 Penelope A. Morel
Penelope A. Morel
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 December 2013
Sec. Molecular Innate Immunity
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00415
This article is part of the Research Topic Dendritic Cell Control of Immune Responses View all 12 articles
Type 1 diabetes (T1D) is a T cell mediated autoimmune disease characterized by immune mediated destruction of the insulin-producing β cells in the islets of Langerhans. Dendritic cells (DC) have been implicated in the pathogenesis of T1D and are also used as immunotherapeutic agents. Plasmacytoid (p)DC have been shown to have both protective and pathogenic effects and a newly described merocytic DC population has been shown to break tolerance in the mouse model of T1D, the non-obese diabetic (NOD) mouse. We have used DC populations to prevent the onset of T1D in NOD mice and clinical trials of DC therapy in T1D diabetes have been initiated. In this review we will critically examine the recent published literature on the role of DC subsets in the induction and regulation of the autoimmune response in T1D.
Type 1 diabetes (T1D) is an autoimmune disease characterized by immune mediated destruction of the insulin-producing β cells in the islets of Langerhans of the pancreas. The pathogenesis of T1D is multifactorial with genetic, immunological, metabolic, and environmental factors all contributing (1). It begins with a loss of self-tolerance to islet-derived self-antigens, which usually occurs early in life. This could occur as a result of a viral infection targeting the pancreas or following pancreatic remodeling. These insults lead to the death of β cells, release of self-antigens, and induction of inflammatory cytokines such as TNF-α and IL-1-β. Dendritic cells (DC) present within the pancreas take up released β-cell-derived antigens and migrate to the draining lymph nodes (LN) and activate naïve islet-specific CD4 and CD8 T cells. Depending on the signals delivered to the T cell by DCs the T cells will differentiate into either inflammatory effector cells such as T helper (Th)1 cells or anti-inflammatory Th2 or regulatory (Treg) cells. Activated islet-specific T cells then migrate to the pancreas where they infiltrate and collect around the islets. The early infiltrates appear to be dominated by Th2 and Treg cells; this is termed peri-insulitis as there is little invasion into the islet. At some point, the infiltrate becomes invasive and begins to enter and destroy the β cells, and the balance between the regulatory and inflammatory T cell populations is lost. The precise factors that trigger both loss of self-tolerance and the development of invasive insulitis are not well understood. DCs play important roles at all stages of the autoimmune response in T1D due to their pivotal role in activating naïve T cells and in maintaining self-tolerance (2). This review will explore recent developments in our understanding of the roles DC play in the pathogenesis, and prevention, of T1D (Figure 1) as well as the therapeutic potential of DCs for the prevention and treatment of T1D. Much of the work in this area has been focused on the non-obese diabetic (NOD) model of the disease. This mouse strain spontaneously develops diabetes and shares many of the genetic and immunological features of the human disease (3). Where possible we will highlight similarities and differences between NOD mouse studies and relevant studies in human T1D patients.
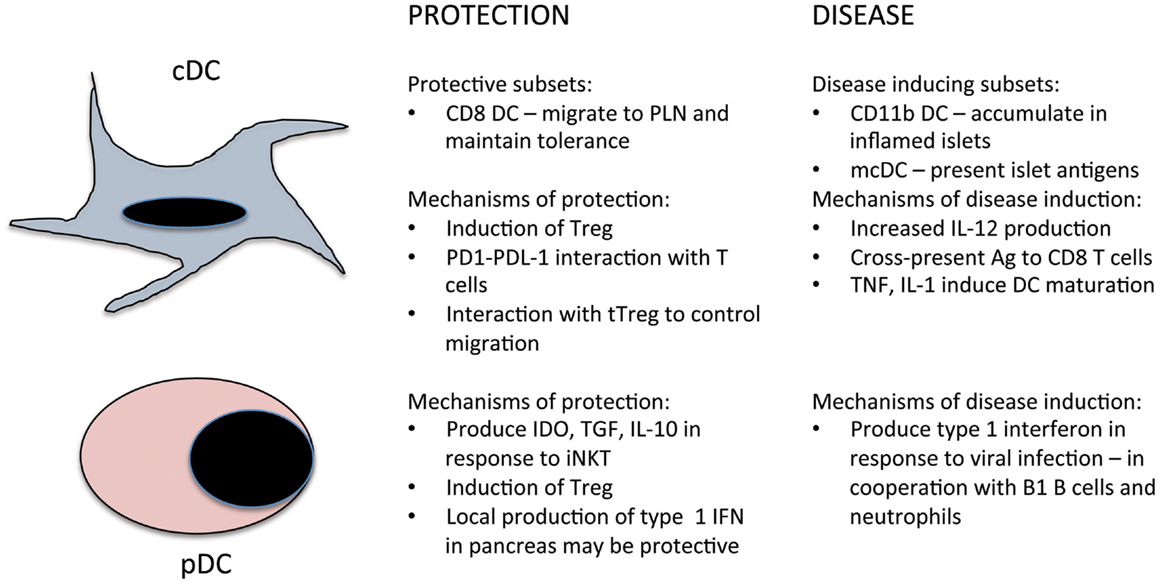
Figure 1. Summary of the roles of cDC and pDC in the prevention and induction of T1D.
Since their discovery by Ralph Steinman in the 1970s (4–6) DCs have been the subject of intense study. DCs are antigen presenting cells (APC) that bridge the innate and adaptive immune systems (7). They act as sentinels of the immune system through their location in peripheral tissues, where they form a dense network, and their ability to respond to pathogens through expression of pathogen recognition receptors such as Toll-like receptors (TLR). While in peripheral tissues DCs have an immature phenotype characterized by low levels of major histocompatibility complex (MHC) and co-stimulatory molecules (CD80, CD86) and a high endocytic rate; the function of tissue DCs is the detection and processing of antigen. Upon interaction with a pathogen DCs become activated, undergo maturation and migrate to the draining LN. This maturation leads to an increase in the expression of MHC, CD80, and CD86, a decrease in endocytosis and a change in the pattern of chemokine receptor expression. Mature DCs entering the T cell zone of the draining lymph node are primed to present antigen to naïve T cells. Even in the absence of pathogen there is a steady state migration of DC from the periphery to the LN and this is necessary for the maintenance of peripheral tolerance (8).
There are two main classes of DC: plasmacytoid DCs (pDCs) and conventional or classical DCs (cDCs) (7). pDCs are primarily located in the blood and lymphoid organs and produce high levels of type 1 interferon (IFN) following engagement of TLRs with foreign nucleic acids (9). pDCs express a restricted set of TLRs, TLR7, and TLR9, that recognize foreign RNA and DNA respectively as well as several unique markers that distinguish them from cDCs. These include BDCA-2 and ILT7 for human pDCs and SiglecH and Bst2 for murine pDCs. In the mouse pDCs are commonly identified as CD11clow B220+ SiglecH+ Bst2+. Human pDCs are CD11c− and in addition to BDCA-2 and ILT7 also express CD123 and BDCA-4 (9). There are two main populations of cDCs in mouse: CD11c+ CD11b− CD8α+ Clec9A+ and CD11c+ CD11b+ CD8α− Clec9A− commonly referred to as CD8 DC and CD11b cDC respectively. Both CD8 and CD11b cDC arise from a common precursor characterized by expression of the C-type lectin receptor DNGR-1, but it appears that pDC arise from a distinct precursor as yet to be defined (10, 11). Recently the CD11b cDC population has been further subdivided on the basis of endothelial cell selective adhesion molecule (ESAM) expression (12). Similar populations can be identified in human such that CD11c+ Clec9A+ BDCA-3+ DCs correspond to CD8 cDC and CD11c+ CD11b+ BDCA-1+ DCs are analogous to CD11b cDC. These populations mainly reside in the secondary lymphoid organs and are present during steady state. Similar populations exist in peripheral tissues but they express a slightly different set of markers (7). CD11c+ CD11b− CD103+ DC and CD11c+ CD11b+ CD103+ DC in the periphery are equivalent to the CD8 cDC and CD11b cDC populations respectively (13).
The two cDC populations also have distinct functions with respect to pathogen recognition, cytokine production, and T cell activation. CD8 and CD103 cDCs express a unique pattern of TLR and other pathogen recognition receptors: they are the only subset to express TLR3 and TLR11 and also uniquely express the C-type lectin Clec9A, DEC205 (CD205), and langerin (CD207) (7, 9). CD8 and CD103 cDC are specialized in the cross-presentation of externally derived antigen on MHC class I for the activation of naïve CD8 T cells. The recent advances in the molecular mechanisms of cross-presentation are covered in a review included in this Research Topic (14). In addition, CD8 cDC appear to preferentially activate CD8 T cells due to the fact that they produce high levels of the cytokines interleukin (IL)-12 and IL-15 that are important for the differentiation and survival of CD8 T cells (15–17). CD8 and CD103 cDCs are the main producers of IL-12, an important cytokine in the differentiation of inflammatory Th1 cells (15, 16, 18). There are also reports suggesting a role for CD8 and CD103 cDCs in the maintenance of peripheral self-tolerance through the deletion of self-reactive T cells (19, 20) and the induction of Treg cells (21). While CD11b cDC are capable of cross-presentation and CD8 T cell activation they appear to be most effective in antigen presentation and activation of CD4 T cells (22) In addition the dermal and intestinal CD11b+ CD103+ DCs are strong inducers of Treg due to their expression of aldehyde dehydrogenase, the enzyme that converts Vitamin A to retinoic acid, necessary for Treg conversion (23, 24). Several comprehensive reviews on DC subsets and their development have recently been published and can be consulted for further details (7, 9, 13).
T and B cell tolerance to self-antigens is acquired during development through clonal deletion of self-reactive cells. DC play an important role in the process of central tolerance in the thymus as they present an array of self-antigens to developing T cells (25). If the T cells have a high affinity for self-antigen they undergo apoptosis and are deleted. Those T cells with a moderate affinity for self are induced to differentiate into forkhead transcription factor (Foxp3)+ suppressor cells (26) known as thymus (t)Treg (27). In contrast, peripheral (p)Treg cells arise in the periphery upon Ag contact and several subsets of pTreg can be induced depending on the signals they encounter (28). These include IL-10 (29), TGF-β (30), retinoic acid or vitamin D3 (31, 32), low Ag dose (33), and specific DC subsets (21, 34, 35). As the study of DC biology has evolved, it has become apparent that it is possible to induce the differentiation of Treg using DCs conditioned by immunomodulatory agents (36–39). For example, the presence of TGF-β during the primary stimulation induces the production and expansion of Foxp3+ Treg (35). In addition it has been possible to induce IL-10-producing T cells with DC generated using a combination of GM-CSF, TNF-α, and IL-10 (39). Studies have shown that DC producing IDO induce the generation of Treg from naïve T cells (40). Other reports have suggested that immature DC induce the development of functional suppressor cells (36) and that immature DC might, therefore, be therapeutically useful in autoimmunity (37, 38). Recent studies using both nominal and self-Ags have demonstrated that targeting Ags to CD8 cDC in the LN or spleen in the absence of an inflammatory stimulus results in the generation of Treg (41, 42). Similar results have been obtained when Ags were targeted to CD8− DCIR2+ DC (21). The CD11b cDC subset has been shown to induce Treg proliferation and expansion in the presence of GM-CSF (43). A recent paper has described a feedback loop in which the number of Tregs in the periphery can be manipulated by altering the relative number of DCs suggesting that DCs play an important role in regulating pTreg numbers (44). We have recently suggested that DC in the periphery may function to maintain tolerance by presenting endogenous self-antigens to naïve T cells thereby inducing pTreg (45).
Early studies of NOD mouse revealed abnormalities in the development of myeloid cells demonstrating a reduced proliferative capacity of the bone marrow in response to GM-CSF and M-CSF (46). Several groups, including our own, have identified defects in the development of specific DC subsets in the spleen and other lymphoid organs (47–49). These defects result in a decrease in the numbers of the CD8 cDC population (50, 51), a population which has been shown to have regulatory function (52–54), with a concomitant increase in the CD11b cDC population. Recent studies have also shown that treatment of NOD with Flt 3 ligand (FL) protects mice from diabetes development and this is correlated with an enhancement in the number of CD8 cDC (55, 56). Our own studies demonstrated that treatment of NOD mice with FL increased the number of CD8 cDC and that transfer of these cells to prediabetic NOD mice could have a partial effect in preventing diabetes (50). However a recent study suggested that the timing of FL treatment was critical since disease would be exacerbated if autoimmune CD8 T cells were already present (57).
Several studies have examined the function of bone marrow-derived and isolated DC populations with respect to cytokine production and T cell differentiation and the results have not been consistent. It has been reported by some investigators that NOD DC produce higher levels of the Th1-driving cytokine IL-12 (58–60) while others failed to find such an association (49, 50, 56). Macrophages also appear to be an important source of IL-12 in NOD mice (51, 61). Adenosine plays an important role in modulating the immune response to tissue inflammation (62, 63) and a recent study found that NOD DC express higher levels of adenosine deaminase (ADA), the enzyme responsible for catabolic degradation of adenosine, and that transfer of ADA deficient DC to NOD mice protected them from diabetes (64). These studies suggest that NOD mice have imbalances in DC subsets and alterations in DC function that may contribute to pathogenesis of T1D.
Dendritic cells are important in both central and peripheral tolerance through the deletion of self-reactive cells and the induction of Treg. NOD mice have defects in central and peripheral tolerance mechanisms (65, 66). Recent studies have shown that the number and/or function of Treg cells decrease as NOD mice age and this is associated with onset of diabetes (67). The development and maintenance of Treg in NOD mice is highly dependent on the presence of co-stimulatory molecules CD80 and CD86 (68). In addition it was recently shown that interaction between the inhibitory molecules PD-1 on T cells and PDL-1 on DC is necessary for the maintenance of tolerance in NOD mice (69). Blockade of this interaction resulted in increased DC/T cell interaction time in the islets and T cell activation leading to the generation of autoreactive effector cells (69). Another study came to the same conclusion by using different antigen constructs to increase the time of antigen presentation (70). IL-2 and signaling via IL-2R are also critical for Treg development and maintenance (71). Recent studies have identified defects in IL-2R signaling in T1D patients (72) and the diabetes susceptibility locus, Idd3, which contains IL-2 was recently shown to control Treg function through an effect on APC (73–75). Several early reports demonstrated that the inflammatory cytokine TNF-α plays an important role in the initiation of T1D (76) and more recently this has been attributed to effects of TNF-α on DC subsets (77). In this study administration of TNF-α to NOD mice was shown to decrease the number of CD8 DC, increase the CD11b DC population, and the DC had a more mature phenotype and activated islet-specific T cells (77) A recent study describing the depletion of Treg in NOD mice showed that the increase in diabetes in these animals was associated with aggressive infiltration of pancreatic islets by DC rather than CD4 T cells (78). These results suggest that tTreg in this context may prevent autoimmunity by controlling the migration of cDC into the islet.
Analysis of the phenotype and function of BM-derived DC in NOD mice have suggested that these cells produce higher levels of IL-12p70, and that this related to increased expression of NFκB (58). Our own studies suggest that BM-derived DC from NOD can produce higher or lower levels of IL-12p70 depending on the culture conditions and activation stimuli used (49, 79, 80). Recent studies using DTR transgenic mice that allow the targeted depletion of macrophages or DC have demonstrated that the CD11b cDC population is responsible for presenting antigen to autoreactive T cells (81). Ablation of these cells protected NOD mice from diabetes development. The interaction of iNKT cells with cDC was shown to lead to either protection via induction of Treg or exacerbation of disease if the interaction occurred at the same time as TLR4 ligation (82).
The role of pDC in diabetes pathogenesis is somewhat more controversial and there are conflicting reports in the literature. Depletion of pDC using the CD11c-DTR system caused accelerated disease (81), suggesting a protective role for pDC. The protection induced by pDC was correlated with increased local production of IDO and increased numbers of NKT cells in the pancreas. In a model of virus infection it was shown that activation of iNKT cells stimulated TGF-β production from pDC, which led to the induction of Treg and protection from diabetes (83). In contrast, another study found that IFN-α could be detected in the pancreas of NOD mice at early ages and that blocking the ability of mice to respond to IFN-α prevented diabetes (84). Furthermore these investigators showed that depletion of pDC using a depleting antibody reduced the incidence of diabetes (85). Another study revealed a crosstalk between B-1a B cells, neutrophils and pDC that contributed to the induction of diabetes via type 1 IFN production (86). This study suggests that DNA from dying β cells together with antibodies from B-1a B cells form complexes that are potentiated by neutrophils and lead to the induction of type 1 IFN by pDC (86). The authors show that this is seen only in NOD mice and speculate that this is related to the defects in the ability of NOD macrophages to clear debris (87). Several studies have examined the role of pDC, type 1 IFN and viral infection in the induction of T1D; reviewed in Swiecki et al. (88). The situation is complex with some studies showing exacerbation of diabetes (89, 90) with viral infection and others showing protection (91, 92). It has been suggested that whether viral infection leads to diabetes induction is related to the tropism of the virus and the local environment in which type 1 IFN is produced (88). Thus a virus infecting the pancreas may induce local type 1 IFN, which protects islets from infection and damage, whereas in other situations the type 1 IFN, produced by pDC following viral infection, could result in activation of autoreactive T cells and diabetes exacerbation (88). It has been reported that pDC also accumulate in the islets of NOD mice (93) at a later time point, around 10 weeks of age. The infiltrating pDC were shown to express IDO, which is characteristic of a more tolerogenic phenotype of DC (94) leading to the speculation that these cells may be attempting to control ongoing T cell activation. Studies in the next few years will hopefully clarify these contrasting results on the role that pDC play in diabetes pathogenesis.
A novel population of cDC, termed merocytic (mc)DC, have recently been identified and shown to be responsible for cross-presentation of islet antigens to CD8 T cells and direct presentation to CD4 T cells (95, 96). The mcDC express CD11c but are negative for both CD11b and CD8, they accumulate in the spleen as NOD mice age and were shown to secrete large amounts of type 1 IFN (95, 96). In addition transfer of mcDC pulsed with irradiated islets to non-diabetic NOD mice accelerated the onset of diabetes (95, 96). The number of mcDC was recently mapped to the Idd13 congenic interval suggesting that the relative number of these cells is genetically determined (97). Further studies are required to determine the significance of this DC population, and whether a counterpart of these cells exists in human.
Dendritic cells are constitutively present within islets of normal mice and have been shown to express peptide-MHC (pMHC) complexes containing peptides derived from islet antigens (98). Two major DC subsets can be found within islets; the CD11b cDC and CD103 cDC (99, 100). CD103 DC depend on FL for their homeostasis whereas islet CD11b DC appear to arise from monocytes and are unaffected by the absence of FL (101). The number of DCs in the islets remains relatively stable but these numbers increase following T cell infiltration and inflammation (98, 99, 102). In addition the phenotype of the DC becomes more inflammatory with increases in the expression of co-stimulatory molecules and MHC (2). The evidence suggests that the initiation of the autoimmune response occurs within the draining pancreatic lymph node (PLN) since removal of the PLN prevents diabetes (103) and several studies have shown that the initial proliferation of islet-specific CD4 and CD8 T cells takes place in the PLN (104, 105). Islet CD103 DC migrate to the PLN where they present islet antigens to specific CD4 and CD8 T cells (2, 101), and when T cells infiltrate the islet they localize to DC-rich areas (2). The movement of DC and T cells within the islet can now be visualized using a novel two-photon imaging technique (106). In addition pancreatic CD103 DC from NOD mice have been shown to express less IL-10 than similar populations from non-diabetic strains suggesting that these have lost their ability to induce tolerance (107). Islet CD11b DC are relatively poor at presenting antigen under steady state conditions but they accumulate as inflammation increases and become more mature. Since CD11b DC do not appear to migrate to PLN their role appears to be in the modulating the local tissue response (101).
Several studies have examined the blood of newly diagnosed T1D patients for the presence of DC subsets. pDCs have been shown to be increased (108) or decreased (109) at the time of diagnosis. A recent detailed longitudinal analysis of immune parameters in newly diagnosed T1D children has revealed that reduced numbers of cDC1s and NKT cells at the time of diagnosis are correlated with reduced residual β cell function 1 year later (110). Another study found decreased numbers of cDCs and pDCs in newly diagnosed pediatric T1D patients and also observed a decrease in the expression of CCR2 on these cells (111). Other studies have found correlations with vitamin D levels and immune cells in T1D patients (112). A study of pancreatic biopsies on a small number of new onset T1D patients revealed the presence of infiltrating macrophages and DC that produce TNF-α (113). A recent description of three cases of fulminant T1D secondary to enterovirus infection revealed marked islet infiltration of activated DC and macrophages and the presence of inflammatory cytokines (114). Thus it is likely that DCs will be shown to contribute to the onset of human T1D.
The fact that DCs play an important role in the induction and maintenance of self-tolerance has made them attractive targets for therapeutic interventions. Three main strategies have been employed and these include the adoptive transfer of specific DC subsets, the in vitro expansion of Tregs with specific DCs and the in vivo targeting of DC subsets (115). As discussed above the complexity of DC subsets and the plasticity of their function have made this a challenging objective, since there is a fine balance between the immunostimulatory and immunoregulatory functions of DC (45, 116, 117). DCs with a so called semi-mature phenotype, which consists of increased MHC and co-stimulatory molecule expression but low inflammatory cytokine production are thought to be most effective in inducing tolerance in the context of autoimmunity (118, 119). This maturation state can be induced by exposing DC to the cytokine TNF-α and DCs treated in this way have been shown to prevent experimental allergic encephalomyelitis (EAE) (118). In general the function of these DCs is to induce the expansion of Treg or other regulatory mechanisms such as Th2 cells. The attraction for using DCs as therapeutic agents is that since they present specific antigen to T cells it is possible to envision the generation of antigen-specific tolerance (120).
Dendritic cells were first used as therapeutic agents to prevent diabetes in NOD mice over 20 years ago by Clare-Salzler et al. (121). In this study DCs isolated from the PLN of NOD mice and transferred to prediabetic NOD mice were able to prevent disease onset whereas DCs from other LN or spleen were not effective. Later studies by the same group have suggested that DC in the PLN are more mature and that this is important for their therapeutic effect (122). We have previously shown that the injection of semi-mature bone marrow-derived DC prior to the onset of destructive insulitis prevents the subsequent development of diabetes (123, 124). The therapeutic DC populations expressed high levels of co-stimulatory molecules (CD80, CD86, and CD40) and produced low levels of IL-12p70 following CD40 ligation (49). Further investigation revealed that the therapeutic DC population changed the cytokine milieu in treated NOD mice. Whereas NOD mice generally exhibit a strong bias toward type 1 cytokine production, injection of DC induced the production of type 2 cytokines (125). In addition we (124) and others (126, 127) showed that injection of DC transduced with the IL-4 gene could effectively prevent diabetes in NOD mice when given at later time points, past the onset of invasive insulitis. We also confirmed, using in vitro studies, that therapeutic DC could drive the differentiation of Th2 cells (79), whereas non-therapeutic DC did not. The therapeutic DCs were not treated with any tolerogenic agents and did not require the addition of islet-derived peptides but the cultures were performed in FBS, which was subsequently shown to induce a Th2 environment that may have been non-specifically contributing to the therapeutic effect (128). In more recent experiments we have shown that the therapeutic DC population induces the expansion of Treg in the presence of low dose antigen (129) and DCs cultured in the absence of FBS are equally able to induce Treg expansion (unpublished observations). In addition it has been shown that DC grown in autologous serum were able to prevent diabetes but only when pulsed with insulin-derived peptides (130).
In order to ensure that DCs maintain a tolerogenic phenotype and drive tolerance rather than immunity several approaches aimed at conditioning the DC have been explored. These have included treating DC with cytokines such as IL-10 (130, 131), IL-10/TGF-β (132), and TSLP (IL-25) (133), pharmacological agents such as dexamethasone and vitamin D3 (134), carbon monoxide (CO) (135), anti-CTLA-4 antibody (136), secretory IgA (137) among others. In general, the treatment of DCs with these agents induces a semi-mature phenotype in the DC characterized by reduced expression of co-stimulatory molecules, reduced inflammatory cytokine production and resistance to further maturation stimuli. The infusion of these modified DC into NOD mice results in the deletion of autoreactive CD4 T cells and the generation of islet-specific Treg (130, 131, 133, 136, 137). One exception to this was the treatment of NOD mice with CO-treated DC, which reduced β1-integrin expression by CD8 T cells and thus inhibited their migration into the islet (135). Another approach has been the use of anti-sense oligonucleotides to down-regulate the expression of the co-stimulatory molecules, CD40, CD80, and CD86 on DC (138). This approach prevented diabetes in NOD mice through the induction of Treg and a phase 1 clinical trial using this approach was initiated (139).
Dendritic cells have also been used in vitro in order to expand Treg cells, and these Treg can be adoptively transferred into individuals with autoimmunity with the hope of curbing or arresting the inflammatory response. An early study expanded Treg, isolated from an islet-specific TCR transgenic NOD mice (BDC2.5), with peptide pulsed DC and IL-2 and showed that these cells could prevent diabetes in prediabetic NOD mice (34). Further studies from this group indicated that DC expressing higher levels of the co-stimulatory molecule, CD86 were the most efficient at inducing Treg expansion (140). The CD11b cDC subset has been shown to induce Treg proliferation and expansion in the presence of GM-CSF (43). A recent study examining the mechanism by which GM-CSF treatment prevents diabetes has further suggested that the expression of OX40L and the Notch3 ligand Jagged1 on DC was necessary for Treg expansion in vitro, although this study did not determine whether these polyclonal expanded Treg were effective in preventing diabetes (141). Some of the approaches to generate tolerogenic DC are being developed for clinical trial in the context of transplantation (142) but these are not yet being fully developed for use in autoimmune disease. A recent study compared the ability of several DC conditioning regimens to induce suppressive Treg in the human system (143). This study found that DC pretreated with IL-10 induced the most suppressive Treg, while TGF-β, rapamycin, and dexamethasone were less effective (143).
In general these types of therapies have involved the use of reagents that deliberately target specific DC subsets but these can also include approaches that, while not directly targeting DC, act by changing the local DC environment. Recent studies using both nominal and self-antigens have demonstrated that targeting antigens to CD8 cDC in the LN or spleen using anti-CD205 antibodies coupled to specific antigens in the absence of an inflammatory stimulus results in the generation of Treg (41, 42). Similar results have been obtained when antigens were targeted to CD11b cDC (21). A recent study used anti-CD205 antibodies coupled to the peptide mimetope for the CD4 BDC2.5 T cell to target DC in NOD mice (144). This resulted in the generation of long-lived and stable BDC2.5 Treg but this had no impact on the development of diabetes. However when the anti-CD205 was coupled to the proinsulin protein diabetes was prevented (144). Similar studies using an anti-CD205 coupled to HA and given to INS-HA/TCR-HA mice also demonstrated efficacy in the prevention of diabetes (145). In this mouse model the foreign antigen HA is expressed in the pancreas under the control of the rat insulin promoter and the T cells are all transgenic for an HA-specific TCR (145). These results suggest that the choice of antigen for targeting will be critical if this approach is to move forward. A similar approach has been used to induce tolerance in islet-specific CD8 T cells. These studies reported initial activation of adoptively transferred islet-specific CD8 cells followed by deletion of these cells, but the impact of these on diabetes development was not reported (146).
It has been known for some time that varying the dose of the stimulating antigen has a profound effect on T cell differentiation (147–149). Interest in this area has been renewed by the observation that the induction of Treg is optimal in situations of low TCR signal strength (33, 129, 150). The induction of Treg by low dose antigen occurs optimally when DC are presenting the antigen (129), and while the addition of TGF-β is not necessary a recent study showed that TGF-β production by T cells is required (151). Interestingly the most effective induction of Treg in vivo occurs when low doses of a high affinity peptide are used, rather than higher doses of low affinity peptides (152). Furthermore it was recently shown that the affinity of the peptide affects the duration of DC/T cell contacts in vivo providing an explanation for why low dose of more potent pMHC complexes are most effective at driving Treg expansion in vivo (153). Several approaches have been attempted to deliver low dose antigen in vivo including the continuous delivery of low dose antigen via osmotic pump (154) and the direct targeting of DC subsets as discussed above (21, 41, 42). In the context of T1D a recent study demonstrated that low doses of a high affinity insulin peptide mimetope was more effective at preventing diabetes than the native peptide (155). These results suggest that long-lasting peripheral tolerance could be achieved by delivering the appropriate dose of antigen to steady state DC in the periphery. The challenge will be translating this to the human situation although a recent paper showed that human insulin-specific Treg could be induced by low dose stimulation (156).
Another way to target DC in vivo is to use microparticles (157). A recent study used microspheres loaded with anti-sense oligonucleotides specific for CD40, CD80, and CD86 and showed prevention of diabetes along with an increase in Treg in treated NOD mice (158). These investigators further showed that repeated treatment with these microspheres could reverse diabetes in newly diagnosed NOD mice. The microspheres were taken up by resident DC in the spleen and these DC showed reduced expression of the co-stimulatory molecules (158).
In a study of combined therapy with oral anti-CD3 and intravenous anti-CD20 antibodies the prevention of diabetes seen in these animals was correlated with an increase in Treg and IL-10-producing T cells. Furthermore a population of IL-27 producing DCs was found to be responsible for the induction of IL-10-producing T cells in this system (159). As discussed above FL treatment of NOD mice in some circumstances prevents T1D (55, 56) but the timing of FL administration has to be before the onset of autoimmunity otherwise disease is exacerbated (57). Treatment of NOD mice with soluble CTLA-4 has been shown to restore the tolerance inducing properties of DC through the IFN-γ dependent activation of IDO. In this model CTLA-A binds to B7 molecules on DC and this stimulates the release of IFN-γ, which activates the immunosuppressive mechanism of tryptophan catabolism (160). A novel approach has been to induce accumulation of thymic DC populations which results in an increase in suppressive Tregs. A recent study identified the delta-like ligand 4 (Dll4)-Notch signaling pathway as important in controlling the number of thymic DCs (161). These investigators found that blockade of the Dll4-Notch pathway resulted in an increase in thymic DC numbers and thymic Treg. Treatment of NOD mice with these blockers not only prevented the establishment of disease in prediabetic animals but also was able to reverse disease in newly diagnosed diabetic animals (161). This protection was dependent on the presence of Treg since treatment of non-diabetic animals with a Treg-depleting antibody reversed the protection (161). Several studies have reported the use of α-GalCer, an agonist for iNKT cells, as a means to prevent disease in NOD mice (162–164). Recent studies have suggested that α-GalCer treatment induces tolerogenic DC populations that induce non-inflammatory islet-specific T cells (165), and novel derivatives of α-GalCer have been developed that have the same effect without some of the profound suppressive effects on iNKT function (166).
As discussed above a phase I clinical trial utilizing autologous monocyte-derived DC treated with anti-sense oligonucleotides to down-modulate co-stimulatory molecules has been conducted. In this trial of 10 patients with established T1D, 7 were given the immunosuppressive DC and 3 were given unmanipulated DC. In this trial no adverse events were observed and there were few changes noted in terms of insulin requirements, immune cell phenotype, and cytokine production (139). Interestingly, further analysis of this clinical trial revealed an increase in certain B cell populations and subsequent in vitro studies revealed that human DC treated with these anti-sense oligonucleotides could induce the proliferation of immunosuppressive B cells (167). A second trial using this same approach is now ongoing but results are not yet available. The field of DC-based vaccines is much more advanced in the setting of cancer and many clinical trials have been conducted; reviewed in the current Research Topic (Butterfileld, under review). Many features of cancer DC vaccines have been studied and it has become apparent that characteristics such as the form of the antigen used to pulse the DC, the conditioning regimen of the DC, and the route of administration all play critical roles in determining the outcome of the DC-based immunization. These features will need to be taken into consideration as more DC-based therapies are proposed for the treatment of T1D. There have been numerous clinical trials in T1D with the aim of inducing antigen-specific tolerance (168). These trials utilize islet-derived antigens, such as insulin GAD65 or hsp 70, and various routes of administration have been tested including oral, nasal, and intradermal. So far these trials have failed to have a significant impact on disease although in some cases evidence for immune tolerance was observed (168). It is to be expected that the success of such trials will depend on the APC, most likely a DC, targeted by these antigen formulations. At the present time not much attention is being paid to the nature of the DC presenting the antigen in these trials but, in view of our increasing understanding of the complexity of DC phenotype and function this is likely to change.
As discussed at the beginning of this review many of the studies that examine the therapeutic potential of DC in the treatment/prevention of T1D have been performed in the NOD mouse. This has been a very useful model for identifying susceptibility genes and understanding the progression to disease but therapeutic approaches that have successful in this mouse model have not translated well to the clinic (169). DCs play a pivotal role in setting the tone of the immune response. On the one hand they contribute to the development and maintenance of self-tolerance and on the other they contribute to the breaking of that tolerance and the initiation of disease. A deeper understanding of how DC can influence the development of autoimmune diabetes will aid in the development of novel therapeutic strategies. A great deal of progress has been achieved in the last several years and we have a better understanding of how DC can both maintain and break self-tolerance. The challenge in the future will be to use this knowledge to achieve the ultimate goal of inducing antigen-specific tolerance to prevent autoimmunity without causing widespread immune suppression.
The author, editor and chief editor declare that while the author Penelope Morel and the editor Lisa Butterfield are currently employed by the same institution, University of Pittsburgh, USA, there has been no conflict of interest during the review and handling of this manuscript, and the manuscript reviewers involved were from other institutions.
1. Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, et al. How does type 1 diabetes develop? The notion of homicide or beta-cell suicide revisited. Diabetes (2011) 60(5):1370–9. doi: 10.2337/db10-1797
2. Calderon B, Unanue ER. Antigen presentation events in autoimmune diabetes. Curr Opin Immunol (2012) 24(1):119–28. doi:10.1016/j.coi.2011.11.005
3. Leiter EH, Prochazka M, Coleman DL. Animal model of human disease, the nonobese diabetic (NOD) mouse. Am J Pathol (1987) 128:380–3.
4. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med (1973) 137(5):1142–62. doi:10.1084/jem.137.5.1142
5. Steinman RM, Lustig DS, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. 3. Functional properties in vivo. J Exp Med (1974) 139(6):1431–45. doi:10.1084/jem.139.6.1431
6. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J Exp Med (1974) 139(2):380–97. doi:10.1084/jem.139.6.1431
7. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol (2013) 31(1):563–604. doi:10.1146/annurev-immunol-020711-074950
8. Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A (2002) 99(1):351–8. doi:10.1073/pnas.231606698
9. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol (2011) 29(1):163–83. doi:10.1146/annurev-immunol-031210-101345
10. Onai N, Kurabayashi K, Hosoi-Amaike M, Toyama-Sorimachi N, Matsushima K, Inaba K, et al. A clonogenic progenitor with prominent plasmacytoid dendritic cell developmental potential. Immunity (2013) 38(5):943–57. doi:10.1016/j.immuni.2013.04.006
11. Schraml Barbara U, van Blijswijk J, Zelenay S, Whitney Paul G, Filby A, Acton Sophie E, et al. Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell (2013) 154(4):843–58. doi:10.1016/j.cell.2013.07.014
12. Lewis Kanako L, Caton Michele L, Bogunovic M, Greter M, Grajkowska Lucja T, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity (2011) 35(5):780–91. doi:10.1016/j.immuni.2011.08.013
13. Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nat Rev Immunol (2013) 13(8):566–77. doi:10.1038/nri3477
14. Nair-Gupta P, Blander JM. An updated view of the intracellualr mechanisms regulating cross-presentation. Front Immunol (2013) 4:401. doi:10.3389/fimmu.2013.00401
15. Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O’Keeffe M. Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J Immunol (2001) 166(9):5448–55.
16. Farrand KJ, Dickgreber N, Stoitzner P, Ronchese F, Petersen TR, Hermans IF. Langerin+ CD8alpha+ dendritic cells are critical for cross-priming and IL-12 production in response to systemic antigens. J Immunol (2009) 183(12):7732–42. doi:10.4049/jimmunol.0902707
17. Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol (2001) 167(3):1179–87.
18. Pulendran B, Lingappa J, Kennedy MK, Smith J, Teepe M, Rudensky A, et al. Developmental pathways of dendritic cells in vivo: distinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand-treated mice. J Immunol (1997) 159(5):2222–31.
19. Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med (2009) 206(3):549–59. doi:10.1084/jem.20082394
20. Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med (2002) 196(8):1091–7. doi:10.1084/jem.20021215
21. Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, et al. CD8+CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory t cells. J Immunol (2008) 181(10):6923–33.
22. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (2007) 315(5808):107–11. doi:10.1126/science.1136080
23. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med (2007) 204(8):1775–85. doi:10.1084/jem.20070602
24. Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med (2007) 204(8):1757–64. doi:10.1084/jem.20070590
25. Proietto AI, van Dommelen S, Zhou P, Rizzitelli A, D’Amico A, Steptoe RJ, et al. Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc Natl Acad Sci U S A (2008) 105(50):19869–74. doi:10.1073/pnas.0810268105
26. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi:10.1126/science.1079490
27. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14(4):307–8. doi:10.1038/ni.2554
28. Wing K, Fehérvári Z, Sakaguchi S. Emerging possibilities in the development and function of regulatory T cells. Int Immunol (2006) 18(7):991–1000. doi:10.1093/intimm/dxl044
29. Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion and characteristics of human T regulatory type 1 cells in co-cultures simulating tumor microenvironment. Cancer Immunol Immunother (2007) 56(9):1429–42. doi:10.1007/s00262-007-0280-9
30. Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J Immunol (2004) 172(9):5149–53.
31. Chambers ES, Hawrylowicz CM. The impact of vitamin D on regulatory T cells. Curr Allergy Asthma Rep (2011) 11(1):29–36. doi:10.1007/s11882-010-0161-8
32. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (2007) 317(5835):256–60. doi:10.1126/science.1145697
33. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol (2005) 6(12):1219–27. doi:10.1038/ni1265
34. Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med (2004) 199(11):1467–77. doi:10.1084/jem.20040180
35. Yamazaki S, Bonito AJ, Spisek R, Dhodapkar M, Inaba K, Steinman RM. Dendritic cells are specialized accessory cells along with TGF− for the differentiation of Foxp3+ CD4+ regulatory T cells from peripheral Foxp3 precursors. Blood (2007) 110(13):4293–302. doi:10.1182/blood-2007-05-088831
36. Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med (2000) 192(9):1213–22. doi:10.1084/jem.192.9.1213
37. Jonuleit H, Schmitt E, Steinbrink K, Enk AH. Dendritic cells as a tool to induce anergic and regulatory T cells. Trends Immunol (2001) 22(7):394–400. doi:10.1016/S1471-4906(01)01952-4
38. Roncarolo MG, Levings MK, Traversari C. Differentiation of T regulatory cells by immature dendritic cells. J Exp Med (2001) 193(2):F5–9. doi:10.1084/jem.193.2.F5
39. Wakkach A, Fournier N, Brun V, Breittmayer JP, Cottrez F, Groux H. Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity (2003) 18(5):605–17. doi:10.1016/S1074-7613(03)00113-4
40. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor {zeta}-chain and induce a regulatory phenotype in naive T cells. J Immunol (2006) 176(11):6752–61.
41. Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med (2002) 196(12):1627–38. doi:10.1084/jem.20021598
42. Kretschmer K, Heng TS, von Boehmer H. De novo production of antigen-specific suppressor cells in vivo. Nat Protoc (2006) 1(2):653–61. doi:10.1038/nprot.2006.105
43. Zou T, Caton AJ, Koretzky GA, Kambayashi T. Dendritic cells induce regulatory T cell proliferation through antigen-dependent and -independent interactions. J Immunol (2010) 185(5):2790–9. doi:10.4049/jimmunol.0903740
44. Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med (2009) 206(9):1853–62. doi:10.1084/jem.20090746
45. Morel PA, Turner MS. Dendritic cells and the maintenance of self-tolerance. Immunol Res (2011) 50(2–3):124–9. doi:10.1007/s12026-011-8217-y
46. Serreze DV, Gaskins HR, Leiter EH. Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J Immunol (1993) 150(6):2534–43.
47. Prasad SJ, Goodnow CC. Cell-intrinsic effects of non-MHC NOD genes on dendritic cell generation in vivo. Int Immunol (2002) 14(6):677–84. doi:10.1093/intimm/dxf034
48. Steptoe RJ, Ritchie JM, Harrison LC. Increased generation of dendritic cells from myeloid progenitors in autoimmune-prone nonobese diabetic mice. J Immunol (2002) 168:5032–41.
49. Feili-Hariri M, Morel PA. Phenotypic and functional characteristics of BM-derived DC from NOD and non diabetes-prone strains. Clin Immunol (2001) 98:133–42. doi:10.1006/clim.2000.4959
50. Vasquez AC, Feili-Hariri M, Tan RJ, Morel PA. Qualitative and quantitative abnormalities in splenic dendritic cell populations in NOD mice. Clin Exp Immunol (2004) 135(2):209–18. doi:10.1111/j.1365-2249.2003.02359.x
51. Marleau AM, Summers KL, Singh B. Differential contributions of APC subsets to T cell activation in nonobese diabetic mice. J Immunol (2008) 180(8):5235–49.
52. den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med (2000) 192(12):1685–96. doi:10.1084/jem.192.12.1685
53. Pettersson A, Wu XC, Ciumas C, Lian H, Chirsky V, Huang YM, et al. CD8alpha dendritic cells and immune protection from experimental allergic encephalomyelitis. Clin Exp Immunol (2004) 137(3):486–95. doi:10.1111/j.1365-2249.2004.02556.x
54. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med (2001) 194(6):769–80. doi:10.1084/jem.194.6.769
55. Chilton PM, Rezzoug F, Fugier-Vivier I, Weeter LA, Xu H, Huang Y, et al. Flt3-ligand treatment prevents diabetes in NOD mice. Diabetes (2004) 53(8):1995–2002. doi:10.2337/diabetes.53.8.1995
56. O’Keeffe M, Brodnicki TC, Fancke B, Vremec D, Morahan G, Maraskovsky E, et al. Fms-like tyrosine kinase 3 ligand administration overcomes a genetically determined dendritic cell deficiency in NOD mice and protects against diabetes development. Int Immunol (2005) 17(3):307–14. doi:10.1093/intimm/dxh210
57. Van Belle TL, Juntti T, Liao J, von Herrath MG. Pre-existing autoimmunity determines type 1 diabetes outcome after Flt3-ligand treatment. J Autoimmun (2010) 34(4):445–52. doi:10.1016/j.jaut.2009.11.010
58. Poligone B, Weaver DJJ, Sen P, Baldwin ASJ, Tisch R. Elevated NF-kappaB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J Immunol (2002) 168:188–96.
59. Weaver DJJ, Poligone B, Bui T, Abdel-Motal UM, Baldwin ASJ, Tisch R. Dendritic cells from nonobese diabetic mice exhibit a defect in NF-kappa B regulation due to a hyperactive I kappa B kinase. J Immunol (2001) 167:1461–8.
60. Marleau AM, Singh B. Myeloid dendritic cells in non-obese diabetic mice have elevated costimulatory and T helper-1-inducing abilities. J Autoimmun (2002) 19(1–2):23–35. doi:10.1006/jaut.2002.0597
61. Liu J, Beller D. Aberrant production of IL-12 by macrophages from several autoimmune-prone mouse strains is characterized by intrinsic and unique patterns of NF-kappa B expression and binding to the IL-12 p40 promoter. J Immunol (2002) 169(1):581–6.
62. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204(6):1257–65. doi:10.1084/jem.20062512
63. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem (2010) 285(10):7176–86. doi:10.1074/jbc.M109.047423
64. Ghaemi Oskouie F, Shameli A, Yang A, Desrosiers MD, Mucsi AD, Blackburn MR, et al. High levels of adenosine deaminase on dendritic cells promote autoreactive T cell activation and diabetes in nonobese diabetic mice. J Immunol (2011) 186(12):6798–806. doi:10.4049/jimmunol.1004222
65. Liston A, Lesage S, Gray DH, O’Reilly LA, Strasser A, Fahrer AM, et al. Generalized resistance to thymic deletion in the NOD mouse; a polygenic trait characterized by defective induction of Bim. Immunity (2004) 21(6):817–30. doi:10.1016/j.immuni.2004.10.014
66. Kreuwel HT, Biggs JA, Pilip IM, Pamer EG, Lo D, Sherman LA. Defective CD8+ T cell peripheral tolerance in nonobese diabetic mice. J Immunol (2001) 167(2):1112–7.
67. Tritt M, Sgouroudis E, d’Hennezel E, Albanese A, Piccirillo CA. Functional waning of naturally occurring CD4+ regulatory T-cells contributes to the onset of autoimmune diabetes. Diabetes (2008) 57(1):113–23. doi:10.2337/db06-1700
68. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12(4):431–40. doi:10.1016/S1074-7613(00)80195-8
69. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol (2009) 10(11):1185–92. doi:10.1038/ni.1790
70. Obst R, van Santen HM, Melamed R, Kamphorst AO, Benoist C, Mathis D. Sustained antigen presentation can promote an immunogenic T cell response, like dendritic cell activation. Proc Natl Acad Sci U S A (2007) 104(39):15460–5. doi:10.1073/pnas.0707331104
71. Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity (2010) 33(2):153–65. doi:10.1016/j.immuni.2010.08.004
72. Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes (2010) 59(2):407–15. doi:10.2337/db09-0694
73. Anderson AC, Chandwaskar R, Lee DH, Kuchroo VK. Cutting edge: the Idd3 genetic interval determines regulatory T cell function through CD11b+CD11c- APC. J Immunol (2008) 181(11):7449–52.
74. Hamilton-Williams EE, Martinez X, Clark J, Howlett S, Hunter KM, Rainbow DB, et al. Expression of diabetes-associated genes by dendritic cells and CD4 T cells drives the loss of tolerance in nonobese diabetic mice. J Immunol (2009) 183(3):1533–41. doi:10.4049/jimmunol.0900428
75. Sgouroudis E, Kornete M, Piccirillo CA. IL-2 production by dendritic cells promotes Foxp3(+) regulatory T-cell expansion in autoimmune-resistant NOD congenic mice. Autoimmunity (2011) 44(5):406–14. doi:10.3109/08916934.2010.536795
76. Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, et al. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med (1994) 180(3):995–1004. doi:10.1084/jem.180.3.995
77. Lee LF, Xu B, Michie SA, Beilhack GF, Warganich T, Turley S, et al. The role of TNF-alpha in the pathogenesis of type 1 diabetes in the nonobese diabetic mouse: analysis of dendritic cell maturation. Proc Natl Acad Sci U S A (2005) 102(44):15995–6000. doi:10.1073/pnas.0508122102
78. Lee MH, Lee WH, Todorov I, Liu CP. CD4+ CD25+ regulatory T cells prevent type 1 diabetes preceded by dendritic cell-dominant invasive insulitis by affecting chemotaxis and local invasiveness of dendritic cells. J Immunol (2010) 185(4):2493–501. doi:10.4049/jimmunol.1001036
79. Feili-Hariri M, Falkner DH, Morel PA. Polarization of naive T cells into Th1 or Th2 by distinct cytokine-driven murine dendritic cell populations: implications for immunotherapy. J Leukoc Biol (2005) 78:656–64. doi:10.1189/jlb.1104631
80. Feili-Hariri M, Flores RR, Vasquez AC, Morel PA. Dendritic cell immunotherapy for autoimmune diabetes. Immunol Res (2006) 36(1–3):167–73. doi:10.1385/IR:36:1:167
81. Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol (2007) 179(8):5041–53.
82. Caielli S, Conforti-Andreoni C, Di Pietro C, Usuelli V, Badami E, Malosio ML, et al. On/off TLR signaling decides proinflammatory or tolerogenic dendritic cell maturation upon CD1d-mediated interaction with invariant NKT cells. J Immunol (2010) 185(12):7317–29. doi:10.4049/jimmunol.1000400
83. Diana J, Brezar V, Beaudoin L, Dalod M, Mellor A, Tafuri A, et al. Viral infection prevents diabetes by inducing regulatory T cells through NKT cell-plasmacytoid dendritic cell interplay. J Exp Med (2011) 208(4):729–45. doi:10.1084/jem.20101692
84. Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A (2008) 105(34):12439–44. doi:10.1073/pnas.0806439105
85. Li Q, McDevitt HO. The role of interferon alpha in initiation of type I diabetes in the NOD mouse. Clin Immunol (2011) 140(1):3–7. doi:10.1016/j.clim.2011.04.010
86. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med (2013) 19(1):65–73. doi:10.1038/nm.3042
87. Creusot RJ. Initiating type I diabetes: new suspects in the lineup. Nat Med (2013) 19(1):18–20. doi:10.1038/nm.3044
88. Swiecki M, McCartney SA, Wang Y, Colonna M. TLR7/9 versus TLR3/MDA5 signaling during virus infections and diabetes. J Leukoc Biol (2011) 90(4):691–701. doi:10.1189/jlb.0311166
89. Alba A, Planas R, Clemente X, Carrillo J, Ampudia R, Puertas MC, et al. Natural killer cells are required for accelerated type 1 diabetes driven by interferon-beta. Clin Exp Immunol (2008) 151(3):467–75. doi:10.1111/j.1365-2249.2007.03580.x
90. Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A (2007) 104(12):5115–20. doi:10.1073/pnas.0700442104
91. von Herrath M. Diabetes: a virus-gene collaboration. Nature (2009) 459(7246):518–9. doi:10.1038/459518a
92. Filippi CM, Estes EA, Oldham JE, von Herrath MG. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J Clin Invest (2009) 119(6):1515–23. doi:10.1172/JCI38503
93. Welzen-Coppens JM, van Helden-Meeuwsen CG, Leenen PJ, Drexhage HA, Versnel MA. The kinetics of plasmacytoid dendritic cell accumulation in the pancreas of the NOD mouse during the early phases of insulitis. PLoS One (2013) 8(1):e55071. doi:10.1371/journal.pone.0055071
94. Grohmann U, Fallarino F, Puccetti P. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol (2003) 24(5):242–8. doi:10.1016/S1471-4906(03)00072-3
95. Katz JD, Janssen EM. Breaking T cell tolerance to beta cell antigens by merocytic dendritic cells. Cell Mol Life Sci (2011) 68(17):2873–83. doi:10.1007/s00018-011-0730-6
96. Katz JD, Ondr JK, Opoka RJ, Garcia Z, Janssen EM. Cutting edge: merocytic dendritic cells break T cell tolerance to beta cell antigens in nonobese diabetic mouse diabetes. J Immunol (2010) 185(4):1999–2003. doi:10.4049/jimmunol.1001398
97. Pelletier AN, Lesage S. The Idd13 congenic interval defines the number of merocytic dendritic cells, a novel trait associated with autoimmune diabetes susceptibility. J Autoimmun (2013) 43:70–7. doi:10.1016/j.jaut.2013.04.004
98. Calderon B, Suri A, Miller MJ, Unanue ER. Dendritic cells in islets of Langerhans constitutively present beta cell-derived peptides bound to their class II MHC molecules. Proc Natl Acad Sci U S A (2008) 105(16):6121–6. doi:10.1073/pnas.0801973105
99. Melli K, Friedman RS, Martin AE, Finger EB, Miao G, Szot GL, et al. Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J Immunol (2009) 182(5):2590–600. doi:10.4049/jimmunol.0803543
100. Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med (2009) 206(13):3115–30. doi:10.1084/jem.20091756
101. Yin N, Xu J, Ginhoux F, Randolph GJ, Merad M, Ding Y, et al. Functional specialization of islet dendritic cell subsets. J Immunol (2012) 188(10):4921–30. doi:10.4049/jimmunol.1103725
102. Calderon B, Carrero JA, Miller MJ, Unanue ER. Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc Natl Acad Sci U S A (2011) 108(4):1561–6. doi:10.1073/pnas.1018973108
103. Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med (2002) 196(3):369–77. doi:10.1084/jem.20011353
104. Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med (1999) 189(2):331–9. doi:10.1084/jem.189.2.331
105. Zhang Y, O’Brien B, Trudeau J, Tan R, Santamaria P, Dutz JP. In situ beta cell death promotes priming of diabetogenic CD8 T lymphocytes. J Immunol (2002) 168(3):1466–72.
106. Coppieters K, Martinic MM, Kiosses WB, Amirian N, von Herrath M. A novel technique for the in vivo imaging of autoimmune diabetes development in the pancreas by two-photon microscopy. PLoS One (2010) 5(12):e15732. doi:10.1371/journal.pone.0015732
107. Welzen-Coppens JM, van Helden-Meeuwsen CG, Leenen PJ, Drexhage HA, Versnel MA. Reduced numbers of dendritic cells with a tolerogenic phenotype in the prediabetic pancreas of NOD mice. J Leukoc Biol (2012) 92(6):1207–13. doi:10.1189/jlb.0312168
108. Allen JS, Pang K, Skowera A, Ellis R, Rackham C, Lozanoska-Ochser B, et al. Plasmacytoid dendritic cells are proportionally expanded at diagnosis of type 1 diabetes and enhance islet autoantigen presentation to T-cells through immune complex capture. Diabetes (2009) 58(1):138–45. doi:10.2337/db08-0964
109. Vuckovic S, Withers G, Harris M, Khalil D, Gardiner D, Flesch I, et al. Decreased blood dendritic cell counts in type 1 diabetic children. Clin Immunol (2007) 123(3):281–8. doi:10.1016/j.clim.2007.03.002
110. Galgani M, Nugnes R, Bruzzese D, Perna F, De Rosa V, Procaccini C, et al. Meta-immunological profiling of children with type 1 diabetes identifies new biomarkers to monitor disease progression. Diabetes (2013) 62(7):2481–91. doi:10.2337/db12-1273
111. Nieminen JK, Vakkila J, Salo HM, Ekstrom N, Harkonen T, Ilonen J, et al. Altered phenotype of peripheral blood dendritic cells in pediatric type 1 diabetes. Diabetes Care (2012) 35(11):2303–10. doi:10.2337/dc11-2460
112. Rose K, Penna-Martinez M, Klahold E, Karger D, Shoghi F, Kahles H, et al. Influence of the vitamin D plasma level and vitamin D-related genetic polymorphisms on the immune status of patients with type 1 diabetes: a pilot study. Clin Exp Immunol (2013) 171(2):171–85. doi:10.1111/cei.12013
113. Uno S, Imagawa A, Okita K, Sayama K, Moriwaki M, Iwahashi H, et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia (2007) 50(3):596–601. doi:10.1007/s00125-006-0569-9
114. Aida K, Nishida Y, Tanaka S, Maruyama T, Shimada A, Awata T, et al. RIG-I- and MDA5-initiated innate immunity linked with adaptive immunity accelerates beta-cell death in fulminant type 1 diabetes. Diabetes (2011) 60(3):884–9. doi:10.2337/db10-0795
115. Mukherjee G, Dilorenzo TP. The immunotherapeutic potential of dendritic cells in type 1 diabetes. Clin Exp Immunol (2010) 161(2):197–207. doi:10.1111/j.1365-2249.2010.04157.x
116. Morel PA, Feili-Hariri M, Coates PT, Thomson AW. Dendritic cells, T cell tolerance and therapy of adverse immune reactions. Clin Exp Immunol (2003) 133(1):1–10. doi:10.1046/j.1365-2249.2003.02161.x
117. Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol (2007) 7(8):610–21. doi:10.1038/nri2132
118. Menges M, Rossner S, Voigtlander C, Schindler H, Kukutsch NA, Bogdan C, et al. Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J Exp Med (2002) 195(1):15–21. doi:10.1084/jem.20011341
119. Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol (2002) 23(9):445–9. doi:10.1016/S1471-4906(02)02281-0
120. Clemente-Casares X, Tsai S, Huang C, Santamaria P. Antigen-specific therapeutic approaches in Type 1 diabetes. Cold Spring Harbor Perspect Med (2012) 2(2):a007773. doi:10.1101/cshperspect.a007773
121. Clare-Salzler MJ, Brooks J, Chai A, Van Herle K, Anderson C. Prevention of diabetes in nonobese diabetic mice by dendritic cell transfer. J Clin Invest (1992) 90(3):741–8. doi:10.1172/JCI115946
122. Naumov YN, Bahjat KS, Gausling R, Abraham R, Exley MA, Koezuka Y, et al. Activation of CD1d-restricted T cells protects NOD mice from developing diabetes by regulating dendritic cell subsets. Proc Natl Acad Sci U S A (2001) 98(24):13838–43. doi:10.1073/pnas.251531798
123. Feili-Hariri M, Dong X, Alber SM, Watkins SC, Salter RD, Morel PA. Immunotherapy of NOD mice with bone marrow-derived dendritic cells. Diabetes (1999) 48:2300–8. doi:10.2337/diabetes.48.12.2300
124. Feili-Hariri M, Falkner DH, Gambotto A, Papworth GD, Watkins SC, Robbins PD, et al. Dendritic cells transduced to express IL-4 prevent diabetes in nonobese diabetic mice with established insulitis. Human Gene Ther (2003) 14:13–23. doi:10.1089/10430340360464679
125. Feili-Hariri M, Falkner DH, Morel PA. Regulatory Th2 response induced following adoptive transfer of dendritic cells in prediabetic NOD mice. Eur J Immunol (2002) 32:2021–30. doi:10.1002/1521-4141(200207)32:7<2021::AID-IMMU2021>3.0.CO;2-J
126. Creusot RJ, Chang P, Healey DG, Tcherepanova IY, Nicolette CA, Fathman CG. A short pulse of IL-4 delivered by DCs electroporated with modified mRNA can both prevent and treat autoimmune diabetes in NOD mice. Mol Ther (2010) 18(12):2112–20. doi:10.1038/mt.2010.146
127. Ruffner MA, Robbins PD. Dendritic cells transduced to express interleukin 4 reduce diabetes onset in both normoglycemic and prediabetic nonobese diabetic mice. PLoS One (2010) 5(7):e11848. doi:10.1371/journal.pone.0011848
128. Haase C, Ejrnaes M, Juedes AE, Wolfe T, Markholst H, von Herrath MG. Immunomodulatory dendritic cells require autologous serum to circumvent nonspecific immunosuppressive activity in vivo. Blood (2005) 106(13):4225–33. doi:10.1182/blood-2005-03-0975
129. Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol (2009) 183(8):4895–903. doi:10.4049/jimmunol.0901459
130. Haase C, Yu L, Eisenbarth G, Markholst H. Antigen-dependent immunotherapy of non-obese diabetic mice with immature dendritic cells. Clin Exp Immunol (2010) 160(3):331–9. doi:10.1111/j.1365-2249.2010.04104.x
131. Tai N, Yasuda H, Xiang Y, Zhang L, Rodriguez-Pinto D, Yokono K, et al. IL-10-conditioned dendritic cells prevent autoimmune diabetes in NOD and humanized HLA-DQ8/RIP-B7.1 mice. Clin Immunol (2011) 139(3):336–49. doi:10.1016/j.clim.2011.03.003
132. Torres-Aguilar H, Sanchez-Torres C, Jara LJ, Blank M, Shoenfeld Y. IL-10/TGF-beta-treated dendritic cells, pulsed with insulin, specifically reduce the response to insulin of CD4+ effector/memory T cells from type 1 diabetic individuals. J Clin Immunol (2010) 30(5):659–68. doi:10.1007/s10875-010-9430-5
133. Besin G, Gaudreau S, Menard M, Guindi C, Dupuis G, Amrani A. Thymic stromal lymphopoietin and thymic stromal lymphopoietin-conditioned dendritic cells induce regulatory T-cell differentiation and protection of NOD mice against diabetes. Diabetes (2008) 57(8):2107–17. doi:10.2337/db08-0171
134. Kleijwegt FS, Jansen DT, Teeler J, Joosten AM, Laban S, Nikolic T, et al. Tolerogenic dendritic cells impede priming of naive CD8(+) T cells and deplete memory CD8(+) T cells. Eur J Immunol (2013) 43(1):85–92. doi:10.1002/eji.201242879
135. Simon T, Pogu S, Tardif V, Rigaud K, Remy S, Piaggio E, et al. Carbon monoxide-treated dendritic cells decrease beta1-integrin induction on CD8(+) T cells and protect from type 1 diabetes. Eur J Immunol (2013) 43(1):209–18. doi:10.1002/eji.201242684
136. Karumuthil-Melethil S, Perez N, Li R, Prabhakar BS, Holterman MJ, Vasu C. Dendritic cell-directed CTLA-4 engagement during pancreatic beta cell antigen presentation delays type 1 diabetes. J Immunol (2010) 184(12):6695–708. doi:10.4049/jimmunol.0903130
137. Diana J, Moura IC, Vaugier C, Gestin A, Tissandie E, Beaudoin L, et al. Secretory IgA induces tolerogenic dendritic cells through SIGNR1 dampening autoimmunity in mice. J Immunol (2013) 191(5):2335–43. doi:10.4049/jimmunol.1300864
138. Machen J, Harnaha J, Lakomy R, Styche A, Trucco M, Giannoukakis N. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J Immunol (2004) 173(7):4331–41.
139. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34(9):2026–32. doi:10.2337/dc11-0472
140. Tarbell KV, Yamazaki S, Steinman RM. The interactions of dendritic cells with antigen-specific, regulatory T cells that suppress autoimmunity. Semin Immunol (2006) 18(2):93–102. doi:10.1016/j.smim.2006.01.009
141. Gopisetty A, Bhattacharya P, Haddad C, Bruno JC Jr, Vasu C, Miele L, et al. OX40L/Jagged1 cosignaling by GM-CSF-induced bone marrow-derived dendritic cells is required for the expansion of functional regulatory T cells. J Immunol (2013) 190(11):5516–25. doi:10.4049/jimmunol.1202298
142. Moreau A, Varey E, Bouchet-Delbos L, Cuturi MC. Cell therapy using tolerogenic dendritic cells in transplantation. Transplant Res (2012) 1(1):13. doi:10.1186/2047-1440-1-13
143. Boks MA, Kager-Groenland JR, Haasjes MSP, Zwaginga JJ, van Ham SM, ten Brinke A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction – a comparative study of human clinical-applicable DC. Clin Immunol (2012) 142(3):332–42. doi:10.1016/j.clim.2011.11.011
144. Petzold C, Riewaldt J, Koenig T, Schallenberg S, Kretschmer K. Dendritic cell-targeted pancreatic beta-cell antigen leads to conversion of self-reactive CD4(+) T cells into regulatory T cells and promotes immunotolerance in NOD mice. Rev Diabet Stud (2010) 7(1):47–61. doi:10.1900/RDS.2010.7.47
145. Bruder D, Westendorf AM, Hansen W, Prettin S, Gruber AD, Qian Y, et al. On the edge of autoimmunity: T-cell stimulation by steady-state dendritic cells prevents autoimmune diabetes. Diabetes (2005) 54(12):3395–401. doi:10.2337/diabetes.54.12.3395
146. Mukhopadhaya A, Hanafusa T, Jarchum I, Chen YG, Iwai Y, Serreze DV, et al. Selective delivery of beta cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in NOD mice. Proc Natl Acad Sci U S A (2008) 105(17):6374–9. doi:10.1073/pnas.0802644105
147. Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K. Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J Exp Med (1995) 182(5):1591–6. doi:10.1084/jem.182.5.1591
148. Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol (1997) 15:297–322. doi:10.1146/annurev.immunol.15.1.297
149. Hosken NA, Shibuya K, Heath AW, Murphy KM, O’Garra A. The effect of antigen dose on CD4+ T helper cell phenotype development in a T cell receptor-alpha beta-transgenic model. J Exp Med (1995) 182(5):1579–84. doi:10.1084/jem.182.5.1579
150. Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A (2008) 105(22):7797–802. doi:10.1073/pnas.0800928105
151. Gabryšová L, Christensen JR, Wu X, Kissenpfennig A, Malissen B, O’Garra A. Integrated T-cell receptor and costimulatory signals determine TGF-β-dependent differentiation and maintenance of Foxp3+ regulatory T cells. Eur J Immunol (2011) 41(5):1242–8. doi:10.1002/eji.201041073
152. Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med (2010) 207(8):1701–11. doi:10.1084/jem.20091999
153. Gottschalk RA, Hathorn MM, Beuneu H, Corse E, Dustin ML, Altan-Bonnet G, et al. Distinct influences of peptide-MHC quality and quantity on in vivo T-cell responses. Proc Natl Acad Sci U S A (2012) 109(3):881–6. doi:10.1073/pnas.1119763109
154. von Boehmer H. Peptide-based instruction of suppressor commitment in naive T cells and dynamics of immunosuppression in vivo. Scand J Immunol (2005) 62(Suppl 1):49–54. doi:10.1111/j.1365-3083.2005.01609.x
155. Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med (2011) 208(7):1501–10. doi:10.1084/jem.20110574
156. Long SA, Rieck M, Tatum M, Bollyky PL, Wu RP, Muller I, et al. Low-dose antigen promotes induction of FOXP3 in human CD4+ T cells. J Immunol (2011) 187(7):3511–20. doi:10.4049/jimmunol.1003880
157. Keselowsky BG, Xia CQ, Clare-Salzler M. Multifunctional dendritic cell-targeting polymeric microparticles: engineering new vaccines for type 1 diabetes. Hum Vaccin (2011) 7(1):37–44. doi:10.4161/hv.7.1.12916
158. Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, et al. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes (2008) 57(6):1544–55. doi:10.2337/db07-0507
159. Hu C, Ding H, Zhang X, Wong FS, Wen L. Combination treatment with anti-CD20 and oral anti-CD3 prevents and reverses autoimmune diabetes. Diabetes (2013) 62(8):2849–58. doi:10.2337/db12-1175
160. Fallarino F, Bianchi R, Orabona C, Vacca C, Belladonna ML, Fioretti MC, et al. CTLA-4-Ig activates forkhead transcription factors and protects dendritic cells from oxidative stress in nonobese diabetic mice. J Exp Med (2004) 200(8):1051–62. doi:10.1084/jem.20040942
161. Billiard F, Lobry C, Darrasse-Jeze G, Waite J, Liu X, Mouquet H, et al. Dll4-Notch signaling in Flt3-independent dendritic cell development and autoimmunity in mice. J Exp Med (2012) 209(5):1011–28. doi:10.1084/jem.20111615
162. Hong S, Wilson MT, Serizawa I, Wu L, Singh N, Naidenko OV, et al. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med (2001) 7(9):1052–6. doi:10.1038/nm0901-1052
163. Sharif S, Arreaza GA, Zucker P, Mi QS, Sondhi J, Naidenko OV, et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med (2001) 7(9):1057–62. doi:10.1038/nm0901-1057
164. Wang B, Geng YB, Wang CR. CD1-restricted NK T cells protect nonobese diabetic mice from developing diabetes. J Exp Med (2001) 194(3):313–20. doi:10.1084/jem.194.3.313
165. Wang J, Cho S, Ueno A, Cheng L, Xu BY, Desrosiers MD, et al. Ligand-dependent induction of noninflammatory dendritic cells by anergic invariant NKT cells minimizes autoimmune inflammation. J Immunol (2008) 181(4):2438–45.
166. Tohn R, Blumenfeld H, Haeryfar SM, Veerapen N, Besra GS, Porcelli SA, et al. Stimulation of a shorter duration in the state of anergy by an invariant natural killer T cell agonist enhances its efficiency of protection from type 1 diabetes. Clin Exp Immunol (2011) 164(1):26–41. doi:10.1111/j.1365-2249.2011.04323.x
167. Di Caro V, Phillips B, Engman C, Harnaha J, Trucco M, Giannoukakis N. Retinoic acid-producing, ex vivo-generated human tolerogenic dendritic cells induce the proliferation of immunosuppressive B-lymphocytes. Clin Exp Immunol (2013) 174(2):302–17. doi:10.1111/cei.12177
168. Coppieters KT, Harrison LC, von Herrath MG. Trials in type 1 diabetes: antigen-specific therapies. Clin Immunol (2013) 149(3):345–55. doi:10.1016/j.clim.2013.02.002
Keywords: dendritic cells, type 1 diabetes, T regulatory cells, autoimmunity, tolerance
Citation: Morel PA (2013) Dendritic cell subsets in type 1 diabetes: friend or foe? Front. Immunol. 4:415. doi: 10.3389/fimmu.2013.00415
Received: 01 October 2013; Paper pending published: 01 November 2013;
Accepted: 13 November 2013; Published online: 06 December 2013.
Edited by:
Lisa Helene Butterfield, University of Pittsburgh, USAReviewed by:
Bruno Laugel, Cardiff University School of Medicine, UKCopyright: © 2013 Morel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Penelope A. Morel, Department of Immunology, University of Pittsburgh, 200 Lothrop Street, BST E1048, Pittsburgh, PA 15261, USA e-mail:bW9yZWxAcGl0dC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.