
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 10 October 2013
Sec. Antigen Presenting Cell Biology
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00321
This article is part of the Research Topic Processing, editing, and presenting the MHC class II peptidome View all 21 articles
 Sue Tsai1†
Sue Tsai1† Pere Santamaria1,2*
Pere Santamaria1,2*Major histocompatibility complex (MHC) genes, also known as human leukocyte antigen genes (HLA) in humans, are the prevailing contributors of genetic susceptibility to autoimmune diseases such as Type 1 Diabetes (T1D), multiple sclerosis, and rheumatoid arthritis, among others (1–3). Although the pathways through which MHC molecules afford autoimmune risk or resistance remain to be fully mapped out, it is generally accepted that they do so by shaping the central and peripheral T-cell repertoires of the host toward autoimmune proclivity or resistance, respectively. Disease-predisposing MHC alleles would both spare autoreactive thymocytes from central tolerance and bias their development toward a pathogenic phenotype. Protective MHC alleles, on the other hand, would promote central deletion of autoreactive thymocytes and skew their development toward non-pathogenic phenotypes. This interpretation of the data is at odds with two other observations: that in MHC-heterozygous individuals, resistance is dominant over susceptibility; and that it is difficult to understand how deletion of one or a few clonal autoreactive T-cell types would suffice to curb autoimmune responses driven by hundreds if not thousands of autoreactive T-cell specificities. This review provides an update on current advances in our understanding of the mechanisms underlying MHC class II-associated autoimmune disease susceptibility and/or resistance and attempts to reconcile these seemingly opposing concepts.
Major histocompatibility complex (MHC) class II molecules are surface heterodimers expressed by thymic epithelial cells and professional antigen-presenting cells (APCs) that present antigenic peptides to T-cell receptors (TCR) on cognate T-cells. A developing thymocyte first encounters a highly heterogeneous array of endogenous (self) peptide-MHC (pMHC) complexes on thymic APCs. Combinations of self-peptides and MHC molecules instruct thymocytes to either survive or perish, based on the affinity/avidity with which their TCRs bind pMHC. Through processes referred to as thymic positive and negative selection, thymocytes that bind self pMHCs with either intermediate-to-low or high avidity are instructed to survive or perish, respectively (4). As a result, the peripheral T-cell repertoire is largely composed of T-cells capable of recognizing foreign peptides in the context of self MHCs or self pMHCs that are expressed exclusively in peripheral tissues. This forms the basis of self vs. non-self discrimination in adaptive immunity, ensuring that the peripheral immune system is populated by a diverse repertoire of T-cells capable of mounting immune responses against an essentially limitless universe of foreign antigens, while minimizing the risk of causing autoimmunity. Nonetheless, thymic negative selection is not an “air-tight” process and, as a result, some autoreactive specificities evade negative selection and populate the periphery (5, 6). As they possess the potential to cause autoimmunity, additional regulatory mechanisms have evolved to keep them in check, such as the induction of anergy or activation-induced cell death by tolerogenic APCs (7, 8), or suppression by regulatory T-cells (Tregs). The latter can dampen autoimmune responses through several mechanisms, including direct inhibition of APCs (9–14).
Antigenic peptides are embedded in the peptide-binding groove of the MHC class II molecule, which consists of two flanking alpha helices atop a beta-pleated sheet. With some exceptions, these peptides are anchored onto the MHC class II binding cleft through their amino acid side chains at four positions, termed pockets 1, 4, 6, and 9. Interestingly, autoimmune disease-promoting MHC class II alleles often differ from disease-non-promoting ones by only a few amino acids that are primarily located at the TCR-MHC interface or in the peptide-binding groove, at times adjacent to key anchoring pockets (15, 16). This has suggested that MHC-linked disease risk is associated with differences in the repertoire of self-peptides that are presented to T-cells. Indeed, substantial evidence from different autoimmune diseases supports this possibility. For instance, HLA-DRB1 alleles affording susceptibility to Rheumatoid arthritis (RA) share a conserved sequence of amino acids at residues 67–74 of the DRβ chain (17), which is situated on one of the alpha helices flanking the peptide-binding groove. Genome-wide SNP analyses of seropositive RA patents vs. healthy controls confirmed the genetic contribution of two of these residues, 71 and 74, as well as three others, all of them located in the peptide-binding groove, to RA susceptibility (18). Available peptide-binding data from RA-predisposing DRB1*0401 and *0404 vs. RA-protective *0402 alleles revealed an association between the repertoire of bound peptides and polymorphisms at position 71 (19), although it remains to be determined how these associations contribute to autoreactivity. Of note, DRB1 residues 71 and 74 appear to be a hotspot for other autoimmune disease-associated polymorphisms as well, including autoimmune thyroiditis (20) and Multiple Sclerosis (MS) (21), owing to their vicinity to the P4 pocket.
A similar phenomenon may underlie the MHC-T1D associations. By comparing disease-promoting and -protective class II molecules in mice and humans, it was found that polymorphisms at DQβ chain positions 56 and 57, located on one of the two alpha helices flanking the peptide-binding groove, strongly correlated with both disease susceptibility and resistance (22, 23). Alleles that encode Asp at position 57 in humans (or Pro56 and Asp57 in mice) afford protection, whereas those that encode Ser or Ala (or His56 and Ser57 in mice) at these positions afford risk (22, 23). In fact, an alignment of protective murine I-Aβ chains with their diabetogenic I-Aβg7 counterpart reveals a striking consensus sequence spanning residues 56–67. The I-Aβg7 His56 and Ser57 residues, unlike the disease-protective Pro56 and Asp57 residues, fail to form a salt bridge and neutralize the positive charge imparted by Arg76 on the I-Aαd chain, resulting in a positively charged P9 pocket that favors the binding peptides carrying acidic amino acids at the C-terminus (24, 25). This positive charge results in the unstable binding of many self-peptides to I-Ag7 and presumably the presentation of a narrower range of epitopes (15, 26–30). Similarly in humans, the predisposing class II variants, DQ2 and DQ8, carry an Ala at the beta chain position 57 and exhibit preference for binding peptides carrying acidic residues at their carboxyterminus (31, 32).
A direct consequence of altered peptide presentation is the undermining of a number of MHC class II-mediated processes. For example, MHC class II molecules that afford autoimmune susceptibility would promote positive selection of pathogenic autoreactive thymocytes in the cortex while failing to trigger their subsequent deletion in the medulla. On the other hand, protective class II would select a repertoire of T-cells endowed with decreased pathogenicity (33). Another process that likely bears the brunt of such alteration is negative selection; this is supported by observations made in animal studies comparing disease-promoting MHCs to their disease-protective counterparts differing by only a few amino acids at positions located in the peptide-binding groove. In these studies, protective class II molecules curbed autoimmunity by promoting the negative selection of certain, in this case class II-promiscuous, autoreactive T-cell specificities (34–38). The latter observations brought forth the idea that protective class II polymorphisms located at or near the peptide-binding groove may be recognized by certain MHC-promiscuous autoreactive TCRs with an avidity and/or affinity above the threshold required for negative selection. We have recently shown that dendritic cells play a key role in this process (38, 39). Figure 1 depicts the proposed relationship between pMHC:TCR interaction strength and its outcome in terms of the selection of pathogenic vs. regulatory T cell clonotypes, and how MHC polymorphisms play into this selective process.
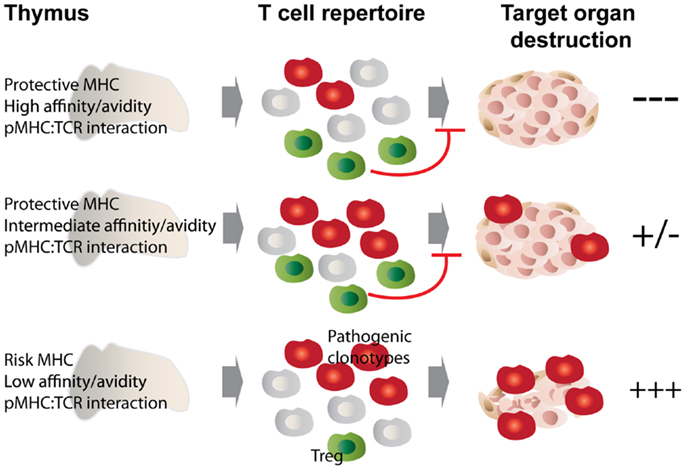
Figure 1. MHC class II polymorphisms afford autoimmune disease resistance through shaping the T cell and Treg repertoire. MHC class II molecules that afford disease risk allow the escape of pathogenic autoreactive T cells from central tolerance, while protective MHC class II molecules confer disease resistance through promoting negative selection as well as autoreactive Treg development. We propose that these processes are governed by the affinity/avidity with which pMHCs are bound by TCRs. Disease-protective pMHC interact with MHC-promiscuous, autoreactive thymocytes with increased affinity/avidity (top and middle panels), leading to enhanced negative selection and agonist selection of Tregs, which then dampen the autoimmune response through various mechanisms. In the bottom scenario, low affinity/avidity interaction between pMHC and autoreactive TCRs leads to defective negative selection and Treg development, with the net result of autoimmunity.
These observations are compatible with the idea that low-avidity presentation of self-peptides by disease-promoting MHCs to potentially pathogenic autoreactive thymocytes would contribute to autoimmune disease susceptibility by impairing their negative selection without compromising positive selection, as the affinity/avidity thresholds for these processes are different (40). Exposure to increased antigenic loads and/or differentially processed epitopes in the periphery (41) could then suffice to fuel the activation of these thymic escapees and their recruitment into an autoimmune response. Our studies with the diabetogenic, MHC-promiscuous (but I-Ag7-restricted) 4.1-TCR are entirely compatible with this view (34–36, 38, 39).
Nevertheless, not all protective class II molecules are equally effective at eliciting central tolerance of diabetogenic TCRs (33, 36, 38, 39, 42), indicating that class II-associated resistance to autoimmunity cannot be solely accounted for by this process (deletion of key autoreactive T-cell specificities). Furthermore, it is difficult to understand how deletion of one or a handful of autoreactive T-cell types might be able to blunt autoimmune responses involving many other specificities. One could argue that enhanced negative selection of a key immunodominant specificity playing a critical role in disease initiation would be sufficient. However, without knowing if a given specificity is capable of singularly initiating disease or swaying its outcome, the consequences of its deletion are hard to predict. Most likely, deleting any single or combination of T-cell specificity(ies) will not alter the course of a disease that is mediated by a polyclonal T-cell repertoire, as we have previously demonstrated (43).
It is therefore likely that enhanced central tolerance of autoreactive T-cells by protective class II molecules cannot be the only mechanism underlying the MHC-associated autoimmune disease resistance. Engagement of pMHCs expressed on thymic medullary epithelial cells or bone marrow-derived APCs by thymocytes can yield different outcomes, depending on the avidity of the interaction. Although the most avid pMHC:TCR interactions invariably promote thymocyte apoptosis, strong interactions that fall below the threshold for deletion can result in an alternative outcome – the generation of Treg cells. This process, also referred to as “agonist selection” of regulatory T-cells (44), occurs within a window of affinity/avidity that spans the thresholds of positive and negative selection (45).
Since many of the early mechanistic studies on this topic predated the discovery of Treg cells by as long as a decade (46), the potential effects of autoimmune disease-associated class II polymorphisms on Treg cell development and/or function were not investigated in detail. However, some of these early studies showed that protective MHC class II molecules induced dominant, T-cell-transferrable tolerance (47, 48).
The observation that certain (albeit not all) protective class II molecules lost their anti-diabetogenic effects in monoclonal (RAG-deficient) autoreactive TCR-transgenic mice led us to suspect a contribution by endogenous (non-TCR-transgenic) autoreactive T-cells to this process (38, 39). We measured the frequency and function of Treg cells in TCR-transgenic and non-transgenic NOD mice co-expressing T1D-protective class II alleles (38, 39). Autoreactive TCR-transgenic NOD mice co-expressing anti-diabetogenic class II alleles harbored increased numbers of autoreactive Treg cells that had superior regulatory activity as compared to Treg cells arising in mice only expressing wild-type H-2g7. In non-TCR-transgenic animals expressing protective class II alleles, increases in Treg numbers were restricted to the target organ and local draining lymph nodes, where there was an enrichment for autoreactive specificities. These observations suggested that protective class II molecules promote the differentiation of potentially pathogenic autoreactive thymocytes into Treg cells, which would then be able to effectively blunt all other autoreactive T-cell responses by suppressing autoantigen presentation in the draining lymph nodes (38, 39). Furthermore, consistent with the model of agonist selection, such increases in MHC class II-induced autoreactive Treg selection coincided with the up-regulation of CD5, CD69, and Nur77 on thymocytes, which are induced by high affinity pMHC:T-cell interactions (49), and were paralleled by negative selection (38). Expression of sub-tolerogenic, but still anti-diabetogenic class II variants in these autoreactive TCR-transgenic NOD mice promoted increases in the in vivo regulatory capacity of the peripheral Treg cell pool, owing to increases in the peripheral frequency of autoreactive regulatory T-cell specificities, further substantiating the above observations (39). Remarkably, all these effects were driven by protective class II types expressed exclusively in DCs, therefore suggesting that class II-driven autoreactive Treg formation occur in the thymic medulla (38, 39). Figure 2 highlights the proposed interactions between disease-promoting and protective MHC class II molecules on thymic APCs and autoreactive TCRs on thymocytes. The nature of such interaction will be further discussed below.
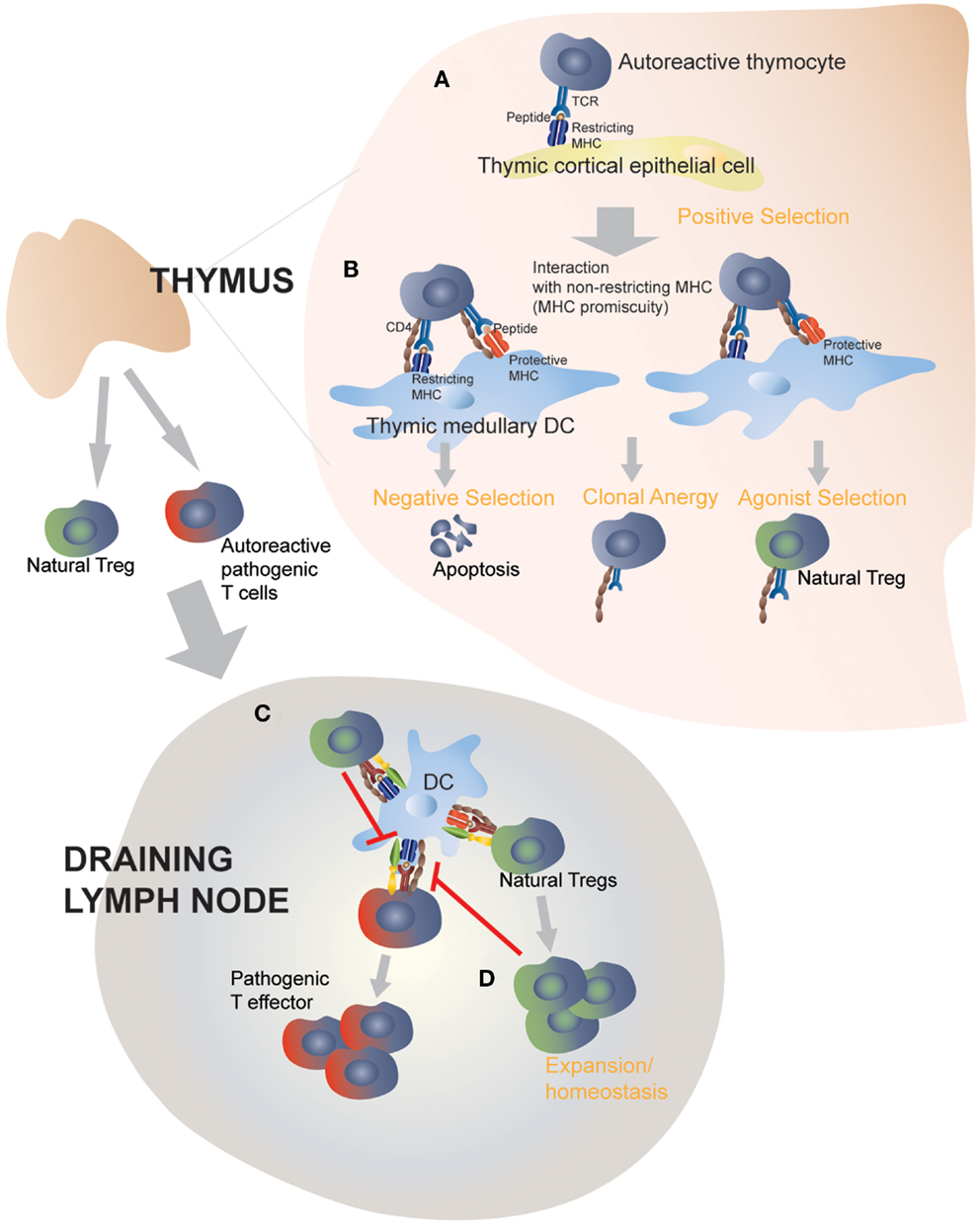
Figure 2. Protective MHC class II molecules mediate central and peripheral tolerance by targeting MHC-promiscuous autoreactive TCRs. Positive selection on I-Ag7 in the thymic cortex determines the MHC restriction of thymocytes (A). In the absence of protective MHC molecules, negative selection is defective and fail to purge the repertoire of pathogenic autoreactive thymocytes (not shown) (B). Transgenic expression of disease-protective MHC class II molecules on dendritic cells leads to enhanced negative selection and clonal anergy of autoreactive, MHC-promiscuous thymocytes, and promotes autoreactive Treg differentiation and functional development (C). Thymic derived Tregs then exit into the periphery and suppress the activation of pathogenic T cells by directly acting on autoantigen-loaded APCs. This step does not require protective MHC class II molecules, although a role of protective MHC class II molecules, expressed on peripheral APCs, in perpetuating autoreactive Tregs or enhancing their homeostasis cannot be ruled out (D).
These observations help us understand how a single class II molecule can blunt an autoantigenically complex T-cell response: agonist-induced development of autoreactive Tregs from thymocytes expressing MHC-promiscuous autoreactive TCRs (Figures 1 and 2). Still, several questions remain as to how protective class II molecules manage to do so. What are the changes in the bound peptide repertoire, and/or the overall pMHC structure that promote enhanced Treg selection? Studies using H-2DM-deficient mice predicted that such a process is peptide-dependent but not necessarily peptide-specific, suggesting a dominant role for MHC residues, as opposed to peptide residues, in driving this outcome (35). Another observation that provides useful insights is the requirement for endogenous TCRs in protective MHC-induced autoreactive Treg formation. Looking at the three different autoreactive TCR/MHC class II combinations that were explored, we can conclude that not all autoreactive TCRs are overtly MHC-promiscuous or promiscuous for all protective MHC types and therefore capable of engaging protective MHC (38, 39). Instead, we hypothesize that the endogenous autoreactive thymocyte repertoire (positively selected and restricted by disease-promoting MHC types) is inherently promiscuous for other (suppressing or even non-disease-promoting) MHC types, and that only those TCRs that can engage the latter with an affinity/avidity above the threshold required for agonist-induced Treg formation will contribute to disease suppression. In other words, MHC alleles affording dominant resistance to a given autoimmune disease are those capable of harnessing the intrinsic MHC promiscuity of pathogenic TCRs to generate autoreactive Treg cells.
Since class II molecules are expressed on all professional APCs, one has to wonder if protective class II types also contribute to autoimmune disease suppression by shaping the post-thymic peripheral T-cell repertoire. It is well accepted that disease-predisposing class II molecules are responsible for presenting self epitopes to autoreactive T-cells, leading to their activation and therefore exerting a direct impact on the effector phase of the autoimmune response. Through antigen presentation, predisposing class II molecules have also been proposed to contribute to autoimmune inflammation by modulating the cytokine milieu produced by T-cells (50). The contribution of disease-protective class II in the periphery, however, is less clear. Certain studies implicated protective class II molecules in mechanisms such as “determinant capture” that would interfere with or compete against self-antigen presentation by disease-predisposing MHCs (51–53). These mechanisms, although plausible, were countered by other reports (54–56). Although we find that autoreactive Treg cells need not have to engage protective pMHC class II complexes in the periphery to effect disease suppression (38, 39), it remains to be determined whether protective MHC class II play a role in their homeostatic survival. Repeated encounters with disease-promoting pMHC in the periphery (57, 58) may be sufficient for both survival and suppression.
The above interpretation of the data implies that certain autoreactive T-cell specificities have the capacity to recognize more than one peptide in the context of more than one type of class II molecule (Figure 2). In fact, it has been established that individual TCRs have the potential to recognize a surprisingly wide array of peptides in the context of a single MHC molecule (59), or multiple MHC molecules presenting one or more peptide(s) (60, 61). Furthermore, we and others have provided evidence that pathogenic autoreactive TCRs are inherently “MHC-promiscuous” (34, 62–64). In this context, MHC diversity, we propose, can be viewed as nature’s guard against T-cell-driven autoimmunity. By exploiting their tendency to “stick” to certain class II molecules, protective MHC types can either eliminate autoreactive T-cells at the get-go and/or, most importantly, instruct them to join the ranks of regulatory T-cells. The molecular basis underlying the interaction between autoreactive TCRs and protective MHCs remains as yet undefined. Structural studies of autoreactive TCRs cloned from relapsing-remitting MS patients or animals induced to develop a similar demyelinating disease (EAE) revealed anomalous characteristics of pMHC binding by these autoreactive TCRs, which engage cognate peptide in the context of disease-predisposing class II molecules in an atypical manner that is distinct from the binding topology of conventional TCRs on foreign pMHC complexes (65–67). Altered binding appeared to be associated with fewer contacts between the TCR and the peptide. In some cases, the TCR/pMHC interaction leaned toward the N-terminal end of the bound peptide, and in others the TCR/pMHC contact was weakened by the altered peptide docking on the MHC, due to the partial occupancy of the peptide-binding groove (65). Whether a pathogenic autoreactive TCR behaves differently in the presence of a disease-protective MHC remains to be determined. As we inferred from functional studies, protective MHC molecules may be recognized by a given autoreactive TCR with higher affinity/avidity than a disease-promoting MHC type, thereby correcting deficiencies in negative selection or Treg development. We propose that disease-protective class II molecules signal the deletion of autoreactive thymocytes as well as their development into Tregs by taking advantage of the atypical binding characteristics of certain autoreactive TCRs, i.e., their intrinsic MHC promiscuity.
The implications of MHC polymorphism evolution vis-à-vis health and disease are manifold. Polymorphic class I and II molecules endow the adaptive immune system with the flexibility to recognize a wide array of pathogens, in terms of both selecting a diverse T-cell repertoire in the thymus, and enabling their activation in the periphery through antigen presentation. In close association to their function in generating TCR diversity through thymic education, MHC molecules also have the important role of discouraging T-cells from brewing autoimmunity. MHC polymorphisms affect the outcome of this educational process, with protective MHC purging harmful clonotypes and disease-promoting MHC permitting their escape. Furthermore, a previously unappreciated role of protective class II molecules in autoimmune disease resistance has been identified: mobilization of self-reactive Treg cells to fight harmful self-reactivity. We propose that protective class II molecules do so by engaging MHC-promiscuous, autoreactive thymocytes in a manner that promotes Treg formation, and that future work on elucidating the interaction between autoreactive TCRs and protective class II molecules will shed light on this very important issue.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to those authors whose contributions we could not mention due to space constraints. S. Tsai is currently a postdoctoral fellow at the Toronto General Research Institute, University of Toronto. P. Santamaria is a scientist of Alberta Innovates – Health Solutions and a scholar of the Instituto de Investigaciones Sanitarias Carlos III. The authors’ work was supported by the Canadian Institutes for Health Research. The Julia McFarlane Diabetes Research Centre is supported by the Canadian Diabetes Association.
1. Todd JA, Wicker LS. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity (2001) 15:387–95. doi: 10.1016/S1074-7613(01)00202-3
2. MacKay K, Eyre S, Myerscough A, Milicic A, Barton A, Laval S, et al. Whole-genome linkage analysis of rheumatoid arthritis susceptibility loci in 252 affected sibling pairs in the United Kingdom. Arthritis Rheum (2002) 46:632–9. doi:10.1002/art.10147
3. Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, et al. Risk alleles for multiple sclerosis identified by a genome wide study. N Engl J Med (2007) 357:851–62. doi:10.1056/NEJMoa073493
4. Palmer E. Negative selection – clearing out the bad apples from the T-cell repertoire. Nat Rev Immunol (2003) 3:383–91. doi:10.1038/nri1085
5. Bouneaud C, Kourilsky P, Bousso P. Impact of negative selection on the T cell repertoire reactive to a self-peptide: a large fraction of T cell clones escapes clonal deletion. Immunity (2000) 13:829–40. doi:10.1016/S1074-7613(00)00080-7
6. Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity (2006) 25:261–70. doi:10.1016/j.immuni.2006.06.009
7. Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol (2002) 2:11–9. doi:10.1038/nri701
8. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol (2003) 21:685–711. doi:10.1146/annurev.immunol.21.120601.141040
9. Bettini M, Vignali DA. Regulatory T cells and inhibitory cytokines in autoimmunity. Curr Opin Immunol (2009) 21:612–8. doi:10.1016/j.coi.2009.09.011
10. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4:1206–12. doi:10.1038/ni1003
11. Serra P, Amrani A, Yamanouchi J, Han B, Thiessen S, Utsugi T, et al. CD40 ligation releases immature dendritic cells from the control of regulatory CD4+CD25+ T cells. Immunity (2003) 19:877–89. doi:10.1016/S1074-7613(03)00327-3
12. Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol (2006) 7:83–92. doi:10.1038/ni1289
13. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110:1225–32. doi:10.1182/blood-2006-12-064527
14. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
15. Latek RR, Suri A, Petzold SJ, Nelson CA, Kanagawa O, Unanue ER, et al. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity (2000) 12:699–710. doi:10.1016/S1074-7613(00)80220-4
16. Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol (2006) 6:271–82. doi:10.1038/nri1805
17. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum (1987) 30:1205–13. doi:10.1002/art.1780301102
18. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet (2012) 44:291–6. doi:10.1038/ng.1076
19. Hammer J, Gallazzi F, Bono E, Karr RW, Guenot J, Valsasnini P, et al. Peptide binding specificity of HLA-DR4 molecules: correlation with rheumatoid arthritis association. J Exp Med (1995) 181:1847–55. doi:10.1084/jem.181.5.1847
20. Menconi F, Monti MC, Greenberg DA, Oashi T, Osman R, Davies TF, et al. Molecular amino acid signatures in the MHC class II peptide-binding pocket predispose to autoimmune thyroiditis in humans and in mice. Proc Natl Acad Sci U S A (2008) 105:14034–9. doi:10.1073/pnas.0806584105
21. Smith KJ, Pyrdol J, Gauthier L, Wiley DC, Wucherpfennig KW. Crystal structure of HLA-DR2 (DRA*0101, DRB1*1501) complexed with a peptide from human myelin basic protein. J Exp Med (1998) 188:1511–20. doi:10.1084/jem.188.8.1511
22. Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class II I-A beta chain is unique. Proc Natl Acad Sci U S A (1987) 84:2435–9.
23. Todd JA, Bell JI, Mcdevitt HO. HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature (1987) 329:599–604. doi:10.1038/329599a0
24. Suri A, Vidavsky I, Van Der Drift K, Kanagawa O, Gross ML, Unanue ER. In APCs, the autologous peptides selected by the diabetogenic I-Ag7 molecule are unique and determined by the amino acid changes in the P9 pocket. J Immunol (2002) 168:1235–43.
25. Suri A, Walters JJ, Rohrs HW, Gross ML, Unanue ER. First signature of islet beta-cell-derived naturally processed peptides selected by diabetogenic class II MHC molecules. J Immunol (2008) 180:3849–56.
26. Carrasco-Marin E, Shimizu J, Kanagawa O, Unanue ER. The class II MHC I-Ag7 molecules from non-obese diabetic mice are poor peptide binders. J Immunol (1996) 156:450–8.
27. Corper AL, Stratmann T, Apostolopoulos V, Scott CA, Garcia KC, Kang AS, et al. A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science (2000) 288:505–11. doi:10.1126/science.288.5465.505
28. Stratmann T, Apostolopoulos V, Mallet-Designe V, Corper AL, Scott CA, Wilson IA, et al. The I-Ag7 MHC class II molecule linked to murine diabetes is a promiscuous peptide binder. J Immunol (2000) 165:3214–25.
29. Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci U S A (2010) 107:10978–83. doi:10.1073/pnas.1006545107
30. Mohan JF, Unanue ER. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat Rev Immunol (2012) 12:721–8. doi:10.1038/nri3294
31. Godkin A, Friede T, Davenport M, Stevanovic S, Willis A, Jewell D, et al. Use of eluted peptide sequence data to identify the binding characteristics of peptides to the insulin-dependent diabetes susceptibility allele HLA-DQ8 (DQ 3.2). Int Immunol (1997) 9:905–11. doi:10.1093/intimm/9.6.905
32. van Lummel M, Van Veelen PA, Zaldumbide A, De Ru A, Janssen GM, Moustakas AK, et al. Type 1 diabetes-associated HLA-DQ8 transdimer accommodates a unique peptide repertoire. J Biol Chem (2012) 287:9514–24. doi:10.1074/jbc.M111.313940
33. Luhder F, Katz J, Benoist C, Mathis D. Major histocompatibility complex class II molecules can protect from diabetes by positively selecting T cells with additional specificities. J Exp Med (1998) 187:379–87. doi:10.1084/jem.187.3.379
34. Schmidt D, Verdaguer J, Averill N, Santamaria P. A mechanism for the major histocompatibility complex-linked resistance to autoimmunity. J Exp Med (1997) 186:1059–75. doi:10.1084/jem.186.7.1059
35. Schmidt D, Amrani A, Verdaguer J, Bou S, Santamaria P. Autoantigen-independent deletion of diabetogenic CD4+ thymocytes by protective MHC class II molecules. J Immunol (1999) 162:4627–36.
36. Thiessen S, Serra P, Amrani A, Verdaguer J, Santamaria P. T-cell tolerance by dendritic cells and macrophages as a mechanism for the major histocompatibility complex-linked resistance to autoimmune diabetes. Diabetes (2002) 51:325–38. doi:10.2337/diabetes.51.2.325
37. Taneja V, Behrens M, Basal E, Sparks J, Griffiths MM, Luthra H, et al. Delineating the role of the HLA-DR4 “shared epitope” in susceptibility versus resistance to develop arthritis. J Immunol (2008) 181:2869–77.
38. Tsai S, Serra P, Clemente-Casares X, Yamanouchi J, Thiessen S, Slattery RM, et al. Antidiabetogenic MHC class II promotes the differentiation of MHC-promiscuous autoreactive T cells into FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A (2013) 110:3471–6. doi:10.1073/pnas.1211391110
39. Tsai S, Serra P, Clemente-Casares X, Slattery RM, Santamaria P. Dendritic cell-dependent in vivo generation of autoregulatory T cells by antidiabetogenic MHC class II. J Immunol (2013) 191:70–82. doi:10.4049/jimmunol.1300168
40. Gascoigne NR, Palmer E. Signaling in thymic selection. Curr Opin Immunol (2011) 23:207–12. doi:10.1016/j.coi.2010.12.017
41. Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol (2010) 11:350–4. doi:10.1038/ni.1850
42. Bohme J, Schuhbaur B, Kanagawa O, Benoist C, Mathis D. MHC-linked protection from diabetes dissociated from clonal deletion of T cells. Science (1990) 249:293–5. doi:10.1126/science.2115690
43. Han B, Serra P, Amrani A, Yamanouchi J, Maree AF, Edelstein-Keshet L, et al. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat Med (2005) 11:645–52. doi:10.1038/nm1250
44. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol (2001) 2:301–6. doi:10.1038/86302
45. Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol (2012) 12:157–67.
46. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
47. Singer SM, Tisch R, Yang XD, Mcdevitt HO. An Abd transgene prevents diabetes in nonobese diabetic mice by inducing regulatory T cells. Proc Natl Acad Sci U S A (1993) 90:9566–70. doi:10.1073/pnas.90.20.9566
48. Singer SM, Tisch R, Yang XD, Sytwu HK, Liblau R, Mcdevitt HO. Prevention of diabetes in NOD mice by a mutated I-Ab transgene. Diabetes (1998) 47:1570–7. doi:10.2337/diabetes.47.10.1570
49. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med (2011) 208:1279–89. doi:10.1084/jem.20110308
50. Mangalam AK, Taneja V, David CS. HLA class II molecules influence susceptibility versus protection in inflammatory diseases by determining the cytokine profile. J Immunol (2013) 190:513–8. doi:10.4049/jimmunol.1201891
51. Deng H, Apple R, Clare-Salzler M, Trembleau S, Mathis D, Adorini L, et al. Determinant capture as a possible mechanism of protection afforded by major histocompatibility complex class II molecules in autoimmune disease. J Exp Med (1993) 178:1675–80. doi:10.1084/jem.178.5.1675
52. Trembleau S, Gregori S, Penna G, Gorny I, Adorini L. IL-12 administration reveals diabetogenic T cells in genetically resistant I-Ealpha-transgenic nonobese diabetic mice: resistance to autoimmune diabetes is associated with binding of Ealpha-derived peptides to the I-A(g7) molecule. J Immunol (2001) 167:4104–14.
53. Ge X, James EA, Reijonen H, Kwok WW. Differences in self-peptide binding between T1D-related susceptible and protective DR4 subtypes. J Autoimmun (2011) 36:155–60. doi:10.1016/j.jaut.2010.12.004
54. Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell (1993) 74:1089–100. doi:10.1016/0092-8674(93)90730-E
55. Hanson MS, Cetkovic-Cvrlje M, Ramiya VK, Atkinson MA, Maclaren NK, Singh B, et al. Quantitative thresholds of MHC class II I-E expressed on hematopoietically derived antigen-presenting cells in transgenic NOD/Lt mice determine level of diabetes resistance and indicate mechanism of protection. J Immunol (1996) 157:1279–87.
56. Mellanby RJ, Phillips JM, Parish NM, Cooke A. Both central and peripheral tolerance mechanisms play roles in diabetes prevention in NOD-E transgenic mice. Autoimmunity (2008) 41:383–94. doi:10.1080/08916930801991021
57. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity (2004) 21:267–77. doi:10.1016/j.immuni.2004.07.009
58. Andersson J, Stefanova I, Stephens GL, Shevach EM. CD4+CD25+ regulatory T cells are activated in vivo by recognition of self. Int Immunol (2007) 19:557–66. doi:10.1093/intimm/dxm021
59. Wooldridge L, Ekeruche-Makinde J, Van Den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem (2011) 287:1168–77. doi:10.1074/jbc.M111.289488
60. Brock R, Wiesmuller KH, Jung G, Walden P. Molecular basis for the recognition of two structurally different major histocompatibility complex/peptide complexes by a single T-cell receptor. Proc Natl Acad Sci U S A (1996) 93:13108–13. doi:10.1073/pnas.93.23.13108
61. Doherty DG, Penzotti JE, Koelle DM, Kwok WW, Lybrand TP, Masewicz S, et al. Structural basis of specificity and degeneracy of T cell recognition: pluriallelic restriction of T cell responses to a peptide antigen involves both specific and promiscuous interactions between the T cell receptor, peptide, and HLA-DR. J Immunol (1998) 161:3527–35.
62. Ranheim EA, Tarbell KV, Krogsgaard M, Mallet-Designe V, Teyton L, Mcdevitt HO, et al. Selection of aberrant class II restricted CD8+ T cells in NOD mice expressing a glutamic acid decarboxylase (GAD)65-specific T cell receptor transgene. Autoimmunity (2004) 37:555–67. doi:10.1080/08916930400020545
63. Serreze DV, Holl TM, Marron MP, Graser RT, Johnson EA, Choisy-Rossi C, et al. MHC class II molecules play a role in the selection of autoreactive class I-restricted CD8 T cells that are essential contributors to type 1 diabetes development in nonobese diabetic mice. J Immunol (2004) 172:871–9.
64. Logunova NN, Viret C, Pobezinsky LA, Miller SA, Kazansky DB, Sundberg JP, et al. Restricted MHC-peptide repertoire predisposes to autoimmunity. J Exp Med (2005) 202:73–84. doi:10.1084/jem.20050198
65. Wucherpfennig KW, Call MJ, Deng L, Mariuzza R. Structural alterations in peptide-MHC recognition by self-reactive T cell receptors. Curr Opin Immunol (2009) 21:590–5. doi:10.1016/j.coi.2009.07.008
66. Wucherpfennig KW, Sethi D. T cell receptor recognition of self and foreign antigens in the induction of autoimmunity. Semin Immunol (2011) 23:84–91. doi:10.1016/j.smim.2011.01.007
Keywords: MHC class II, autoimmune diseases, susceptibility genes, resistance genes, type 1 diabetes, autoreactive T cells, T regulatory cells
Citation: Tsai S and Santamaria P (2013) MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front. Immunol. 4:321. doi: 10.3389/fimmu.2013.00321
Received: 31 July 2013; Accepted: 22 September 2013;
Published online: 10 October 2013.
Edited by:
Laura Santambrogio, Albert Einstein College of Medicine, USACopyright: © 2013 Tsai and Santamaria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pere Santamaria, University of Calgary, 3330 Hospital Drive, Calgary, AB T2N 4N1, Canada e-mail:cHNhbnRhbWFAdWNhbGdhcnkuY2E=
†Present address: Sue Tsai, Department of Advanced Diagnostics-Metabolism, Toronto General Research Institute, University Health Networks, Toronto, ON, Canada
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.