 Antoine Marçais1,2,3,4,5
Antoine Marçais1,2,3,4,5 Sébastien Viel1,2,3,4,5,6
Sébastien Viel1,2,3,4,5,6 Morgan Grau1,2,3,4,5
Morgan Grau1,2,3,4,5 Thomas Henry1,2,3,4,5
Thomas Henry1,2,3,4,5 Jacqueline Marvel1,2,3,4,5
Jacqueline Marvel1,2,3,4,5 Thierry Walzer1,2,3,4,5*
Thierry Walzer1,2,3,4,5*- 1CIRI, International Center for Infectiology Research, Université de Lyon, Lyon, France
- 2U1111, INSERM, Lyon, France
- 3Ecole Normale Supérieure de Lyon, Lyon, France
- 4Centre International de Recherche en Infectiologie, Université Lyon 1, Lyon, France
- 5UMR5308, CNRS, Lyon, France
- 6Laboratoire d’Immunologie, Hospices Civils de Lyon, Centre Hospitalier Lyon Sud, Lyon, France
Natural Killer (NK) cells are innate lymphocytes with an important role in the early defense against intracellular pathogens and against tumors. Like other immune cells, almost every aspects of their biology are regulated by cytokines. Interleukin (IL)-15 is pivotal for their development, homeostasis, and activation. Moreover, numerous other activating or inhibitory cytokines such as IL-2, IL-4, IL-7, IL-10, IL-12, IL-18, IL-21, Transforming growth factor-β (TGFβ) and type I interferons regulate their activation and their effector functions at different stages of the immune response. In this review we summarize the current understanding on the effect of these different cytokines on NK cell development, homeostasis, and functions during steady-state or upon infection by different pathogens. We try to delineate the cellular sources of these cytokines, the intracellular pathways they trigger and the transcription factors they regulate. We describe the known synergies or antagonisms between different cytokines and highlight outstanding questions in this field of investigation. Finally, we discuss how a better knowledge of cytokine action on NK cells could help improve strategies to manipulate NK cells in different clinical situations.
Natural killer (NK) cells are Innate Lymphoid Cells (ILC) involved in the immuno-surveillance of cancers and in the early control of infections by intracellular pathogens (1). They can kill cells recognized as targets through a battery of surface receptors (2) and produce large amounts of IFN-γ upon activation (1). Recently, the growing ILC family has been reclassified into three groups according to the pattern of cytokine they secrete. In this classification, NK cells are part of the group 1 ILC subset (3). In mice, NK cells mainly develop in the bone marrow (BM) (4, 5). If the earliest committed NK cell progenitor (pre-pro NK) does not express CD122 (6) which is the β subunit of the IL-2/IL-15 receptor, expression of this molecule is acquired soon after at the NK precursor (NKP) stage (7). The expression of this receptor is thereafter conserved and is a hallmark of the NK cell population. This underlines the fact that the various aspects of NK cell development, homeostasis, and function are conditioned by IL-15. If this cytokine is fundamental, a variety of other cytokines have been shown to influence the behavior of NK cells, alone or in synergy. In this review, we aim to describe the complex interplay between the molecular pathways triggered by these cytokines. We restricted our field of investigation to the direct effects of cytokines on NK cells. We believe that a good understanding of these pathways is essential to the rational design of drugs targeting the various aspects of NK cell functions.
The γc Family of Cytokines
The central role of cytokines sharing the γc subunit as a co-receptor (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21) in the constitution of the NK cell pool was appreciated some 20 years ago. Indeed, X-linked severe combined immunodeficiency patients presenting mutations leading to loss of γc function (8) and mice with targeted γc deletion (9) both presented a quasi absence of NK cells indicating that these cells relied on one or more of these cytokines. Absence of mature NK cells in mice genetically deficient in IL-2/15Rβ chain restricted the list of candidates to IL-2 and IL-15 (10). Finally, genetic ablation of IL-15 (11) or IL-15Rα (12), leading to a very similar phenotype with a near complete absence of mature NK cells, formally demonstrated the paramount importance of this cytokine for the generation of NK cells. In contrast, mice deficient for IL-2, IL-4, and IL-7 had normal NK cell numbers under homeostatic conditions (13). Importantly and despite expression of IL-2/15Rβ, NKPs appeared to be independent of γc signaling since they were present in normal numbers in γc deficient animals, suggesting that IL-15 only becomes important for subsequent maturation steps (13). Other γc cytokines are also involved in NK cell homeostasis and activation as summarized in Figure 1.
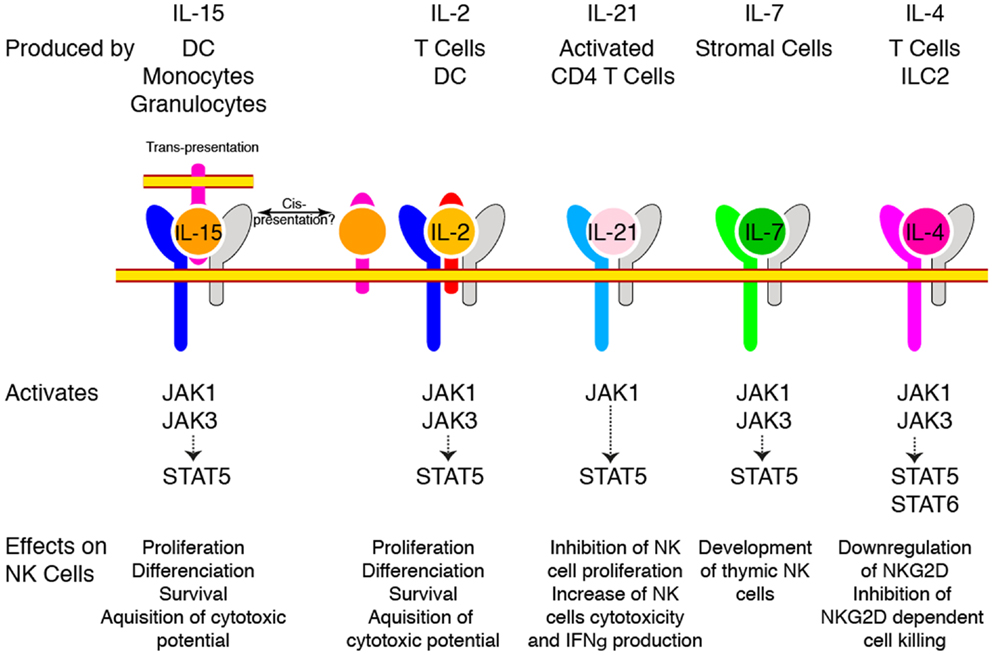
Figure 1. γc Cytokines regulate NK cell homeostasis and function. Different cytokines use γc as a co-receptor to regulate NK cell biology. The cellular source of these cytokines is indicated on the top. The different receptor chains and the major signaling pathways they trigger are shown. The effect of each cytokine on NK cells is also mentioned at the bottom.
IL-15
Discovery/Receptors/Trans-Presentation
IL-15 was discovered as a result of its “IL-2-like” stimulatory activity since it was able to support proliferation of IL-2 dependent cell lines (14, 15). Structurally, IL-15 is a 14–15 kDa protein presenting sparse sequence similarity (19%) but extensive 3-dimensional analogy with IL-2 and belonging to the same four α-helix bundle cytokine family (15). IL-15 and IL-2 both interact with receptor complexes containing the common gamma chain (γc) (16) and IL-2/15Rβ chain (15, 17). The γc subunit also takes part in the formation of the receptors for IL-4, IL-7, IL-9, and IL-21 (18) whereas IL-2/15Rβ is only used for signaling by IL-2 and IL-15. IL-2 and IL-15 receptors only differ by their α chain, IL-2Rα (CD25) being dedicated to IL-2 and IL-15Rα to IL-15 (19). In contrast to IL-2Rα, IL-15Rα alone displays a high affinity of binding for IL-15 (1.4 × 10−11M), equivalent to that of the heterotrimeric IL-2R for IL-2 (19). This last property is fundamental to understand the physiology of IL-15. Indeed, this high affinity, coupled to the fact that IL-15 and IL-15Rα are co-expressed by the same cells, allows intracellular binding of IL-15 to IL-15Rα in the endoplasmic reticulum. The complex is then shuttled to the cell membrane and presented to activate neighboring cells expressing IL-2/15Rβ/γc. This mechanism was called trans-presentation (20, 21) and proposed to explain the counter-intuitive fact that expression of IL-15Rα was not needed on responding NK cells to maintain their homeostasis, as would have been expected for a classical cytokine response scheme, but on neighboring cells (22–26). We can hypothesize that such a mechanism allows a very precise delivery of the cytokine stimulus perhaps coupled to other stimulating molecules. As NK cells rely on IL-15 for different purposes, trans-presentation would confine IL-15 signal to selective niches where it would sustain needs of specific NK cell sub-populations. Moreover, systemic availability of this cytokine could be detrimental as exemplified by the development of fatal leukemia in IL-15 transgenic animals (27, 28). The debate whether cis-presentation (i.e., autocrine presentation of IL-15) also occurs was recently revived by a paper showing that following bacterial challenge, NK cells produce and present IL-15 in a time frame and quantities matching DCs and that this cis-presentation was as important as DC trans-presentation to elicit NK cell IFN-γ production (29).
Role
The key role of IL-15 in NK cell biology is underlined by the complete absence of these cells in mice deficient in components of the IL-15 signaling axis (11, 12). However, the role of IL-15 in NK cell physiology is not limited to development. Indeed, this cytokine controls as well survival of mature NK cells in the periphery (24, 25, 30, 31), an effect that is probably mediated by up-regulation of anti-apoptotic Bcl2 family members and down regulation of apoptotic ones (31, 32). Moreover, resting NK cells are poor effectors and need to be primed beforehand to express their full effector capacity. This priming step is also controlled by IL-15, presented by dendritic cells, or monocytes (33–38). Mechanistically, IL-15 signals NK cells to constitute stocks of the effector proteins GzmB and Perforin, absent from unprimed NK cells (33). IL-15, synergizing with IL-12, is also mandatory for IFN-γ expression by NK cells (29, 34, 35). Finally, IL-15 controls NK cell homeostatic proliferation (25, 36, 39) as well as proliferation induced following bacterial, viral, or fungal infections (21, 35, 40, 41). How IL-15 can mediate such a wide range of effects, some homeostatic (differentiation, survival), and some context-dependent (priming, IFN-γ secretion) is still unresolved. One possibility would be that varying IL-15 concentration triggers different responses on NK cells. In line with this idea, decreasing γc expression levels results in reduction of the peripheral NK cell pool (42, 43). This suggests that maximal expression of this receptor and hence maximal signal transduction is necessary for optimal transduction of the IL-15 signal. A recent study tested this model in vivo using mouse strains deficient for IL-15Rα or bearing chimeric IL-15Rα either as transgene or knocked in the IL-15Rα locus (44). This strategy allowed the authors to study NK cell populations exposed to five different levels of IL-15 trans-presentation (from null to normal levels). This disclosed the fact that on one hand, constituting a normal peripheral NK cell pool, relying on high proliferation rate in the BM, requires a high level of IL-15 trans-presentation. On the other hand, maturation is much less demanding. The impact of these different levels of IL-15 on the different signaling pathways downstream of the IL-15R has not been analyzed.
Regulation
How is IL-15 regulated at the basal state remains largely unknown. IRF1, a transcription factor involved in type I IFN (IFN I)-induced IL-15 production, probably plays a role in this process. Indeed, expression of this factor is necessary on hematopoietic as well as non-hematopoietic cells for NK cell generation (45). IL-15 mRNA is expressed in vivo by a number of tissues and cell types, from hematopoietic (radiosensitive in chimera experiments) and non-hematopoietic origin (radio-resistant) (24, 46, 47). Chimera experiments have suggested that IL-15 trans-presentation by cells of the hematopoietic system is the most efficient since limiting IL-15Rα expression to the hematopoietic system is sufficient to generate normal NK cell numbers in the BM and only slightly decreased numbers in the periphery (26, 39). In line with its dual function in NK cell homeostasis and activation, IL-15 is expressed at low level under homeostatic conditions in monocytes/macrophages but this expression can be considerably enhanced by several pro-inflammatory agents like LPS (48), poly(I:C), or IFN I (49). More recently, using a transgenic mouse line in which emerald GFP (EmGFP) is expressed under the control of endogenous Il15 regulatory elements, Lefrançois and collaborators have tracked the cell subsets expressing IL-15 mRNA under homeostatic or inflammatory conditions (50, 51). They confirmed the expression of this cytokine mRNA by a broad distribution of myeloid cells including monocytes, neutrophils, eosinophils, mast cells, and dendritic cells, the strongest expression being observed in basophils. More surprisingly, they described high transcription of IL-15 by Hematopoietic Stem Cells (HSC) and its progressive down regulation during T cell differentiation (51). The significance of this last result awaits further confirmation and functional tests. In addition, IL-15 expression is regulated at several steps including the post-transcriptional level. How much of this regulation is conserved in this reporter remains to be tested. It is however worth noting that these results perfectly correlate with the transcriptomic data available at the Immgen Consortium website (www.immgen.org) for the cell types analyzed (52).
Signaling
In terms of signaling, most of our knowledge was generated by studies focused on the IL-2-IL-2 receptor interaction (Figure 2). Given the shared receptor and the similarity of effect of IL-2 and IL-15 on cultured cells, it was inferred that IL-15 stimulation would lead to activation of the same pathways. And indeed, most of the experiments conducted so far suggested a remarkable conservation. However, these two cytokines are not functionally redundant as exemplified by the divergent immunological outcomes of IL-2 or IL-15 treatment (53). A recent in vitro study aiming at understanding these differences evidenced subtle changes in the gene transcription induced in CD8 T cells stimulated with IL-2 or IL-15 (54). This observation opens up the possibility that some differences exist in the signaling pathways downstream of the IL-2 or IL-15 receptors. In this context, the exact contribution of the different signaling pathways during NK cell development and activation is poorly understood. Upon IL-2 binding to its receptor, signaling is triggered by Janus Kinases (Jak) 1 and 3, bound to IL-15Rβ and γc (55–58). These kinases phosphorylate tyrosine residues of IL-15Rβ, which serve as docking sites for phosphotyrosine binding proteins such as the Shc adapter protein, Insulin Receptor Substrate (IRS) proteins, and STAT5a and b transcription factors and lead to the activation of three main transduction pathways: the Jak-STAT pathway, the phosphoinositide 3-kinase (PI3K)/Akt pathway, and the Mitogen Activated Protein Kinase (MAPK) pathway. Given its very proximal role in signal transduction, deficiency in Jak3 results in the absence of NK cells (59). In an attempt to dissect the importance of the different pathways stemming from the IL-2/15Rβ subunit, complementation of Il2/15rβ−/− mice with IL-2/15Rβ transgenes deleted for different cytoplasmic domains was undertaken (60). This approach demonstrated the necessity of the membrane distal H-region (Figure 2) containing tyrosine 392 and 510 and known to recruit STAT3 and 5 and potentially the p85 subunit of PI3K for the generation of a normal NK cell pool (60, 61). In contrast, the truncated protein generated after deletion of the membrane proximal A-region, known to interact with Lck, the p85 subunit of PI3K and Shc, was able to perfectly complement the IL-2/15Rβ deficient mice and to restore NK cell homeostasis and functional response to IL-15 and seems thus dispensable. The importance of the STAT5 pathway was confirmed by several studies (62, 63). Indeed, NK cell number was reduced in STAT5b and to a lesser extend in STAT5a deficient animals and this was associated with a decreased response to IL-2 and IL-15 (63). NK cell specific deletion of both STAT5 factors lead to the complete disappearance of this population resulting from survival defects, probably associated with a block of differentiation at the NKP stage (62). In accordance with the putative role of the H-region of IL-2/15Rβ to recruit p85 via phosphorylated Y392 is a series of studies dissecting the role of the PI3K pathway in NK cell development and functions (64–66). Class IA PI3Ks comprise a p110 catalytic subunit associated with a p85, p55, or p50 regulatory subunit. The p110 catalytic subunits are encoded by three genes Pik3ca, Pik3cb, and Pik3cd. Class IB PI3Ks consist of the catalytic subunit p110γ (encoded by the Pik3cg) associated with the regulatory subunits p101 or p84. NK cells express all catalytic p110 (66), however only the role of p110γ and δ were examined using mice deficient for these proteins (65, 66) or bearing a catalytically inactive form of p110δ (64). Defect in p110γ or δ signaling lead to a decrease in peripheral NK cell number (64–66), combined defect in p110γ∕δ prevented terminal NK cell maturation (66). Moreover, defect in PI3K signaling lead to impaired proliferative (64–66) and cytotoxic (64, 66) responses to IL-2. Importantly, PI3K signaling can be triggered by diverse stimuli including cytokines other than IL-2 or IL-15 (67) chemokines (68) and NK activating receptors (69). The phenotype described in the PI3K deficient NK cells can thus be the consequence of the impairment of responses to other stimuli and not only IL-15. Moreover, direct activation of PI3K by IL-15 was not assessed in these studies neither in vitro nor in vivo leaving the question of a direct effect unanswered. The generation of phosphatidylinositol triphosphate by PI3K recruits a vast number of targets to the plasma membrane and leads to their activation. However, the downstream targets important for NK cell differentiation and activation have not been investigated. We recently discovered that the kinase mechanistic Target Of Rapamycin (mTOR), which can be activated downstream of PI3K, is a key signaling node activated by IL-15 and responsible for NK cell maturation and activation by pro-inflammatory signals (Marçais et al. manuscript in preparation). mTOR is an evolutionarily conserved serine/threonine kinase integrating various extracellular cues: metabolite and growth factors but also antigenic and inflammatory signals as recently described for T cells (70). mTOR takes part in two complexes: mTORC1 and mTORC2 differing by their constituting members and the targets they phosphorylate. We followed phosphorylation of key mTOR targets by flow cytometry and showed that mTOR activity is developmentally regulated with a progressive shut down upon differentiation and BM egress. In contrast, mTOR activity is strongly induced when NK cells are exposed to pro-inflammatory signals triggered by poly(I:C) injection. Of note, mTOR activation necessitated high IL-15 concentrations; instead, STAT5 phosphorylation was readily triggered by low doses of IL-15. This could provide a first molecular basis to explain the dual effect of IL-15 on NK cells. We also showed that IL-15 controls mTOR activity both in vitro and in vivo. This is confirmed by the observation that NK cells harvested from mice with NK cell specific mTOR deletion are arrested at the immature CD11blow CD27high stage and their activation in response to poly(I:C) or IL-15 is severely impaired. Interestingly, survival of mTOR deficient NK cells is not affected in accordance with previous studies suggesting that the pro-survival signals given by IL-15 are mediated by STAT5 (62). Preliminary results suggest that only a fraction of mTOR activity is controlled via PI3K. This would fit with the fact that the phenotype of mTOR deficient NK cells is much stronger than PI3K deficient cells. The identity of the relevant mTORC1 or mTORC2 downstream targets remains to be addressed.
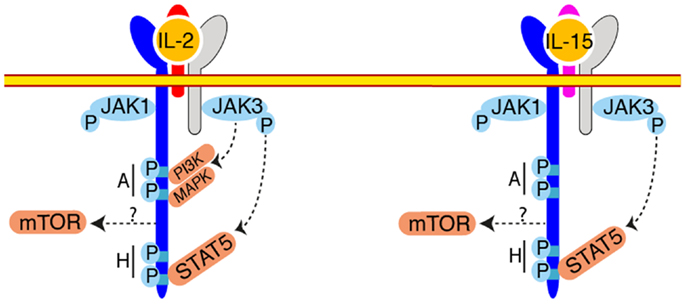
Figure 2. Signaling downstream IL-2/IL-15Rβ. Upon engagement of IL-2 or IL-15 different tyrosine residues are phosphorylated in the IL-2/15Rβ intracytoplasmic regions, across two main regions (“A” and “H”). These phosphorylations lead to the recruitment and downstream phosphorylation of STAT5 by JAK1 and JAK3. The mTOR pathway takes also part to IL-15 signaling but the mode of mTOR activation is currently unknown. Activation of mTOR is essential to increase NK cell metabolism. PI3K and MAPK are also activated upon IL-2 engagement and probably also upon IL-15 engagement but this requires formal demonstration.
Apart from STAT5, the transcription factors, downstream of IL-15 are not characterized. It is worth mentioning the fact that upon MCMV infection, NK cells activate the E2F pathway, a phenomenon that can be blocked using blocking IL-2/15Rβ antibodies (71). As IL-2 has been shown to mediate its proliferative effects through E2F activation (72), we can hypothesize that E2F is also involved in IL-15 induced proliferation. This has however not been formally tested. Moreover IL-15 stimulation leads to NF-κB p65-mediated increase in Myc expression in a context of IL-15 driven leukemia (28).
IL-2
As discussed above, a number of IL-15 effects are recapitulated by in vitro treatment with IL-2. In particular, it has long been known that activated cytotoxic NK cells from BM culture can be generated after exposure to IL-2 (73, 74). However, when IL-2 and IL-15 were compared, 10 to 50 times more IL-2 than IL-15 was needed to activate NK cells (37). This is due to the fact that NK cells do not express the high affinity IL-2 receptor due to their lack of IL-2Rα expression at steady-state. Instead, trans-presentation of IL-15 allows sensing of nanomolar quantities by cells expressing only IL-2/15Rβ and γc (54). Whether IL-2 in vitro effects are relevant in vivo is difficult to evaluate. Indeed, effect on NK cells of mutations affecting IL-2 signaling are difficult to interpret since they result in overt auto-immunity due to the role of IL-2 in the generation and maintenance of Treg cells. To avoid this caveat, mice deficient both for IL-2 and T cells have been generated. NK cell differentiation and numbers are normal in these Rag−/−Il2−/− double deficient mice. IL-2 is thus not needed for maintenance of NK cell homeostasis in the absence of T and B cells (13). This is concordant with the fact that, unlike IL-15, which is produced under homeostatic conditions, IL-2 production mainly results from stimulation of the immune system. This cytokine could nevertheless play a role during NK cell priming following inflammatory challenge. Two studies even suggested that IL-2 could play a non-redundant role in this process (75, 76). Indeed, Granucci et al. described a non-redundant role for IL-2 produced by DCs in the first hours following bacterial challenge (76). In this study, DC-derived IL-2 was important for the induction of IFN-γ secretion by NK cells while induction of cytotoxicity was independent of IL-2. A direct contact between the NK and the DC was needed, suggesting that other molecules were involved. This interaction was functionally relevant for bacterial clearance and anti-tumor response. One caveat of this study is however the use of IL-2 deficient DCs differentiated from BM harvested from IL-2 deficient hosts in which T cell dependent auto-immunity develops. Another study described that, following Leishmania major infection, the T cell-derived IL-2 was necessary for the induction of IFN-γ secretion by NK cells (75). These findings are challenged by the fact that NK cell priming does not happen in the absence of IL-15 (29, 34, 35). A possibility to reconcile these studies would be to imagine cooperation between the two cytokines, both being needed. In this context, cytotoxicity induction would be under IL-15 control specifically since IL-2 only impacts IFN-γ production in these models (75, 76). The fact that 2 cytokines signaling through the same IL-2Rβ/γc-STAT5-mTOR axis lead to such dissociated effects could be due to difference in the spatio-temporal availability as well as the mode of delivery of these cytokines to NK cells. In any case, this issue remains open for further investigations.
Interest for the IL-2 dependent control of NK cells has recently been renewed by a series of studies proposing that NK cell activity was kept in check by Treg cells buffering excess IL-2 produced by activated T cells (77, 78). In these studies, the authors show that systemic Treg cell depletion leads to an increase in CD4 T cell derived IL-2. This increased IL-2 availability was correlated to increased NK cell IFN-γ production (78) and cytotoxicity toward missing-self targets (77) and could be abrogated by blocking IL-2 antibody treatment. Similar findings have been reported upon transfer of in vitro pre-activated NK cells in an irradiated host (79). Mechanistically, Gasteiger et al. linked this increased responsiveness to a better capacity to generate conjugates with target cells after even very short-term exposure to IL-2. Interestingly, this increased cytotoxic capacity was only evidenced against missing-self targets, IL-2 being unable to increase cytotoxicity toward cells expressing both inhibitory and activating ligands. The same group also described phenotypical changes of the NK cell population with the progressive emergence of a CD127+ immature NK cell population following Treg depletion (80). A similar population also accumulated in tumor-bearing or chronically infected animals. This population was able to up-regulate IL-2Rα upon low dose IL-12 stimulation confirming previous findings (81). The authors interpreted this result as an increase in the capacity to sense and use IL-2. However, expression of IL-2Rα also renders cells more sensitive to IL-15 (54), the cytokine involved in the homeostasis of this population is thus debatable.
At this stage, we can conclude that DCs and monocytes probably have a prominent role in NK cell activation, through IL-15 trans-presentation. However, this does not exclude the fact that other closely related cytokines like IL-2 and other cell types like T cells or NK cells themselves can contribute and in some conditions replace the IL-15 priming.
IL-21
IL-21R was discovered independently by two groups, it is homolog to IL-2Rβ and its ligand, IL-21, homolog to IL-2, IL-4, and IL-15 with the strongest similarity with the latest (82, 83). Upon IL-21 binding, IL-21R pairs with γc and signals through JAK1 and STAT5 (82). IL-21 is expressed by activated CD4 T cells while IL-21R is found on lymphoid cells (83, 84). Its first effects described on human NK cells were a potentiation of differentiation from BM progenitors and activation of mature NK cells (83). These effects were in accordance with further studies showing that IL-21 inhibits IL-15 effects on NK cell proliferation but potentiates IL-15 driven NK cell terminal differentiation, i.e., cytotoxicity and IFN-γ secretion (84–87). Forced expression of IL-21 in vivo by hydrodynamic plasmid delivery decreases, in an NK cell dependent manner, the number of lung metastasis obtained after tumor cell lines injection (85). Given that IL-21 boosts T cell proliferation in vitro, it was suggested that its production by activated T cells could help shutdown the NK cell response once adaptive immunity was functional (84). However no in vivo data came to confirm this hypothesis. IL-21 produced by CD4 T cells is essential to prevent CD8 T cell exhaustion during chronic viral infections (88–90). In addition to direct positive effects on antiviral T cells, IL-21 restricts virus-driven Treg cell expansion and their suppressive effect on CD8 T cells (91). Whether IL-21 also controls the function of NK cells during chronic infections remains to be formally tested even though ex vivo treatment of NK cells from HIV-infected patients with IL-21 improves their effector function (92, 93).
IL-7
IL-7, another member of the γc family of cytokines signaling via STAT5, is well known for its role during early steps of B and T cell development in the BM and thymus respectively (94). The fact that early pre-pro NK cells and immature NK cells express high levels of IL-7Rα (6) is puzzling since NK cell development and acquisition of effector functions is perfectly normal in the absence of IL-7 (13, 95). It should be noted that IL-7Rα is also used in combination with the Cytokine Receptor-like factor 2 to form the thymic stromal lymphopoietin receptor (TSLPR). Whether this cytokine plays a role in early stages of NK cell physiology is unknown. It has however been shown to regulate CD8 T cell viability (96). A peculiar thymic IL-7-dependent NK cell subset has also been described (97). This subset is present in minute amount in mice thymi (between 10,000 and 100,000 cells), is virtually absent in Il7−/− animals, expresses IL-7Rα and depends on GATA-3 in contrast to BM derived NK cells. Functionally, these NK cells are poorly cytolytic but secrete higher amount of cytokines than conventional NK cells. Given the very low abundance of this population, their function has not been investigated.
IL-4
IL-4 is also a member of the γc family of cytokines, well known for its pro-Th2 effects during T cell differentiation. Its absence does not affect NK cell generation and homeostasis (13). However, NK cells express the IL-4 receptor as evidenced by their sensitivity to IL-4 treatment in vitro (86). Of note is the strong ability of IL-4 to repress some key NK effector functions, such as cytokine production or cytotoxicity. Indeed, it has been demonstrated that IL-4 suppresses the inflammatory cytokine (IFN-γ, TNFα, and GM-CSF) production-increase that is induced following IL-12 treatment in human NK cells (98). Similarly, in mouse NK cells, IL-4 treatment induces a decrease in the cytokine-induced-cytolytic-activity toward tumor cells or immature DC. The mode of action of IL-4 did not involve a down regulation of perforin or granzyme-B by NK cells but could be mediated through NKG2D down regulation (86, 98). In line with these observations, the capacity of NK cells to shape the adaptive immune response and favor a polarized Th1 response through interactions with DCs is abrogated when NK cells are pretreated with IL-4 (99). IL-4 treated NK cells are unable to induce DC maturation and favor tolerogenic or Th2 responses (99).
As mentioned above, another measurable effect of IL-4 was to down regulate NKG2D and other NK cell markers expression in vitro and in vivo and as a result to decrease NKG2D dependent cell killing (86). Similarly IL-4 treatment has been shown to down regulate NKG2D and CCL5 expression by memory CD8 T cells (100–102). This is in contrast to the promotion, by IL-4, of innate memory-like CD8 T cells generation that has been recently described (103). In the mouse strain Balb/c, high frequency of IL-4-secreting PLZF+ NKT cells is associated with increased proportion of memory phenotype CD8 T cells compared to C57Bl/6 mice (104). The generation of this memory population has been shown to be dependent on IL-4, as revealed by the lack of memory phenotype CD8 T cells in Cd1d−/− and Il4r−/− mice. IL-4 produced during the course of a Th2 response is thought to act in a similar way, as it induces a strong proliferation of memory phenotype as well as naive CD8 T cells (105). The stimulation of memory CD8 T cells by IL-4 induces a strong up-regulation of the Eomes transcription factor (104). Given the implication of Eomes in NK cell differentiation, it is tempting to speculate that IL-4 could have far reaching effects on NK cell biology.
IL-12 Family
The IL-12 family of cytokines is constituted by heterodimeric cytokines presenting a four α-helix bundle structure and belonging to the IL-6 super-family (106). The constituting heterodimers of this family are formed by combination of two possible β-chains and three possible α-chains. Indeed, the cytokine β-chains are components of two cytokines (p40 of IL-12 and IL-23 and Ebi3 of IL-27 and IL-35) while α-chains include p35, components of IL-12 and IL-35, p19 component of IL-23, and p28 component of IL-27. On the receptor side, IL-12 is recognized by an IL-12Rβ1/β2 receptor, IL-23 by an IL-12Rβ1/IL-23R heterodimer, IL-27 by an IL-27R/gp130 complex, and IL-35 by a gp130/IL-12Rβ2. They have activating as well as inhibitory roles on the immune system, IL-12 and IL-23 being seen as more pro-inflammatory while IL-27 and IL-35 have been more described as inhibitory. Moreover, given the receptors and ligands promiscuity, some members can compete with others generating a complexity far from being understood.
IL-12
Discovery/Receptors
IL-12 was purified at the end of the 80s from the supernatant of EBV immortalized B cell lines and named NK cell stimulating factor (NKSF) for its ability to induce IFN-γ, cytotoxic activity, and proliferation of NK cells in vitro (107). NKSF was later renamed IL-12 and is constituted of two polypeptides: IL-12p40 and p35 covalently linked by disulfide bonds, and binding to a heterodimeric receptor composed of IL-12Rβ1 and β2. Importantly, NK cell constitutively express both chains of the IL-12R (108).
Regulation/Production
IL-12 is produced by several types of Antigen Presenting Cells, including DCs (109, 110) and activated macrophages (48, 111). In vivo, IL-12 is produced early after viral infection (112). The inducing signals include pathogen derivative (113), sensed by TLRs and the RIG I pathway, but also molecules expressed by activated T cells like CD40L (109), or the NK cell derived IFN-γ (114–116). The exact mechanism leading to IL-12 production can be more complex as is the case following CpG stimulation. Indeed, upon CpG injection in vivo, IL-12 is induced by IL-15 after a cross-talk between conventional DCs and plasmacytoid DCs (117). Interestingly, IL-12 delivery seems to involve the formation of a synapse between the NK cell and the DC involving the polarization of the DC’s secretory apparatus toward the NK cell (118).
Signaling
Upon binding, IL-12Rβ2 becomes tyrosine phosphorylated and provides docking sites for the kinases tyk2 and Jak2 leading to the phosphorylation and activation of STAT4 (119–121). The importance of tyk2 and STAT4 in NK cells is manifested by the sharp decrease of IFN-γ production in mice deficient for these molecules (40, 121). One report also suggests that some of IL-12 effects are mediated by PKCθ (122), however no further study has confirmed this point.
Role
The major role of IL-12 in vivo is to induce IFN-γ production, while a marginal effect was described at first on early proliferation and development of cytotoxicity (40, 112, 123). Indeed, IFN-γ production is reduced 20-fold in IL-12p35 deficient mice while cytotoxicity is intact (40), antibody mediated blocking of the cytokine leads to similar results (112). Moreover, microscopy studies have shown a perfect time- and location-dependent correlation between IL-12 production by DCs and IFN-γ production by NK cells in Listeria induced granuloma (124). As IFN-γ can itself promote IL-12p40 expression, it generates a positive feedback loop promoting inflammation and the differentiation of monocytes into DCs (114, 124). More recently, it has been shown that IL-12 in combination with another cytokine, IL-18, helps optimal NK cell expansion (125, 126) an effect which is not dependent on IFN-γ secretion. Moreover, in some settings, IL-12/IL-18 can drive an IL-15 independent response of NK cells (127, 128). However these results are controversial, indeed, another study found no or only a very minor role of IL-18 and IL-12 for the promotion of the expansion of Ly49H+ NK cells during MCMV infection in vivo (129). Moreover, the settings used in one study are very peculiar since they involve adoptive transfer of WT NK cells in Il15−/− × Il15rα−/−mice to unmask the IL-12 dependent proliferative effects (128).
At the molecular level, induction of IFN-γ by IL-12 secretion relies on increased transcription as determined by run-on experiments (130). This transcriptional response is abrogated in tyk2 deficient mice (121). IFN-γ mRNA, like many other cytokine messengers, bears AU rich elements in its 3′UTR, which renders it unstable (131) moreover, it is also a target for miRNA mediated regulation (132). Hence, a large part of its expression relies on post-transcriptional regulation. In this context, it has been shown that IL-18 stabilizes the IFN-γ mRNA through the activation of a MAPK p38 dependent pathway (133) (Figure 3). This effect explains at least in part the formidable synergy existing between IL-12 and IL-18 on induction of IFN-γ secretion (134). Interestingly, IL-12 in combination with IL-18 can also trigger IFN-γ secretion by memory T cells in vivo in an antigen independent fashion underlying acquisition of “innate-like” capacities by these cells (38, 135). Conversely, several groups have been able to induce long-term survival of an NK cell population by transferring NK cells briefly activated in vitro in the presence of IL-12 and IL-18 in sublethally irradiated (79) or Rag−/− hosts (136). This echoes the concept of NK cell memory proposed in 2006 by the group of von Andrian (137). Indeed, under particular circumstances, a fraction of the activated NK pool survives the resolution of the response and is able to mount recall responses (126). This property is severely impaired in the absence of IL-12 (126). How exposure to these cytokines imprints long-term survival onto NK cells in these particular settings is not understood and could constitute fertile ground for further discoveries.
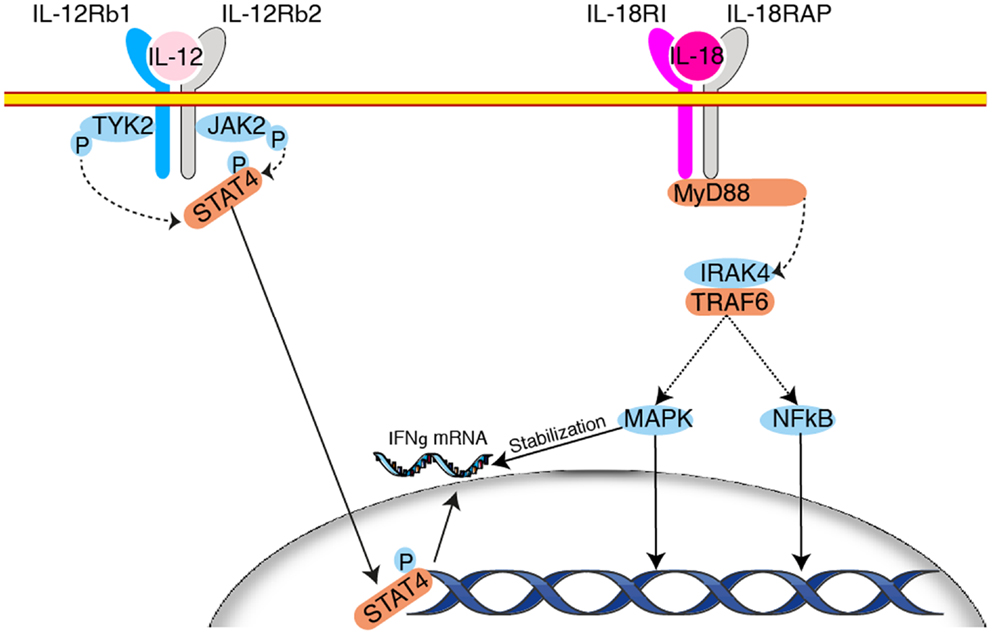
Figure 3. Synergy between IL-12 and IL-18 for the induction of IFN-γ production. Engagement of the heterodimeric IL-12 receptor leads to STAT4 phosphorylation after recruitment of the kinases Tyk2 and JAK2. STAT4 transctivates IFN-γ transcription. Upon binding of IL-18 to its receptors there is activation of the MAPK pathway downstream the adapter MyD88. This leads to the stabilization of IFN-γ mRNA and enhances IFN-γ secretion by NK cells. Other mechanisms may also contribute to the synergy between IL-12 and IL-18.
As previously mentioned, IL-12, in synergy with IL-18, has the capacity to induce IL-2Rα on NK cells (81, 86), the CD127+ population of NK cells being extremely sensitive to this stimulation (80). This property may help to explain the sustained IL-2-dependent proliferation of IL-15/IL-12/IL-18 pre-activated NK cells after in vivo transfer (79). IL-18 is not the only cytokine synergizing with IL-12. Indeed, early studies described a synergy between IL-12 and IL-2 (107). Similarly to IL-18, the synergy between IL-12 and IL-2 also involves regulation of IFN-γ mRNA half-life (130).
IL-18
IL-18 is a cytokine that was originally identified as an IFN-γ inducible factor (IGIF) (138) and it appears to share its biologic functions with IL-12, including enhancement of the NK cell activity (139). Its absence leads to decreased NK cell response in a variety of models (140, 141). It is part of the IL-1 family which comprises 11 members (142). It is produced as an inactive pro-IL-18 precursor. In contrast to other IL-1 family member such as IL-1β, proIL-18 is constitutively expressed (143). ProIL-18 requires cleavage by active caspase-1 in the inflammasome complex to generate biologically active IL-18 (144, 145). Other proteases [caspase-8 (146, 147), proteinase-3 (148), granzyme-B (149)] have been reported to cleave proIL-18 and generate bioactive cytokine. The physiologic producers of IL-18 include myeloid cells such as activated macrophages (138, 150), dendritic cells (151), neutrophils (152), and Ly6C+CCR2+ inflammatory monocytes (38). In addition, IL-18 is expressed in numerous non-hematopoietic lineages (153). IL-18 mature form is a leaderless protein secreted via a poorly understood mechanism (154). The ability of IL-18 released by macrophages or DCs to activate the production of IFN-γ by NK cells is dependent on cell-to-cell contact (155–159). In agreement with this cell-to-cell contact requirement, IL-18 secretion by DC is polarized and occurs at the immunological synapse formed between the DC and the NK cell (160). Furthermore, another regulatory step may include the presence of a membrane-bound form of IL-18 as an intermediate between the cytosolic pro IL-18 and the mature soluble IL-18 (161). Finally, IL-18 signaling can be antagonized by IL-18BP, which is limiting the systemic effects of this cytokine (162).
Upon IL-18 binding, its primary receptor, IL-18R1, dimerizes with a second receptor subunit: IL-18R accessory protein (IL-18RAP). This recruits myeloid differentiation primary response protein 88 (MyD88) and initiates signaling through IL-1R-associated kinase 4 (IRAK4) and TNFR-associated factor 6 (TRAF6), leading to activation of the NF-κB and MAPK pathways. The importance of IL-18 for NK cells is underlined by the fact that at steady state, NK cells are the only hematopoietic cells analyzed by the Immunological Genome Project to contain consequent levels of transcripts for IL-18R1 and IL-18RAP (www.immgen.org). This expression grants them with exquisite sensitivity to IL-18 stimulation (150). Moreover, NK from IL-18R1-deficient mice have decreased IFN-γ secretion and cytotoxic capacities (163). A similar phenotype is observed in IRAK4 deficient NK cells (164). Other pathways may also be activated as suggested by impaired IFN-γ secretion in response to IL-12/18 stimulation in p110γ or δ deficient NK cells (65, 66). IL-18 is critical for IFN-γ production by NK cells during numerous bacterial (165), fungal (166), parasites (127), and viral (167) infections. In addition to regulating IFN-γ production, IL-18 takes part to the priming of NK cells (29, 168), and leads to the acquisition of novel migratory function through up-regulation of CCR7 (155, 169). It has also been shown that IL-18 induces the release of CC chemokine Ligand 3 (CCL3) by NK cells, which in turns recruits inflammatory monocytes in the intestine and contribute to local inflammation (170).
The role of IL-18 in regulating the anti-tumoral activity of NK cells is unclear and might be highly dependent of the other signals received by NK cells concomitantly to IL-18. Indeed IL-18 has been shown to promote tumor immuno-suppression and tumor growth by converting Kit− NK cells into Kit+ NK cells. NK cells from this Kit+ subset have the potential to lyse DCs leading to a reduction in the tumor immuno-surveillance (171). In contrast, others studies have demonstrated an anti-tumor effect of IL-18 (172, 173) in part through the generation of “helper” NK cells producing CCL3 and CCL4, which results to the local recruitment of DCs and effector CD8 T cells (174).
As detailed above, the main effect of IL-18 on NK cells is to synergize with IL-12 to induce IFN-γ production. However, in some systems, IL-12 can be dispensable while IL-18 is not (29, 175). Importantly, if systemic IFN-γ production depends on IL-12/IL-18 synergy, local IFN-γ production in the liver can be preserved in the absence of IL-18 and be sufficient to allow host survival upon MCMV infection (167).
Transforming Growth Factor-β
Production
Transforming growth factor-β is a cytokine with a pivotal role in the regulation of the immune system. TGFβ1 is the predominant TGFβ isoform expressed in the immune system. TGFβ associates non-covalently with the latency-associated protein (LAP), forming a complex called the small latent complex (SLC). The SLC can be secreted as such or in association with latent TGFβ-binding protein (LTBP) as a large latent complex (LLC). TGFβ must be released from the complexes to bind to TGFβ receptors (176). This can be achieved through different mechanisms that remain mostly unclear. TGFβ binding proteins control ligand access but can also act as ligand reservoirs (177). Virtually all cells of the immune system can produce TGFβ. TGFβ is regulated at transcriptional, post-transcriptional, and post-translational levels. It is therefore difficult to precisely determine the sources of active TGFβ during immune responses.
Signaling
Transforming growth factor-β mediates its biological functions through binding to type I and II transmembrane serine/threonine kinase receptors. TGFβ1 signals mostly via TGFBR1 (type I receptor) and TGFBR2 (type II receptor). TGFBR1 is not necessary for binding to TGFβ but initiates signaling. Two signaling pathways have been described that are dependent or not on smad transcription factors. Receptor-associated smads (mostly R-smad 2 and 3 in the immune system) are sequestered in the cytoplasm in the absence of signaling. Upon phosphorylation by TGFBR1, R-smad 2 and 3 interact with the common mediator smad-4 and are translocated into the nucleus. Smad complexes recruit other transcription factors to activate or repress the expression of a wide range of genes. Various Smad-independent TGFβ signaling pathways operate in a context-dependent manner and contribute to cell-specific biological responses. TGFβ may thus activate small GTPases, MAP kinases, and the PI3K pathway (177). In T cells, a JNK-c-Jun pathway has been shown to suppress the expression of Eomes in Th17 cells in response to TGFβ (178). In NK cells, smad-independent TGFβ signaling pathways have not been addressed.
Impact of TGFβ on NK Cells
The addition of recombinant TGFβ in cultures of mouse spleen cells or human PBMC with IL-2 in vitro has long been shown to reduce NK cell proliferation and cytotoxicity (179–182). Administration of TGFβ also depresses NK cell activity in mice (183), and reduces their proliferation during antiviral responses (184) while blocking TGFβ increases NK cell cytotoxic activity (185). It was later found that TGFβ also counteracts IL-12 mediated cytokine production by mouse NK cells (186, 187) and human NK cells (188, 189). TGFβ not only counteracts the effects of IL-2 and IL-12 but also reduces IFN-γ production in response to the engagement of the Fc receptor CD16 on human NK cells (190). Finally, TGFβ also shapes the NK cell surface by reducing the expression of NK cell receptors NKG2D and NKp30 (191, 192) and changes their trafficking properties by modulating the expression of chemokine receptors (193).
When does TGFβ act on NK cells in vivo? Early studies show that TGFβ is produced during viral infections, especially by T cells at late stages of infection, which could help limiting NK cell cytotoxicity (194). More recent studies show that the transgenic expression of a dominant negative form of the TGFBR2 receptor in CD11c positive cells (including dendritic cells and NK cells) dramatically increases the number of mature NK cells (195), suggesting that TGFβ negatively regulates NK cell development and maturation at steady-state, especially during infancy (196). The smad-dependent pathway has been shown to be important to limit NK cell IFN-γ production by repressing the expression of T-bet (189). Whether this pathway also limits NK cell proliferation and cytotoxicity induced by pro-inflammatory cytokines remains to be determined. Early studies have suggested that TGFβ acts very rapidly, perhaps in a smad3 independent manner to decrease tyrosine phosphorylation induced by IL-2 (197).
IL-10
IL-10 was described as a Th2 cytokine that inhibited Th1 cytokine synthesis (198). It is now known to be produced by macrophages, DCs, B cells, various subsets of T cells, and NK cells themselves (199–202). NK cells constitutively express both chains of IL-10 receptor (Immgen data). Several diverging effects of IL-10 on NK cells have been described (203–207). Most of these effects seem to be indirect, indeed, IL-10 in vitro treatment of purified NK cells does not have noticeable effects (86). However, to the best of our knowledge, this has not been thoroughly tested using chimeras or transfer of IL-10R deficient or sufficient NK cells in IL-10 sufficient hosts. The experiments of in vivo blockade using antibodies being non-informative about the responding cell type (208, 209).
IFN I
IFN-α/β or type I IFN (IFN I) were originally identified as proteins responsible for induction of cellular resistance to viral infections. They are produced by various immune and non-immune cell types. According to a recent study, the capacity of mononuclear phagocytes (i.e., DCs and macrophages) to express IFN I and subsequently IL-15 after microbial challenge could be imprinted by previous contact with the microbial flora (210). However this results still awaits confirmation. IFN I effects on NK cells have been known for a long time. Indeed, IFN I induce NK cell proliferation and cytotoxicity (211). However, IFN I receptor (IFNAR) deficiency can be compensated by recombinant IL-15 injection (40). The bulk of IFN I effects are thus probably mediated through release of IL-15 as recently confirmed by a systems biology analysis of the response to MCMV viral infection (71). In this study, the authors show that IFN-stimulated genes were not strongly up regulated by NK cells, suggesting that, at least at the transcriptional level, these cells were not the primary targets of IFN I. This lower responsiveness was associated with a lower expression of STAT1 by NK cells. IFN I dependent effects on NK cells are however not absent and some studies have evidenced a direct role of IFN I on the induction of NK cell cytotoxicity (212, 213).
Concluding Remarks
Natural killer cell development and function depend on a multiplicity of cytokines which have complementary as well as overlapping functions. A complete understanding of their action will require the precise identification of the cell types producing them, the time window during which they are produced and the signaling events that their receptors engage in NK cells. These various parameters and the outcome on NK cells could be very different depending on the infectious agent. The role of some cytokines such as IL-2 may thus be important only with particular pathogens and efforts should therefore be made to diversify the models of infection. Moreover, many cytokine effects are only seen when combining them, as best exemplified by IL-12 and IL-18. It is therefore essential to define the relevant cytokine combinations in different niches and to delineate the signaling pathways they induce as well as their combined effects on NK cells. How NK cells integrate signals from activating and inhibitory cytokines and which molecules act as “integrators” are important issues to address. Recent technological advances such as mass cytometry will be instrumental for this purpose in that they allow the simultaneous measurement of up to 100 parameters using very low cell numbers. The latter technique can be applied to the study of cell signaling using phospho-specific antibodies raised against various molecules of the transduction machinery. Recently generated NK-specific Cre-expressing mouse lines will also be important to discriminate between direct vs. indirect effects of various cytokines on in vivo NK cell physiology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9:503–10. doi: 10.1038/ni1582
2. Diefenbach A, Raulet DH. Natural killer cells: stress out, turn on, tune in. Curr Biol (1999) 9:R851–3. doi:10.1016/S0960-9822(00)80044-5
3. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13:145–9. doi:10.1038/nri3365
4. Haller O, Wigzell H. Suppression of natural killer cell activity with radioactive strontium: effector cells are marrow dependent. J Immunol (1977) 1950(118):1503–6.
5. Seaman WE, Gindhart TD, Greenspan JS, Blackman MA, Talal N. Natural killer cells, bone, and the bone marrow: studies in estrogen-treated mice and in congenitally osteopetrotic (mi/mi) mice. J Immunol (1979) 1950(122):2541–7.
6. Carotta S, Pang SHM, Nutt SL, Belz GT. Identification of the earliest NK-cell precursor in the mouse BM. Blood (2011) 117:5449–52. doi:10.1182/blood-2010-11-318956
7. Rosmaraki EE, Douagi I, Roth C, Colucci F, Cumano A, Santo JPD. Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol (2001) 31:1900–9. doi:10.1002/1521-4141(200106)31:6<1900::AID-IMMU1900>3.0.CO;2-M
8. Noguchi M, Yi H, Rosenblatt HM, Filipovich AH, Adelstein S, Modi WS, et al. Interleukin-2 receptor γ chain mutation results in X-linked severe combined immunodeficiency in humans. Cell (1993) 73:147–57. doi:10.1016/0092-8674(93)90167-O
9. DiSanto JP, Müller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci U S A (1995) 92:377–81. doi:10.1073/pnas.92.2.377
10. Suzuki H, Duncan GS, Takimoto H, Mak TW. Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor β chain. J Exp Med (1997) 185:499–506. doi:10.1084/jem.185.3.499
11. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med (2000) 191:771–80. doi:10.1084/jem.191.5.771
12. Lodolce JP, Boone DL, Chai S, Swain RE, Dassopoulos T, Trettin S, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity (1998) 9:669–76. doi:10.1016/S1074-7613(00)80664-0
13. Vosshenrich CAJ, Ranson T, Samson SI, Corcuff E, Colucci F, Rosmaraki EE, et al. Roles for common cytokine receptor gamma-chain-dependent cytokines in the generation, differentiation, and maturation of NK cell precursors and peripheral NK cells in vivo. J Immunol (2005) 174:1213–21.
14. Burton JD, Bamford RN, Peters C, Grant AJ, Kurys G, Goldman CK, et al. A lymphokine, provisionally designated interleukin T and produced by a human adult T-cell leukemia line, stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc Natl Acad Sci U S A (1994) 91:4935–9. doi:10.1073/pnas.91.11.4935
15. Grabstein KH, Eisenman J, Shanebeck K, Rauch C, Srinivasan S, Fung V, et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science (1994) 264:965–8. doi:10.1126/science.8178155
16. Giri JG, Ahdieh M, Eisenman J, Shanebeck K, Grabstein K, Kumaki S, et al. Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. EMBO J (1994) 13:2822.
17. Bamford RN, Grant AJ, Burton JD, Peters C, Kurys G, Goldman CK, et al. The interleukin (IL) 2 receptor beta chain is shared by IL-2 and a cytokine, provisionally designated IL-T, that stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc Natl Acad Sci U S A (1994) 91:4940–4. doi:10.1073/pnas.91.11.4940
18. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γc family cytokines. Nat Rev Immunol (2009) 9:480–90. doi:10.1038/nri2580
19. Giri JG, Kumaki S, Ahdieh M, Friend DJ, Loomis A, Shanebeck K, et al. Identification and cloning of a novel IL-15 binding protein that is structurally related to the alpha chain of the IL-2 receptor. EMBO J (1995) 14:3654–63.
20. Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Rα recycles and presents IL-15 in trans to neighboring cells. Immunity (2002) 17:537–47. doi:10.1016/S1074-7613(02)00429-6
21. Mortier E, Woo T, Advincula R, Gozalo S, Ma A. IL-15R chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J Exp Med (2008) 205:1213–25. doi:10.1084/jem.20071913
22. Burkett PR, Koka R, Chien M, Chai S, Boone DL, Ma A. Coordinate expression and trans presentation of interleukin (IL)-15Rα and IL-15 supports natural killer cell and memory CD8+ T cell homeostasis. J Exp Med (2004) 200:825–34. doi:10.1084/jem.20041389
23. Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med (2009) 206:25–34. doi:10.1084/jem.20082013
24. Koka R, Burkett PR, Chien M, Chai S, Chan F, Lodolce JP, et al. Interleukin (IL)-15R -deficient natural killer cells survive in normal but not IL-15R -deficient mice. J Exp Med (2003) 197:977–84. doi:10.1084/jem.20021836
25. Prlic M, Blazar BR, Farrar MA, Jameson SC. In vivo survival and homeostatic proliferation of natural killer cells. J Exp Med (2003) 197:967–76. doi:10.1084/jem.20021847
26. Schluns KS, Nowak EC, Cabrera-Hernandez A, Puddington L, Lefrançois L, Aguila HL. Distinct cell types control lymphoid subset development by means of IL-15 and IL-15 receptor α expression. Proc Natl Acad Sci U S A (2004) 101:5616–21. doi:10.1073/pnas.0307442101
27. Fehniger TA, Suzuki K, Ponnappan A, VanDeusen JB, Cooper MA, Florea SM, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med (2001) 193:219–31. doi:10.1084/jem.193.2.219
28. Mishra A, Liu S, Sams GH, Curphey DP, Santhanam R, Rush LJ, et al. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell (2012) 22:645–55. doi:10.1016/j.ccr.2012.09.009
29. Zanoni I, Spreafico R, Bodio C, Di Gioia M, Cigni C, Broggi A, et al. IL-15 cis presentation is required for optimal NK cell activation in lipopolysaccharide-mediated inflammatory conditions. Cell Rep (2013) 4(6):1235–49. doi:10.1016/j.celrep.2013.08.021
30. Cooper MA. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood (2002) 100:3633–8. doi:10.1182/blood-2001-12-0293
31. Ranson T. IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood (2003) 101:4887–93. doi:10.1182/blood-2002-11-3392
32. Huntington ND, Puthalakath H, Gunn P, Naik E, Michalak EM, Smyth MJ, et al. Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat Immunol (2007) 8:856–63. doi:10.1038/ni1487
33. Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, et al. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity (2007) 26:798–811. doi:10.1016/j.immuni.2007.04.010
34. Koka R, Burkett P, Chien M, Chai S, Boone DL, Ma A. Cutting edge: murine dendritic cells require IL-15Rα to prime NK cells. J Immunol (2004) 173:3594–8. doi:10.1084/jem.20041389
35. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity (2007) 26:503–17. doi:10.1016/j.immuni.2007.03.006
36. Mortier E, Advincula R, Kim L, Chmura S, Barrera J, Reizis B, et al. Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity (2009) 31:811–22. doi:10.1016/j.immuni.2009.09.017
37. Puzanov IJ, Bennett M, Kumar V. IL-15 can substitute for the marrow microenvironment in the differentiation of natural killer cells. J Immunol (1996) 157:4282–5.
38. Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8+ T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity (2012) 37:549–62. doi:10.1016/j.immuni.2012.05.029
39. Castillo EF, Stonier SW, Frasca L, Schluns KS. Dendritic cells support the in vivo development and maintenance of NK cells via IL-15 trans-presentation. J Immunol (2009) 183:4948–56. doi:10.4049/jimmunol.0900719
40. Nguyen KB, Salazar-Mather TP, Dalod MY, Deusen JBV, Wei X, Liew FY, et al. Coordinated and distinct roles for IFN-αβ, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol (2002) 169:4279–87.
41. Tran P, Ahmad R, Xu J, Ahmad A, Menezes J. Host’s innate immune response to fungal and bacterial agents in vitro: up-regulation of interleukin-15 gene expression resulting in enhanced natural killer cell activity. Immunology (2003) 109:263–70. doi:10.1046/j.1365-2567.2003.01659.x
42. Orr SJ, Quigley L, McVicar DW. In vivo expression of signaling proteins in reconstituted NK cells. J Immunol Methods (2009) 340:158–63. doi:10.1016/j.jim.2008.10.014
43. Orr SJ, Roessler S, Quigley L, Chan T, Ford JW, O’Connor GM, et al. Implications for gene therapy-limiting expression of IL-2R c delineate differences in signaling thresholds required for lymphocyte development and maintenance. J Immunol (2010) 185:1393–403. doi:10.4049/jimmunol.0903528
44. Lee GA, Liou Y-H, Wang S-W, Ko K-L, Jiang S-T, Liao N-S. Different NK cell developmental events require different levels of IL-15 trans-presentation. J Immunol (2011) 187:1212–21. doi:10.4049/jimmunol.1100331
45. Ogasawara K, Hida S, Azimi N, Tagaya Y, Sato T, Yokochi-Fukuda T, et al. Requirement for IRF-1 in the microenvironment supporting development of natural killer cells. Nature (1998) 391:700–3. doi:10.1038/35636
46. Burkett PR, Koka R, Chien M, Chai S, Chan F, Ma A, et al. IL-15Rα expression on CD8+ T cells is dispensable for T cell memory. Proc Natl Acad Sci U S A (2003) 100:4724–9. doi:10.1073/pnas.0737048100
47. Schluns KS, Klonowski KD, Lefrançois L. Transregulation of memory CD8 T-cell proliferation by IL-15Rα+ bone marrow-derived cells. Blood (2004) 103:988–94. doi:10.1182/blood-2003-08-2814
48. Doherty TM, Seder RA, Sher A. Induction and regulation of IL-15 expression in murine macrophages. J Immunol (1996) 156:735–41.
49. Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity (1998) 8:591–9. doi:10.1016/S1074-7613(00)80564-6
50. Colpitts SL, Stoklasek TA, Plumlee CR, Obar JJ, Guo C, Lefrançois L. Cutting edge: the role of IFN-α receptor and MyD88 signaling in induction of IL-15 expression in vivo. J Immunol (2012) 188:2483–7. doi:10.4049/jimmunol.1103609
51. Colpitts SL, Stonier SW, Stoklasek TA, Root SH, Aguila HL, Schluns KS, et al. Transcriptional regulation of IL-15 expression during hematopoiesis. J Immunol (2013) 191:3017–24. doi:10.4049/jimmunol.1301389
52. Heng TSP, Painter MW, Immunological Genome, Project Consortium. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol (2008) 9:1091–4. doi:10.1038/ni1008-1091
53. Oh S, Berzofsky JA, Burke DS, Waldmann TA, Perera LP. Coadministration of HIV vaccine vectors with vaccinia viruses expressing IL-15 but not IL-2 induces long-lasting cellular immunity. Proc Natl Acad Sci U S A (2003) 100:3392–7. doi:10.1073/pnas.0630592100
54. Ring AM, Lin J-X, Feng D, Mitra S, Rickert M, Bowman GR, et al. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat Immunol (2012) 13(12):1187–95. doi:10.1038/ni.2449
55. Boussiotis VA, Barber DL, Nakarai T, Freeman GJ, Gribben JG, Bernstein GM, et al. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science (1994) 266:1039–42. doi:10.1126/science.7973657
56. Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu ZJ, et al. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science (1994) 266:1045–7. doi:10.1126/science.7973659
57. Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science (1994) 266:1042–5. doi:10.1126/science.7973658
58. Zhu M, Berry JA, Russell SM, Leonard WJ. Delineation of the regions of interleukin-2 (IL-2) receptor β chain important for association of Jak1 and Jak3 Jak1-INDEPENDENT FUNCTIONAL RECRUITMENT OF Jak3 TO IL-2Rβ. J Biol Chem (1998) 273:10719–25. doi:10.1074/jbc.273.17.10719
59. Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity (1995) 3:771–82. doi:10.1016/1074-7613(95)90066-7
60. Fujii H. Functional dissection of the cytoplasmic subregions of the IL-2 receptor beta c chain in primary lymphocyte populations. EMBO J (1998) 17:6551–7. doi:10.1093/emboj/17.22.6551
61. Truitt KE, Mills GB, Turck CW, Imboden JB. SH2-dependent association of phosphatidylinositol 3’-kinase 85-kDa regulatory subunit with the interleukin-2 receptor beta chain. J Biol Chem (1994) 269:5937–43.
62. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood (2011) 117:1565–73. doi:10.1182/blood-2010-06-291633
63. Imada K, Bloom ET, Nakajima H, Horvath-Arcidiacono JA, Udy GB, Davey HW, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med (1998) 188:2067–74. doi:10.1084/jem.188.11.2067
64. Guo H, Samarakoon A, Vanhaesebroeck B, Malarkannan S. The p110 of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J Exp Med (2008) 205:2419–35. doi:10.1084/jem.20072327
65. Kim N, Saudemont A, Webb L, Camps M, Ruckle T, Hirsch E, et al. The p110delta catalytic isoform of PI3K is a key player in NK-cell development and cytokine secretion. Blood (2007) 110:3202–8. doi:10.1182/blood-2007-02-075366
66. Tassi I, Cella M, Gilfillan S, Turnbull I, Diacovo TG, Penninger JM, et al. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity (2007) 27:214–27. doi:10.1016/j.immuni.2007.07.014
67. Polak R, Buitenhuis M. The PI3K/PKB signaling module as key regulator of hematopoiesis: implications for therapeutic strategies in leukemia. Blood (2012) 119:911–23. doi:10.1182/blood-2011-07-366203
68. Saudemont A, Garçon F, Yadi H, Roche-Molina M, Kim N, Segonds-Pichon A, et al. p110γ and p110δ isoforms of phosphoinositide 3-kinase differentially regulate natural killer cell migration in health and disease. Proc Natl Acad Sci U S A (2009) 106:5795–800. doi:10.1073/pnas.0808594106
69. Bryceson YT, March ME, Ljunggren H-G, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev (2006) 214:73–91. doi:10.1111/j.1600-065X.2006.00457.x
70. Zeng H, Chi H. mTOR and lymphocyte metabolism. Curr Opin Immunol (2013) 25:347–55. doi:10.1016/j.coi.2013.05.002
71. Baranek T, Manh T-PV, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe (2012) 12:571–84. doi:10.1016/j.chom.2012.09.002
72. Brennan P, Babbage JW, Burgering BM, Groner B, Reif K, Cantrell DA. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity (1997) 7:679–89. doi:10.1016/S1074-7613(00)80388-X
73. Grimm EA, Robb RJ, Roth JA, Neckers LM, Lachman LB, Wilson DJ, et al. Lymphokine-activated killer cell phenomenon. III. Evidence that IL-2 is sufficient for direct activation of peripheral blood lymphocytes into lymphokine-activated killer cells. J Exp Med (1983) 158:1356–61. doi:10.1084/jem.158.4.1356
74. Strausser JL, Rosenberg SA. In vitro growth of cytotoxic human lymphocytes I. Growth of cells sensitized in vitro to alloantigens. J Immunol (1978) 121:1491–5.
75. Bihl F, Pecheur J, Bréart B, Poupon G, Cazareth J, Julia V, et al. Primed antigen-specific CD4+ T cells are required for NK cell activation in vivo upon Leishmania major infection. J Immunol (2010) 1950(185):2174–81. doi:10.4049/jimmunol.1001486
76. Granucci F, Zanoni I, Pavelka N, Dommelen SLH, van Andoniou CE, Belardelli F, et al. A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J Exp Med (2004) 200:287–95. doi:10.1084/jem.20040370
77. Gasteiger G, Hemmers S, Firth MA, Floc’h AL, Huse M, Sun JC, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med (2013) 103:5460. doi:10.1084/jem.20122462
78. Sitrin J, Ring A, Garcia KC, Benoist C, Mathis D. Regulatory T cells control NK cells in an insulitic lesion by depriving them of IL-2. J Exp Med (2013) 93:2260. doi:10.1084/jem.20122248
79. Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med (2012) 209:2351–65. doi:10.1084/jem.20120944
80. Gasteiger G, Hemmers S, Bos PD, Sun JC, Rudensky AY. IL-2-dependent adaptive control of NK cell homeostasis. J Exp Med (2013) 33:119. doi:10.1084/jem.20122571
81. Lee S-H, Fragoso MF, Biron CA. Cutting edge: a novel mechanism bridging innate and adaptive immunity: IL-12 induction of CD25 to form high-affinity IL-2 receptors on NK cells. J Immunol (2012) 1950(189):2712–6. doi:10.4049/jimmunol.1201528
82. Ozaki K, Kikly K, Michalovich D, Young PR, Leonard WJ. Cloning of a type I cytokine receptor most related to the IL-2 receptor β chain. Proc Natl Acad Sci U S A (2000) 97:11439–44. doi:10.1073/pnas.200360997
83. Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature (2000) 408:57–63. doi:10.1038/35040504
84. Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity (2002) 16:559–69. doi:10.1016/S1074-7613(02)00295-9
85. Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol (2004) 172:2048–58.
86. Brady J, Carotta S, Thong RPL, Chan CJ, Hayakawa Y, Smyth MJ, et al. The interactions of multiple cytokines control NK cell maturation. J Immunol (2010) 185:6679–88. doi:10.4049/jimmunol.0903354
87. Strengell M, Matikainen S, Sirén J, Lehtonen A, Foster D, Julkunen I, et al. IL-21 in synergy with IL-15 or IL-18 enhances IFN-γ production in human NK and T cells. J Immunol (2003) 170:5464–9.
88. Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science (2009) 324:1569–72. doi:10.1126/science.1174182
89. Fröhlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science (2009) 324:1576–80. doi:10.1126/science.1172815
90. Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science (2009) 324:1572–6. doi:10.1126/science.1175194
91. Schmitz I, Schneider C, Fröhlich A, Frebel H, Christ D, Leonard WJ, et al. IL-21 restricts virus-driven Treg cell expansion in chronic LCMV infection. PLoS Pathog (2013) 9:e1003362. doi:10.1371/journal.ppat.1003362
92. Iannello A, Boulassel M-R, Samarani S, Tremblay C, Toma E, Routy J-P, et al. IL-21 enhances NK cell functions and survival in healthy and HIV-infected patients with minimal stimulation of viral replication. J Leukoc Biol (2010) 87:857–67. doi:10.1189/jlb.1009701
93. Strbo N, de Armas L, Liu H, Kolber MA, Lichtenheld M, Pahwa S. IL-21 augments natural killer effector functions in chronically HIV-infected individuals. AIDS (2008) 22:1551–60. doi:10.1097/QAD.0b013e3283089367
94. Freeden-Jeffry U, von Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med (1995) 181:1519–26. doi:10.1084/jem.181.4.1519
95. He YW, Malek TR. Interleukin-7 receptor alpha is essential for the development of gamma delta + T cells, but not natural killer cells. J Exp Med (1996) 184:289–93. doi:10.1084/jem.184.1.289
96. Rochman Y, Leonard WJ. The role of thymic stromal lymphopoietin in CD8+ T cell homeostasis. J Immunol (2008) 181:7699–705.
97. Vosshenrich CAJ, García-Ojeda ME, Samson-Villéger SI, Pasqualetto V, Enault L, Goff OR-L, et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol (2006) 7:1217–24. doi:10.1038/ni1395
98. Marcenaro E, Della Chiesa M, Bellora F, Parolini S, Millo R, Moretta L, et al. IL-12 or IL-4 prime human NK cells to mediate functionally divergent interactions with dendritic cells or tumors. J Immunol (2005) 1950(174):3992–8.
99. Agaugué S, Marcenaro E, Ferranti B, Moretta L, Moretta A. Human natural killer cells exposed to IL-2, IL-12, IL-18, or IL-4 differently modulate priming of naive T cells by monocyte-derived dendritic cells. Blood (2008) 112:1776–83. doi:10.1182/blood-2008-02-135871
100. Marçais A, Tomkowiak M, Walzer T, Coupet C-A, Ravel-Chapuis A, Marvel J. Maintenance of CCL5 mRNA stores by post-effector and memory CD8 T cells is dependent on transcription and is coupled to increased mRNA stability. Eur J Immunol (2006) 36:2745–54. doi:10.1002/eji.200636259
101. Marçais A, Coupet C-A, Walzer T, Tomkowiak M, Ghittoni R, Marvel J. Cell-autonomous CCL5 transcription by memory CD8 T cells is regulated by IL-4. J Immunol (2006) 177:4451–7.
102. Ventre E, Brinza L, Schicklin S, Mafille J, Coupet C-A, Marcais A, et al. Negative regulation of NKG2D expression by IL-4 in memory CD8 T cells. J Immunol (2012) 189:3480–9. doi:10.4049/jimmunol.1102954
103. Lee YJ, Jameson SC, Hogquist KA. Alternative memory in the CD8 T cell lineage. Trends Immunol (2011) 32:50–6. doi:10.1016/j.it.2010.12.004
104. Weinreich MA, Odumade OA, Jameson SC, Hogquist KA. T cells expressing the transcription factor PLZF regulate the development of memory-like CD8+ T cells. Nat Immunol (2010) 11:709–16. doi:10.1038/ni.1898
105. Morris SC, Heidorn SM, Herbert DR, Perkins C, Hildeman DA, Khodoun MV, et al. Endogenously produced IL-4 nonredundantly stimulates CD8+ T cell proliferation. J Immunol (2009) 182:1429–38.
106. Vignali DAA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol (2012) 13:722–8. doi:10.1038/ni.2366
107. Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med (1989) 170:827–45. doi:10.1084/jem.170.3.827
108. Hyodo Y, Matsui K, Hayashi N, Tsutsui H, Kashiwamura S, Yamauchi H, et al. IL-18 Up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J Immunol (1999) 162:1662–8.
109. Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med (1996) 184:747–52. doi:10.1084/jem.184.2.747
110. Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol (1995) 154:5071–9.
111. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (1993) 260:547–9. doi:10.1126/science.8097338
112. Orange JS, Biron CA. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol (1996) 156:4746–56.
113. Sousa CRE, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, et al. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med (1997) 186:1819–29. doi:10.1084/jem.186.11.1819
114. Goldszmid RS, Caspar P, Rivollier A, White S, Dzutsev A, Hieny S, et al. NK cell-derived interferon-gamma; orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity (2012) 36:1047–59. doi:10.1016/j.immuni.2012.03.026
115. Ma X, Chow JM, Gri G, Carra G, Gerosa F, Wolf SF, et al. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J Exp Med (1996) 183:147–57. doi:10.1084/jem.183.1.147
116. Mailliard RB, Son Y-I, Redlinger R, Coates PT, Giermasz A, Morel PA, et al. Dendritic cells mediate NK cell help for Th1 and CTL responses: two-signal requirement for the induction of NK cell helper function. J Immunol (2003) 171:2366–73.
117. Kuwajima S, Sato T, Ishida K, Tada H, Tezuka H, Ohteki T. Interleukin 15-dependent crosstalk between conventional and plasmacytoid dendritic cells is essential for CpG-induced immune activation. Nat Immunol (2006) 7:740–6. doi:10.1038/ni1348
118. Borg C, Jalil A, Laderach D, Maruyama K, Wakasugi H, Charrier S, et al. NK cell activation by dendritic cells (DCs) requires the formation of a synapse leading to IL-12 polarization in DCs. Blood (2004) 104:3267–75. doi:10.1182/blood-2004-01-0380
119. Bacon CM, McVicar DW, Ortaldo JR, Rees RC, O’Shea JJ, Johnston JA. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: differential use of Janus family tyrosine kinases by IL-2 and IL-12. J Exp Med (1995) 181:399–404. doi:10.1084/jem.181.1.399
120. Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, et al. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med (1995) 181:1755–62. doi:10.1084/jem.181.5.1755
121. Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, et al. Tyk2 plays a restricted role in IFNα signaling, although it is required for IL-12-mediated T cell function. Immunity (2000) 13:561–71. doi:10.1016/S1074-7613(00)00055-8
122. Page KM, Chaudhary D, Goldman SJ, Kasaian MT. Natural killer cells from protein kinase C θ-/- mice stimulated with interleukin-12 are deficient in production of interferon-γ. J Leukoc Biol (2008) 83:1267–76. doi:10.1189/jlb.1107745
123. Orange JS, Wang B, Terhorst C, Biron CA. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J Exp Med (1995) 182:1045–56. doi:10.1084/jem.182.4.1045
124. Kang S-J, Liang H-E, Reizis B, Locksley RM. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity (2008) 29:819–33. doi:10.1016/j.immuni.2008.09.017
125. Andrews DM, Scalzo AA, Yokoyama WM, Smyth MJ, Degli-Esposti MA. Functional interactions between dendritic cells and NK cells during viral infection. Nat Immunol (2003) 4:175–81. doi:10.1038/ni880
126. Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, Lanier LL. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J Exp Med (2012) 209:947–54. doi:10.1084/jem.20111760
127. Haeberlein S, Sebald H, Bogdan C, Schleicher U. IL-18, but not IL-15, contributes to the IL-12-dependent induction of NK-cell effector functions by Leishmania infantum in vivo. Eur J Immunol (2010) 40:1708–17. doi:10.1002/eji.200939988
128. Sun JC, Ma A, Lanier LL. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J Immunol (2009) 183:2911–4. doi:10.4049/jimmunol.0901872
129. French AR, Sjölin H, Kim S, Koka R, Yang L, Young DA, et al. DAP12 signaling directly augments proproliferative cytokine stimulation of NK cells during viral infections. J Immunol (2006) 177:4981–90.
130. Chan SH, Kobayashi M, Santoli D, Perussia B, Trinchieri G. Mechanisms of IFN-gamma induction by natural killer cell stimulatory factor (NKSF/IL-12). Role of transcription and mRNA stability in the synergistic interaction between NKSF and IL-2. J Immunol (1992) 148:92–8.
131. Ogilvie RL, SternJohn JR, Rattenbacher B, Vlasova IA, Williams DA, Hau HH, et al. Tristetraprolin mediates interferon-γ mRNA decay. J Biol Chem (2009) 284:11216–23. doi:10.1074/jbc.M901229200
132. Steiner DF, Thomas MF, Hu JK, Yang Z, Babiarz JE, Allen CDC, et al. MicroRNA-29 regulates T-box transcription factors and interferon-gamma; production in helper T cells. Immunity (2011) 35(2):169–81. doi:10.1016/j.immuni.2011.07.009
133. Mavropoulos A, Sully G, Cope AP, Clark AR. Stabilization of IFN-γ mRNA by MAPK p38 in IL-12- and IL-18-stimulated human NK cells. Blood (2005) 105:282–8. doi:10.1182/blood-2004-07-2782
134. Robinson D, Shibuya K, Mui A, Zonin F, Murphy E, Sana T, et al. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-γ production and activates IRAK and NFκB. Immunity (1997) 7:571–81. doi:10.1016/S1074-7613(00)80378-7
135. Berg RE, Crossley E, Murray S, Forman J. Relative contributions of NK and CD8 T cells to IFN-γ mediated innate immune protection against Listeria monocytogenes. J Immunol (2005) 175:1751–7.
136. Cooper MA, Elliott JM, Keyel PA, Yang L, Carrero JA, Yokoyama WM. Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A (2009) 106:1915–9. doi:10.1073/pnas.0813192106
137. O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol (2006) 7:507–16. doi:10.1038/ni1332
138. Okamura H, Tsutsui H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature (1995) 378:88–91. doi:10.1038/378088a0
139. Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity (1998) 8:383–90. doi:10.1016/S1074-7613(00)80543-9
140. Wang Y, Chaudhri G, Jackson RJ, Karupiah G. IL-12p40 and IL-18 play pivotal roles in orchestrating the cell-mediated immune response to a poxvirus infection. J Immunol (2009) 183:3324–31. doi:10.4049/jimmunol.0803985
141. Wei X, Leung BP, Niedbala W, Piedrafita D, Feng G, Sweet M, et al. Altered immune responses and susceptibility to Leishmania major and Staphylococcus aureus infection in IL-18-deficient mice. J Immunol (1999) 163:2821–8.
142. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol (2010) 10:89–102. doi:10.1038/nri2691
143. Gracie JA, Robertson SE, McInnes IB. Interleukin-18. J Leukoc Biol (2003) 73:213–24. doi:10.1189/jlb.0602313
144. Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase-1 processes IFN-γ-inducing factor and regulates LPS-induced IFN-γ production. Nature (1997) 386:619–23. doi:10.1038/386619a0
145. Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science (1997) 275:206–9. doi:10.1126/science.275.5297.206
146. Bossaller L, Chiang P-I, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VAK, et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol (2012) 1950(189):5508–12. doi:10.4049/jimmunol.1202121
147. Pierini R, Perret M, Djebali S, Juruj C, Michallet M-C, Förster I, et al. ASC controls IFN-γ levels in an IL-18-dependent manner in caspase-1-deficient mice infected with Francisella novicida. J Immunol (2013) 1950(191):3847–57. doi:10.4049/jimmunol.1203326
148. Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A, et al. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol (2001) 1950(167):6568–75.
149. Omoto Y, Yamanaka K, Tokime K, Kitano S, Kakeda M, Akeda T, et al. Granzyme B is a novel interleukin-18 converting enzyme. J Dermatol Sci (2010) 59:129–35. doi:10.1016/j.jdermsci.2010.05.004
150. Kastenmüller W, Torabi-Parizi P, Subramanian N, Lämmermann T, Germain RN. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell (2012) 150:1235–48. doi:10.1016/j.cell.2012.07.021
151. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest (2012) 122:1082–96. doi:10.1172/JCI61029
152. Spörri R, Joller N, Hilbi H, Oxenius A. A novel role for neutrophils as critical activators of NK cells. J Immunol (2008) 181:7121–30.
153. Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF Jr, Foley E, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol (1999) 1950(162):6829–35.
154. Carta S, Lavieri R, Rubartelli A. Different members of the IL-1 family come out in different ways: DAMPs vs. cytokines? Front Immunol (2013) 4:123. doi:10.3389/fimmu.2013.00123
155. Bellora F, Castriconi R, Dondero A, Reggiardo G, Moretta L, Mantovani A, et al. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc Natl Acad Sci U S A (2010) 107:21659–64. doi:10.1073/pnas.1007654108
156. Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med (2002) 195:327–33. doi:10.1084/jem.20010938
157. Humann J, Lenz LL. Activation of naive NK cells in response to Listeria monocytogenes requires IL-18 and contact with infected dendritic cells. J Immunol (2010) 1950(184):5172–8. doi:10.4049/jimmunol.0903759
158. Lapaque N, Walzer T, Méresse S, Vivier E, Trowsdale J. Interactions between human NK cells and macrophages in response to Salmonella infection. J Immunol (2009) 1950(182):4339–48. doi:10.4049/jimmunol.0803329
159. Piccioli D, Sbrana S, Melandri E, Valiante NM. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J Exp Med (2002) 195:335–41. doi:10.1084/jem.20010934
160. Semino C, Angelini G, Poggi A, Rubartelli A. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood (2005) 106:609–16. doi:10.1182/blood-2004-10-3906
161. Bellora F, Castriconi R, Doni A, Cantoni C, Moretta L, Mantovani A, et al. M-CSF induces the expression of a membrane-bound form of IL-18 in a subset of human monocytes differentiating in vitro toward macrophages. Eur J Immunol (2012) 42:1618–26. doi:10.1002/eji.201142173
162. Novick D, Kim S-H, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity (1999) 10:127–36. doi:10.1016/S1074-7613(00)80013-8
163. Hoshino K, Tsutsui H, Kawai T, Takeda K, Nakanishi K, Takeda Y, et al. Cutting edge: generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J Immunol (1999) 162:5041–4.
164. Suzuki N, Chen N-J, Millar DG, Suzuki S, Horacek T, Hara H, et al. IL-1 receptor-associated kinase 4 is essential for IL-18-mediated NK and Th1 cell responses. J Immunol (2003) 170:4031–5.
165. Rowland CA, Lertmemongkolchai G, Bancroft A, Haque A, Lever MS, Griffin KF, et al. Critical role of type 1 cytokines in controlling initial infection with Burkholderia mallei. Infect Immun (2006) 74:5333–40. doi:10.1128/IAI.02046-05
166. Kawakami K, Koguchi Y, Qureshi MH, Miyazato A, Yara S, Kinjo Y, et al. IL-18 contributes to host resistance against infection with Cryptococcus neoformans in mice with defective IL-12 synthesis through induction of IFN-gamma production by NK cells. J Immunol (2000) 1950(165):941–7.
167. Pien GC, Satoskar AR, Takeda K, Akira S, Biron CA. Cutting edge: selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J Immunol (2000) 1950(165):4787–91.
168. Chaix J, Tessmer MS, Hoebe K, Fuséri N, Ryffel B, Dalod M, et al. Cutting edge: priming of NK cells by IL-18. J Immunol (2008) 1950(181):1627–31.
169. Mailliard RB, Alber SM, Shen H, Watkins SC, Kirkwood JM, Herberman RB, et al. IL-18-induced CD83+CCR7+ NK helper cells. J Exp Med (2005) 202:941–53. doi:10.1084/jem.20050128
170. Schulthess J, Meresse B, Ramiro-Puig E, Montcuquet N, Darche S, Bègue B, et al. Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity (2012) 37(1):108–21. doi:10.1016/j.immuni.2012.05.013
171. Terme M, Ullrich E, Aymeric L, Meinhardt K, Coudert JD, Desbois M, et al. Cancer-induced immunosuppression: IL-18-elicited immunoablative NK cells. Cancer Res (2012) 72:2757–67. doi:10.1158/0008-5472.CAN-11-3379
172. Allen IC, TeKippe EM, Woodford R-MT, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med (2010) 207:1045–56. doi:10.1084/jem.20100050
173. Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai R-M, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med (2010) 207:1625–36. doi:10.1084/jem.20100199
174. Wong JL, Berk E, Edwards RP, Kalinski P. IL-18-primed helper NK cells collaborate with dendritic cells to promote recruitment of effector CD8+ T cells to the tumor microenvironment. Cancer Res (2013) 73:4653–62. doi:10.1158/0008-5472.CAN-12-4366
175. Reading PC, Whitney PG, Barr DP, Wojtasiak M, Mintern JD, Waithman J, et al. IL-18, but not IL-12, regulates NK cell activity following intranasal herpes simplex virus type 1 infection. J Immunol (2007) 179:3214–21.
176. Li MO, Wan YY, Sanjabi S, Robertson A-KL, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol (2006) 24:99–146. doi:10.1146/annurev.immunol.24.021605.090737
177. Weiss A, Attisano L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol (2013) 2:47–63. doi:10.1002/wdev.86
178. Ichiyama K, Sekiya T, Inoue N, Tamiya T, Kashiwagi I, Kimura A, et al. Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-β is mediated by suppression of eomesodermin. Immunity (2011) 34:741–54. doi:10.1016/j.immuni.2011.02.021
179. Espevik T, Figari IS, Ranges GE, Palladino MA Jr. Transforming growth factor-beta 1 (TGF-beta 1) and recombinant human tumor necrosis factor-alpha reciprocally regulate the generation of lymphokine-activated killer cell activity. Comparison between natural porcine platelet-derived TGF-beta 1 and TGF-beta 2, and recombinant human TGF-beta 1. J Immunol (1988) 1950(140):2312–6.
180. Grimm EA, Crump WL III, Durett A, Hester JP, Lagoo-Deenadalayan S, Owen-Schaub LB. TGF-beta inhibits the in vitro induction of lymphokine-activated killing activity. Cancer Immunol Immunother (1988) 27:53–8. doi:10.1007/BF00205758
181. Mulé JJ, Schwarz SL, Roberts AB, Sporn MB, Rosenberg SA. Transforming growth factor-beta inhibits the in vitro generation of lymphokine-activated killer cells and cytotoxic T cells. Cancer Immunol Immunother (1988) 26:95–100.
182. Rook AH, Kehrl JH, Wakefield LM, Roberts AB, Sporn MB, Burlington DB, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol (1986) 1950(136):3916–20.
183. Wallick SC, Figari IS, Morris RE, Levinson AD, Palladino MA. Immunoregulatory role of transforming growth factor beta (TGF-beta) in development of killer cells: comparison of active and latent TGF-beta 1. J Exp Med (1990) 172:1777–84. doi:10.1084/jem.172.6.1777
184. Su HC, Leite-Morris KA, Braun L, Biron CA. A role for transforming growth factor-beta 1 in regulating natural killer cell and T lymphocyte proliferative responses during acute infection with lymphocytic choriomeningitis virus. J Immunol (1991) 1950(147):2717–27.
185. Arteaga CL, Hurd SD, Winnier AR, Johnson MD, Fendly BM, Forbes JT. Anti-transforming growth factor (TGF)-beta antibodies inhibit breast cancer cell tumorigenicity and increase mouse spleen natural killer cell activity. Implications for a possible role of tumor cell/host TGF-beta interactions in human breast cancer progression. J Clin Invest (1993) 92:2569–76.
186. Bellone G, Aste-Amezaga M, Trinchieri G, Rodeck U. Regulation of NK cell functions by TGF-beta 1. J Immunol (1995) 1950(155):1066–73.
187. Hunter CA, Bermudez L, Beernink H, Waegell W, Remington JS. Transforming growth factor-beta inhibits interleukin-12-induced production of interferon-gamma by natural killer cells: a role for transforming growth factor-beta in the regulation of T cell-independent resistance to Toxoplasma gondii. Eur J Immunol (1995) 25:994–1000. doi:10.1002/eji.1830250420
188. Meadows SK, Eriksson M, Barber A, Sentman CL. Human NK cell IFN-gamma production is regulated by endogenous TGF-beta. Int Immunopharmacol (2006) 6:1020–8. doi:10.1016/j.intimp.2006.01.013
189. Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, et al. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity (2006) 24:575–90. doi:10.1016/j.immuni.2006.03.016
190. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol (2008) 1950(181):3784–92.
191. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A (2003) 100:4120–5. doi:10.1073/pnas.0730640100
192. Lee J-C, Lee K-M, Kim D-W, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol (2004) 1950(172):7335–40.
193. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol (2013) 1950(190):5321–8. doi:10.4049/jimmunol.1202693
194. Su HC, Ishikawa R, Biron CA. Transforming growth factor-beta expression and natural killer cell responses during virus infection of normal, nude, and SCID mice. J Immunol (1993) 1950(151):4874–90.
195. Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol (2005) 6:600–7. doi:10.1038/ni1197
196. Marcoe JP, Lim JR, Schaubert KL, Fodil-Cornu N, Matka M, McCubbrey AL, et al. TGF-β is responsible for NK cell immaturity during ontogeny and increased susceptibility to infection during mouse infancy. Nat Immunol (2012) 13:843–50. doi:10.1038/ni.2388
197. Ortaldo JR, Mason AT, O’Shea JJ, Smyth MJ, Falk LA, Kennedy IC, et al. Mechanistic studies of transforming growth factor-beta inhibition of IL-2-dependent activation of CD3- large granular lymphocyte functions. Regulation of IL-2R beta (p75) signal transduction. J Immunol (1991) 1950(146):3791–8.
198. Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med (1989) 170:2081–95. doi:10.1084/jem.170.6.2081
199. Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galán JE, et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter Knockin tiger mouse. Immunity (2006) 25:941–52. doi:10.1016/j.immuni.2006.09.013
200. Lee S-H, Kim K-S, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J Exp Med (2009) 206:2235–51. doi:10.1084/jem.20082387
201. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. doi:10.1146/annurev.immunol.19.1.683
202. Perona-Wright G, Mohrs K, Szaba FM, Kummer LW, Madan R, Karp CL, et al. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe (2009) 6:503–12. doi:10.1016/j.chom.2009.11.003
203. Cai G, Kastelein RA, Hunter CA. IL-10 enhances NK cell proliferation, cytotoxicity and production of IFN-γ when combined with IL-18. Eur J Immunol (1999) 29:2658–65. doi:10.1002/(SICI)1521-4141(199909)29:09<2658::AID-IMMU2658>3.3.CO;2-7
204. Carson WE, Lindemann MJ, Baiocchi R, Linett M, Tan JC, Chou CC, et al. The functional characterization of interleukin-10 receptor expression on human natural killer cells. Blood (1995) 85:3577–85.
205. Chiu B-C, Stolberg VR, Chensue SW. Mononuclear phagocyte-derived IL-10 suppresses the innate IL-12/IFN-γ axis in lung-challenged aged mice. J Immunol (2008) 181:3156–66.
206. Qian C, Jiang X, An H, Yu Y, Guo Z, Liu S, et al. TLR agonists promote ERK-mediated preferential IL-10 production of regulatory dendritic cells (diffDCs), leading to NK-cell activation. Blood (2006) 108:2307–15. doi:10.1182/blood-2006-03-005595
207. Shibata Y, Foster LA, Kurimoto M, Okamura H, Nakamura RM, Kawajiri K, et al. Immunoregulatory roles of IL-10 in innate immunity: IL-10 inhibits macrophage production of IFN-γ-inducing factors but enhances NK cell production of IFN-γ. J Immunol (1998) 161:4283–8.
208. Lassen MG, Lukens JR, Dolina JS, Brown MG, Hahn YS. Intrahepatic IL-10 maintains NKG2A+Ly49- liver NK cells in a functionally hyporesponsive state. J Immunol (2010) 184:2693–701. doi:10.4049/jimmunol.0901362
209. Stacey MA, Marsden M, Wang ECY, Wilkinson GWG, Humphreys IR. IL-10 restricts activation-induced death of NK cells during acute murine cytomegalovirus infection. J Immunol (2011) 187:2944–52. doi:10.4049/jimmunol.1101021
210. Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity (2012) 37:171–86. doi:10.1016/j.immuni.2012.05.020
211. Gidlund M, Orn A, Wigzell H, Senik A, Gresser I. Enhanced NK cell activity in mice injected with interferon and interferon inducers. Nature (1978) 273:759–61. doi:10.1038/273759a0
212. Beuneu H, Deguine J, Bouvier I, Santo JPD, Albert ML, Bousso P. Cutting edge: a dual role for type I IFNs during polyinosinic-polycytidylic acid-induced NK cell activation. J Immunol (2011) 187:2084–8. doi:10.4049/jimmunol.1004210
Keywords: natural killer cells, cytotoxicity, interferons, signal transduction, interleukin-15, interleukin-12, interleukin-18, TGF-beta
Citation: Marçais A, Viel S, Grau M, Henry T, Marvel J and Walzer T (2013) Regulation of mouse NK cell development and function by cytokines. Front. Immunol. 4:450. doi: 10.3389/fimmu.2013.00450
Received: 16 October 2013; Paper pending published: 04 November 2013;
Accepted: 27 November 2013; Published online: 12 December 2013.
Edited by:
Aurore Saudemont, University College London, UKReviewed by:
Marc Dalod, Centre National de la Recherche Scientifique, FranceSebastian Carotta, Walter & Eliza Hall Institute of Medical Research, Australia
Copyright: © 2013 Marçais, Viel, Grau, Henry, Marvel and Walzer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thierry Walzer, U1111, Centre International de Recherche en Infectiologie (CIRI), INSERM – CNRS UMR5308, Université Lyon 1, ENS de Lyon, 21 Avenue Tony Garnier – 69365 Lyon Cedex 07, France e-mail:dGhpZXJyeS53YWx6ZXJAaW5zZXJtLmZy