 Annalisa Ciabattini
Annalisa Ciabattini Elena Pettini
Elena Pettini Donata Medaglini
Donata Medaglini
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 December 2013
Sec. Mucosal Immunity
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00421
This article is part of the Research Topic Expert Opinions in Mucosal Vaccines View all 4 articles
T cell priming is a critical event in the initiation of the immune response to vaccination since it deeply influences both the magnitude and the quality of the immune response induced. CD4+ T cell priming, required for the induction of high-affinity antibodies and immune memory, represents a key target for improving and modulating vaccine immunogenicity. A major challenge in the study of in vivo T cell priming is due to the low frequency of antigen-specific T cells. This review discusses the current knowledge on antigen-specific CD4+ T cell priming in the context of vaccination, as well as the most advanced tools for the characterization of the in vivo T cell priming and the opportunities offered by the application of systems biology.
T cell priming is an essential event for the induction of the adaptive immune response to vaccination. T cell priming is influenced by the type of vaccine formulation (antigen, adjuvant, delivery system), the dose and the route of administration. The characterization of T cell priming induced by a vaccination strategy is therefore critical in order to develop optimal prime-boost combinations capable of eliciting the type of immune response required to fight a specific pathogen.
The efficacy of most preventive vaccines relies on antibody response to block pathogen infection and generation of immune memory cells capable of rapid and effective reactivation following pathogen re-exposure (1, 2). In this context, primary activation of T-helper cells that are required for the induction of high-affinity antibodies and immune memory is essential (2). Furthermore, CD4+ T cell priming has been shown to be an early predictor of vaccine immunogenicity in humans (3, 4).
A limitation in the study of in vivo T cell priming is due to the low frequency of antigen-specific T cells. This has been overcome by the application of technologies such as adoptive transfer of transgenic antigen-specific T cells into recipient mice and major histocompatibility complexes (MHCs) class II tetramers (5, 6). It is particularly attractive to also consider systems biology approaches that have been recently applied to vaccinology to model T cell priming and develop tools to predict vaccine responsiveness and efficacy (7–9).
Here we review the current knowledge on antigen-specific CD4+ T cell priming in the context of prophylactic vaccination. Immunological events following primary vaccination by systemic and mucosal routes and their relevance for the rational development of prime-boost strategies are addressed. Moreover, the methodologies for studying in vivo CD4+ T cell priming and the potential of applying systems biology for its modeling are discussed.
CD4+ T cell priming represents a key step in the vaccination process due to the close relationship between CD4+ T cells and both long-term humoral immunity and protective antibodies. CD4+ T cell priming is influenced by several factors such as the local pro-inflammatory environment, the nature and the dose of the antigen, the vaccine formulation including the type of adjuvant and the route of immunization (10, 11). A schematic representation of the T cell priming event in the context of vaccination is reported in Figure 1. Generation of primed T-helper cells requires contact between antigen-bearing dendritic cells (DCs) and specific CD4+ T cells within the T zone of the lymph node (LN) closest to the site of vaccination (2, 12). The process of CD4+ T cell priming begins when naïve cells, that constantly transit between the circulatory and lymphatic systems, bind their T cell antigen receptors (TCRs) to foreign peptides loaded on MHCs class II molecules presented by antigen presenting cells (APCs), thus leading to T cell proliferation (13). Antigen persistence and duration of peptide presentation by APCs influence the magnitude of the primary T cell response (14, 15). The very early interaction between antigen-specific T cells and peptide-MHC-bearing APCs within the LN has been described with static and dynamic imaging methods and movies (13, 16, 17). Interaction between APCs and antigen-specific naïve T cells takes place within the first 8–20 h and is dependent on the presence of the antigen (13). Activated T cells begin to proliferate and finally, in a later and antigen-independent phase, they expand and differentiate into various functionally defined subsets of effector cells that, depending on the nature of the cytokine milieu generated by innate cells, express specific master transcription factors (18, 19). Polarization of the distinct effector T cell subsets is indeed regulated by the strength of antigenic stimulation, as well as by the cytokines present during priming (20). These polarizing cytokines are derived from the APCs, the responding T cells or bystander cells. Effector T cells can be emigrant lymphocytes such as Th1, Th2, or Th17 that exit the LNs and move to inflamed tissues, regulatory cells (Treg), or T follicular helper (Tfh) cells that relocate to B-T cell borders and interfollicular regions (21–23). Tfh cells are specialized to regulate multiple stages of antigen-specific B cell immunity through cognate cell contact and the secretion of cytokines (21). In the extra-follicular reaction, some antigen-primed B cells, after cognate contact of Tfh cells, undergo a process of rapid differentiation in short-lived plasma cells producing low-affinity antibodies such as IgM and IgG that appear in serum at low concentration a few days after immunization (2, 21). Interaction of Tfh cells with B cells drives the formation of germinal center (GC) a dynamic micro-anatomical structure that supports the generation of B cell activation, antibody class switch recombination and affinity maturation (22, 24). Tfh cells that localize in GCs are referred to as GC-Tfh cells. A fraction of B cells matured during the GC reaction acquires the capacity to migrate toward long-term survival niches located within the bone marrow (BM) from where they can release vaccine antibodies for extended periods. Another fraction includes class-specific affinity-matured memory B cells that are able to rapidly expand and differentiate into plasma cells after antigen re-challenge (25).
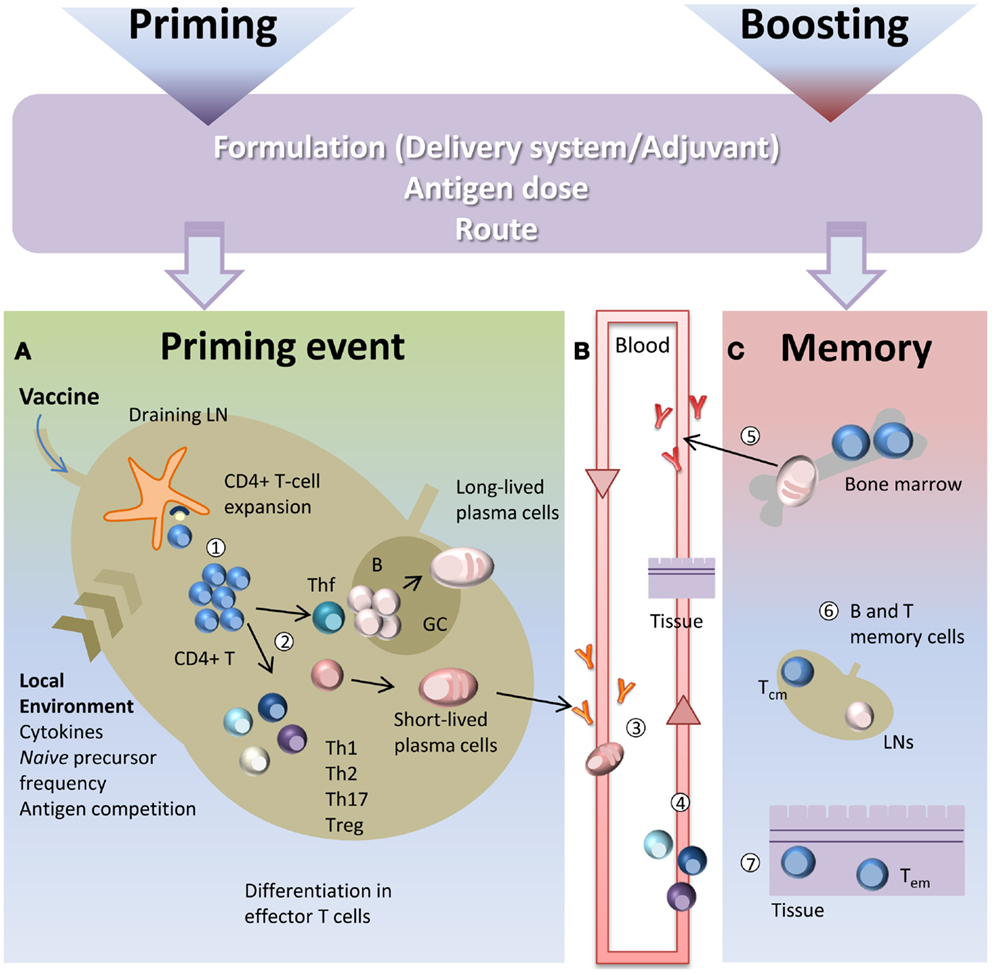
Figure 1. Immune response triggered by vaccination. Primary immune response triggered by vaccine administration is influenced by several factors, such as the vaccine formulation (including delivery systems and/or adjuvants), the nature and the dose of the antigen, and the route of immunization. (A) After vaccine administration, DCs mature and migrate to the T cell zone of draining LNs. DCs express vaccine epitopes on their MHC class II molecules, thus engaging naïve antigen-specific CD4+ T cells and inducing their proliferation and differentiation into effector T-helper cells (1). The local environment deeply influences the T cell priming event and the polarization of distinct effector T cell subsets (2). Effector T cells differentiate into subpopulations, such as Th1, Th2, Th17, Treg that mainly exert their function outside the LN. Some primed CD4+ T cells differentiate into Tfh that relocate to B-T cell borders. Cognate contact between Tfh cells and antigen-primed B cells in the extra-follicular regions of the lymph nodes is required for clonal expansion and antibody class switching (without affinity maturation) of short-lived plasma cells. GC-Tfh drives the GC reaction, in which B cells undergo clonal expansion, isotype switching, affinity maturation, and differentiate into long-lived plasma cells. (B) Low-affinity IgM and IgG antibodies produced by short-lived plasma cells during the extra-follicular reaction, appear at low levels in the serum within a few days after immunization (3). Effector Th1, Th2, and Th17 subpopulations exit the LN and through the blood disseminate toward other LNs and toward the inflamed tissue (in this context, the site of vaccine inoculation) where exert their effector function (4). (C) The long-lived plasma cells exit the LN at the end of the GC reaction and migrate to survival niches mostly located in the bone marrow (BM) where they survive through signals provided by supporting stromal cells and continue to release hypermutated antibodies (5). Another fraction of B cells, matured during the GC reaction, develop a memory phenotype and disseminate into the extra-follicular areas of the LN where they persist as resting cells until booster immunization or pathogen encounter (6). Memory T cells traffic through T cell areas of secondary LNs and BM (Tcm) (6), or localize within tissue (Tem) (7). Booster immunization induces a rapid reactivation of memory B and T cells, with proliferation and differentiation into effector cells. Memory B cells mature into plasma cells secreting large amounts of high-affinity antibodies that may be detected in serum within a few days after boosting.
Upon primary activation most of the antigen-experienced CD4+ T cells are short-lived and undergo apoptotic contraction leaving only a small fraction of competent memory precursor cells to migrate into the BM where they differentiate into long-lived memory cells. The frequency of memory T cells reflects therefore the magnitude of the initial T cell expansion and of its subsequent contraction. Two types of memory T cells have been identified based on their phenotype and function (26). Effector memory T cells (Tem) are circulating or tissue-resident cells and exert their immediate effector function after antigen encounter and mediate site-specific protection, while central memory T cells (Tcm) preferentially traffic through T cell areas of secondary LNs and BM and have a high proliferative potential (26). Their role is to recognize antigens transported by activated DCs into LNs and to rapidly undergo massive proliferation generating a delayed, but very large, wave of effector cells (26, 27). During a primary response memory Tfh cells are also generated. These cells are retained within draining lymphoid sites together with antigen-specific memory B cells and persistent complexes of peptide-MHC II (28). During a booster immunization, vaccine antigens restimulate memory T and B cells that rapidly activate a secondary immune response.
In the context of vaccination strategies, T cell priming can be evaluated as a target for improving the immune response during vaccination as well as a tool for modulating the quality of the immune response. The nature and the dose of the vaccine antigen, the adjuvant or the vaccine delivery used, the route of immunization and the local environment are all factors that deeply affect the primary activation of CD4+ T cells (10, 11).
The development of distinct effector CD4+ T cell subsets is determined to a great extent by cytokines present during the T cell priming event that act as powerful polarizing factors (10). APCs express toll-like receptors (TLRs) that recognize distinct and highly conserved pathogen-associated molecules, thus activating a signaling cascade that dramatically impacts the quality and the quantity of the T cell response. This has encouraged the use of TLRs ligands as promising adjuvants (10, 29–33), that can influence the effector fate of antigen-specific primed CD4+ T cells (10). CD4+ T cell priming has been studied for characterizing the mechanism of action of adjuvants such as alum (34), the CpG ODN (35), the lipopolysaccharide (36) or its derivative-like monophosphoryl lipid A (37), cholera toxin (38), or its B subunit (CTB) (39, 40).
Another aspect to consider is the selection of the route of administration of the vaccine that affects the quality and the localization of the T cell response (41, 42). CD4+ T cell priming following immunization by different mucosal routes has been characterized in the murine model (35, 38, 43–45) as discussed in the next section. Recently, we have also demonstrated that the route used for priming, but not for booster immunization, influences the skewing of the CD4+ T effector response toward Th1 or Th2 with a stronger Th1 polarization upon nasal administration compared to the systemic one (46).
The development of vaccination approaches aimed at enhancing Tfh primary response is particularly attractive. The interaction of T-B cells is stabilized by adhesion molecules, such as the signaling lymphocytic activation molecule (SLAM) family, that initiate intracellular signaling via recruitment of specific adapters such as the SLAM/associated protein (SAP) family (47). Targeted manipulation of the SAP/SLAM family has been employed recently as strategy for shaping and strengthening the immune response during vaccination (47, 48). The employment of a nanoparticle delivery system has also recently been shown to promote robust GC formation and enhance the expansion of vaccine antigen-specific Tfh cells leading to an enhanced humoral response (49).
The role of CD4+ T cells in developing durable functional neutralizing antibody responses, via Tfh cells, is considered of key importance for the development of vaccines against pathogens for which no vaccine is currently available, such as HIV (50). Despite the central role of T-helper cells in vaccine immunity, the specific contribution of HIV-specific CD4+ T cells in HIV infection is largely unknown and these cells have mostly been excluded from HIV vaccine design strategies because they can be infected by the virus itself (50). Strikingly, in simian immunodeficiency virus (SIV)-infected non-human primates, Tfh cells did not seem to be preferentially infected by the virus, and their frequency in LNs correlated with the magnitude of the SIV-specific IgG response, the avidity of the SIV-specific antibodies and the generation of the GCs (51).
Studies of H5N1 influenza vaccination of healthy adults have shown an increase in the frequency of virus-specific CD4+ T cells measured 22 days after the first dose. This increase predicted a rise in neutralizing antibody concentrations after boosting as well as their maintenance 6 months later (3, 52), thus suggesting that primary CD4+ T cell response can be considered a predictor marker of the secondary immune response. Similarly, CD4+ T cell expansion has shown to predict neutralizing antibody response to monovalent inactivated influenza A H1N1 vaccine (4).
Targeting mucosal sites by vaccination is an important goal considering that over 90% of infections occur at or through mucosal surfaces. The induction of mucosal immune responses requires the presence of a mucosa-associated lymphoid tissue that provides a continuous source of B and T cells to mucosal effector sites (53). A schematic representation of T cell priming in different mucosal sites following mucosal vaccination is reported in Figure 2. Inductive sites for mucosal immunity consist of organized mucosa-associated lymphoid tissue as well as local and regional draining LNs, whereas the effector sites include different compartments mainly consisting of the lamina propria of various mucose (54). Inductive sites in the gastro-intestinal and respiratory tracts have been well defined, and are composed by aggregated lymphoid tissues (gut-, nasal-, and bronchial-associated lymphoid tissues, respectively) and mucosa-associated LNs (mesenteric and mediastinal LNs). On the contrary, the vaginal mucosa is devoid of histologically demonstrable organized mucosa-associated lymphoid tissue and the role of inductive site is played directly by draining iliac LNs (55). Moreover, antigen-uptake across the vaginal mucosa barrier and immune responses in the genital tract are greatly regulated and influenced by the hormonal state and estrus phase (56). Female genital tract has therefore some unique features that should be taken in consideration in the development of a vaccination strategy.
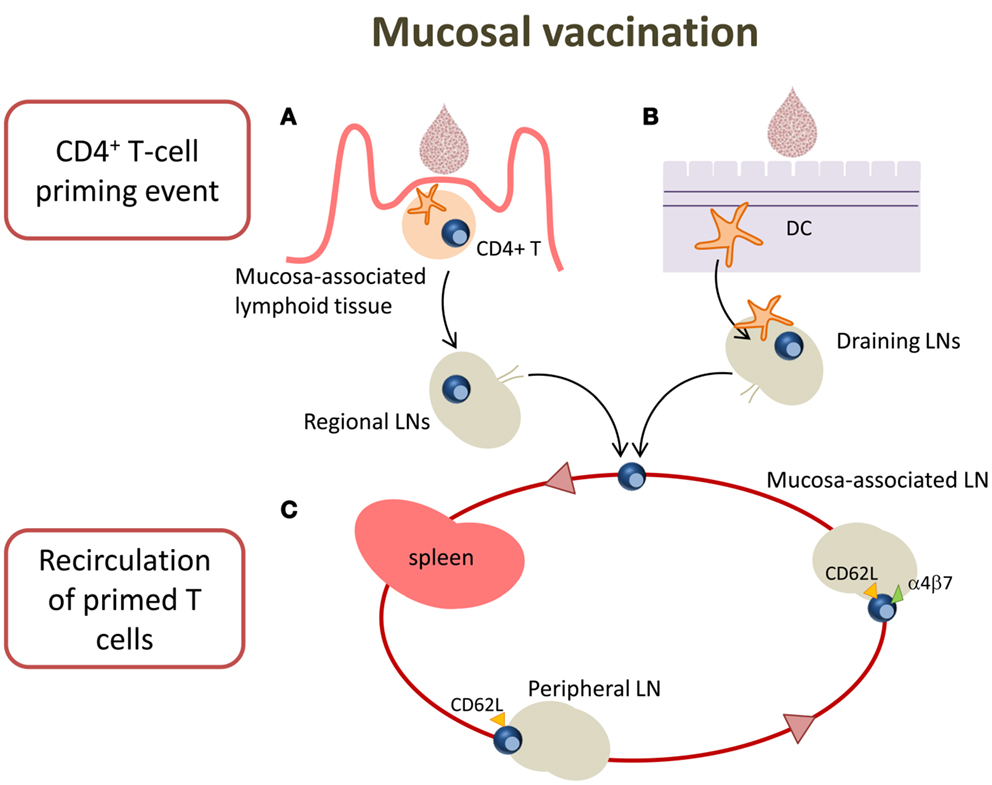
Figure 2. T cell priming in different mucosal sites following mucosal vaccination. Mucosal vaccination targets the epithelium that covers mucosal surfaces. (A) In many mucosal sites, such as the gastro-intestinal and respiratory tracts, underneath the epithelium barrier inductive sites are present, constituted by organized lymphoid tissue. Following vaccination, antigen is sampled by local DCs and transported into the inductive site where antigen-specific T cell priming occurs. Activated T cells migrate from the inductive site toward the regional draining LNs and then enter into the circulatory torrent through the lymphatic system. (B) Vaginal mucosa is devoid of histologically demonstrable organized mucosa-associated lymphoid tissue, therefore after immunization, the antigen is sampled by tissue-resident DCs and transported into the draining iliac LNs that constitute the inductive site. Primed T cells exit the LNs and migrate through the lymphatic system into the blood. (C) A fraction of mucosally primed T cells transiently circulates through the blood into the spleen and disseminates into non-draining LNs; the entry into peripheral LNs is CD62L-dependent, while into mesenteric LNs depends on both CD62L and α4β7 expression.
By using the adoptive transfer system (described in the next section), our laboratory has deeply analyzed the CD4+ T cell priming following nasal and vaginal immunization in the mouse model. Intranasal immunization with the recombinant vaccine vector Streptococcus gordonii (57–62), elicited an early clonal expansion of antigen-specific CD4+ T cells in the nasal-associated lymphoid tissue (NALT), and cervical and mediastinal LNs 3 days after immunization (43–45). Proliferated T cells were CD44hiCD45RBlo and expressed CD69 molecule within the early cell generations (44). Divided T cells disseminated in the respiratory (44), genital, and intestinal tracts (43) where they become detectable 5 days after priming. Similar results of antigen-specific clonal expansion and dissemination were observed immunizing with soluble ovalbumin (OVA) plus the adjuvant CpG ODN (35, 46). We also demonstrated that homing of nasally primed T cells into distal peripheral LNs was CD62L-dependent, while entry into mesenteric LNs depended on both CD62L and α4β7 expression (35) (Figure 2).
T cell priming was also studied following vaginal immunization in hormone synchronized mice, showing a very efficient activation of CD4+ T cells (38, 63). Antigen-specific CD4+ T cell clonal expansion was indeed detected in iliac LNs, and proliferated T cells disseminated toward distal LNs and spleen, similarly to what observed following nasal immunization (38). These data show that vaginal immunization is efficient in eliciting CD4+ T priming despite the absence of an organized mucosa-associated inductive site in the genital tract (Figure 2).
Characterization of the magnitude and quality of the T cell priming elicited by a vaccine formulation is critically important for the rational development of prime-boost vaccine combinations. An interesting approach to vaccination is indeed the heterologous prime-boost strategy that primes the immune system to a target antigen delivered by a vector and then selectively boosts the secondary response only to the vaccine antigen by using a different vaccine formulation. The heterologous prime-boost approach is specifically aimed at the generation and enrichment of high avidity T cells specific for the target antigen (64). Boosting with a different vector carrying the same antigen has been shown to be more efficient in inducing the immune responses compared to boosting with the same vector. The heterologous prime-boost approach is currently exploited in human studies aimed at developing vaccines against pathogens such as HIV (65), tuberculosis (66), and malaria (67). Furthermore, mucosal and parenteral routes can be combined in a vaccination prime-boost strategy to induce immune responses in both the local and systemic compartments. This approach has shown to be as strong or stronger than those resulting from homologous mucosal or parenteral vaccination alone (68–71). Recently, we have demonstrated that the polarization of CD4+ T effector cells is affected by the route used for priming but not for boosting, while local effector responses are mainly dependent on booster route (46).
Recent studies in the mouse model have also assessed the role of peptide-based priming on the subsequent B cell response elicited by whole protein boosting (72) or by infection with the pathogen (73) or with an attenuated viral vaccine (74). These studies showed that CD4+ T cell help is quite selective for the subsequent antibody production. CD4+ T cells specific for an epitope provide the appropriate help mainly to the protein-specific B cells, indicating a deterministic linkage between antibodies and CD4+ T cell responses (73, 74), even if discordant results have been recently reported (75).
Understanding the priming mechanisms of a vaccine formulation and optimizing its priming properties is therefore of critical relevance for the informed design of next generation prime-boost strategies.
Antigen-specific primary activation has been mostly analyzed in animal models, within LNs draining the inoculation site or in the spleen. Primed CD4+ T cells can be detected in draining LNs within a few days after immunization, with a peak after 5–7 days (13, 43, 76). In humans, primed T cells can be studied in peripheral blood starting from 7 days following vaccination (4).
Several procedures have been employed to characterize antigen-specific primed T cells, including assays of helper cell activity using carrier/hapten systems (77), and the commonly used proliferation and cytokine production assays. These methods measure functional parameters as a read-out for T cells which react to the specific antigen challenge in vitro. A major limitation of these assays is that the phenotypic and functional properties of the reactive cells may be altered by the in vitro antigenic restimulation (78).
To overcome this limitation, technologies such as the adoptive transfer of TCR-transgenic T cells in mice (79) and, more recently, MHC class II tetramers (6) have been developed to allow the ex vivo analysis of primed T cells (see below). A summary of the most used assays for studying T cell priming in human and animal studies, with their main advantages and disadvantages, is reported in Table 1.
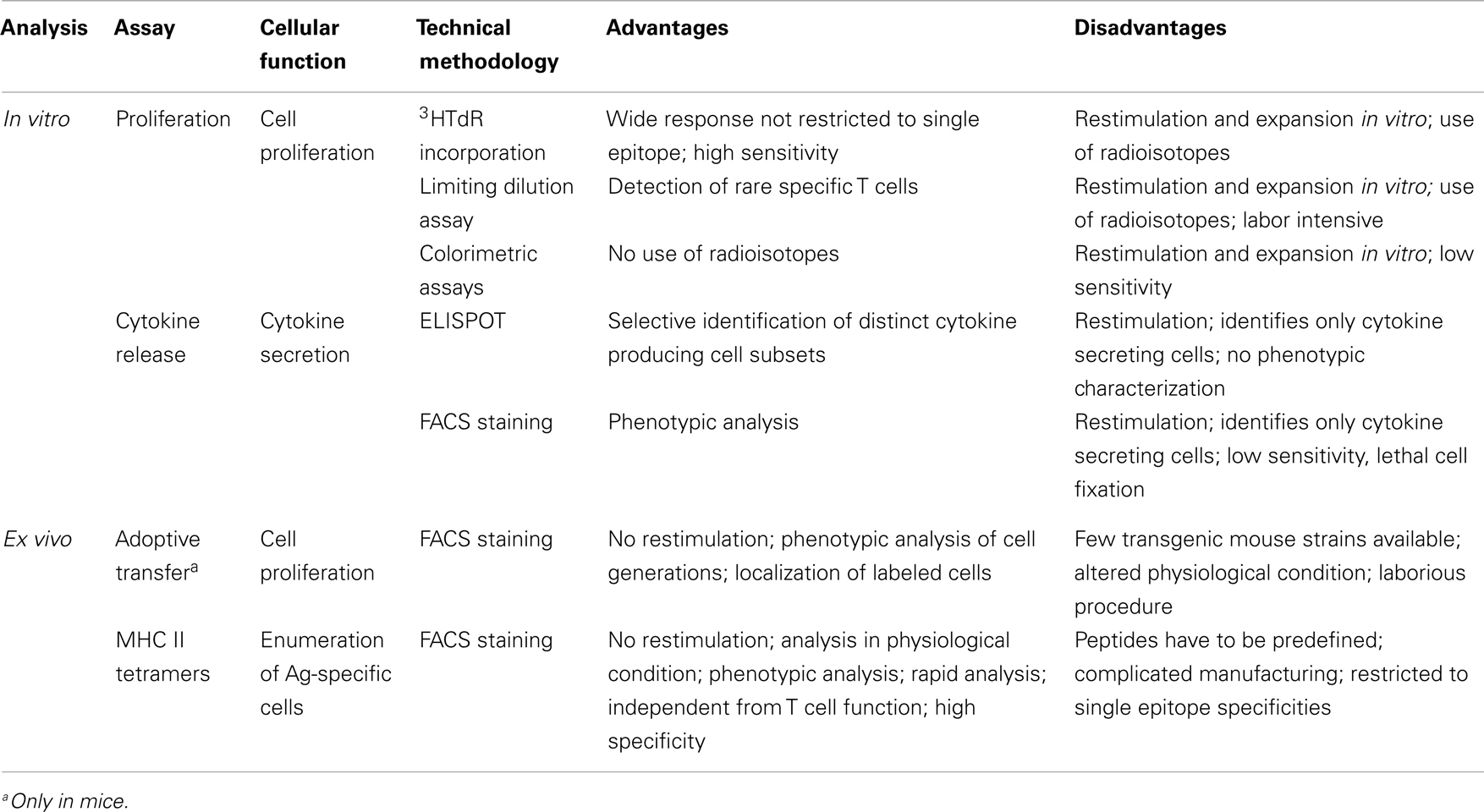
Table 1. Methods used for studying antigen-specific T cell priming in humans and animals.
In order to overcome the limitation of the low frequency of antigen-specific T cells in vivo, Jenkins and colleagues developed the adoptive transfer model of antigen-specific transgenic T cells into recipient mice (79). This system largely increases the number of antigen-specific naïve CD4+ T cells in vivo by employing TCR-transgenic mice that express a TCR specific for a defined peptide/MHC complex on most T cells, and thus allows the ex vivo analysis of their clonal expansion following antigenic stimulation (5). In order to track their proliferation, transgenic T lymphocytes are labeled with the vital dye 5-(and -6)-carboxyfluorescein diacetate succinimidyl ester (CFSE) (80) and then injected intravenously into immunocompetent recipient mice. Following vaccine administration, the T cell proliferation in the secondary lymphoid organs can be studied by flow cytometric analysis of CFSE dilution in the single-cell generations. The adoptive transfer method has proven to be a powerful tool for studying T cell primary responses to parenteral and mucosal immunization (35, 38–40, 43–45, 81–86), the role of the microenvironment for initiating T cell response in secondary lymphoid tissues (87) and the impact of aging on cellular immunity (88). Transgenic mice extensively used for studying the development of CD4+ T cell primary activation following immunization include DO11.10 (89) and OT-II (90) strains that contain rearranged TCR-Vα and -Vβ genes in the germline DNA encoding a TCR specific for chicken OVA peptide323–339 bound to I-A molecules in a context of H-2d and H-2b haplotype, respectively. Other transgenic models have been developed, such as SM1 RAG-2 deficient mice, that allow the visualization of Salmonella flagellin-specific CD4+ T cell responses (91), SMARTA transgenic mice, that produce CD4+ T cells expressing Va2 and Vb8.3 TCR specific for the lymphocytic choriomeningitis virus (LCMV) epitope gp61–80 (92), and Ag85B241–255 TCR-transgenic mice that allow to characterize tuberculosis-specific immune response (86).
Despite the important results obtained with this system, it has the limitation that the high number of naïve antigen-specific T cells transferred into recipient mice alters the physiologic conditions and can influence the immune response observed. Moreover, the study of the antigen-specific primary response is limited by the availability of the transgenic mouse strains with the TCR specific for a given model antigen.
The limitations of the methodology described above have been overcome with the development of MHC-peptide complexes. In 1996, the first work describing the use of a peptide-MHC class I complex for the identification and characterization of antigen-specific T lymphocytes was published (93). Initially developed for the study of CD8+ T cells, the technology has been extended to the class II system in the context of CD4+ T cells (6, 94) and has been applied to the study of human and murine T cells (95–97). This tool has provided an invaluable way to monitor T cell mediated immune responses and quantify the development of an antigen-dependent response. The technology allows identification of antigen-specific T cells based on the specificity of their surface TCR for particular MHC-peptide complexes. Since the affinity of TCR for a single peptide-loaded MHC molecule is generally low, multimerization of the peptide-MHC complexes is necessary for achieving much higher avidities for the TCR (93, 97). Today, the most prevalent multimer used consists of biotin-labeled peptide-MHC complexes bound to streptavidin molecules forming tetravalent structures (96). The peptide of interest can be covalently linked to the β-chain of the MHC molecule for the generation of MHC molecule-peptide complexes or it can be loaded on empty soluble class II molecules (95). Tetramer technology offers the advantage of phenotyping the antigen-specific cells by combining surface marker labeling and allows for the simultaneous detection of different antigen-specific CD4+ T cells by using multiple tetramers conjugated to different fluorescent molecules. The major limitations in the use of tetramers are that immunodominant peptides have to be predefined and that humans have very diverse HLA class II molecules. Moreover, the low frequency of antigen-specific CD4+ T cells in blood (generally 1/3000–30000) and the low avidity of TCR-MHC-peptide complex recognition are challenging issues for the tetramer technology. One strategy developed to overcome the first problem is the selection of the tetramer-positive cells by sorting with magnetic beads, so the antigen-specific population could be enriched as much as 10,000-fold (76, 98).
By using distinct MHC class II tetramers, Jenkins and colleagues have analyzed in the mouse model the primary response of CD4+ T cells specific for three different peptides [the protein FliC427–441 of Salmonella typhimurium, the OVA323–339, and the 2W1S variant of I-Eα protein52–68 (2W)] following intravenous immunization and correlated the primary response to the frequency of the respective naïve population size by combining the tetramer staining to the magnetic bead enrichment (76). Since the frequency of the naïve pool of 2W-specific CD4+ T cells in C57BL/6 mice, was the highest among the three peptides assessed, this peptide has been selected for its expression on Listeria monocytogenes and Leishmania major in order to track, by mean of the 2W-MHC class II tetramer, the peptide-specific T cell primary response following acute infection (99, 100). MHC class II tetramers have been used for identifying CD4+ T cell epitopes of Porphyromonas gingivalis proteins following oral infection of mice and as a tool for tracking and phenotyping specific effector and memory CD4+ T cells (101). In other murine studies, the magnitude and quality of the CD4+ T cell response induced by oral immunization with lipid-formulated BCG has been analyzed by using Mycobacterium tuberculosis Ag85B280–294-specific MHC class II tetramers, and compared with that induced by the subcutaneous immunization with BCG (102).
Mathematical and computational modeling can be employed as tool for integrating experimental data into a quantitative analysis of immune responses to antigens. Application of systems biology in vaccinology, named systems vaccinology, has recently been proposed as new powerful tool to model and characterize immune responses to vaccination and to predict vaccine immunogenicity and efficacy (8, 9). Systems vaccinology aims to model the immunological network, from molecules to cells to tissues, in order to predict vaccine immunogenicity. The identification of molecular signatures induced early after vaccination which correlate with and predict the later development of protective immune responses, represents a strategy to prospectively determine vaccine efficacy. Systems biology approaches provide a detailed level of investigation to better and fully analyze the network of interactions within vaccine-specific innate and adaptive immunity. All this information is expected to high impact on rational vaccine development, providing molecular prediction markers of vaccine immunogenicity, uncovering new correlates of vaccine efficacy, as well as guiding the design of new vaccine formulations and prime-boost strategies (7).
Systems biology represents therefore an attractive tool for studying T cell priming and modeling the initiation of the immune response following vaccination and predicting the priming properties of different vaccine formulations. The application of mathematical models can indeed be highly relevant to analyze antigen-specific T cell primary clonal expansion, based on the dilution of the CFSE dye. Quantitative analysis of T cell proliferation through mathematical models has been previously employed for in vitro studies of lymphocyte proliferation (103–105). On the contrary, the application to in vivo analysis raises several difficulties, mainly due to the fact that a LN is not an “isolated” site but is part of the complex immunological system.
Our group has recently employed a Multi-type Galton–Watson branching process with immigration (63, 106) to model in vivo CD4+ T cell priming and estimate the probabilities of a cell to enter in division, rest in quiescence or migrate/dye. This model has been successfully applied to analyze CD4+ T cell priming in mice immunized by different mucosal routes, such as vaginal or nasal, and has allowed the estimation of the probability of CD4+ T cells to enter into division within the draining LNs (63). Ongoing work is focused on modeling lymphocyte trafficking within the lymphatic systems, including both draining and distal LNs and spleen, in order to obtain further quantitative information and generate a model capable of predicting the amount and distribution of primed CD4+ T cells.
CD4+ T cell priming is an early biomarker of vaccine immunogenicity that should be considered as a critical parameter in the evaluation of vaccination strategies. The advances in understanding CD4+ T cell priming and the availability of latest generation technologies for its study open the way for its use in the rational design of vaccine formulations and prime-boost combinations. CD4+ T cell priming can be also considered an important biomarker for early prediction of vaccine immunogenicity and individual responsiveness to vaccination. The application of systems biology and mathematical modeling to the study of CD4+ T cell priming offers further opportunities to identify early signatures of vaccine immunogenicity and guide the design of next generation vaccines.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The research leading to these results has received funding from the European Union’s Seventh Framework Programme [FP7/2007–2013] under Grant Agreement No: 280873 “Advanced Immunization Technologies” (ADITEC).
1. Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol (2010) 17:1055–65. doi:10.1128/CVI.00131-10
2. Siegrist CA. Vaccine immunology. In: Plotkin AA, Orenstein WA, Offit PA editors. Vaccines. Philadelphia: Elsevier Inc (2008). p. 17–36.
3. Galli G, Medini D, Gorgogni E, Zedda L, Bardelli M, Malzone C, et al. Adjuvanted H5N1 vaccine induces early CD4+ T cell response that predicts long-term persistence of protective antibody levels. Proc Natl Acad Sci U S A (2009) 106:3877–82. doi:10.1073/pnas.0813390106
4. Nayak JL, Fitzgerals TF, Richards KA, Yang H, Treanor JJ, Sant AJ. CD4+ T-cell expansion predicts neutralizing antibody responses to monovalent, inactivated 2009 pandemic influenza A(H1N1) virus subtype H1N1 vaccine. J Infect Dis (2013) 207:297–305. doi:10.1093/infdis/jis684
5. Pape KA, Kearney ER, Khoruts A, Mondino A, Merica R, Chen ZM, et al. Use of adoptive transfer of T-cell-antigen-receptor-transgenic T cell for the study of T-cell activation in vivo. Immunol Rev (1997) 156:67–78. doi:10.1111/j.1600-065X.1997.tb00959.x
6. McHeyezer-Williams MG, Altman JD, Davis MM. Enumeration and characterization of memory cells in the TH compartment. Immunol Rev (1996) 150:5–21. doi:10.1111/j.1600-065X.1996.tb00693.x
7. Buonaguro L, Pulendran B. Immunogenomics and systems biology of vaccines. Immunol Rev (2011) 239:197–208. doi:10.1111/j.1600-065X.2010.00971.x
8. Li S, Nakaya HI, Kazmin DA, Oh JZ, Pulendran B. Systems biological approaches to measure and understand vaccine immunity in humans. Semin Immunol (2013) 25(3):209–18. doi:10.1016/j.smim.2013.05.003
9. Pulendran B, Li S, Nakaya HI. Systems vaccinology. Immunity (2010) 33:516–29. doi:10.1016/j.immuni.2010.10.006
10. Jelley-Gibbs DM, Strutt TM, McKinstry KK, Swain SL. Influencing the fates of CD4 T cells on the path to memory: lessons from influenza. Immunol Cell Biol (2008) 86:343–52. doi:10.1038/icb.2008.13
11. Lanzavecchia A, Sallusto F. Understanding the generation and function of memory T cell subsets. Curr Opin Immunol (2005) 17:326–32. doi:10.1016/j.coi.2005.04.010
12. Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell (2001) 106:263–6. doi:10.1016/S0092-8674(01)00455-X
13. Mempel TR, Henrickson S, von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427:154–9. doi:10.1038/nature02238
14. Obst R, van Santen HM, Mathis D, Benoist C. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. J Exp Med (2005) 201:1555–65. doi:10.1084/jem.20042521
15. Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity (2003) 19:47–57. doi:10.1016/S1074-7613(03)00175-4
16. Germain RN, Jenkins MK. In vivo antigen presentation. Curr Opin Immunol (2004) 16:120–5. doi:10.1016/j.coi.2003.11.001
17. Catron DM, Itano AA, Pape KA, Mueller DL, Jenkins MK. Visualizing the first 50 hr of the primary immune response to a soluble antigen. Immunity (2004) 21:341–7. doi:10.1016/j.immuni.2004.08.007
18. Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol Rev (2013) 252:12–23. doi:10.1111/imr.12032
19. Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol (2012) 24:297–302. doi:10.1016/j.coi.2012.01.014
20. Fazilleau N, McHeyezer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol (2009) 10:375–84. doi:10.1038/ni.1704
21. McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of B cell memory. Nat Rev Immunol (2012) 12:24–34. doi:10.1038/nri3128
22. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
23. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med (2012) 209:1241–53. doi:10.1084/jem.20120994
24. Pepper M, Jenkins MK. Origins of CD4(+) effector and central memory T cells. Nat Immunol (2011) 12:467–71. doi:10.1038/ni.2038
25. Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dörner T, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol (2006) 6:741–50. doi:10.1038/nri1886
26. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:763. doi:10.1146/annurev.immunol.22.012703.104702
27. Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature (2001) 410:101–5. doi:10.1038/35065111
28. Fazilleau N, Eisenbraun MD, Malherbe L, Ebright JN, Pogue-Caley RR, McHeyezer-Williams LJ, et al. Lymphoid reservoirs of antigen-specific memory T helper cells. Nat Immunol (2007) 8:753–61. doi:10.1038/ni1472
29. van Duin D, Medzhitov R, Shaw AC. Triggering TLR signaling in vaccination. Trends Immunol (2006) 27:49–55. doi:10.1016/j.it.2005.11.005
30. Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: distinct cytokine responses of B cells and dendritic cells. Eur J Immunol (2007) 37:3040–53. doi:10.1002/eji.200636483
31. Pulendran B. Modulating vaccine responses with dendritic cells and toll-like receptors. Immunol Rev (2004) 199:227–50. doi:10.1111/j.0105-2896.2004.00144.x
32. Harandi AM, Medaglini D. Mucosal adjuvants. Curr HIV Res (2010) 8:330–5. doi:10.2174/157016210791208695
33. Harandi AM, Medaglini D, Shattock RJ, Working Group convened by EUROPRISE. Vaccine adjuvants: a priority for vaccine research. Vaccine (2010) 28:2363–6. doi:10.1016/j.vaccine.2009.12.084
34. McKee AS, Burchill MA, Munks MW, Jin L, Kappler JW, Friedman RS, et al. Host DNA released in response to aluminum adjuvant enhances MHC class II-mediated antigen presentation and prolongs CD4 T-cell interactions with dendritic cells. Proc Natl Acad Sci U S A (2013) 110:E1122–31. doi:10.1073/pnas.1300392110
35. Ciabattini A, Pettini E, Fiorino F, Prota G, Pozzi G, Medaglini D. Distribution of primed T cells and antigen-loaded antigen presenting cells following intranasal immunization in mice. PLoS One (2011) 6:e19346. doi:10.1371/journal.pone.0019346
36. McAleer JP, Vella AT. Educating CD4 T cells with vaccine adjuvants: lessons from lipopolysaccharide. Trends Immunol (2010) 31:429–35. doi:10.1016/j.it.2010.08.005
37. Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TM. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science (2007) 316:1628–32. doi:10.1126/science.1138963
38. Marks EM, Helgeby A, Andersson JO, Schön K, Lycke NY. CD4+ T-cell immunity in the female genital tract is critically dependent on local mucosal immunization. Eur J Immunol (2011) 41:2642–53. doi:10.1002/eji.201041297
39. Luci C, Hervouet C, Rousseau D, Holmgreen J, Czerkinsky C, Anjuere F. Dendritic cell-mediated induction of mucosal cytotoxic responses following intravaginal immunization with the nontoxic B subunit of cholera toxin. J Immunol (2006) 176:2749–57.
40. Pettini E, Ciabattini A, Pozzi G, Medaglini D. Adoptive transfer of transgenic T cells to study mucosal adjuvants. Methods (2009) 49:340–5. doi:10.1016/j.ymeth.2009.03.026
41. Mohanan D, Slütter B, Henriksen-Lacey M, Bouwstra JA, Perrie Y, Kundig TM, et al. Administration routes affect the quality of immune responses: a cross-sectional evaluation of particulate antigen-delivery systems. J Control Release (2010) 147:342–9. doi:10.1016/j.jconrel.2010.08.012
42. Alkadah A, Thiam F, Mounier M, Charpilienne A, Poncet D, Kohli E, et al. Different profile and distribution of antigen specific T cells induced by intranasal and intrarectal immunization with rotavirus 2/6-VLP with and without LT-R192G. Vaccine (2013) 31:1924–30. doi:10.1016/j.vaccine.2013.02.019
43. Ciabattini A, Pettini E, Arsenijevic S, Pozzi G, Medaglini D. Intranasal immunization with vaccine vector Streptococcus gordonii elicits primed CD4+ and CD8+ cells in the genital and intestinal tracts. Vaccine (2010) 28:1226–33. doi:10.1016/j.vaccine.2009.11.021
44. Ciabattini A, Pettini E, Andersen P, Pozzi G, Medaglini D. Primary activation of antigen-specific naive CD4+ and CD8+ T cells following intranasal vaccination with recombinant bacteria. Infect Immun (2008) 76:5817–25. doi:10.1128/IAI.00793-08
45. Medaglini D, Ciabattini A, Cuppone AM, Costa C, Ricci S, Costalonga M, et al. In vivo activation of naive CD4+ T cells in nasal mucosa-associated lymphoid tissue following intranasal immunization with recombinant Streptococcus gordonii. Infect Immun (2006) 74:2760–6. doi:10.1128/IAI.74.5.2760-2766.2006
46. Fiorino F, Pettini E, Pozzi G, Medaglini D, Ciabattini A. Prime-boost strategies in mucosal immunization affect local IgA production and the type of Th response. Front Immunol (2013) 4:128. doi:10.3389/fimmu.2013.00128
47. Hu J, Havenar-Daughton C, Crotty S. Modulation of SAP dependent T:B cell interactions as a strategy to improve vaccination. Curr Opin Virol (2013) 3:363–70. doi:10.1016/j.coviro.2013.05.015
48. Aldhamen YA, Appledorn DM, Seregin SS, Liu CJ, Schuldt NJ, Godbehere S, et al. Expression of the SLAM family of receptors adapter EAT-2 as a novel strategy for enhancing beneficial immune responses to vaccine antigens. J Immunol (2011) 186:722–32. doi:10.4049/jimmunol.1002105
49. Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A (2012) 109:1080–5. doi:10.1073/pnas.1112648109
50. Streeck H, D’Souza MP, Littman DR, Crotty S. Harnessing CD4+ T cell responses in HIV vaccine development. Nat Med (2013) 19:143–9. doi:10.1038/nm.3054
51. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest (2012) 122:3281–94. doi:10.1172/JCI63039
52. Spensieri F, Borgogni E, Zedda L, Bardelli M, Buricchi F, Volpini G, et al. Human circulating influenza-CD4+ ICOS1+IL-21+ T cells expand after vaccination, exert helper function, and predict antibody responses. Proc Natl Acad Sci U S A (2013) 110:14330–5. doi:10.1073/pnas.1311998110
53. Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol (2006) 6:148–58. doi:10.1038/nri1777
54. Brandtzaeg P, Pabst R. Let’s go mucosal: communication on slippery ground. Trends Immunol (2005) 25:570–7. doi:10.1016/j.it.2004.09.005
55. Pavot V, Rochereau N, Genin C, Verrier B, Paul S. New insights in mucosal vaccine development. Vaccine (2012) 30:142–54. doi:10.1016/j.vaccine.2011.11.003
56. Wira CR, Fahey JV, Sentman CL, Pioli PA, Shen L. Innate and adaptive immunity in female genital tract: cellular responses and interactions. Immunol Rev (2005) 206:306–35. doi:10.1111/j.0105-2896.2005.00287.x
57. Ciabattini A, Cuppone AM, Pulimeno R, Iannelli F, Pozzi G, Medaglini D. Stimulation of human monocytes with the gram-positive vaccine vector Streptococcus gordonii. Clin Vaccine Immunol (2006) 13:1037–43. doi:10.1128/CVI.00110-06
58. Oggioni MR, Pozzi G. A host-vector system for heterologous gene expression in Streptococcus gordonii. Gene (1996) 169:85–90. doi:10.1016/0378-1119(95)00775-X
59. Pozzi G, Oggioni MR. The human oral commensal Streptococcus gordonii as live vector for vaccines. In: Totolian A editor. Pathogenic Streptococci: Present and Future. St. Petersburg: Lancer Publications (1994). p. 163–5.
60. Ricci S, Medaglini D, Marcotte H, Olsen A, Bjorck L. Immunoglobulin-binding domains of peptostreptococcal protein L enhance vaginal colonization of mice by Streptococcus gordonii. Microb Pathog (2001) 30:229–35. doi:10.1006/mpat.2000.0427
61. Medaglini D, Ricci S, Maggi T, Rush CM, Manganelli R, Oggioni MR, et al. Recombinant gram-positive bacteria as vehicles of vaccine antigens. Biotechnol Annu Rev (1997) 3:297–312. doi:10.1016/S1387-2656(08)70038-3
62. Ciabattini A, Giomarelli B, Parigi R, Chiavolini D, Pettini E, Arico B, et al. Intranasal immunization of mice with recombinant Streptococcus gordonii expressing NadA of Neisseria meningitidis induces systemic bactericidal antibodies and local IgA. Vaccine (2008) 26:4244–50. doi:10.1016/j.vaccine.2008.05.049
63. Pettini E, Prota G, Ciabattini A, Boianelli A, Fiorino F, Pozzi G, et al. Vaginal immunisation to elicit primary T-cell activation and dissemination. PLoS One (Forthcoming 2013). doi:10.1371/journal.pone.0080545
64. Woodland D. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol (2004) 25:98–104. doi:10.1016/j.it.2003.11.009
65. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med (2009) 361:2209–20. doi:10.1056/NEJMoa0908492
66. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet (2013) 381:1021–8. doi:10.1016/S0140-6736(13)60177-4
67. Sheehy SH, Duncan CJ, Elias SC, Biswas S, Collins KA, O’Hara GA, et al. Phase Ia clinical evaluation of the safety and immunogenicity of the Plasmodium falciparum blood-stage antigen AMA1 in ChAd63 and MVA vaccine vectors. PLoS One (2012) 7:e31208. doi:10.1371/journal.pone.0031208
68. McCluskie MJ, Weeratna RD, Payette PJ, Davis HL. Parenteral and mucosal prime-boost immunization strategies in mice with hepatitis B surface antigen and CpG DNA. FEMS Immunol Med Microbiol (2002) 32:179–85. doi:10.1111/j.1574-695X.2002.tb00551.x
69. Glynn A, Roy CJ, Powell BS, Adamovicz JJ, Freytag LC, Clements JD. Protection against aerosolized Yersinia pestis challenge following homologous and heterologous prime-boost with recombinant plague antigens. Infect Immun (2005) 73:5256–61. doi:10.1128/IAI.73.8.5256-5261.2005
70. Mapletoft J, Latimer L, Babiuk LA, van Drunen Littel-van den Hurk S. Intranasal immunization of mice with a bovine respiratory syncytial virus vaccine induces superior immunity and protection compared to those by subcutaneous delivery or combinations of intranasal and subcutaneous prime-boost strategies. Clin Vaccine Immunol (2010) 17:23–35. doi:10.1128/CVI.00250-09
71. Pattani A, McKay PF, Garland MJ, Curran RM, Migalska K, Cassidy CM, et al. Microneedle mediated intradermal delivery of adjuvanted recombinant HIV-1 CN54gp140 effectively primes mucosal boost inoculations. J Control Release (2012) 162:529–37. doi:10.1016/j.jconrel.2012.07.039
72. Steede NK, Rust BJ, Hossain MM, Freytag LC, Robinson JE, Landry SJ. Shaping T cell – B cell collaboration in the response to human immunodeficiency virus type 1 envelope glycoprotein gp120 by peptide priming. PLoS One (2013) 8:e65748. doi:10.1371/journal.pone.0065748
73. Alam S, Knowlden ZAG, Sangster MY, Sant AJ. CD4 T cell help is limiting and selective during the primary B cell response to influenza infection. J Virol (Forthcoming 2013). doi:10.1128/JVI.02077-13
74. Sette A, Moutaftsi M, Moyron-Quiroz J, McCausland MM, Davies DH, Johmston RJ, et al. Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity (2008) 28:847–58. doi:10.1016/j.immuni.2008.04.018
75. Yin L, Calvo-Calle JM, Cruz J, Newman FK, Frey SE, Ennis FA, et al. CD4+ T cells provide intermolecular help to generate robust antibody responses in vaccinia virus-vaccinated humans. J Immunol (2013) 190:6023–33. doi:10.4049/jimmunol.1202523
76. Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity (2007) 27:203–13. doi:10.1016/j.immuni.2007.07.007
77. Dunkley ML, Husband AJ. Distribution and functional characteristics of antigen-specific helper T cells arising after Peyer’s patch immunization. Immunology (1987) 61:475–82.
78. Bacher P, Scheffold A. Flow-cytometric analysis of rare antigen-specific T cells. Cytometry A (2013) 83:692–701. doi:10.1002/cyto.a.22317
79. Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity (1994) 1:327–39. doi:10.1016/1074-7613(94)90084-1
80. Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods (2000) 243:147–54. doi:10.1016/S0022-1759(00)00231-3
81. Srinivasan A, Foley J, McSorley SJ. Massive number of antigen-specific CD4 T cells during vaccination with live attenuated Salmonella causes interclonal competition. J Immunol (2004) 172:6884–93.
82. Rowe HM, Lopes L, Ikeda Y, Bailey R, Barde I, Zenke M, et al. Immunization with a lentiviral vector stimulates both CD4 and CD8 T cell responses to an ovalbumin transgene. Mol Ther (2006) 13:310–9. doi:10.1016/j.ymthe.2005.08.025
83. Buhler OT, Wiedig CA, Schmid Y, Grassl GA, Bohn E, Autenrieth IB. The Yersinia enterocolitica invasin protein promotes major histocompatibility complex class I- and class II-restricted T-cell responses. Infect Immun (2006) 74:4322–9. doi:10.1128/IAI.00260-06
84. Loeffler DI, Schoen CU, Goebel W, Pilgrim S. Comparison of different live vaccine strategies in vivo for delivery of protein antigen or antigen-encoding DNA and mRNA by virulence-attenuated Listeria monocytogenes. Infect Immun (2006) 74:3946–57. doi:10.1128/IAI.00112-06
85. Rush C, Mitchell T, Garside P. Efficient priming of CD4+ and CD8+ T cells by DNA vaccination depends on appropriate targeting of sufficient levels of immunologically relevant antigen to appropriate processing pathways. J Immunol (2002) 169:4951–60.
86. Henriksen-Lacey M, Christensen D, Bramwell VW, Lindenstrom T, Agger EM, Andersen P, et al. Liposomal cationic charge and antigen adsorption are important properties for the efficient deposition of antigen at the injection site and ability of the vaccine to induce a CMI response. J Control Release (2010) 145:102–8. doi:10.1016/j.jconrel.2010.03.027
87. Lee Y, Eo SK, Rouse RD, Rouse BT. Influence of CCR7 ligand DNA preexposure on the magnitude and duration of immunity. Virology (2003) 312:169–80. doi:10.1016/S0042-6822(03)00199-5
88. Lefebvre JS, Maue AC, Eaton SM, Lanthier PA, Tighe M, Haynes L. The aged microenvironment contributes to the age-related functional defects of CD4 T cells in mice. Aging Cell (2012) 11:732–40. doi:10.1111/j.1474-9726.2012.00836.x
89. Murphy KM, Heimberger AM, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+ CD8+ TCRlo thymocytes in vivo. Science (1990) 250:1720–3. doi:10.1126/science.2125367
90. Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol (1998) 76:34–40. doi:10.1046/j.1440-1711.1998.00709.x
91. McSorley SJ, Asch S, Costalonga M, Reinhardt RL, Jenkins MK. Tracking salmonella-specific CD4 T cells in vivo reveals a local mucosal response to a disseminated infection. Immunity (2002) 16:365–77. doi:10.1016/S1074-7613(02)00289-3
92. Oxenius A, Bachmann MF, Zinkernagel RM, Hengartner H. Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur J Immunol (1998) 28:390–400. doi:10.1002/(SICI)1521-4141(199801)28:01<390::AID-IMMU390>3.0.CO;2-O
93. Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science (1996) 274:94–6. doi:10.1126/science.274.5284.94
94. Crawford F, Kozono H, White J, Marrack P, Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity (1998) 8:675–82. doi:10.1016/S1074-7613(00)80572-5
95. Vollers SS, Stern LJ. Class II major histocompatibility complex tetramer staining: progress, problems, and prospects. Immunology (2008) 123:305–13. doi:10.1111/j.1365-2567.2007.02801.x
97. Cochran JR, Cameron TO, Stern LJ. The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity (2000) 12:241–50. doi:10.1016/S1074-7613(00)80177-6
98. Scriba TJ, Purbhoo M, Day CL, Robinson N, Fidler S, Fox J, et al. Ultrasensitive detection and phenotyping of CD4+ T cells with optimized HLA class II tetramer staining. J Immunol (2005) 175:6334–43.
99. Ertelt JM, Rowe JH, Johanns TM, Lai JC, McLachlan JB, Way SS. Selective priming and expansion of antigen-specific Foxp3- CD4+ T cells during Listeria monocytogenes infection. J Immunol (2009) 182:3032–8. doi:10.4049/jimmunol.0803402
100. Pagan AJ, Peters NC, Debrabant A, Ribeiro-Gomes F, Pepper M, Karp CL, et al. Tracking antigen-specific CD4+ T cells throughout the course of chronic Leishmania major infection in resistant mice. Eur J Immunol (2013) 43:427–38. doi:10.1002/eji.201242715
101. Bittner-Eddy PD, Fischer LA, Costalonga M. Identification of gingipain-specific I-Ab -restricted CD4+ T cells following mucosal colonization with Porphyromonas gingivalis in C57BL/6 mice. Mol Oral Microbiol (2013) 28:452–66. doi:10.1111/omi.12038
102. Ancelet LR, Aldwell FE, Rich FJ, Kirman JR. Oral vaccination with lipid-formulated BCG induces a long-lived, multifunctional CD4(+) T cell memory immune response. PLoS One (2012) 7:e45888. doi:10.1371/journal.pone.0045888
103. Yates A, Chan C, Strid J, Moon S, Callard R, George A, et al. Reconstruction of cell population dynamics using CFSE. BMC Bioinformatics (2007) 8:196. doi:10.1186/1471-2105-8-196
104. Yakovlevi AY, Yanevi MN. Relative frequencies in multitype branching processes. Ann Appl Probab (2009) 19:1–14. doi:10.1214/08-AAP539
Keywords: T cell priming, vaccination, CD4+ T cells, mucosal immunity, adoptive transfer, MHC class II tetramers
Citation: Ciabattini A, Pettini E and Medaglini D (2013) CD4+ T cell priming as biomarker to study immune response to preventive vaccines. Front. Immunol. 4:421. doi: 10.3389/fimmu.2013.00421
Received: 27 September 2013; Accepted: 20 November 2013;
Published online: 04 December 2013.
Edited by:
Mats Bemark, University of Gothenburg, SwedenReviewed by:
Margaret Lorraine Dunkley, Hunter Immunology Ltd., AustraliaCopyright: © 2013 Ciabattini, Pettini and Medaglini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annalisa Ciabattini and Donata Medaglini, Laboratorio di Microbiologia Molecolare e Biotecnologia (LA.M.M.B.), Dipartimento di Biotecnologie Mediche, Università di Siena, Policlinico Le Scotte, V lotto piano 1, Viale Bracci, Siena 53100, Italy e-mail:YW5uYWxpc2EuY2lhYmF0dGluaUB1bmlzaS5pdA==;ZG9uYXRhLm1lZGFnbGluaUB1bmlzaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.