted in colitis-prone phenotype of recipient wild-type mice. Similarly, mice deficient in the inflammasome adaptor Asc harbored a colitogenic gut microbiota that was transmissible to co-housed WT mice (79). Wild-type mice exhibited increased colonic Il-6 levels compared to single-housed wild-type mice when they were co-housed with either Nlrp6- or Asc-deficient mice. Of note, the microbiota-mediated transmissible cell proliferation and tumor formation were abrogated when either a neutralizing anti-IL-6 receptor antibody was administered or intestinal IL-6 receptor was conditionally deleted in intestinal epithelial cells. Recently, the role of Nlrp6 for colonic health was extended to the small intestine (80). In a mouse model for small-bowel inflammation, stress-mediated release of corticotropin-releasing hormone (CRH) inhibited intestinal Nlrp6 (but not Nlrp3) expression and altered the composition of the intestinal microbiota, which ultimately led to intestinal inflammation (80). Together, these studies point to a critical role for the NLR-forming inflammasomes, in particular Nlrp6, in modulating intestinal homeostasis via an influence on microbiota composition. It must be emphasized that anti-IL-1 treatment, despite having an effect in DSS colitis, lacks efficacy in IBD. With the above knowledge in mind, this field is now at a point where the translation into the human situation is desperately needed.
Conclusion
We are beginning to realize that maintaining the long-term stability of the co-evolved human gut microbe communities is an important mechanism for maintaining human health. The ecology of intestinal microbiota is not only necessary for digestion of nutrients and the delivery of local metabolites (e.g., butyrate) to intestinal epithelial cells but also critically shapes immune responses of the host. An important fact for future studies will be to delineate how this interaction licenses migratory immune cells for functions in other organ systems such as the brain. It must be emphasized that most of our knowledge of the role of NLRs for this symbiotic relationship originates from animal studies in rodents and that there are also conflicting results in terms of the extent of the influence of single NLRs in this context. Beyond the biology of NLRs, these results teach us two things: (i) we have to reassess how we set up our immunological animal models in the future. From findings that susceptibility to provocation models may be transmissible by genetically determined microbiota to wild-type animals, it is clear that a regular F2 intercross with littermate housing may not be an ideal scenario. On the other hand, drift in microbiota in separated lines over generations may exert bigger effects than the actual genotype. This is a dilemma we have to solve. (ii) We have to be careful how we interpret the findings. In the end only the transfer into the human situation will help us to understand the factual influence of microbiota on traits in health and disease.
Taken together, NLRs represent watchful guardians at the intestinal barrier, which help to maintain immunological homeostasis in an organ system facing strong environmental influences. This environment has changed drastically over the past 100 years and some NLR family members are clearly involved in chronic inflammatory diseases associated with this lifestyle. Decoding the exact molecular signals of NLRs that contribute to the resilience of microbial communities on mucosal surfaces may provide approaches to prevent or ameliorate this range of human diseases. More than 10 years into NLR research we are still far away from understanding how these molecules actually exert their function and how we can target them in therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors apologize to the authors of important contributions, which have been left out unintended and due to space constraints. Philip Rosenstiel is supported by the DFG EXC306 and the grant Ro 2994 3-1.
References
1. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature (2010) 464:59–65. doi:10.1038/nature08821
2. Holzapfel WH, Haberer P, Snel J, Schillinger U, Huis in’t Veld JH. Overview of gut flora and probiotics. Int J Food Microbiol (1998) 41:85–101. doi:10.1016/S0168-1605(98)00044-0
3. Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol (2009) 9:313–23. doi:10.1038/nri2515
4. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature (2012) 489:231–41. doi:10.1038/nature11551
5. Rosenstiel P, Philipp EE, Schreiber S, Bosch TC. Evolution and function of innate immune receptors – insights from marine invertebrates. J Innate Immun (2009) 1:291–300. doi:10.1159/000211193
6. Schreiber S, Rosenstiel P, Albrecht M, Hampe J, Krawczak M. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat Rev Genet (2005) 6:376–88. doi:10.1038/nrg1607
7. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411:603–6. doi:10.1038/35079114
8. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
9. Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet (2001) 357:1925–8. doi:10.1016/S0140-6736(00)05063-7
10. Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology (2003) 124:140–6. doi:10.1053/gast.2003.50019
11. Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci U S A (2003) 100:3455–60. doi:10.1073/pnas.0530276100
12. Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest (2008) 118:545–59. doi:10.1172/JCI33145
13. Perez LH, Butler M, Creasey T, Dzink-Fox J, Gounarides J, Petit S, et al. Direct bacterial killing in vitro by recombinant Nod2 is compromised by Crohn’s disease-associated mutations. PLoS One (2010) 5:e10915. doi:10.1371/journal.pone.0010915
14. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16:90–7. doi:10.1038/nm.2069
15. Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim YG, Magalhães JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11:55–62. doi:10.1038/ni.1823
16. Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, et al. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut (2011) 60:1229–35. doi:10.1136/gut.2010.228908
17. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39:207–11. doi:10.1038/ng1954
18. Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent, anti-bacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology (2010) 13:13. doi:10.1053/j.gastro.2010.07.006
19. Billmann-Born S, Lipinski S, Bock J, Till A, Rosenstiel P, Schreiber S. The complex interplay of NOD-like receptors and the autophagy machinery in the pathophysiology of Crohn disease. Eur J Cell Biol (2011) 90:593–602. doi:10.1016/j.ejcb.2010.10.015
20. Bokoch GM, Knaus UG. NADPH oxidases: not just for leukocytes anymore! Trends Biochem Sci (2003) 28:502–8. doi:10.1016/S0968-0004(03)00194-4
21. Silverman DH, Wu H, Karnovsky ML. Muramyl peptides and serotonin interact at specific binding sites on macrophages and enhance superoxide release. Biochem Biophys Res Commun (1985) 131:1160–7. doi:10.1016/0006-291X(85)90212-8
22. Lipinski S, Till A, Sina C, Arlt A, Grasberger H, Schreiber S, et al. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J Cell Sci (2009) 122:3522–30. doi:10.1242/jcs.050690
23. Legrand-Poels S, Kustermans G, Bex F, Kremmer E, Kufer TA, Piette J. Modulation of Nod2-dependent NF-kappaB signaling by the actin cytoskeleton. J Cell Sci (2007) 120:1299–310. doi:10.1242/jcs.03424
24. Eitel J, Krull M, Hocke AC, N’Guessan PD, Zahlten J, Schmeck B, et al. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J Immunol (2008) 181:2664–71.
25. Kufer TA, Kremmer E, Adam AC, Philpott DJ, Sansonetti PJ. The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol (2008) 10:477–86. doi:10.1111/j.1462-5822.2007.01062.x
26. McDonald C, Chen FF, Ollendorff V, Ogura Y, Marchetto S, Lecine P, et al. A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. J Biol Chem (2005) 280:40301–9. doi:10.1074/jbc.M508538200
27. Barnich N, Aguirre JE, Reinecker H-C, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J Cell Biol (2005) 170:21–6. doi:10.1083/jcb.200502153
28. Lipinski S, Grabe N, Jacobs G, Billmann-Born S, Till A, Hasler R, et al. RNAi screening identifies mediators of NOD2 signaling: implications for spatial specificity of MDP recognition. Proc Natl Acad Sci U S A (2012) 109:21426–31. doi:10.1073/pnas.1209673109
29. Nimmo ER, Stevens C, Phillips AM, Smith A, Drummond HE, Noble CL, et al. TLE1 modifies the effects of NOD2 in the pathogenesis of Crohn’s disease. Gastroenterology (2011) 141(972–981):e971–2. doi:10.1053/j.gastro.2011.05.043
30. Yamamoto-Furusho JK, Barnich N, Hisamatsu T, Podolsky DK. MDP-NOD2 stimulation induces HNP-1 secretion, which contributes to NOD2 antibacterial function. Inflamm Bowel Dis (2010) 16:736–42. doi:10.1002/ibd.21144
31. Voss E, Wehkamp J, Wehkamp K, Stange EF, Schröder JM, Harder J. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J Biol Chem (2006) 281:2005–11. doi:10.1074/jbc.M511044200
32. Rosenstiel P, Sina C, End C, Renner M, Lyer S, Hellmig S, et al. DMBT1 is a downstream target of NOD2/CARD15 and negatively regulates TLR4-mediated NF-kB-activation. J Immunol (2006) 178:8203–11.
33. Madsen J, Mollenhauer J, Holmskov U. Review: Gp-340/DMBT1 in mucosal innate immunity. Innate Immun (2010) 16:160–7. doi:10.1177/1753425910368447
34. Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A (2005) 102:18129–34. doi:10.1073/pnas.0505256102
35. Perminow G, Beisner J, Koslowski M, Lyckander LG, Stange E, Vatn MH, et al. Defective Paneth cell-mediated host defense in pediatric ileal Crohn’s disease. Am J Gastroenterol (2010) 105:452–9. doi:10.1038/ajg.2009.643
36. Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut (2003) 52:1591–7. doi:10.1136/gut.52.11.1591
37. Wehkamp J, Harder J, Weichenthal M, Schwab M, Schäffeler E, Schlee M, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut (2004) 53:1658–64. doi:10.1136/gut.2003.032805
38. Bevins CL, Stange EF, Wehkamp J. Decreased Paneth cell defensin expression in ileal Crohn’s disease is independent of inflammation, but linked to the NOD2 1007fs genotype. Gut (2009) 58:882–3; discussion 883–4.
39. Simms LA, Doecke JD, Walsh MD, Huang N, Fowler EV, Radford-Smith GL. Reduced alpha-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut (2008) 57:903–10. doi:10.1136/gut.2007.142588
40. Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (2005) 307:731–4. doi:10.1126/science.1104911
41. Shanahan MT, Carroll IM, Grossniklaus E, White A, von Furstenberg RJ, Barner R, et al. Mouse Paneth cell antimicrobial function is independent of Nod2. Gut (2013). doi:10.1136/gutjnl-2012-304190
42. Van Limbergen J, Geddes K, Henderson P, Russell RK, Drummond HE, Satsangi J, et al. Paneth cell marker CD24 in NOD2 knockout organoids and in inflammatory bowel disease (IBD). Gut (2013). doi:10.1136/gutjnl-2013-305077
43. Rosenstiel P, Hellmig S, Hampe J, Ott S, Till A, Fischbach W, et al. Influence of polymorphisms in the NOD1/CARD4 and NOD2/CARD15 genes on the clinical outcome of Helicobacter pylori infection. Cell Microbiol (2006) 8:1188–98. doi:10.1111/j.1462-5822.2006.00701.x
44. Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A (2010) 107:14739–44. doi:10.1073/pnas.1003363107
45. Kim JG, Lee SJ, Kagnoff MF. Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by toll-like receptors. Infect Immun (2004) 72:1487–95. doi:10.1128/IAI.72.3.1487-1495.2004
46. Geddes K, Rubino S, Streutker C, Cho JH, Magalhaes JG, Le Bourhis L, et al. Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun (2010) 78:5107–15. doi:10.1128/IAI.00759-10
47. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A (2009) 106:15813–8. doi:10.1073/pnas.0907722106
48. Till A, Rosenstiel P, Brautigam K, Sina C, Jacobs G, Oberg HH, et al. A role for membrane-bound CD147 in NOD2-mediated recognition of bacterial cytoinvasion. J Cell Sci (2008) 121:487–95. doi:10.1242/jcs.016980
49. Rehman A, Sina C, Gavrilova O, Häsler R, Ott S, Baines J, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut (2010) 60:1345–62. doi:10.1136/gut.2010.216259
50. Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis (2011) 17:179–84. doi:10.1002/ibd.21339
51. Mondot S, Barreau F, Al Nabhani Z, Dussaillant M, Le Roux K, Dore J, et al. Altered gut microbiota composition in immune-impaired Nod2(-/-) mice. Gut (2012) 61:634–5. doi:10.1136/gutjnl-2011-300478
52. Smith P, Siddharth J, Pearson R, Holway N, Shaxted M, Butler M, et al. Host genetics and environmental factors regulate ecological succession of the mouse colon tissue-associated microbiota. PLoS One (2012) 7:e30273. doi:10.1371/journal.pone.0030273
53. Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest (2013) 123:700–11. doi:10.1172/JCI62236
54. Robertson SJ, Zhou JY, Geddes K, Rubino SJ, Cho JH, Girardin SE, et al. Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes (2013) 4:222–31. doi:10.4161/gmic.24373
55. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
56. McGovern DP, Hysi P, Ahmad T, van Heel DA, Moffatt MF, Carey A, et al. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet (2005) 14:1245–50. doi:10.1093/hmg/ddi135
57. Huebner C, Ferguson LR, Han DY, Philpott M, Barclay ML, Gearry RB, et al. Nucleotide-binding oligomerization domain containing 1 (NOD1) haplotypes and single nucleotide polymorphisms modify susceptibility to inflammatory bowel diseases in a New Zealand Caucasian population: a case-control study. BMC Res Notes (2009) 2:52. doi:10.1186/1756-0500-2-52
58. Zouali H, Lesage S, Merlin F, Cezard JP, Colombel JF, Belaiche J, et al. CARD4/NOD1 is not involved in inflammatory bowel disease. Gut (2003) 52:71–4. doi:10.1136/gut.52.1.71
59. Franke A, Ruether A, Wedemeyer N, Karlsen TH, Nebel A, Schreiber S. No association between the functional CARD4 insertion/deletion polymorphism and inflammatory bowel diseases in the German population. Gut (2006) 55:1679–80. doi:10.1136/gut.2006.104646
60. Van Limbergen J, Nimmo ER, Russell RK, Drummond HE, Smith L, Anderson NH, et al. Investigation of NOD1/CARD4 variation in inflammatory bowel disease using a haplotype-tagging strategy. Hum Mol Genet (2007) 16:2175–86. doi:10.1093/hmg/ddm169
61. Lu WG, Zou YF, Feng XL, Yuan FL, Gu YL, Li X, et al. Association of NOD1 (CARD4) insertion/deletion polymorphism with susceptibility to IBD: a meta-analysis. World J Gastroenterol (2010) 16:4348–56. doi:10.3748/wjg.v16.i34.4348
62. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med (2010) 16:228–31. doi:10.1038/nm.2087
63. Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol (2004) 5:1166–74. doi:10.1038/ni1131
64. Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G, et al. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol (2011) 186:4872–80. doi:10.4049/jimmunol.1003761
65. Chen GY, Shaw MH, Redondo G, Nunez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res (2008) 68:10060–7. doi:10.1158/0008-5472.CAN-08-2061
66. Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature (2008) 456:507–10. doi:10.1038/nature07450
67. Natividad JM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1-/-; Nod2-/- mice. Inflamm Bowel Dis (2012) 18:1434–46. doi:10.1002/ibd.22848
68. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol (2009) 27:229–65. doi:10.1146/annurev.immunol.021908.132715
69. Franchi L, Núñez G. Immunology. Orchestrating inflammasomes. Science (2012) 337:1299–300. doi:10.1126/science.1229010
70. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity (2012) 36:464–76. doi:10.1016/j.immuni.2012.02.001
71. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi:10.1038/nature11582
72. Rubino SJ, Selvanantham T, Girardin SE, Philpott DJ. Nod-like receptors in the control of intestinal inflammation. Curr Opin Immunol (2012) 24:398–404. doi:10.1016/j.coi.2012.04.010
73. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A (2011) 108:9601–6. doi:10.1073/pnas.1100981108
74. Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol (2011) 186:7187–94. doi:10.4049/jimmunol.1100412
75. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature (2012) 491:259–63. doi:10.1038/nature11535
76. Annand PK, Kanneganti TD. NLRP6 in infection and inflammation. Microbes Infect (2013) 15:661–8. doi:10.1016/j.micinf.2013.06.009
77. Kuehbacher T, Rehman A, Lepage P, Hellmig S, Fölsch UR, Schreiber S, et al. Intestinal TM7 bacterial phylogenies in active inflammatory bowel disease. J Med Microbiol (2008) 57:1569–76. doi:10.1099/jmm.0.47719-0
78. Lucke K, Miehlke S, Jacobs E, Schuppler M. Prevalence of bacteroides and Prevotella spp. in ulcerative colitis. J Med Microbiol (2006) 55:617–24.
79. Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci U S A (2013) 110:9862–7. doi:10.1073/pnas.1307575110
Keywords: NLR, Crohn disease, intestinal mucosa, mucosal immunity, gut microbiota, inflammation, symbiosis
Citation: Lipinski S and Rosenstiel P (2013) Debug your bugs – how NLRs shape intestinal host-microbe interactions. Front. Immunol. 4:479. doi: 10.3389/fimmu.2013.00479
Received: 23 August 2013; Paper pending published: 20 September 2013;
Accepted: 09 December 2013; Published online: 27 December 2013.
Edited by:
Thomas A. Kufer, University of Cologne, GermanyReviewed by:
Martin Rottenberg, Karolinska Institutet, SwedenGuntram A. Grassl, Christian-Albrechts-University of Kiel, Kiel, Germany
Copyright: © 2013 Lipinski and Rosenstiel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip Rosenstiel, Institute of Clinical Molecular Biology, Christian-Albrechts-University of Kiel, Schittenhelmstr. 12, Kiel D-24105, Germany e-mail:cC5yb3NlbnN0aWVsQG11Y29zYS5kZQ==