 Catherine Chaput
Catherine Chaput Leif Erik Sander
Leif Erik Sander Norbert Suttorp
Norbert Suttorp Bastian Opitz
Bastian Opitz- Department of Internal Medicine/Infectious Diseases and Pulmonary Medicine, Charité – Universitätsmedizin Berlin, Berlin, Germany
The lung is a particularly vulnerable organ at the interface of the body and the exterior environment. It is constantly exposed to microbes and particles by inhalation. The innate immune system needs to react promptly and adequately to potential dangers posed by these microbes and particles, while at the same time avoiding extensive tissue damage. Nucleotide-binding oligomerization domain-like receptors (NLRs) represent a group of key sensors for microbes and damage in the lung. As such they are important players in various infectious as well as acute and chronic sterile inflammatory diseases, such as pneumonia, chronic obstructive pulmonary disease (COPD), acute lung injury/acute respiratory distress syndrome, pneumoconiosis, and asthma. Activation of most known NLRs leads to the production and release of pro-inflammatory cytokines, and/or to the induction of cell death. We will review NLR functions in the lung during infection and sterile inflammation.
Introduction
The respiratory tract constitutes a large surface of the body with the outside environment that is exposed to high volume airflow and large numbers of inhaled microbes and particles. The microflora of the upper respiratory tract consists of non-pathogenic bacteria but also frequently comprises potential pathogens such as Streptococcus pneumoniae and Staphylococcus aureus (1). The distal bronchi and the alveoli have long been considered sterile, however more recently microbes that are either aspirated from the upper respiratory tract (2) or constantly reside in the lower airways (3, 4) have been found by culture-independent approaches.
Inflammatory disorders of the respiratory tract involving the innate immune system include both infectious and non-infectious diseases. Lower respiratory tract infections, or pneumonia, generally develop when facultative pathogenic microbes that colonize the upper respiratory tract are aspirated or airborne pathogens are inhaled. The World Health Organization (WHO) estimates 429 million cases of acute lower respiratory tract infections in 2004, making it the third leading cause of death world-wide (5). Moreover, non-infectious and chronic lung diseases substantially contribute to morbidity. Chronic obstructive pulmonary disease (COPD) is mainly caused by tobacco smoke and can exacerbate during acute infections, ranks as the number four leading cause of death in most industrialized countries (5). Acute lung injury (ALI) and its severest form, called acute respiratory distress syndrome (ARDS), can develop after infectious as well as non-infectious insults (6). Another potentially life-threatening disorder is allergic asthma, which is characterized by airway hyperresponsiveness due to allergen-triggered airway inflammation causing chronic recurrent airflow obstruction. Long-term exposure to silica, asbestos, or coal particles can cause chronic occupational lung disease called pneumoconiosis.
The innate immune system is a key player in various infectious and non-infectious disorders of the lung (7–9). It senses infections, sterile tissue damage, and probably any disturbance of host cell and tissue integrity by so-called pattern-recognition receptors (PRRs). PRRs comprise different protein families such as the transmembrane Toll-like receptors (TLRs) and the intracellularly located nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) (10–12). PRRs recognize conserved microbial molecules, referred to as pathogen-associated molecular patterns (PAMPs) (13). However, recent evidence suggests that recognition of disturbed host cell integrity and danger signals might also play a role in the immune responses to invading pathogens (see below). In contrast to the original paradigm (13) it is now well-accepted that PRRs also sense non-microbial ligands generated during sterile tissue damage, often called damage-associated molecular patterns (DAMPs) (14, 15). Moreover, some PRRs can additionally respond to large particles and therefore appear to be key mediators in pneumoconiosis (16–18).
The NLR family comprises 22 members in humans and even more in mice. Most NLRs share common structural characteristics including a C-terminal leucine-rich repeat (LRR) domain, often involved in ligand recognition, a central NOD, and a variable N-terminal effector domain (10). Based on the type of effector domains that is either a caspase recruitment domain (CARD), a pyrin domain (PYD), or a baculoviral inhibitor of apoptosis protein repeat (BIR) domain, the NLR family can be further divides into five subfamilies. The NLRA subfamily consist of only one member, the transcription factor CIITA, of which at least one splice variant expresses a CARD (Figure 1). CIITA is involved in transcriptional activation of genes encoding major histocompatibility complex class II [for detailed discussion of this unique NLR protein we refer to Ref. (19)]. The NLRB group of NLRs expresses a BIR domain and consists of NAIP1–7 in mice and NAIP in humans. The NLRC subfamily includes the CARD-containing molecules NOD1, NOD2, and NLRC3–5, whereas the 14 known NLRP proteins (NLRP1–14) express a PYD. NLRX1 is the only member of the NLRX subgroup, and the only NLR protein that is localized in mitochondria (10, 20). Whereas some NLR proteins function as bona fide PRRs, other family members act as adaptor molecules or regulators of signal transduction.
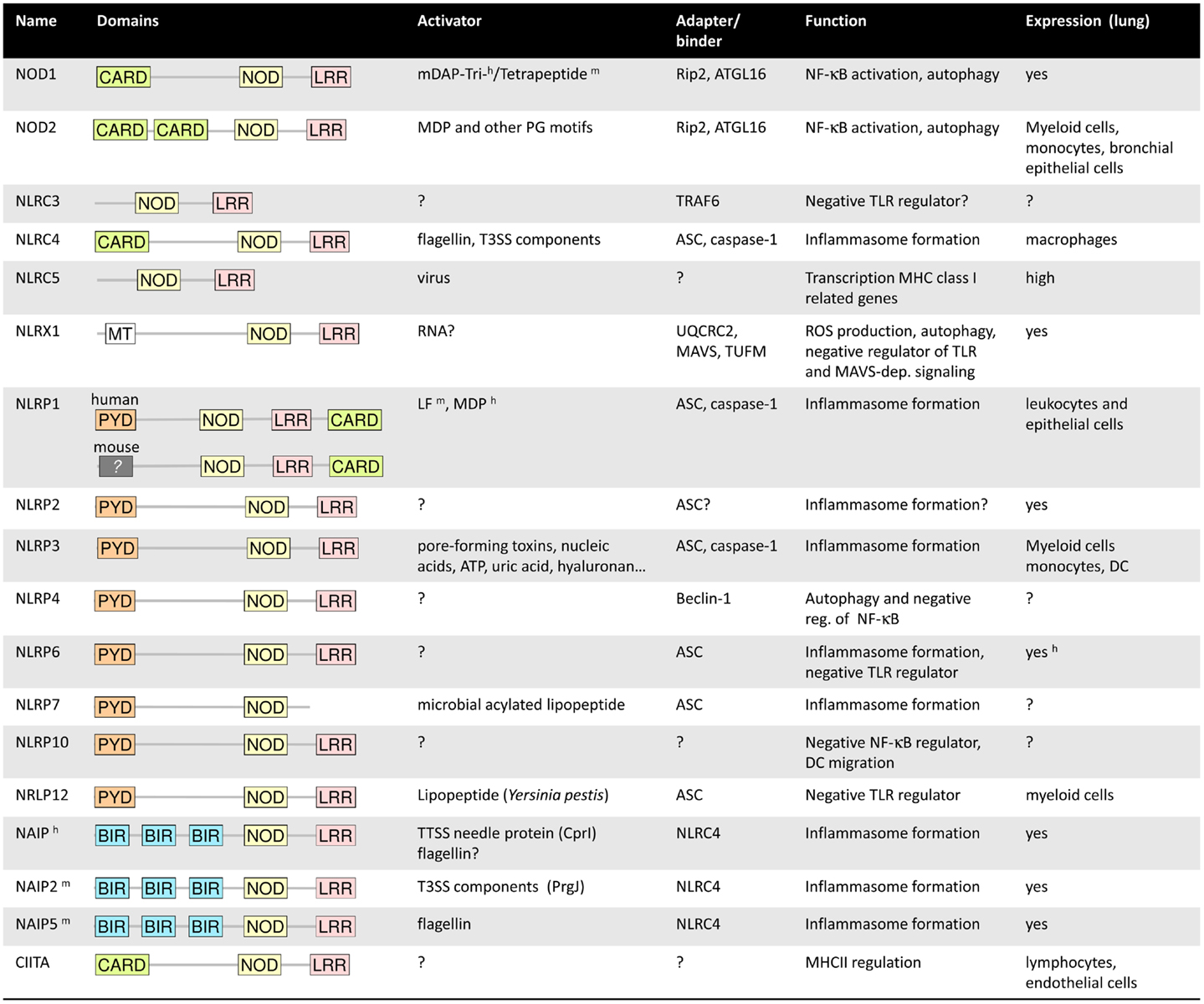
Figure 1. Summary of the main characteristics of the NLRs. h and m symbolized a characteristic specific to human or mouse. For more details, refer to the main text.
In this review article we discuss the current knowledge about NLR expression and function in the lung in different pulmonary diseases. We have grouped the NLRs based on functional similarities and summarize major pathways and common principles of function.
NOD1 and NOD2
NOD1 and NOD2 were the first NLR proteins to be discovered (21–25). In the lung, NOD1 is expressed in various cell types including lung epithelial cells, endothelial cells, human airway smooth muscle cells, and different types of leukocytes (26–29). NOD2 has been found in alveolar macrophages, neutrophils, and bronchial epithelial cells (30–32). NOD1 responds to bacterial cell wall peptidoglycan containing meso-diaminopimelic acid found predominantly in Gram-negative bacteria (33, 34). NOD2 recognizes the muramyl-dipeptide (MDP) MurNAc-l-Ala-d-isoGln, which is conserved in peptidoglycans of the majority of bacteria (35, 36). Other peptidoglycan motifs can be recognized by NOD1 and NOD2, for details refer to review (37).
Ligand recognition by both receptors leads to signal transduction through Rip2 kinase with downstream activation of MAP kinases and the transcription factor NF-κB, leading to activation of genes encoding different cytokines, chemokines (e.g., IL-8), and antimicrobial peptides. Both NOD signaling cascades are regulated by small GTPases such as Rac1, however conflicting evidence exists as to whether this regulation enhances or reduces NOD-dependent NF-κB activation (38–40). A recent study suggested that Rac1 is activated upstream of NOD1, and that NOD1 essentially senses GTPase activation rather than the peptidoglycan fragments directly (41). NOD1 and NOD2 can recruit the GTPase ATG16L1 and subsequently stimulate autophagy, a highly conserved bulk degradation system with antimicrobial activity against intracellular pathogens (42).
Among the studied lung pathogens, NOD1 responds to Chlamydophila pneumoniae, Legionella pneumophila, Klebsiella pneumoniae, Haemophilus influenzae, and Pseudomonas aeruginosa (32, 40, 43–47), whereas NOD2 senses S. pneumoniae, S. aureus, Escherichia coli, C. pneumoniae, and Mycobacterium tuberculosis (30–32, 44, 48–50) (Figure 2). Accordingly, Rip2−/− mice – and to a lesser extend also Nod1−/− and Nod2−/− mice – display impaired chemokine production, neutrophil recruitment, and reduced antibacterial defense in response to pulmonary C. pneumoniae or L. pneumophila infection (32, 44). NOD2 is also required for efficient antibacterial innate and adaptive immunity in the chronic phase of pulmonary M. tuberculosis infection (51), and polymorphisms in the human NOD2 gene have been associated with resistance or susceptibility to tuberculosis (52). Of note, mycobacteria express N-glycolylated MDP that has a stronger NOD2-activating potential compared to the MDP (53). NOD2 controls inflammatory responses to S. aureus pneumonia (49), and it is also required to clear pneumococcal colonization of the upper respiratory tract by CCR2-dependently recruited monocytes/macrophages. It was shown that professional phagocytes produce CCL2 after LysM-mediated bacterial digestion and subsequent NOD2-dependent detection of S. pneumoniae-derived peptidoglycan (48). Similarly, NOD1 controls neutrophil-dependent clearance of nasopharyngeal colonization with encapsulated H. influenzae in mice, whereas it is redundant for non-encapsulated strains (46). NOD1 might critically regulate microbial competition in the upper respiratory tract as H. influenzae derived peptidoglycan fragments activate NOD1, which instructs neutrophils to clear co-colonizing S. pneumoniae (54). Finally, one study implicated NOD2 in antiviral immunity to RSV and influenza virus infections (55).
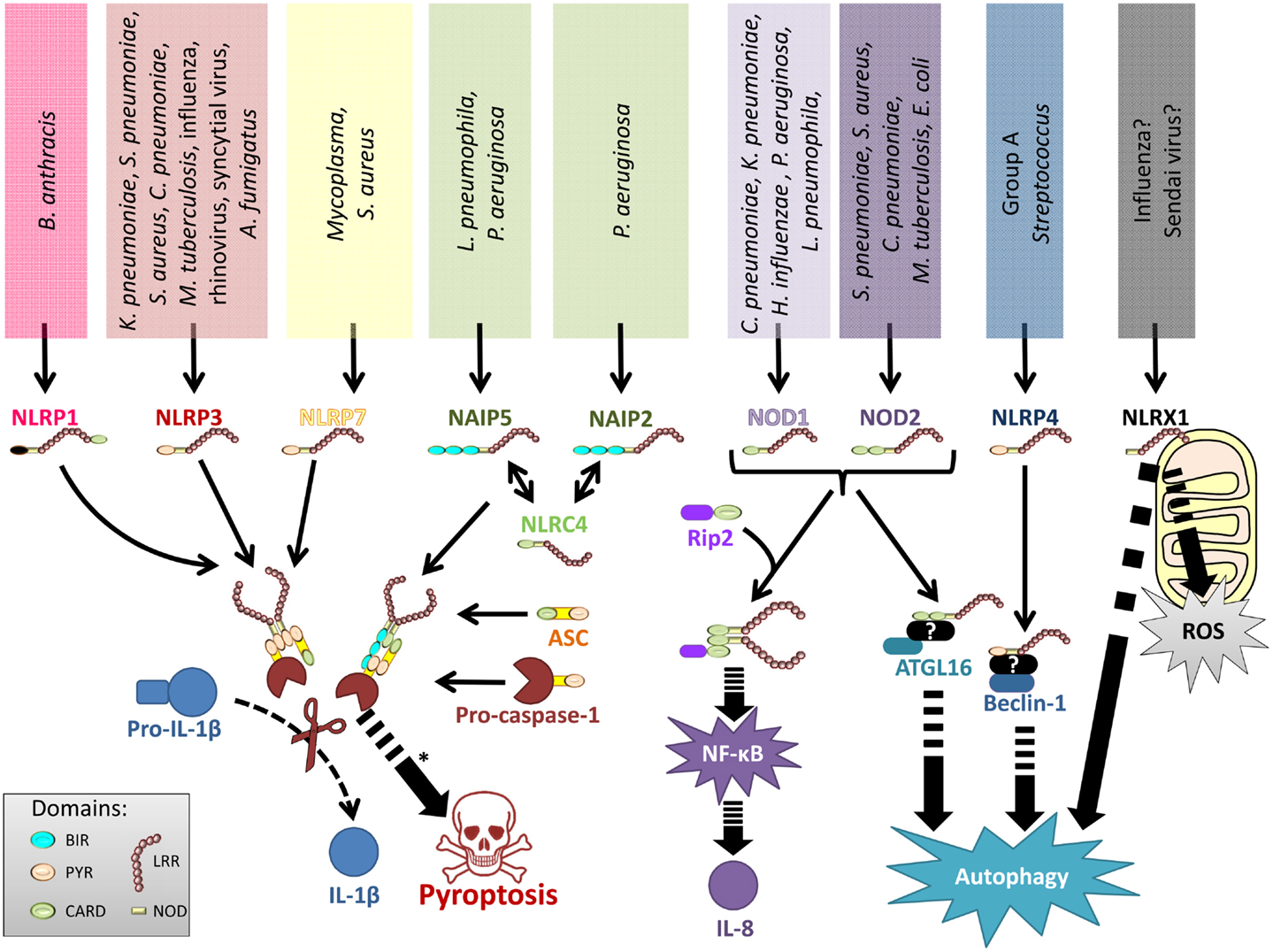
Figure 2. Representation of NLRs involved in lung infections. Various lung pathogens can be sensed by different NLR. These NLRs either form inflammasomes to regulate IL-1 family cytokines and pyroptotic cell death, stimulate production of NF-κB-dependent inflammatory mediators, regulate autophagy, or control ROS production.
Importantly, NOD proteins might also indirectly regulate immune responses in the respiratory tract. An elegant study by Weiser and colleagues showed than intestinal microbiota-derived NOD1 ligands translocate into the circulation and the bone marrow, where it enhances protective neutrophil functions in the periphery. This NOD1 induced neutrophil activation is required for efficient clearance of S. pneumoniae or S. aureus from the respiratory tract (56). NOD2 regulates the composition of the intestinal microbiota in mice (57, 58) and one might speculate about a similar function in shaping microbial communities in the upper respiratory tract.
Finally, NOD1 and NOD2 have been implicated in granulomatous and allergic lung diseases. For example, a genetic variation in NOD1 was found to be associated with increased susceptibility to sarcoidosis in a Japanese cohort (59), and NOD2 polymorphisms were associated with severe pulmonary sarcoidosis in Caucasian patients (60). NOD1 as well as NOD2 polymorphisms have been associated with increased risk of developing allergy and allergic asthma (61–64). Moreover, intranasal delivery of NOD2 ligands was shown to inhibit airway tolerance to antigens by modulating the Treg/Th2-cell balance (65). However, the function of NOD1/2 in these diseases remains ill-defined as compared to their well-established role in host defense.
NLRP Proteins
The NLRP subgroup of NLRs comprises 14 proteins of which NLRP1, NLRP3, NLRP6, NLRP7, and NLRP12 form multiprotein complexes termed inflammasomes, consisting of one or two NLR proteins, the adapter molecule ASC and pro-caspase-1 (20). Inflammasomes serve as platforms for autocatalytic caspase-1 activation, which in turn critically regulates IL-1β and IL-18 production by processing their zymogens proIL-1β and proIL-18, and induce an inflammatory form of cell death called pyroptosis. Inflammasome activation has also been implicated in the production of eicosanoids (66). A number of NLRPs, such as NLRP6 and NLRP12 exert inflammasome-independent functions, like negative regulation of innate immune signaling pathways (as discussed below).
NLRP1
NLRP1 was the first NLR protein to be described as forming an inflammasome (67). In humans, NLRP1 is abundantly expressed in myeloid cells, lymphocytes, and respiratory epithelial cells (68). A biochemical study showed that purified human NLRP1 can form an active inflammasome with ASC and caspase-1 in presence of MDP and ribonucleoside triphosphates (69). Nevertheless, it has so far not been clearly confirmed that MDP can trigger NLRP1 inflammasome formation in human cells.
Mice possess three genes encoding NLRP1, which are present in tandem on chromosome 11: NLRP1a, 1b, and 1c (70). Depending on the genetic background, one, two, or three of these NLRP1s can be expressed. Mouse NLRP1b senses lethal toxin (LT) of Bacillus anthracis, leading to inflammasome activation (70, 71). Using cells from wild-type and NLRPP1−/− mice, it was shown that LT but not MDP could trigger the NLRP1 inflammasome assembly (72). LT consists of two components: protective antigen (PA) and lethal factor (LF). PA mediates cytosolic uptake of LF, which has endopeptidase activity and cleaves several MAPK kinases [reviewed in Ref. (73, 74)]. This way B. anthracis blocks early immune responses by abrogating TLRs and NOD2 signaling (75). The expression of NLRP1b and potentially NLRP1a in macrophages, depending on the mouse background, leads to resistance to LT (70, 72, 75, 76).
It is unclear if NLRP1 mediated LT resistance exists in human cells, however it was mentioned in a recent review that the authors had never observed LT resistance in macrophages isolated from healthy human subjects (77). Studies in rats and mice, which also present cell death induced by LT depending on NLRP1, showed that LF-mediated cleavage of the N-terminal domain of NLRP1 leads to caspase-1 activation and IL-1β release (71, 78, 79). Interestingly, a recent study indicated that a direct cleavage of murine NLRP1b is sufficient to induce inflammasome activation in the absence of LF, and proposed that NLRP1 might function as a sensor of protease activity of multiple pathogens (79).
NLRP3
Expression of NLRP3 is strongly induced by inflammatory cytokines and TLR agonists in myeloid cells (68, 80). Moreover, low level expression has also been found in human bronchial epithelial cells (81). Similar to the other inflammasomes, the NLRP3 inflammasome mediates caspase-1-dependent processing of proIL-1β as well as proIL-18 into their mature forms and stimulates pyroptosis (20).
The NLRP3 inflammasome responds to a broad range of microbial and non-microbial agents. Among lung pathogenic microorganisms, K. pneumoniae, S. pneumoniae, S. aureus, C. pneumoniae, M. tuberculosis, L. pneumophila, influenza virus, human rhinovirus, RSV, and Aspergillus fumigatus have been shown to induce NLRP3 activation (82–99). It is generally accepted that those microbes or their molecules do not directly interact with NLRP3, but instead microbe-induced disruption of host cell physiology is sensed by NLRP3. The exact nature of the NLRP3 activating signal remains somewhat elusive although production of reactive oxygen species (ROS) (16, 100), mitochondrial dysfunction (101, 102), potassium efflux (103, 104), calcium mobilization (105), have been implicated in NLRP3 inflammasome activation during infection. Most pathogens stimulate ROS production in host phagocytes, which might be involved in NLRP3 activation (85, 93). Furthermore, S. aureus, S. pneumoniae, M. tuberculosis, and influenza virus disturb the cell membrane and/or the intracellular ionic concentrations by their pore-forming toxins, secretion apparatus or ion channel proteins (82, 86–88, 96, 106). Other pathogens might activate NLRP3 through an incompletely defined mechanism upstream of NLRP3 that senses microbial RNA (107), and Gram-negative bacteria stimulate a non-canonical caspase-11 inflammasome (108–111). Interestingly, NLRP3 inflammasome activation by non-pathogenic bacteria that do not actively disrupt host cell integrity is dependent on bacterial viability. Live but not dead bacteria contain significant amounts of mRNA, the recognition of which triggers NLRP3 inflammasome formation. This response requires the adaptor protein TRIF, but it remains unclear whether prokaryotic mRNA can directly activate NLRP3 or if it is the result of a proximal signaling event. Detection of bacterial mRNA is a key mechanism employed by the host immune system to sense the presence of viable and thus infectious microbes and thereby to scale the level of infectious threat (112, 113). These findings underscore the role of NLRP3 as a sensor of microbial (and non-microbial) danger signals.
Several in vivo infection models have highlighted the central role of NLRP3 in host defense. NLRP3 was required for efficient antimicrobial responses against S. pneumoniae, K. pneumoniae, and influenza A virus in vivo (84, 85, 87, 88). Interestingly, the known susceptibility of aged mice toward influenza infection has been attributed to a reduced expression of NLRP3, ASC, and caspase-1 (114). It remains to be studied whether a similar mechanism contributes to the elevated susceptibility of elder humans to community-acquired pneumonia (115). Importantly, NLRP3 activation may also contribute to ALI, which was observed in a mouse model of S. aureus pneumonia (96). The net effect of NLRP3 during pneumonia might thus dependent on the pathogen load, the virulence of the pathogen and/or the expression of inflammasome components, as well as the susceptibility of the patient to pulmonary damage.
Of note, NLRP3 (and even more pronounced the NLRP6; see below) inflammasome activation in the gut shapes the intestinal microbiota (116). The commensal microflora in turn induces expression of NLRP3, proIL-1β, and proIL-18 in the lung (117). This microflora driven host gene regulation is beneficial since antibiotic depletion of the resident microbiota resulted in markedly elevated susceptibility to influenza A virus infection in mice (117).
Importantly, the NLRP3 inflammasome responds to a vast range of sterile stimuli, particularly so-called DAMPs released by dying cells including ATP, uric acid metabolites, biglycan as well as hyaluronan (106, 118–121). Experimental studies in mice suggest activation of NLRP3 by some of those DAMPs might have important functions in the pathogenesis of ALI/ARDS, COPD/emphysema, and lung fibrosis.
Efficient pulmonary gas exchange critically depends on the integrity of the fragile lung barrier composed of the alveolar epithelium and the endothelium of the pulmonary microvasculature. ALI and ARDS can develop in the course of pneumonia, sepsis, as a result of mechanical ventilation and hyperoxia, aspiration of gastric content, or major trauma (6). ALI and ARDS are characterized by a disrupted lung barrier, resulting in interstitial and alveolar edema, impaired gas exchange, and in severe cases organ failure and death. In addition, lung fibrosis may develop as a long term consequence of ALI/ARDS. Bleomycin treatment as a mouse model of acute inflammation and fibrosis results in uric acid- and ATP-release by dying cells that stimulated NLRP3 activation and IL-1β production and IL-1R-mediated inflammation, remodeling, and fibrosis (122–124). Bleomycin induced inflammation and fibrosis can be rescued by treatment with IL-1R antagonist (Anakinra) (123), allopurinol (impairs uric acid synthesis), uricase (degrades uric acid) (122), and apyrase (degrades ATP) (124). Moreover, it has been suggested that hyperoxia leads to NLRP3 inflammasome activation, secretion of pro-inflammatory cytokines, epithelial barrier dysfunction, and cell death (125, 126). Mechanical ventilation was shown to enhance IL-18 levels in the lung and serum, and inhibition of caspase-1 or IL-18 reduced ventilation-induced lung injury (127). Human ARDS patients express increased mRNA levels of inflammasome-related genes and IL-18 protein in their peripheral blood (127).
NLRP3 inflammasomes might also contribute to pathogenesis of chronic pulmonary disorders such as COPD and emphysema. Concentrations of uric acid is increased in broncho-alveolar fluid (BALF) of smokers and individuals with COPD as compared to healthy controls (128). COPD patients also have reduced levels of IL-1R antagonist (IL1RA) compared to controls (129). The mouse model of elastase-induced emphysema depends on uric acid, NLRP3, ASC, IL-1R, and MyD88 as critical mediators of inflammation, alveolar wall destruction, and fibrosis (130). Conflicting data exist regarding the contribution of the NLRP3 pathway in tobacco smoke-induced pulmonary inflammation. Whereas the study by Doz et al. indicated that smoke-induced inflammation is mediated by TLRs, the purinergic receptor P2X7, caspase-1, and IL-1R (131, 132), Pauwels et al. reported in another study that smoke-induced pulmonary inflammation occurs independently of the NLRP3 inflammasome (133). Transgenic overexpression of mature IL-1β in the lung epithelium of mice evokes a phenotype that closely resembles COPD, including inflammation, emphysema, airway fibrosis, and mucus cell metaplasia (134). Finally, H. influenzae infection induces NLRP3 expression and activation in human lung tissue, which might be a mechanism of infection-triggered COPD exacerbations (135). These studies together indicate an important role of caspase-1 and IL-1β in COPD and emphysema.
Conflicting evidence exists regarding the role of NLRP3 inflammasome-dependent IL-1β production in experimental asthma. Whereas ovalbumin-induced airway inflammation requires NLRP3 and IL-1β, house dust mite allergens induce pathology in an NLRP3-independent fashion (136, 137).
Pneumoconiosis is an occupational lung disease resulting from long-term exposure to silica, asbestos, or coal particles. It is characterized by pulmonary inflammation as well as fibrosis, which may be driven by NLRP3 inflammasome activation. It has been shown that engulfment of silica or asbestos crystals by resident macrophages leads to NLRP3 inflammasome activation and IL-1β production (16–18). It was suggested that crystal-induced inflammasome formation is a consequence of phagolysosomal disruption and leakage of enzymes such as cathepsin B into the cytoplasm (18). Nlrp3−/− and Asc−/− mice are protected from silica or asbestos-induced granuloma formation and fibrosis (16, 17). In contrast, mesothelioma development, a serious long term consequence of asbestosis, appears to be independent of NLRP3 (138). A recent case-control study in a Chinese population suggested that a NLRP3 polymorphisms may confer increased risk for coal workers’ pneumoconiosis (139).
Taken together, NLRP3 is a key sensor of disturbed cell and tissue integrity during infectious and non-infectious pulmonary disorders.
NLRP4
NLRP4 has been proposed to be involved in reproduction in mammals (140–142). Nevertheless, NLRP4 expression in humans is found in various organs including the lung (142–144). In vitro, this NLR has the feature of a negative regulator of inflammatory responses by lowering NF-κB activation and IFNβ production (143, 144). Another particularity of NLRP4 is that its PYD is structurally different compared to the one in other NLRs, leading to the absence of interaction of NLRP4 with ASC (145). Besides, it has been described that NLRP4 negatively controls autophagy during group A streptococcal infection by interacting with the autophagy regulator Beclin-1 (146). However, in the absence of conditional gene-targeted mice it is hard to predict a functional contribution of NLRP4 to pathologies in the lung.
NLRP6
NLRP6 has been indicated to fulfill anti-inflammatory functions by inhibiting NF-κB signaling downstream of, e.g., TLRs in macrophages and mouse (147). Moreover, elegant studies by the Flavell’s laboratory showed that NLRP6 can form an inflammasome in intestinal epithelial cells that appears to sense components of the gut microflora and in turn regulates the composition of this flora through IL-18 (116). Related or unrelated to these mechanisms, NLRP6 has also been implicated in wound healing of the intestinal mucosa (148). NLRP6 have so far been mainly described in intestinal epithelial cells, neutrophils, and macrophages (116, 147) but our own unpublished data show expression of this protein also in activated murine alveolar epithelial cells (data not shown). The function of NLRP6 in the lung has, however, not been studied yet.
NLRP7
The NLRP7 gene is only present in humans and it is expressed in peripheral blood mononuclear cells (PBMCs) upon LPS and IL-1β stimulation (149). Gene silencing experiments in human monocytes and macrophages recently indicated that NLRP7 responds to bacterial lipopeptides and Mycoplasma as well as S. aureus infections by forming an inflammasome (150). Its precise function during bacterial infections remains unknown, and no data is available regarding its role in pulmonary physiology.
NLRP12
NLRP12 is expressed mainly in myeloid cells (151–153). Its expression is reduced by TLR stimulation and TNFα (151, 154) NLRP12 has been described as a negative regulator of classical and non-classical NF-κB activation downstream of TLR or cytokine receptors, by interacting with IKK and NIK (151, 155, 156). Furthermore, NLRP12 has been indicated to form an inflammasome, however this has so far only been observed upon Yersinia pestis infection (157). Finally, NLRP12 might play a role in adaptive immunity by controlling migration of DCs to the draining lymph nodes (153). NLRP12−/− mice did not respond differently to M. tuberculosis and K. pneumoniae lung infections and allergic airway inflammation wild-type mice in (158, 159), suggesting a functional redundancy with other NLRs, or a minor contribution of NLRP12 to inflammatory processes in the lung.
NLRC4 and NAIP Proteins
NAIP5 and NLRC4 are expressed in the cytosol of bone-marrow and alveolar macrophages. A polymorphism in NAIP5 (also called Bircle) has long been known to affect resistance of inbred mice toward L. pneumophila (160, 161). Whereas most mice strains are resistant against L. pneumophila infection due to a functional NAIP5, A/J mice expressing a NAIP5 that differs in 14 amino acids or NAIP5−/− mice allow L. pneumophila replication (162–164). This NAIP5-mediated resistance against L. pneumophila is dependent on detection of flagellin (162, 165), and on pyroptosis of the infected macrophage as well as effects on the trafficking of the Legionella-containing vacuole (166, 167). Similarly, NLRC4 is well known for mediating caspase-1-dependent responses to L. pneumophila and other flagellated bacteria (168–171). NLRC4, however, also respond to bacteria that express a type 3 secretion system (T3SS) including, for example, P. aeruginosa (172–176). Of note, the Pseudomonas T3SS effector protein ExoU can inhibit this inflammasome activation (176). One study suggested that NLCR4 is partially involved in the production of IL-1β and inflammasome-independent cytokines upon K. pneumoniae infection in vivo (177), even though K. pneumoniae does neither express flagella nor T3SS.
It is now clear that murine NLRC4 forms together with either NAIP5 (and possibly NAIP6) or NAIP2 two different inflammasomes that recognize flagellin or T3SS rod proteins, respectively (178, 179). These inflammasomes appear to regulate IL-1β/IL-18 through ASC and pyroptosis independently of ASC (175, 180, 181).
The exact function of human NAIP is currently incompletely understood. We and others suggested that hNAIP can detect and restrict flagellated Legionella (182–184), whereas others indicated recognition of bacterial T3SS needle proteins by hNAIP (178).
NLRC3 and 5
NLRC3 is expressed in macrophages as well as lymphocytes and has been suggested to function as a negative regulator of the early TLR signaling (185). NLRC5 is highly expressed in mouse and human lung tissue. Its expression can be further induced by IFNγ and LPS in macrophages (186–189). NLRC5 protein has been described to inhibit IL-1β, TNFα, and IL-6 productions in macrophages upon viral infection. Its most prominent role is probably in adaptive immunity by enhancing the transcription of MHC class I related genes (186, 187, 189–197). In light of these data, NLRC5 involvement in pulmonary infection such as influenza infection will be an interesting area of study.
NLRX1
NLRX1 is ubiquitously expressed and located at the mitochondria due to an N-terminal mitochondrial targeting sequence, although the precise location (matrix or outer membrane) is still controversial (198, 199). The C-terminal LRR domain has been shown to bind to RNA but not to DNA by (200). Silencing of NLRX1 expression or knockout at the exons 4–5 in mice leads to exacerbated immune responses in vivo upon TLR stimulation and influenza or Sendai virus infections (201, 202). Knockout of the first four exons or exon 3, however, had no influence on the immune response to Sendai and influenza virus infections (203, 204). Instead, NLRX1 might function as an inducer of mitochondrial ROS production (205). This is consistent with the finding of two different groups that NLRX1 interacts with a protein of the mitochondrial respiratory chain (198, 203). Finally, NLRX1 has been indicated to regulate autophagy (206). Further work is clearly needed to clarify the function and exact mode of action of this unique NLR protein in general and in the lung in particular.
Concluding Remarks
Nucleotide-binding oligomerization domain-like receptors proteins are without doubt key players in the innate immune responses to infectious and sterile inflammatory diseases of the lung, although many functions of several NLR family members, particularly in the lung, are still unknown. Many NLRs respond in functional cooperation with other innate sensors to invading microbes, particles, and endogenous danger signals after tissue damage. In similarity to possibly most immune receptors they can exert beneficial or detrimental functions, depending on the magnitude and the context of their activation. Increasing knowledge on specific activators and inhibitors of these pathways might help to manipulate them therapeutically in the not-so-distant future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the lab for helpful discussions. Research Support: own work described in the text was supported by grants given by the Deutsche Forschungsgemeinschaft (SFB-TR84 to Catherine Chaput, Norbert Suttorp, Bastian Opitz; OP 86/7-2 to Bastian Opitz; SA1940/2-1 to Leif Erik Sander).
References
1. Lemon KP, Klepac-Ceraj V, Schiffer HK, Brodie EL, Lynch SV, Kolter R. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. MBio (2010) 1:e00129–10. doi:10.1128/mBio.00129-10
2. Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med (2011) 184:957–63. doi:10.1164/rccm.201104-0655OC
3. Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One (2011) 6:e16384. doi:10.1371/journal.pone.0016384
4. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One (2010) 5:e8578. doi:10.1371/journal.pone.0008578
6. Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol (2011) 6:147–63. doi:10.1146/annurev-pathol-011110-130158
7. Opitz B, van Laak V, Eitel J, Suttorp N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med (2010) 181:1294–309. doi:10.1164/rccm.200909-1427SO
8. dos Santos G, Kutuzov MA, Ridge KM. The inflammasome in lung diseases. Am J Physiol Lung Cell Mol Physiol (2012) 303:L627–33. doi:10.1152/ajplung.00225.2012
9. Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol (2011) 45:189–201. doi:10.1165/rcmb.2011-0011RT
10. Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol (2009) 4:365–98. doi:10.1146/annurev.pathol.4.110807.092239
11. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol (2012) 28:137–61. doi:10.1146/annurev-cellbio-101011-155745
12. Magalhaes JG, Sorbara MT, Girardin SE, Philpott DJ. What is new with Nods? Curr Opin Immunol (2011) 23:29–34. doi:10.1016/j.coi.2010.12.003
13. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54(Pt 1):1–13. doi:10.1101/SQB.1989.054.01.003
14. Matzinger P. The danger model: a renewed sense of self. Science (2002) 296:301–5. doi:10.1126/science.1071059
15. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol (2010) 28:321–42. doi:10.1146/annurev-immunol-030409-101311
16. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (2008) 320:674–7. doi:10.1126/science.1156995
17. Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A (2008) 105:9035–40. doi:10.1073/pnas.0803933105
18. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9:847–56. doi:10.1038/ni.1631
19. Zika E, Ting JP. Epigenetic control of MHC-II: interplay between CIITA and histone-modifying enzymes. Curr Opin Immunol (2005) 17:58–64. doi:10.1016/j.coi.2004.11.008
20. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi:10.1016/j.cell.2010.01.040
21. Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem (1999) 274:14560–7. doi:10.1074/jbc.274.21.14560
22. Bertin J, Nir WJ, Fischer CM, Tayber OV, Errada PR, Grant JR, et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappaB. J Biol Chem (1999) 274:12955–8. doi:10.1074/jbc.274.19.12955
23. Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem (2001) 276:4812–8. doi:10.1074/jbc.M008072200
24. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411:603–6. doi:10.1038/35079114
25. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411:599–603. doi:10.1038/35079107
26. Slevogt H, Seybold J, Tiwari KN, Hocke AC, Jonatat C, Dietel S, et al. Moraxella catarrhalis is internalized in respiratory epithelial cells by a trigger-like mechanism and initiates a TLR2- and partly NOD1-dependent inflammatory immune response. Cell Microbiol (2007) 9:694–707. doi:10.1111/j.1462-5822.2006.00821.x
27. Opitz B, Puschel A, Beermann W, Hocke AC, Forster S, Schmeck B, et al. Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J Immunol (2006) 176:484–90.
28. Barton JL, Berg T, Didon L, Nord M. The pattern recognition receptor Nod1 activates CCAAT/enhancer binding protein beta signalling in lung epithelial cells. Eur Respir J (2007) 30:214–22. doi:10.1183/09031936.00143906
29. Månsson Kvarnhammar A, Tengroth L, Adner M, Cardell L-O. Innate immune receptors in human airway smooth muscle cells: activation by TLR1/2, TLR3, TLR4, TLR7 and NOD1 agonists. PLoS One (2013) 8:e68701. doi:10.1371/journal.pone.0068701
30. Opitz B, Puschel A, Schmeck B, Hocke AC, Rosseau S, Hammerschmidt S, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem (2004) 279:36426–32. doi:10.1074/jbc.M403861200
31. Theivanthiran B, Batra S, Balamayooran G, Cai S, Kobayashi K, Flavell RA, et al. NOD2 signaling contributes to host defense in the lungs against Escherichia coli infection. Infect Immun (2012) 80:2558–69. doi:10.1128/IAI.06230-11
32. Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, Slepenkin AV, et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog (2009) 5:e1000379. doi:10.1371/journal.ppat.1000379
33. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science (2003) 300:1584–7. doi:10.1126/science.1084677
34. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol (2003) 4:702–7. doi:10.1038/ni945
35. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278:8869–72. doi:10.1074/jbc.C200651200
36. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem (2003) 278:5509–12. doi:10.1074/jbc.C200673200
37. Kuo CC, Lee A, Campbell LA. Cleavage of the N-linked oligosaccharide from the surfaces of Chlamydia species affects attachment and infectivity of the organisms in human epithelial and endothelial cells. Infect Immun (2004) 72:6699–701. doi:10.1128/IAI.72.11.6699-6701.2004
38. Eitel J, Krull M, Hocke AC, N’Guessan PD, Zahlten J, Schmeck B, et al. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J Immunol (2008) 181:2664–71.
39. Legrand-Poels S, Kustermans G, Bex F, Kremmer E, Kufer TA, Piette J. Modulation of Nod2-dependent NF-kappaB signaling by the actin cytoskeleton. J Cell Sci (2007) 120:1299–310. doi:10.1242/jcs.03424
40. Regueiro V, Moranta D, Frank CG, Larrarte E, Margareto J, March C, et al. Klebsiella pneumoniae subverts the activation of inflammatory responses in a NOD1-dependent manner. Cell Microbiol (2011) 13:135–53. doi:10.1111/j.1462-5822.2010.01526.x
41. Keestra AM, Winter MG, Auburger JJ, Frassle SP, Xavier MN, Winter SE, et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature (2013) 496:233–7. doi:10.1038/nature12025
42. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11:55–62. doi:10.1038/ni.1823
43. Berrington WR, Iyer R, Wells RD, Smith KD, Skerrett SJ, Hawn TR. NOD1 and NOD2 regulation of pulmonary innate immunity to Legionella pneumophila. Eur J Immunol (2010) 40:3519–27. doi:10.1002/eji.201040518
44. Frutuoso MS, Hori JI, Pereira MS, Junior DS, Sonego F, Kobayashi KS, et al. The pattern recognition receptors Nod1 and Nod2 account for neutrophil recruitment to the lungs of mice infected with Legionella pneumophila. Microbes Infect (2010) 12:819–27. doi:10.1016/j.micinf.2010.05.006
45. Opitz B, Forster S, Hocke AC, Maass M, Schmeck B, Hippenstiel S, et al. Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ Res (2005) 96:319–26. doi:10.1161/01.RES.0000155721.83594.2c
46. Zola TA, Lysenko ES, Weiser JN. Mucosal clearance of capsule-expressing bacteria requires both TLR and nucleotide-binding oligomerization domain 1 signaling. J Immunol (2008) 181:7909–16.
47. Travassos LH, Carneiro LA, Girardin SE, Boneca IG, Lemos R, Bozza MT, et al. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J Biol Chem (2005) 280:36714–8. doi:10.1074/jbc.M501649200
48. Davis KM, Nakamura S, Weiser JN. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Invest (2011) 121:3666–76. doi:10.1172/JCI57761
49. Kapetanovic R, Jouvion G, Fitting C, Parlato M, Blanchet C, Huerre M, et al. Contribution of NOD2 to lung inflammation during Staphylococcus aureus-induced pneumonia. Microbes Infect (2010) 12:759–67. doi:10.1016/j.micinf.2010.05.003
50. Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, de Jong DJ, Langenberg DM, et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog (2005) 1:e34. doi:10.1371/journal.ppat.0010034
51. Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol (2008) 181:7157–65.
52. Austin CM, Ma X, Graviss EA. Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J Infect Dis (2008) 197:1713–6. doi:10.1086/588384
53. Coulombe F, Divangahi M, Veyrier F, de Leseleuc L, Gleason JL, Yang Y, et al. Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J Exp Med (2009) 206:1709–16. doi:10.1084/jem.20081779
54. Lysenko ES, Clarke TB, Shchepetov M, Ratner AJ, Roper DI, Dowson CG, et al. Nod1 signaling overcomes resistance of S. pneumoniae to opsonophagocytic killing. PLoS Pathog (2007) 3:e118. doi:10.1371/journal.ppat.0030118
55. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol (2009) 10:1073–80. doi:10.1038/ni.1782
56. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med (2010) 16:228–31. doi:10.1038/nm.2087
57. Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut (2011) 60:1354–62. doi:10.1136/gut.2010.216259
58. Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A (2009) 106:15813–8. doi:10.1073/pnas.0907722106
59. Tanabe T, Ishige I, Suzuki Y, Aita Y, Furukawa A, Ishige Y, et al. Sarcoidosis and NOD1 variation with impaired recognition of intracellular Propionibacterium acnes. Biochim Biophys Acta (2006) 1762:794–801. doi:10.1016/j.bbadis.2006.07.006
60. Sato H, Williams HR, Spagnolo P, Abdallah A, Ahmad T, Orchard TR, et al. CARD15/NOD2 polymorphisms are associated with severe pulmonary sarcoidosis. Eur Respir J (2010) 35:324–30. doi:10.1183/09031936.00010209
61. Kabesch M, Peters W, Carr D, Leupold W, Weiland SK, von Mutius E. Association between polymorphisms in caspase recruitment domain containing protein 15 and allergy in two German populations. J Allergy Clin Immunol (2003) 111:813–7. doi:10.1067/mai.2003.1336
62. Weidinger S, Klopp N, Rummler L, Wagenpfeil S, Novak N, Baurecht HJ, et al. Association of NOD1 polymorphisms with atopic eczema and related phenotypes. J Allergy Clin Immunol (2005) 116:177–84. doi:10.1016/j.jaci.2005.02.034
63. Weidinger S, Klopp N, Rummler L, Wagenpfeil S, Baurecht HJ, Gauger A, et al. Association of CARD15 polymorphisms with atopy-related traits in a population-based cohort of Caucasian adults. Clin Exp Allergy (2005) 35:866–72. doi:10.1111/j.1365-2222.2005.02269.x
64. Hysi P, Kabesch M, Moffatt MF, Schedel M, Carr D, Zhang Y, et al. NOD1 variation, immunoglobulin E and asthma. Hum Mol Genet (2005) 14:935–41. doi:10.1093/hmg/ddi087
65. Duan W, Mehta AK, Magalhaes JG, Ziegler SF, Dong C, Philpott DJ, et al. Innate signals from Nod2 block respiratory tolerance and program T(H)2-driven allergic inflammation. J Allergy Clin Immunol (2010) 126(1284-93):e10. doi:10.1016/j.jaci.2010.09.021
66. von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature (2012) 490:107–11. doi:10.1038/nature11351
67. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi:10.1016/S1097-2765(02)00599-3
68. Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, et al. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem (2007) 55:443–52. doi:10.1369/jhc.6A7101.2006
69. Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25:713–24. doi:10.1016/j.molcel.2007.01.032
70. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet (2006) 38:240–4. doi:10.1038/ng1724
71. Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog (2012) 8:e1002638. doi:10.1371/journal.ppat.1002638
72. Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, et al. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol (2012) 189:2006–16. doi:10.4049/jimmunol.1201065
73. Abrami L, Reig N, van der Goot FG. Anthrax toxin: the long and winding road that leads to the kill. Trends Microbiol (2005) 13:72–8. doi:10.1016/j.tim.2004.12.004
74. Tonello F, Montecucco C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol Aspects Med (2009) 30:431–8. doi:10.1016/j.mam.2009.07.006
75. Terra JK, Cote CK, France B, Jenkins AL, Bozue JA, Welkos SL, et al. Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J Immunol (2010) 184:17–20. doi:10.4049/jimmunol.0903114
76. Sastalla I, Crown D, Masters SL, McKenzie A, Leppla SH, Moayeri M. Transcriptional analysis of the three Nlrp1 paralogs in mice. BMC Genomics (2013) 14:188. doi:10.1186/1471-2164-14-188
77. Moayeri M, Sastalla I, Leppla SH. Anthrax and the inflammasome. Microbes Infect (2012) 14:392–400. doi:10.1016/j.micinf.2011.12.005
78. Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, et al. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS One (2012) 7:e49741. doi:10.1371/journal.pone.0049741
79. Chavarria-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog (2013) 9:e1003452. doi:10.1371/journal.ppat.1003452
80. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183:787–91. doi:10.4049/jimmunol.0901363
81. Hirota JA, Hirota SA, Warner SM, Stefanowicz D, Shaheen F, Beck PL, et al. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J Allergy Clin Immunol (2012) 129(1116-25):e6. doi:10.1016/j.jaci.2011.11.033
82. Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol (2009) 183:3942–8. doi:10.4049/jimmunol.0900729
83. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med (2009) 206:79–87. doi:10.1084/jem.20081667
84. Thomas PG, Dash P, Aldridge JR Jr, Ellebedy AH, Reynolds C, Funk AJ, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity (2009) 30:566–75. doi:10.1016/j.immuni.2009.02.006
85. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity (2009) 30:556–65. doi:10.1016/j.immuni.2009.02.005
86. Carlsson F, Kim J, Dumitru C, Barck KH, Carano RA, Sun M, et al. Host-detrimental role of Esx-1-mediated inflammasome activation in mycobacterial infection. PLoS Pathog (2010) 6:e1000895. doi:10.1371/journal.ppat.1000895
87. McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog (2010) 6:e1001191. doi:10.1371/journal.ppat.1001191
88. Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol (2011) 187:434–40. doi:10.4049/jimmunol.1003143
89. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol (2009) 183:2008–15. doi:10.4049/jimmunol.0900138
90. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol (2010) 12:1046–63. doi:10.1111/j.1462-5822.2010.01450.x
91. He X, Mekasha S, Mavrogiorgos N, Fitzgerald KA, Lien E, Ingalls RR. Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. J Immunol (2010) 184:5743–54. doi:10.4049/jimmunol.0903937
92. Eitel J, Meixenberger K, van Laak C, Orlovski C, Hocke A, Schmeck B, et al. Rac1 regulates the NLRP3 inflammasome which mediates IL-1beta production in Chlamydophila pneumoniae infected human mononuclear cells. PLoS One (2012) 7:e30379. doi:10.1371/journal.pone.0030379
93. Said-Sadier N, Padilla E, Langsley G, Ojcius DM. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One (2010) 5:e10008. doi:10.1371/journal.pone.0010008
94. Shimada K, Crother TR, Karlin J, Chen S, Chiba N, Ramanujan VK, et al. Caspase-1 dependent IL-1beta secretion is critical for host defense in a mouse model of Chlamydia pneumoniae lung infection. PLoS One (2011) 6:e21477. doi:10.1371/journal.pone.0021477
95. Segovia J, Sabbah A, Mgbemena V, Tsai SY, Chang TH, Berton MT, et al. TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One (2012) 7:e29695. doi:10.1371/journal.pone.0029695
96. Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, et al. Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis (2012) 205:807–17. doi:10.1093/infdis/jir846
97. Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L, et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol (2012) 42:374–84. doi:10.1002/eji.201141548
98. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol (2013). doi:10.1165/rcmb.2013-0032OC
99. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A (2013) 110:1851–6. doi:10.1073/pnas.1211521110
100. Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem (2007) 282:2871–9. doi:10.1074/jbc.M608083200
101. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi:10.1038/nature09663
102. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi:10.1016/j.immuni.2012.01.009
103. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ (2007) 14:1583–9. doi:10.1038/sj.cdd.4402195
104. Franchi L, Kanneganti TD, Dubyak GR, Nunez G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem (2007) 282:18810–8. doi:10.1074/jbc.M610762200
105. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A (2012) 109:11282–7. doi:10.1073/pnas.1117765109
106. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440:228–32. doi:10.1038/nature04515
107. Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature (2006) 440:233–6. doi:10.1038/nature04517
108. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi:10.1038/nature10558
109. Shin S, Brodsky IE. Caspase-11: the noncanonical guardian of cytosolic sanctity. Cell Host Microbe (2013) 13:243–5. doi:10.1016/j.chom.2013.02.011
110. Ng TM, Monack DM. Revisiting caspase-11 function in host defense. Cell Host Microbe (2013) 14:9–14. doi:10.1016/j.chom.2013.06.009
111. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science (2013) 341:1246–9. doi:10.1126/science.1240248
112. Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol (2012) 12:215–25. doi:10.1038/nri3167
113. Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, et al. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature (2011) 474:385–9. doi:10.1038/nature10072
114. Stout-Delgado HW, Vaughan SE, Shirali AC, Jaramillo RJ, Harrod KS. Impaired NLRP3 inflammasome function in elderly mice during influenza infection is rescued by treatment with nigericin. J Immunol (2012) 188:2815–24. doi:10.4049/jimmunol.1103051
115. Ruuskanen O, Lahti E, Jennings LC, Murdoch DR. Viral pneumonia. Lancet (2011) 377:1264–75. doi:10.1016/S0140-6736(10)61459-6
116. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
117. Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A (2011) 108:5354–9. doi:10.1073/pnas.1019378108
118. Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem (2009) 284:24035–48. doi:10.1074/jbc.M109.014266
119. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A (2009) 106:20388–93. doi:10.1073/pnas.0908698106
120. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440:237–41. doi:10.1038/nature04516
121. Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem (2009) 284:12762–71. doi:10.1074/jbc.M806084200
122. Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med (2009) 179:903–13. doi:10.1164/rccm.200808-1274OC
123. Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest (2007) 117:3786–99. doi:10.1172/JCI32285
124. Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med (2010) 182:774–83. doi:10.1164/rccm.201003-0359OC
125. Kolliputi N, Shaik RS, Waxman AB. The inflammasome mediates hyperoxia-induced alveolar cell permeability. J Immunol (2010) 184:5819–26. doi:10.4049/jimmunol.0902766
126. Fukumoto J, Fukumoto I, Parthasarathy PT, Cox R, Huynh B, Ramanathan GK, et al. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol (2013) 305:C182–9. doi:10.1152/ajpcell.00086.2013
127. Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med (2012) 185:1225–34. doi:10.1164/rccm.201201-0003OC
128. Wanderer AA. Interleukin-1beta targeted therapy in severe persistent asthma (SPA) and chronic obstructive pulmonary disease (COPD): proposed similarities between biphasic pathobiology of SPA/COPD and ischemia-reperfusion injury. Isr Med Assoc J (2008) 10:837–42.
129. Sapey E, Ahmad A, Bayley D, Newbold P, Snell N, Rugman P, et al. Imbalances between interleukin-1 and tumor necrosis factor agonists and antagonists in stable COPD. J Clin Immunol (2009) 29:508–16. doi:10.1007/s10875-009-9286-8
130. Couillin I, Vasseur V, Charron S, Gasse P, Tavernier M, Guillet J, et al. IL-1R1/MyD88 signaling is critical for elastase-induced lung inflammation and emphysema. J Immunol (2009) 183:8195–202. doi:10.4049/jimmunol.0803154
131. Doz E, Noulin N, Boichot E, Guenon I, Fick L, Le Bert M, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol (2008) 180:1169–78.
132. Eltom S, Stevenson CS, Rastrick J, Dale N, Raemdonck K, Wong S, et al. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS One (2011) 6:e24097. doi:10.1371/journal.pone.0024097
133. Pauwels NS, Bracke KR, Dupont LL, Van Pottelberge GR, Provoost S, Vanden Berghe T, et al. Role of IL-1alpha and the Nlrp3/caspase-1/IL-1beta axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J (2011) 38:1019–28. doi:10.1183/09031936.00158110
134. Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol (2005) 32:311–8. doi:10.1165/rcmb.2004-0309OC
135. Rotta Detto Loria J, Rohmann K, Droemann D, Kujath P, Rupp J, Goldmann T, et al. Haemophilus Influenzae infection upregulates the NLRP3 inflammasome and leads to caspase-1-dependent secretion of interleukin-1beta – a possible pathway of exacerbations in COPD. PLoS One (2013) 8:e66818. doi:10.1371/journal.pone.0066818
136. Besnard AG, Guillou N, Tschopp J, Erard F, Couillin I, Iwakura Y, et al. NLRP3 inflammasome is required in murine asthma in the absence of aluminum adjuvant. Allergy (2011) 66:1047–57. doi:10.1111/j.1398-9995.2011.02586.x
137. Kool M, Willart MA, van Nimwegen M, Bergen I, Pouliot P, Virchow JC, et al. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity (2011) 34:527–40. doi:10.1016/j.immuni.2011.03.015
138. Chow MT, Tschopp J, Moller A, Smyth MJ. NLRP3 promotes inflammation-induced skin cancer but is dispensable for asbestos-induced mesothelioma. Immunol Cell Biol (2012) 90:983–6. doi:10.1038/icb.2012.46
139. Ji X, Hou Z, Wang T, Jin K, Fan J, Luo C, et al. Polymorphisms in inflammasome genes and risk of coal workers’ pneumoconiosis in a Chinese population. PLoS One (2012) 7:e47949. doi:10.1371/journal.pone.0047949
140. Chang BH, Liu X, Liu J, Quan FS, Guo ZK, Zhang Y. Developmental expression and possible functional roles of mouse Nlrp4e in preimplantation embryos. In vitro Cell Dev Biol Anim (2013) 49:548–53. doi:10.1007/s11626-013-9638-9
141. Tian X, Pascal G, Monget P. Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol Biol (2009) 9:202. doi:10.1186/1471-2148-9-202
142. Zhang P, Dixon M, Zucchelli M, Hambiliki F, Levkov L, Hovatta O, et al. Expression analysis of the NLRP gene family suggests a role in human preimplantation development. PLoS One (2008) 3:e2755. doi:10.1371/journal.pone.0002755
143. Cui J, Li Y, Zhu L, Liu D, Songyang Z, Wang HY, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol (2012) 13:387–95. doi:10.1038/ni.2239
144. Fiorentino L, Stehlik C, Oliveira V, Ariza ME, Godzik A, Reed JC. A novel PAAD-containing protein that modulates NF-kappa B induction by cytokines tumor necrosis factor-alpha and interleukin-1beta. J Biol Chem (2002) 277:35333–40. doi:10.1074/jbc.M200446200
145. Eibl C, Grigoriu S, Hessenberger M, Wenger J, Puehringer S, Pinheiro AS, et al. Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry (2012) 51:7330–41. doi:10.1021/bi3007059
146. Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol (2011) 186:1646–55. doi:10.4049/jimmunol.1001654
147. Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature (2012) 488:389–93. doi:10.1038/nature11250
148. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A (2011) 108:9601–6. doi:10.1073/pnas.1100981108
149. Radian AD, de Almeida L, Dorfleutner A, Stehlik C. NLRP7 and related inflammasome activating pattern recognition receptors and their function in host defense and disease. Microbes Infect (2013) 15:630–9. doi:10.1016/j.micinf.2013.04.001
150. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity (2012) 36:464–76. doi:10.1016/j.immuni.2012.02.001
151. Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem (2005) 280:39914–24. doi:10.1074/jbc.M502820200
152. Lich JD, Ting JP. Monarch-1/PYPAF7 and other CATERPILLER (CLR, NOD, NLR) proteins with negative regulatory functions. Microbes Infect (2007) 9:672–6. doi:10.1016/j.micinf.2007.01.018
153. Arthur JC, Lich JD, Ye Z, Allen IC, Gris D, Wilson JE, et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol (2010) 185:4515–9. doi:10.4049/jimmunol.1002227
154. Lord CA, Savitsky D, Sitcheran R, Calame K, Wright JR, Ting JP, et al. Blimp-1/PRDM1 mediates transcriptional suppression of the NLR gene NLRP12/Monarch-1. J Immunol (2009) 182:2948–58. doi:10.4049/jimmunol.0801692
155. Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, et al. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J Immunol (2007) 178:1256–60.
156. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity (2012) 36:742–54. doi:10.1016/j.immuni.2012.03.012
157. Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity (2012) 37:96–107. doi:10.1016/j.immuni.2012.07.006
158. Allen IC, Lich JD, Arthur JC, Jania CM, Roberts RA, Callaway JB, et al. Characterization of NLRP12 during the development of allergic airway disease in mice. PLoS One (2012) 7:e30612. doi:10.1371/journal.pone.0030612
159. Allen IC, McElvania-TeKippe E, Wilson JE, Lich JD, Arthur JC, Sullivan JT, et al. Characterization of NLRP12 during the in vivo host immune response to Klebsiella pneumoniae and Mycobacterium tuberculosis. PLoS One (2013) 8:e60842. doi:10.1371/journal.pone.0060842
160. Wright EK, Goodart SA, Growney JD, Hadinoto V, Endrizzi MG, Long EM, et al. Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr Biol (2003) 13:27–36. doi:10.1016/S0960-9822(02)01359-3
161. Diez E, Lee SH, Gauthier S, Yaraghi Z, Tremblay M, Vidal S, et al. Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat Genet (2003) 33:55–60. doi:10.1038/ng1065
162. Molofsky AB, Byrne BG, Whitfield NN, Madigan CA, Fuse ET, Tateda K, et al. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med (2006) 203:1093–104. doi:10.1084/jem.20051659
163. Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vance RE, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol (2006) 7:318–25. doi:10.1038/ni1305
164. Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol (2008) 9:1171–8. doi:10.1038/ni.1646
165. Ren T, Zamboni DS, Roy CR, Dietrich WF, Vance RE. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog (2006) 2:e18. doi:10.1371/journal.ppat.0020018
166. Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog (2009) 5:e1000361. doi:10.1371/journal.ppat.1000361
167. Fortier A, de Chastellier C, Balor S, Gros P. Birc1e/Naip5 rapidly antagonizes modulation of phagosome maturation by Legionella pneumophila. Cell Microbiol (2007) 9:910–23. doi:10.1111/j.1462-5822.2006.00839.x
168. Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozoren N, Brady G, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem (2006) 281:35217–23. doi:10.1074/jbc.M604933200
169. Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem (2001) 276:28309–13. doi:10.1074/jbc.C100250200
170. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol (2006) 7:569–75. doi:10.1038/ni1344
171. Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nat Immunol (2006) 7:576–82. doi:10.1038/ni1346
172. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A (2010) 107:3076–80. doi:10.1073/pnas.0913087107
173. Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog (2007) 3:e111. doi:10.1371/journal.ppat.0030111
174. Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci U S A (2008) 105:2562–7. doi:10.1073/pnas.0712183105
175. Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Nunez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol (2007) 37:3030–9. doi:10.1002/eji.200737532
176. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med (2007) 204:3235–45. doi:10.1084/jem.20071239
177. Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S. NLRC4 inflammasome-mediated production of IL-1beta modulates mucosal immunity in the lung against gram-negative bacterial infection. J Immunol (2012) 188:5623–35. doi:10.4049/jimmunol.1200195
178. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature (2011) 477:596–600. doi:10.1038/nature10510
179. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature (2011) 477:592–5. doi:10.1038/nature10394
180. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe (2010) 8:471–83. doi:10.1016/j.chom.2010.11.007
181. Case CL, Roy CR. Asc modulates the function of NLRC4 in response to infection of macrophages by Legionella pneumophila. mBio (2011) 2:e00117–11. doi:10.1128/mBio.00117-11
182. Vinzing M, Eitel J, Lippmann J, Hocke AC, Zahlten J, Slevogt H, et al. NAIP and Ipaf control Legionella pneumophila replication in human cells. J Immunol (2008) 180:6808–15.
183. Boniotto M, Tailleux L, Lomma M, Gicquel B, Buchrieser C, Garcia S, et al. Population variation in NAIP functional copy number confers increased cell death upon Legionella pneumophila infection. Hum Immunol (2012) 73:196–200. doi:10.1016/j.humimm.2011.10.014
184. Katagiri N, Shobuike T, Chang B, Kukita A, Miyamoto H. The human apoptosis inhibitor NAIP induces pyroptosis in macrophages infected with Legionella pneumophila. Microbes Infect (2012) 14:1123–32. doi:10.1016/j.micinf.2012.03.006
185. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat Immunol (2012) 13:823–31. doi:10.1038/ni.2378
186. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A (2010) 107:13794–9. doi:10.1073/pnas.1008684107
187. Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J Immunol (2010) 185:1681–91. doi:10.4049/jimmunol.0903900
188. Kuenzel S, Till A, Winkler M, Hasler R, Lipinski S, Jung S, et al. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol (2010) 184:1990–2000. doi:10.4049/jimmunol.0900557
189. Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell (2010) 141:483–96. doi:10.1016/j.cell.2010.03.040
190. Tong Y, Cui J, Li Q, Zou J, Wang HY, Wang RF. Enhanced TLR-induced NF-kappaB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res (2012) 22:822–35. doi:10.1038/cr.2012.53
191. Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol (2011) 186:1333–7. doi:10.4049/jimmunol.1003111
192. Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hosel M, et al. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem (2010) 285:26223–32. doi:10.1074/jbc.M110.109736
193. Biswas A, Meissner TB, Kawai T, Kobayashi KS. Cutting edge: impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J Immunol (2012) 189:516–20. doi:10.4049/jimmunol.1200064
194. Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res (2012) 22:836–47. doi:10.1038/cr.2012.56
195. Robbins GR, Truax AD, Davis BK, Zhang L, Brickey WJ, Ting JP. Regulation of class I major histocompatibility complex (MHC) by nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins. J Biol Chem (2012) 287:24294–303. doi:10.1074/jbc.M112.364604
196. Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, et al. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol (2012) 188:4951–8. doi:10.4049/jimmunol.1103160
197. Neerincx A, Rodriguez GM, Steimle V, Kufer TA. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J Immunol (2012) 188:4940–50. doi:10.4049/jimmunol.1103136
198. Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci (2009) 122:3161–8. doi:10.1242/jcs.051193
199. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature (2008) 451:573–7. doi:10.1038/nature06501
200. Hong M, Yoon SI, Wilson IA. Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity (2012) 36:337–47. doi:10.1016/j.immuni.2011.12.018
201. Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity (2011) 34:843–53. doi:10.1016/j.immuni.2011.02.022
202. Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappaB signaling pathways. Immunity (2011) 34:854–65. doi:10.1016/j.immuni.2011.03.026
203. Rebsamen M, Vazquez J, Tardivel A, Guarda G, Curran J, Tschopp J. NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell Death Differ (2011) 18:1387. doi:10.1038/cdd.2011.64
204. Soares F, Tattoli I, Wortzman ME, Arnoult D, Philpott DJ, Girardin SE. NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun (2013) 19:438–48. doi:10.1177/1753425912467383
205. Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep (2008) 9:293–300. doi:10.1038/sj.embor.7401161
Keywords: NOD-like receptors, inflammasome, lung, pneumonia, lung injury
Citation: Chaput C, Sander LE, Suttorp N and Opitz B (2013) NOD-like receptors in lung diseases. Front. Immunol. 4:393. doi: 10.3389/fimmu.2013.00393
Received: 07 August 2013; Paper pending published: 30 September 2013;
Accepted: 07 November 2013; Published online: 21 November 2013.
Edited by:
Thomas A. Kufer, University of Cologne, GermanyReviewed by:
Dario S. Zamboni, Universidade de São Paulo, BrazilJose A. Bengoechea, Queen’s University Belfast, UK
Copyright: © 2013 Chaput, Sander, Suttorp and Opitz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bastian Opitz, Department of Internal Medicine/Infectious Diseases and Pulmonary Medicine, Charité – Universitätsmedizin Berlin, Augustenburger Platz 1, 13353 Berlin, Germany e-mail:YmFzdGlhbi5vcGl0ekBjaGFyaXRlLmRl