



94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 October 2013
Sec. B Cell Biology
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00302
This article is part of the Research TopicImmune system modeling and analysisView all 14 articles
The function of antibodies (Abs) involves specific binding to antigens (Ags) and activation of other components of the immune system to fight pathogens. The six hypervariable loops within the variable domains of Abs, commonly termed complementarity determining regions (CDRs), are widely assumed to be responsible for Ag recognition, while the constant domains are believed to mediate effector activation. Recent studies and analyses of the growing number of available Ab structures, indicate that this clear functional separation between the two regions may be an oversimplification. Some positions within the CDRs have been shown to never participate in Ag binding and some off-CDRs residues often contribute critically to the interaction with the Ag. Moreover, there is now growing evidence for non-local and even allosteric effects in Ab-Ag interaction in which Ag binding affects the constant region and vice versa. This review summarizes and discusses the structural basis of Ag recognition, elaborating on the contribution of different structural determinants of the Ab to Ag binding and recognition. We discuss the CDRs, the different approaches for their identification and their relationship to the Ag interface. We also review what is currently known about the contribution of non-CDRs regions to Ag recognition, namely the framework regions (FRs) and the constant domains. The suggested mechanisms by which these regions contribute to Ag binding are discussed. On the Ag side of the interaction, we discuss attempts to predict B-cell epitopes and the suggested idea to incorporate Ab information into B-cell epitope prediction schemes. Beyond improving the understanding of immunity, characterization of the functional role of different parts of the Ab molecule may help in Ab engineering, design of CDR-derived peptides, and epitope prediction.
Antibodies (Abs) have two distinct functions: one is to bind specifically to their target antigens (Ags); the other is to elicit an immune response against the bound Ag by recruiting other cells and molecules. The association between an Ab and an Ag involves myriad of non-covalent interactions between the epitope – the binding site on the Ag, and the paratopes – the binding site on the Ab. The ability of Abs to bind virtually any non-self surface with exquisite specificity and high affinity is not only the key to immunity but has also made Abs an enormously valuable tool in experimental biology, biomedical research, diagnostics and therapy. The diversity of their binding capabilities is particularly striking given the high structural similarity between all Abs. The availability of increasing amounts of structural data in recent years now allows for a much better understanding of the structural basis of Ab function in general, and of Ag recognition in particular. This review surveys the recent developments and the current gaps and challenges in this field. We focus specifically on the current understanding of the determinants within the Ab structure that contribute to Ag binding. We first discuss the motivations for, and applications of, the study of the structural basis of Ag recognition. Then we describe and discuss the Ab-Ag interface, with specific focus on the paratopes and the complementarity determining regions (CDRs), and their role in Ag binding. The last part focuses on the contribution of the non-CDRs parts of the Ab [i.e., framework regions (FRs) and the constant domains] to Ag binding and on the recent suggestions regarding non-local and allosteric effects in Ab function. Over the last few years numerous reviews have addressed issues that are related or tangential to the topics we review here. This includes reviews of the engineering of Abs (1), their stability (2), affinity maturation (3), and isotype selection (4). While these important topics are relevant to the findings and ideas we review here, they are beyond the scope of this review.
The adaptive immune response involves two types of lymphocytes: T cells, which recognizes Ags that have been processed and their fragments are presented by MHC molecules, and B cells which produce soluble Abs that can identify also the intact Ag in its native form. While the way in which T cells recognize their epitopes has been extensively studied to a level that enables the successful prediction of T-cell epitopes (5, 6), the rules that govern Ab-Ag recognition, including which parts of the Ab structure underlie Ag recognition and how and why certain determinants on the Ag are selected as epitopes, are not as well characterized. Understanding the mechanisms that underlie Ab-Ag recognition, therefore, is crucial for understanding immunity.
The immune system enables Abs to distinguish between foreign and self molecules (7). Autoimmune diseases are characterized by the inappropriate response to self-Ags. It is not always clear what role is played by Abs and what role is played by other components of the immune system in autoimmunity. A variety of molecular mechanisms have been proposed, including sequestered Ags, molecular mimicry, and polyclonal B-cell activation (8). Better understanding of the underpinnings of Ab-Ag recognition may also shed light on these questions.
A fundamental characteristic of the immune system is its ability to continuously generate novel protein recognition sites. Ab-Ag interfaces, therefore, are often considered a model system for elucidating the principles governing biomolecular recognition (9–13). For example, Keskin (14) and McCoy et al. (15) used X-ray crystallographic structures of Ab-Ag complexes to elucidate principles of the molecular architecture of protein–protein interfaces. Other studies, however, view Ab-Ag interfaces as a specific case that may not allow for generalization to all types of protein–protein interfaces (16). Thus, large scale studies of protein–protein interactions often exclude Ab-Ag complexes from the dataset analyzed (16–19). It is, therefore, important to determine to what extent Ab-Ag complexes could serve as a general model for protein–protein interactions.
The specificity of the Ab molecule to its cognate Ag has been exploited for the development of a variety of immunoassays, vaccinations, and therapeutics. Ab engineering may offer to expand the application of Abs by permitting improvements of affinity (20, 21) and specificity (22, 23). Understanding of the role each structural element in the Ab plays in Ag recognition is essential for successful engineering of better binders. The engineering of Abs is also important for the clinical use of Abs from non-human sources. Early studies on the use of rodent Abs in humans determined that they can be immunogenic (24). Humanization by grafting of the CDRs from a mouse Ab to a human FR is a commonly used engineering strategy for reducing immunogenicity (25, 26). In most cases, the successful design of high-affinity, CDR-grafted, Abs requires that key residues in the human acceptor FRs that are crucial for preserving the functional conformation of the CDRs will be back-mutated to the amino acids of the original murine Ab (26, 27). Several groups (28–30) used the experimentally determined 3-D structures of Ab-Ag complexes in the Protein Data Bank (PDB) (31) to determine which residues participate in Ag recognition and binding. Such knowledge can be exploited to identify residues that are important for the function of the Ab in general and for Ag recognition in particular and may guide Ab engineering (32, 33). Residues that help maintain the functional conformation of the CDRs, for example, can be used to improve Ab humanization efforts by CDR-grafting.
Antibody epitopes (sometimes referred to as B-cell epitopes) are the molecular structures within an Ag that make specific contacts with the Ab paratope. B-cell epitopes are used in the development of vaccines and in immunodiagnostics. Correct identification of B-cell epitopes within an antigenic protein, may open the door for the design of molecules (biologic or synthetic) that mimic potentially protective epitopes and could be used to raise specific Abs or be used as a prophylactic or therapeutic vaccines. Identification of B-cell epitopes could promote protective immunity in the context of emerging and re-emerging infectious diseases and potential bioterrorist threats. This may be achieved by choosing from among the putative epitopes those that may provide immunity (e.g., by eliciting Abs that hamper the molecular function of pathogenic Ags). The choice of such epitopes is believed to be relevant for understanding and controlling protective immunity. In the case of the vaccinia virus, for example, which was used as smallpox vaccine and is the only vaccine that has led to the complete eradication of an infectious disease from the human population, individuals possessing a high frequency of memory B-cells specific for major neutralizing Ags of the vaccinia virus are better protected from smallpox than individuals with a memory B-cell pool dominated by specificities for non-protective Ags (34). Thus, understanding the way in which an Ab recognizes its cognate epitope is of particular interest for vaccine design and disease prevention (35). Existing tools for identification of Ab epitopes (such as X-ray crystallography, pepscan, phage display, expressed fragments, partial proteolysis, mass spectrometry, and mutagenesis analysis) are not only expensive, laborious, and time consuming but also fail to identify many epitopes (36). When talking about protein Ags, most of these methods typically identify linear stretches as epitopes, while, arguably, most of the epitopes on protein Ags are conformational and even discontinuous. As for computational approaches, despite more than 30 years of efforts (37), existing B-cell epitope prediction methods are not accurate enough (38, 39) and are, therefore, not widely used. This is exemplified in Figure 1, in which the structure of hen egg lysozyme (HEL) Ag and three Abs that bind it are shown (Figures 1A,B), as well as the epitopes predicted by three different methods (Figure 1C).
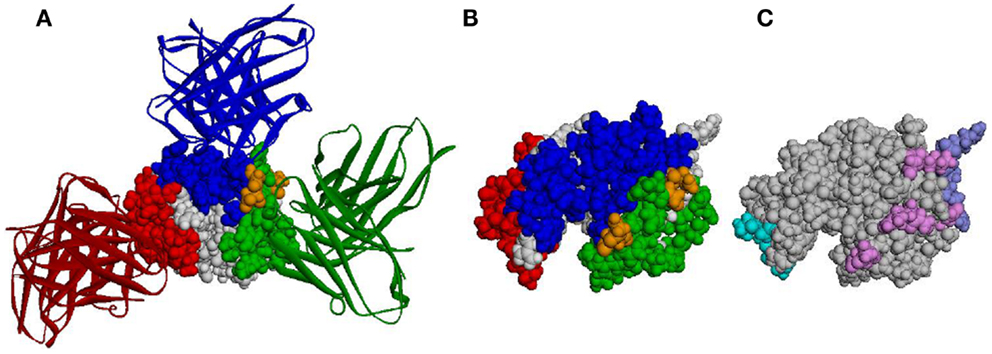
Figure 1. Predicted epitopes vs. the actual epitopes of HEL. (A) The 3-D structure of HEL (CPK representation) together with three Abs (ribbon representation). PDB IDs 1JHL, 3D9A, and 1MLC were superimposed according to HEL structure. Epitope residues are colored blue, green, and red according to the corresponding Ab. Residues that are common to two epitopes are colored orange. (B) The structure of HEL colored according to the same three epitopes as in (A), presented in a different orientation. (C) The structure of HEL colored according to the epitopes predicted by Discotope (light blue), ellipro (purple), and seppa (pink). Note, not all predicted residues of Discotope and ellipro are observable in the presented orientation.
In general, current methods are trying to identify epitopic residues based on the presence of features associated with residues that bind the Ab (40–50). One possible explanation for the failure of these methods is that the differences between epitopes and other residues are not substantial. Indeed, several analyses (51–53) have shown that the amino-acid composition of epitopes is essentially indistinguishable from that of other surface-exposed non-epitopic residues.
This lack of intrinsic properties that clearly differentiate between epitopic and non-epitopic residues and the fact (demonstrated in Figure 1) that most of the Ag surface may become a part of an epitope under some circumstances (54–57) suggest that epitopes depend, to a great extent, on the Abs that recognize them. This is exemplified in Figure 1: most of the HEL surface residues are part of an epitope of at least one Ab (Figures 1A,B), even though this figure shows only three Abs (out of dozens known to bind HEL). Almost all the residues predicted to be epitopic may be considered as correct predictions as they bind some Ab (Figure 1C) but also as false predictions as they don’t bind the others. Similarly, predicting that a residue is not in an epitope may be either a true negative or a false negative, depending on the Ab considered. It has recently been suggested by us (Sela-Culang et al., submitted) and by others (58–60) that predicting epitopes should be done for a certain Ab. A similar concept was successfully applied in the case of T-cell epitope prediction methods: these methods do not examine the Ag for general features. Rather, different predictions are made, dependent on the specific MHC molecule binding and presenting the epitope to T cells.
As shown in Figure 2, Abs are all-beta proteins consisting of four polypeptide chains: two identical heavy (H) chains and two identical light (L) chains (61). The light and heavy chains are linked by disulfide bonds to form the arms of a Y-shaped structure, each arm is known as a Fab (61). The Fab is composed of two variable domains (VH in the heavy chain and VL in the light chain) and two constant domains (CH1 and CL) (62). In the pairing of light and heavy chains, the two variable domains dimerize to form the Fv fragment which contains the Ag binding site. Within each variable domain lie six hypervariable loops (63), three in the light chain (L1, L2, and L3) and three in the heavy chain (H1, H2, and H3), supported by a conserved FR of β-sheets. The light and heavy variable domains fold in a manner that brings the hypervariable loops together to create the Ag binding site or paratope. Two additional domains of the heavy chain, CH2, and CH3, compose the Fc region which is responsible for mediating the biological activity of the Ab molecule.
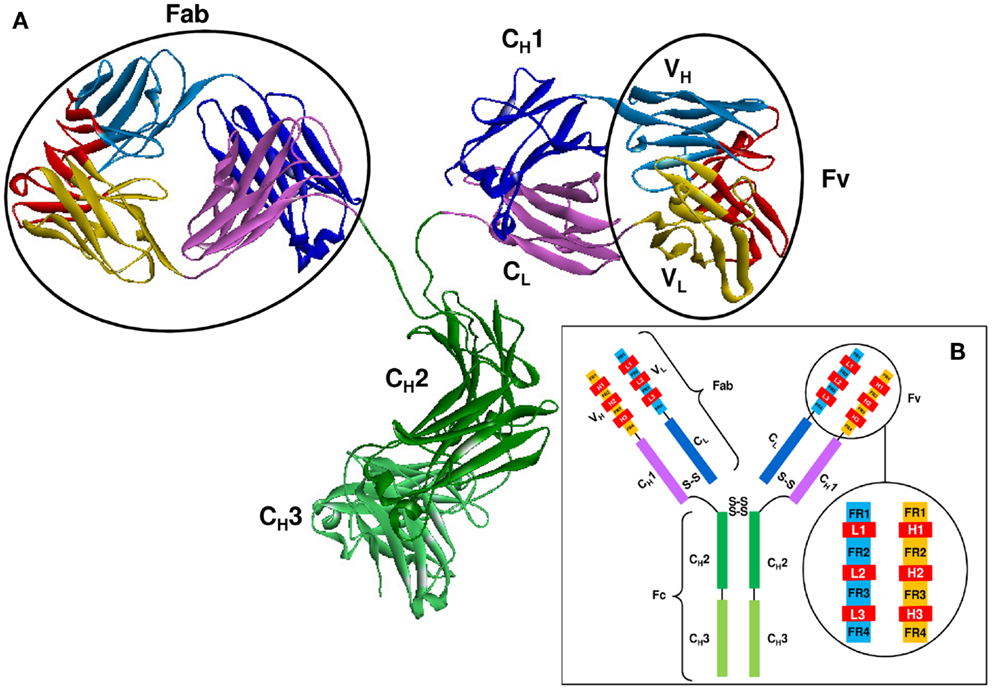
Figure 2. The structure of an Ab molecule. (A) The 3-D structure of an Ab molecule (PDB ID: 1IGT). (B) A schematic representation of the Ab scaffold.
As indicated by their names, CDRs are believed to account for the recognition of the Ag. Therefore, a major focus in analyzing the structural basis for Ag recognition has been in identifying the exact boundaries of the CDRs in a given Ab. It is a common practice to identify paratopes through the identification of CDRs. Kabat and co-authors (63, 64) were the first to introduce a systematic approach to identify CDRs in newly sequenced Abs. It was based on the assumption that CDRs are the most variable regions between Abs. Therefore, they aligned the (fairly limited) set of Ab sequences available at that time and identified the most variable positions. Based on the alignment, they introduced a numbering scheme for the residues in the hypervariable regions and determined which positions mark the beginning and the end of each CDR. As structural data became available, Chothia and Lesk (65, 66) manually analyzed a small number of experimentally solved 3-D structures and determined the structural location of the loop regions. The boundaries of the FRs and CDRs were determined and the latter have been shown to adopt a restricted set of conformations, based on the presence of certain residues at key positions in the CDRs and the flanking FRs. Their finding that Kabat’s definitions of L1 and H1 are structurally incorrect led to the introduction of the Chothia numbering scheme. With the increase of available structural data, they ran their analysis anew and introduced a new definition of L1 (66) in 1989. In 1997 (67), however, they concluded that this correction was erroneous, and reverted to their original 1987 numbering scheme. While the Kabat and Chothia schemes treated separately the different families of immunoglobulin domains, Lefranc and colleagues (68, 69) proposed a unified numbering scheme (referred to as IMGT numbering scheme) for immunoglobulin variable domain genomic sequences, including Ab light and heavy variable domains, as well as T-cell receptor variable domains. To correlate between the sequence, structure, and domain folding behavior of all immunoglobulin variable domains, the Aho numbering scheme spatially aligned known 3-D structures of immunoglobulins and unified their numbering (70).
A drawback of the Kabat, Chothia, and IMGT numbering schemes is that CDRs length variability takes into account only the most common loop lengths; While both Kabat and Chothia schemes accommodate insertions with insertion letters (e.g., 27A), the IMGT scheme avoids the use of insertion codes for all but the least common very long loops, and the Aho numbering scheme places insertions and deletions symmetrically around a key position. However, Abs with unusually long insertions may be hard to annotate using these methods and, as a result, their CDRs may not be identified correctly. For instance, the recently determined 3-D crystal structure of two bovine Abs (71) reveal exceptionally long H3 CDRs ( >60 residues), with long insertions which these methods cannot accommodate and thus cannot identify the CDRs of these Abs.
While identification of paratopes is often done through identification of CDRs, not all the residues within the CDRs bind the Ag. In fact, an early analysis of the 3-D structures of Abs suggested that only 20–33% of the residues within the CDRs participate in Ag binding (72). In 1996, MacCallum and colleagues (73) performed a detailed residue-level analysis of Ag contacts. They suggested that contacting residues are more common at CDRs residues which are located at the center of the Ag combining site, and that non-contacting residues within the CDRs correspond with residues that are important for maintaining the structural conformations of the hypervariable loops and not necessarily for recognition of the Ag. Thus, they introduced a mapping of Ag-contacting propensities for each Ab position and proposed a new definition for CDRs based on these propensities. Padlan and co-workers (28) utilized Abs sequence and structure data to perform a by-position summary of Ag contacts. They found that the residues that are directly involved in the interaction with the Ag are also, in general, the most variable ones. They suggested that the residues that interact with the Ag should be called Specificity Determining Residues (SDRs).
The number of publicly available structures of Ab-Ag complexes increased in recent years to a level that enabled large-scale analyses. In a recent analysis (29) we utilized all available protein-Ab complexes in the PDB to identify the structural regions in which Ag binding actually occurs. This approach was implemented into a method dubbed Paratome (30, 74) that is based on a multiple structure alignment (MSTA) of all available Ab-Ag complexes in the PDB. The MSTA revealed regions of structural consensus where the pattern of structural positions that bind the Ag is highly similar among all Abs. These regions of structural binding consensus were termed antigen binding regions (ABRs). While CDRs, as identified by methods such as Kabat (63), Chothia (65), and IMGT (69), may miss ∼20% of the Ag binding residues, ABRs cover ∼96% of the residues that actually bind the Ag (30). To avoid confusions and cumbersome nomenclature, herein we generically refer to CDRs, SDRs, and ABRs as “CDRs” unless otherwise specified. Figure 3 shows an example of CDRs as identified by Kabat, Chothia, IMGT, and Paratome for one Ab (anti-IL-15, PDB ID: 2XQB), compared to the actual Ag binding residues. It can be seen that in this example, some of the CDRs (e.g., L3, H3) identified by the four methods are almost identical, while in other CDRs (e.g., L2, H1, and H2) there are substantial differences between the methods. The MSTA of Abs with known 3-D structure also confirmed previous observations that there are structural positions within the CDRs in which none, or only a small percentage of the Abs contact the Ag. This is shown in Figure 4 where an example of such a position is marked by a green arrow.

Figure 3. Comparison of different CDR identification methods. The light (A) and heavy (B) chains of PDB ID 2XQB were numbered according to Kabat (colored green) and Chothia (colored red) using the Abnum tool (www.bioinf.org.uk/abs/abnum) and CDRs were extracted according to the CDR definitions table (www.bioinf.org.uk/abs/#cdrs). CDRs according to IMGT (colored orange) were identified using the IMGT-gap tool (www.imgt.org/3Dstructure-DB/cgi/DomainGapAlign.cgi). ABRs according to Paratome (colored blue) were identified using the Paratome server (www.ofranlab.org/paratome). Contacts (colored purple) between the Ab and IL-15 were defined using a 6-Å cutoff value.
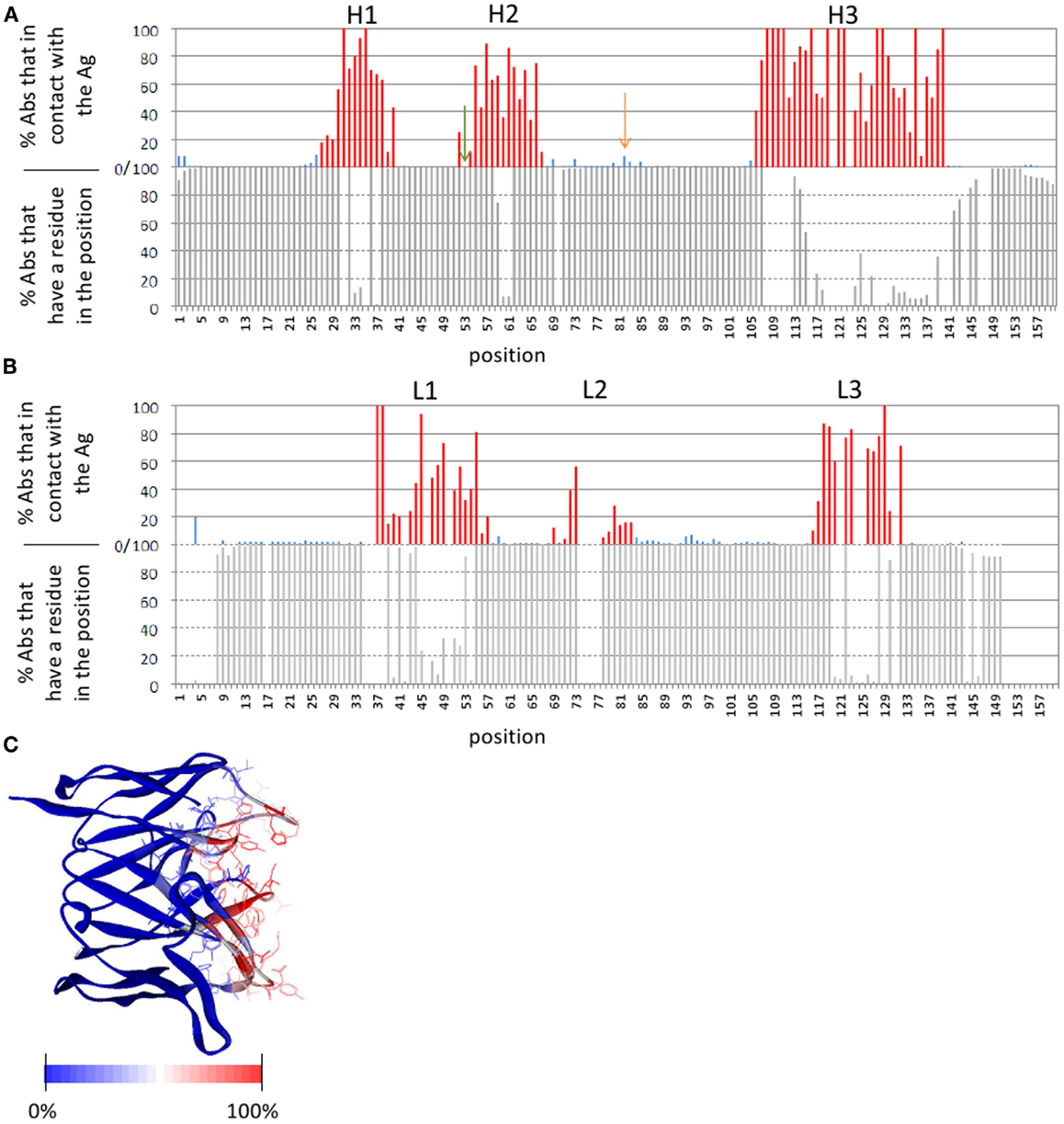
Figure 4. Ab positions that contact the Ag. (A,B) The lower graphs show the percentage of Abs with known 3-D structure that have a residue in a given position (i.e., in other Abs there is a gap in the MSTA in that position). The upper graphs show the percentage of Abs that contact the Ag out of those Abs that have a residue in that position. (A) Depicts the heavy chain and (B) depicts the light chain. In the upper graphs, the ABRs are colored red and the FRs are colored blue. An example of a position within an ABR that is not in contact with the Ag in any of the Abs, is marked by a green arrow. An example of a position in the FRs that is in contact with the Ag in many (8%) of the Abs is marked by an orange arrow. (C) The Ab Fv domain (PDB ID: 1QFU) is colored according to the percentage of all Abs with known 3-D structure in which the residue in that position is in contact with the Ag: from red (100% of the Abs) to blue (0%). ABR residues are presented as lines. The definition of the ABRs is according to the Paratome server. A 6-Å cutoff value was used to define residues in contact. Percentages of contacts were calculated based on an MSTA of all protein Ab-Ag complexes in the PDB (30).
Designed systems are often characterized as either modular or integral. In a modular system different components, or modules, function independent of the function of other modules. The generation of Abs in the immune system is based on combining different elements, in a way that may be considered modular where each component is capable of binding the Ag regardless of the others. However, some analyses suggest that Ag binding warrants a more integrative view of the relationships between the different components of the Ab.
The binding-sites of interacting proteins are usually composed of surface patches that have good shape and electrostatic complementary (15, 75, 76). It has been shown that CDRs are characterized by an amino-acid composition that is different from that of other protein loops (77) and also from other types of protein–protein interfaces (58). Thus, one would expect that epitopes, just like paratopes, should have a distinct amino-acid composition. However, several recent analyses (51, 53) have shown that this is not the case: while epitopes differ from other types of interfaces (10, 29, 60), their amino-acid composition is virtually the same as that of non-epitopic surface residues.
Several studies have shown that each CDR has its own unique amino-acid composition, different from the composition of the other CDRs (52, 58, 78). Additionally, we have shown that each CDR has a unique set of contact preferences, therefore, favoring certain amino-acids over others (52). Dividing epitope residues into six subsets according to the CDR they bind, we found that each of the subsets has a distinct amino-acid composition, distinguishable from non-epitope surface (52). In other words, when the six subsets of epitope residues are considered together the unique composition of each subset disappears so that the overall amino-acid composition of the entire epitope is indistinguishable from the rest of the surface. Pathogenic epitopes may have evolved to resemble Ag surface to escape recognition. On the other hand, the integration of the six CDRs together, each with its own unique amino-acid composition and contact preferences, could be the evolutionary response of the immune system that enables Abs to recognize virtually any surface patch on the Ag.
Despite this integrated effect of the CDRs, Abs can be also considered as a modular system, composed of different elements (such as the Fab, VH and VL, or the six CDRs), which may bind the Ag on their own. Such smaller Ab fragments that retain Ag binding affinity and specificity, hold a great potential for drug design (79–81) as they have improved pharmacokinetics, tissue and tumor penetration, and can be produced more economically (80, 81). They may also be combined with other fragments to yield better binders. Although such smaller fragments cannot induce effector function such as complement activation (due to the lack of the constant domains), they may neutralize the targeted Ag. Fab and single-chain variable (scFv) fragments usually maintain specific binding to the Ag (82). VH and VL fragments usually show sticky behavior, low solubility, and reduced Ag binding affinity (83–85), although, they sometime retain specificity to the Ag (83, 85–87).
The CDRs may provide additional level of modularity. According to the commonly accepted hotspot hypothesis, the binding energy of two proteins is largely determined by a very small number of critical interface residues (12, 88–90). Thus, one may wonder whether an individual CDR could bind the Ag on its own provided that it harbors hotspots. Several linear peptides containing one or more of the CDRs that retained Ag specificity have been reported (91–98). Although their affinity was usually in the micromolar range, it could be significantly improved by introducing relatively minor modifications (91, 99). However, many attempts to isolate and design such CDR derived peptides failed (100, 101). One possible reason is that a CDR, on its own, may not fold to the same conformation as in the context of the entire Fab, which may be crucial for binding. Cyclizing the CDR by adding Cys residues at its edges was suggested as a solution for this problem (96, 102–104). Another reason might lie in the fact that many attempts for the design of CDR-derived peptides are made based on CDR-H3, as it is considered to be the most important CDR for Ag binding (67, 105–107). However, the median length of ABR-H2 is substantially longer than that of H3, and both typically form the same number of interactions with the Ag (52). In addition, while ABR-H3 was shown to have the highest contribution to Ag binding energy on average (52), there are individual cases in which other CDRs are the dominant ones (52, 102). It is also possible that in some cases the binding depends on specific contacts from residues in different CDRs, which may preclude the design of CDR-derived peptides that maintain specificity. We have shown (102) that CDRs that are able to bind the Ag on their own have unique characteristics and, thus, can be computationally identified given the Ab-Ag complex structure. This may enhance the design of CDR-derived peptides that are not necessarily based on CDR-H3.
Within the variable domain, the CDRs are believed to be responsible for Ag recognition, while the FR residues are considered a scaffold for the CDRs. However, it is now well established that some of the FR residues may play an important role in Ag binding (32, 108). As mentioned above, many such FR residues were identified during the process of Ab humanization by CDR grafting. While grafting only the CDRs usually results in a significant drop or a complete loss of binding, the binding affinity can be retained by back mutating some of the FR residues to the original murine sequence, emphasizing their role in Ag binding (26, 109–115).
Framework region residues that affect Ag binding can be divided into two categories. The first are FR residues that contact the Ag, thus are part of the binding-site (108, 109, 111, 116–123). Some of these residues are close in sequence to the CDRs (in fact they may be within the boundaries of CDRs according to some CDR identification methods, but not according to others, as shown in Figure 3). Other residues are those that are far from the CDRs in sequence, but are in close proximity to it in the 3-D structure. In particular, a loop in the heavy chain FR-3, sometimes referred to as CDR-H4, accounts for 1.3% of human Ab-Ag contacts (78, 124). This CDR-H4 is also enriched (in human Abs) in somatic hypermutations (Burkovitz et al., submitted). Figure 4 shows positions that are not in the CDRs but are in contact with the Ag in many Abs [e.g., the one marked by an orange arrow (4A), which corresponds to CDR-H4].
In the second category of FR residues that affect Ag binding, are residues that are not in contact with the Ag, but affect Ag binding indirectly (108, 109, 120, 121). These residues can be further divided to those that are in spatial proximity to the CDRs, and those that are not. The former are assumed to affect binding by providing a structural support to the CDRs, enabling them to adopt the right conformation and orientation, shaping the binding-site required for Ag binding (32). For example, it has been suggested that a certain position in heavy chain FR-3, close in structure but not in sequence to CDR-H1 and CDR-H2, affects the orientation of CDR-H2 relative to CDR-H1 in such a way that a large side-chain packs between them and separates them while a small side-chain allows them to be closer to each other (109, 120). Nevertheless, this is not always true, as was shown in the case of the anti-lysozyme D1.3 Ab: while mutating Lys in this position to either Val, Ala, or Arg resulted in affinity difference, no structural change was observed (121).
Framework region residues that are more distant from the paratope are suggested to play a role in maintaining the overall structure of the Fv domains (32). However, these FR residues may also affect the Ag binding-site itself, by directing the relative orientation of the VH vs. the VL, and thus the orientation of the CDRs relative to each other (125–128). In particular, FR-2 residues were shown to play an important role in VH-VL interaction (129). Moreover, Masuda et al. (130) pointed to a specific position in the FR-2 loop, which controls the strength of the VH-VL interaction as well as its dependence on Ag binding. We have shown that the conformation of this loop changes upon Ag binding more than other residues in the FRs, and that the binding related conformational changes in this loop are similar in their magnitude to those of the CDRs (107). The potential role of the VH-VL interface in Ag binding is further supported by the observation that residues that are in the VH-VL interface (and are not a part of the Ab-Ag interface), are more likely to be mutated during the somatic hypermutation process, than residues that are not in either of these interfaces (Burkovitz et al., submitted).
Understanding the role of FR residues in Ag binding is crucial for efficient Ab design in general and for humanization in particular. Specifically, knowing in advance which FR residues may affect Ag binding, one may consider back-mutating these residues into their murine sequence, to improve affinity during CDR grafting. To this end, attempts were made to identify positions that contribute to Ag binding in multiple cases (32, 113, 119). For example, Haidar et al. (32) used a non-redundant dataset of Ab-Ag complex structures to identify positions that frequently contact the CDRs, and combined these positions with those that were back-mutated frequently in the humanization literature. The 17 FR positions they identified were successfully used to design a combinatorial library for Ab humanization. Additional Abs, for which structures of both wild-type and a mutant(s) are available, may reveal the structural mechanisms by which each FR position affects Ag binding.
Until recently, Ab constant domains were considered responsible for the isotype and for effector function, such as complement activation, Fc receptor binding, avidity, and serum half-life (131). However, many studies now provide a strong evidence for a role for the constant region in Ag binding (131–147). There are many examples of Abs with identical variable domains but different isotypes that bind the same Ag with a different affinity or specificity (134–146). For instance, two Abs sharing identical variable domains but expressing different isotypes were shown to bind tubulin with significantly different affinities (135). Consistent with these studies, it has been shown that the complex of HEL and the Fv version of the HyHEL-10 Ab has an order of magnitude lower dissociation constant than the complex of HEL with the Fab version of this Ab (147). A probable explanation for this phenomenon would be an allosteric influence of the constant domains on the structure of the variable domains. Indeed, several structural studies provided some evidence for such structural effects (133, 146, 147). For example, Janda et al. (133) analyzed by Circular Dichroism (CD) spectra four different Ab isotypes of the 3E5 family that share identical variable domains, and showed that the different isotypes undergo different structural changes upon binding a common Ag. Similar results were obtained for anti-nuclear Abs as well: Xia et al. (146) compared four different isotypes of the PL9–11 anti-nuclear Ab sharing the same variable region, and found that the changes in secondary structure content (as revealed by CD analysis) as well as the wave length shifts of tryptophan fluorescence emission, upon Ag binding, are both isotype dependent. Recently, Tudor et al. (144) showed that this allosteric effect may control not only Ag binding affinity and specificity, but also the epitope recognized. They showed that two anti-HIV-1 IgG1 and IgA2 Abs with identical variable regions, recognize only partially overlapping epitopes.
Differences in affinity and specificity of Abs with the same variable region but different isotypes may play a role in autoimmunity if they occur in a self-reactive Ag. For example, different isotypes have been shown to be associated with different clinical outcomes for lupus erythematosus: a set of anti-PL9–11 Abs sharing the same variable domain but different isotypes were shown to bind DNA and chromatin, as well as the renal Ags, with different affinities that were associated with significant differences in renal pathogenicity in vivo and survival (148).
Several studies have suggested that allosteric effects in Abs may occur on the other direction as well: structural changes in the variable region caused by Ag binding may be transferred into the constant domains, potentially influencing effector activation and cellular response (131, 149–151). For example, Oda et al. (149) showed that the binding of staphylococcal protein A (SPA) or streptococcal protein G (SPG) to the constant region was inhibited by hapten binding in several Abs. A different example was provided by Horgan et al. (151) who observed differences in complement activation of two Abs which differ only in their VH domain.
An allosteric effect in Abs is further supported by a systematic computational analysis we have performed on all available free and Ag-bound pairs of structures (107). Many of the Ag-binding-related structural changes occur distant from the Ag binding-site, including changes in the relative orientation of the heavy and light chains in both the variable and constant domains as well as a change in the elbow angle between the variable and the constant domains. Moreover, the most consistent and substantial conformational change outside of the binding site was found in a loop in the heavy chain constant domain, which is a part of the CH1-CL interface, and is involved in complement binding (152).
What could be the mechanism for these allosteric effects? Changes in the constant domains sequence (different isotypes of the same Ab) or in its conformation (e.g., by effector binding) may lead to a rearrangement of the constant domains relative to each other and relative to the variable domains, which may result in a change to the VH-VL relative orientation (72), thus re-shaping the Ag binding-site (153–155).
The potential influence of the constant region on Ag affinity or specificity suggests that the process of class-switch may be considered, in combination with somatic hypermutations, as a mechanism for Ab diversity (131, 132). Engineering of an Ab of interest is usually associated with the optimization of its affinity to the Ag. Since the constant region may affect this affinity, the isotype selected should be carefully considered. Moreover, the constant region should be taken into account in vaccine design as well since different isotypes may bind the pathogenic Ag with different affinities, thus affecting the response to infection. For example, the anti HIV-1 IgG1 and IgA2 Abs mentioned above share the same variable region, nevertheless, they have been shown to block HIV-1 infection differently (144). While IgA2 blocked HIV-1 transcytosis and CD4+ cell infection more efficiently, IgG1 and IgA2 act synergistically to block HIV-1 transfer from Langerhans cells to T cells. Thus, it has been suggested that a mucosal IgA-based vaccine response should complement an IgG-based vaccine response in blocking HIV-1 transmission.
As Abs are one of the most versatile naturally occurring biosensors, it is of high importance to decipher the structural and molecular mechanisms by which they recognize and bind their Ags. Such knowledge is crucial for understanding immunity, may enable better prediction of Ab epitopes, and assist in Ab engineering.
While the commonly accepted view has been that CDRs hold the key for Ab-Ag recognition, recent findings indicate that not all the positions in the traditionally defined CDRs are important for binding. Furthermore, it has been shown that many positions that contribute critically to the binding energy reside outside of the transitional CDRs. Moreover, different CDR identification methods may often identify radically different stretches as “CDRs,” indicating that CDRs are not well defined and thus are not necessarily a good proxy for the binding site. The hypervariable loops that accommodate the CDRs differ significantly from each other on various aspects. Understanding the way in which their binding preferences are integrated to yield the overall specificity of the Ab is an intriguing structural and biophysical challenge.
Accumulation of recent data suggests that elements that may be spatially distant from the Ag binding site also play a crucial role in Ag recognition. The unorthodox suggestion that non-local and even allosteric effects influence epitope recognition warrants additional analysis and research.
Addressing the open questions regarding the structural basis of Ag recognition requires additional structural data in the form of crystal structures of Abs bound to Ags of different types (proteins, peptides, nucleic acids, and haptens). Large-scale analysis of such structures will allow for the generation and testing of new hypotheses regarding the way in which Abs find and bind their epitopes.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are supported in part by NIAID contract N01-AI-90048C (http://www.niaid.nih.gov/).
1. Vincent KJ, Zurini M. Current strategies in antibody engineering: Fc engineering and pH-dependent antigen binding, bispecific antibodies and antibody drug conjugates. Biotechnol J (2012) 7:1444–50. doi:10.1002/biot.201200250
2. Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci (2007) 96:1–26. doi:10.1002/jps.20727
3. Sheedy C, MacKenzie CR, Hall JC. Isolation and affinity maturation of hapten-specific antibodies. Biotechnol Adv (2007) 25:333–52. doi:10.1016/j.biotechadv.2007.02.003
4. Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol (2007) 25:1369–72. doi:10.1038/nbt1207-1369
5. Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes – a framework for modelling, testing, and applications. Methods (2004) 34:436–43. doi:10.1016/j.ymeth.2004.06.006
6. Kim Y, Sette A, Peters B. Applications for T-cell epitope queries and tools in the Immune Epitope Database and Analysis Resource. J Immunol Methods (2011) 374:62–9. doi:10.1016/j.jim.2010.10.010
7. Frank SA. Specificity and cross-reactivity (Chapter 4). In: Immunology and Evolution of Infectious Disease. Princeton, NJ: Princeton University Press (2002). p. 33–54.
8. Rose NR, MacKay IR, editors. T-cells and autoimmunity. In: The Autoimmune Diseases, 4th ed. San Diego: Elsevier Academic Press (2006). 1160 p.
9. Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci U S A (1996) 93:13–20. doi:10.1073/pnas.93.1.13
10. Jones S, Thornton JM. Analysis of protein-protein interaction sites using surface patches. J Mol Biol (1997) 272:121–32. doi:10.1006/jmbi.1997.1234
11. Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol (1999) 285:2177–98. doi:10.1006/jmbi.1998.2439
12. Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol (1998) 280:1–9. doi:10.1006/jmbi.1998.1843
13. Chakrabarti P, Janin J. Dissecting protein-protein recognition sites. Proteins (2002) 47:334–43. doi:10.1002/prot.10085
14. Keskin O. Binding induced conformational changes of proteins correlate with their intrinsic fluctuations: a case study of antibodies. BMC Struct Biol (2007) 7:31. doi:10.1186/1472-6807-7-31
15. McCoy AJ, Chandana Epa V, Colman PM. Electrostatic complementarity at protein/protein interfaces. J Mol Biol (1997) 268:570–84. doi:10.1006/jmbi.1997.0987
16. Neuvirth H, Raz R, Schreiber G. ProMate: a structure based prediction program to identify the location of protein-protein binding sites. J Mol Biol (2004) 338:181–99. doi:10.1016/j.jmb.2004.02.040
17. Ma B, Elkayam T, Wolfson H, Nussinov R. Protein–protein interactions: structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc Natl Acad Sci U S A (2003) 100:5772–7. doi:10.1073/pnas.1030237100
18. Bordner AJ, Abagyan R. Statistical analysis and prediction of protein-protein interfaces. Proteins (2005) 60:353–66. doi:10.1002/prot.20433
19. Ofran Y, Rost B. Analysing six types of protein-protein interfaces. J Mol Biol (2003) 325:377–87. doi:10.1016/S0022-2836(02)01223-8
20. Marks JD, Griffiths AD, Malmqvist M, Clackson TP, Bye JM, Winter G. By-passing immunization: building high affinity human antibodies by chain shuffling. Biotechnology (N Y) (1992) 10:779–83. doi:10.1038/nbt0792-779
21. Soderlind E, Ohlin M, Carlsson R. Complementarity-determining region (CDR) implantation: a theme of recombination. Immunotechnology (1999) 4:279–85.
22. Hemminki A, Niemi S, Hautoniemi L, Soderlund H, Takkinen K. Fine tuning of an anti-testosterone antibody binding site by stepwise optimisation of the CDRs. Immunotechnology (1998) 4:59–69. doi:10.1016/S1380-2933(98)00002-5
23. Ohlin M, Owman H, Mach M, Borrebaeck CA. Light chain shuffling of a high affinity antibody results in a drift in epitope recognition. Mol Immunol (1996) 33:47–56. doi:10.1016/0161-5890(95)00123-9
24. Mirick GR, Bradt BM, Denardo SJ, Denardo GL. A review of human anti-globulin antibody (HAGA, HAMA, HACA, HAHA) responses to monoclonal antibodies. Not four letter words. Q J Nucl Med Mol Imaging (2004) 48:251–7.
25. Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature (1986) 321:522–5. doi:10.1038/321522a0
26. Queen C, Schneider WP, Selick HE, Payne PW, Landolfi NF, Duncan JF, et al. A humanized antibody that binds to the interleukin 2 receptor. Proc Natl Acad Sci U S A (1989) 86:10029–33. doi:10.1073/pnas.86.24.10029
28. Padlan EA, Abergel C, Tipper JP. Identification of specificity-determining residues in antibodies. FASEB J (1995) 9:133–9.
29. Ofran Y, Schlessinger A, Rost B. Automated identification of complementarity determining regions (CDRs) reveals peculiar characteristics of CDRs and B cell epitopes. J Immunol (2008) 181:6230–5.
30. Kunik V, Peters B, Ofran Y. Structural consensus among antibodies defines the antigen binding site. PLoS Comput Biol (2012) 8:e1002388. doi:10.1371/journal.pcbi.1002388
31. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res (2000) 28:235–42. doi:10.1093/nar/28.1.235
32. Haidar JN, Yuan QA, Zeng L, Snavely M, Luna X, Zhang H, et al. A universal combinatorial design of antibody framework to graft distinct CDR sequences: a bioinformatics approach. Proteins (2012) 80:896–912. doi:10.1002/prot.23246
33. Hanf KJ, Arndt JW, Chen LL, Jarpe M, Boriack-Sjodin PA, Li Y, et al. Antibody humanization by redesign of complementarity-determining region residues proximate to the acceptor framework. Methods (2013). doi:10.1016/j.ymeth.2013.06.024
34. Amanna IJ, Slifka MK, Crotty S. Immunity and immunological memory following smallpox vaccination. Immunol Rev (2006) 211:320–37. doi:10.1111/j.0105-2896.2006.00392.x
35. Yang XD, Yu XL. An introduction to epitope prediction methods and software. Rev Med Virol (2009) 19:77–96. doi:10.1002/rmv.602
36. Xu XL, Sun J, Liu Q, Wang XJ, Xu TL, Zhu RX, et al. Evaluation of spatial epitope computational tools based on experimentally-confirmed dataset for protein antigens. Chin Sci Bull (2010) 55:2169–74. doi:10.1007/s11434-010-3199-z
37. Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci U S A (1981) 78:3824–8. doi:10.1073/pnas.78.6.3824
38. Ponomarenko JV, Bourne PE. Antibody-protein interactions: benchmark datasets and prediction tools evaluation. BMC Struct Biol (2007) 7:64. doi:10.1186/1472-6807-7-64
39. Blythe MJ, Flower DR. Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci (2005) 14:246–8. doi:10.1110/ps.041059505
40. Ansari HR, Raghava GP. Identification of conformational B-cell epitopes in an antigen from its primary sequence. Immunome Res (2010) 6:6. doi:10.1186/1745-7580-6-6
41. Kulkarni-Kale U, Bhosle S, Kolaskar AS. CEP: a conformational epitope prediction server. Nucleic Acids Res (2005) 33:W168–71. doi:10.1093/nar/gki460
42. Liang SD, Zheng DD, Zhang C, Zacharias M. Prediction of antigenic epitopes on protein surfaces by consensus scoring. BMC Bioinformatics (2009) 10:302. doi:10.1186/1471-2105-10-302
43. Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics (2008) 9:514. doi:10.1186/1471-2105-9-514
44. Rubinstein ND, Mayrose I, Pupko T. A machine-learning approach for predicting B-cell epitopes. Mol Immunol (2009) 46:840–7. doi:10.1016/j.molimm.2008.09.009
45. Rubinstein ND, Mayrose I, Martz E, Pupko T. Epitopia: a web-server for predicting B-cell epitopes. BMC Bioinformatics (2009) 10:287. doi:10.1186/1471-2105-10-287
46. Sun J, Wu D, Xu T, Wang X, Xu X, Tao L, et al. SEPPA: a computational server for spatial epitope prediction of protein antigens. Nucleic Acids Res (2009) 37:W612–6. doi:10.1093/nar/gkp417
47. Sweredoski MJ, Baldi P. PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics (2008) 24:1459–60. doi:10.1093/bioinformatics/btn199
48. Ambroise J, Giard J, Gala JL, Macq B. Identification of relevant properties for epitopes detection using a regression model. IEEE/ACM Trans Comput Biol Bioinform (2011) 8:1700–7. doi:10.1109/TCBB.2011.77
49. Liang S, Zheng D, Standley DM, Yao B, Zacharias M, Zhang C. EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results. BMC Bioinformatics (2010) 11:381. doi:10.1186/1471-2105-11-381
50. Zhang W, Xiong Y, Zhao M, Zou H, Ye X, Liu J. Prediction of conformational B-cell epitopes from 3D structures by random forests with a distance-based feature. BMC Bioinformatics (2011) 12:10. doi:10.1186/1471-2105-12-341
51. Janin J, Chothia C. The structure of protein-protein recognition sites. J Biol Chem (1990) 265:16027–30.
52. Kunik V, Ofran Y. The indistinguishability of epitopes from protein surface is explained by the distinct binding preferences of each of the six antigen-binding loops. Protein Eng Des Sel (2013). doi:10.1093/protein/gzt027
53. Kringelum JV, Nielsen M, Padkjær SB, Lund O. Structural analysis of B-cell epitopes in antibody:protein complexes. Mol Immunol (2013) 53:24–34. doi:10.1016/j.molimm.2012.06.001
54. Benjamin DC, Berzofsky JA, East IJ, Gurd FR, Hannum C, Leach SJ, et al. The antigenic structure of proteins: a reappraisal. Annu Rev Immunol (1984) 2:67–101. doi:10.1146/annurev.iy.02.040184.000435
55. Greenbaum JA, Andersen PH, Blythe M, Bui HH, Cachau RE, Crowe J, et al. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J Mol Recognit (2007) 20:75–82. doi:10.1002/jmr.815
56. Novotný J, Handschumacher M, Haber E, Bruccoleri RE, Carlson WB, Fanning DW, et al. Antigenic determinants in proteins coincide with surface regions accessible to large probes (antibody domains). Proc Natl Acad Sci U S A (1986) 83:226–30. doi:10.1073/pnas.83.2.226
57. Thornton JM, Edwards MS, Taylor WR, Barlow DJ. Location of “continuous” antigenic determinants in the protruding regions of proteins. EMBO J (1986) 5:409–13.
58. Zhao L, Li JY. Mining for the antibody-antigen interacting associations that predict the B cell epitopes. BMC Struct Biol (2010) 10(Suppl 1):S6. doi:10.1186/1472-6807-10-S1-S6
59. Zhao L, Wong L, Li JY. Antibody-specified b-cell epitope prediction in line with the principle of context-awareness. IEEE/ACM Trans Comput Biol Bioinform (2011) 8:1483–94. doi:10.1109/TCBB.2011.49
60. Soga S, Kuroda D, Shirai H, Kobori M, Hirayama N. Use of amino acid composition to predict epitope residues of individual antibodies. Protein Eng Des Sel (2010) 23:441–8. doi:10.1093/protein/gzq014
61. Edelman GM, Benacerraf B. On structural and functional relations between antibodies and proteins of the gamma-system. Proc Natl Acad Sci U S A (1962) 48:1035–42. doi:10.1073/pnas.48.6.1035
62. Putnam FW, Liu YS, Low TL. Primary structure of a human IgA1 immunoglobulin. IV. Streptococcal IgA1 protease, digestion, Fab and Fc fragments, and the complete amino acid sequence of the alpha 1 heavy chain. J Biol Chem (1979) 254:2865–74.
63. Wu TT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med (1970) 132:211–50. doi:10.1084/jem.132.2.211
64. Kabat EA, Wu TT, Bilofsky H, Reid-Miller M, Perry H. Sequence of Proteins of Immunological Interest. Bethesda: National Institute of Health (1983).
65. Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol (1987) 196:901–17. doi:10.1016/0022-2836(87)90412-8
66. Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, et al. Conformations of immunoglobulin hypervariable regions. Nature (1989) 342:877–83. doi:10.1038/342877a0
67. Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol (1997) 273:927–48. doi:10.1006/jmbi.1997.1354
68. Giudicelli V, Chaume D, Bodmer J, Muller W, Busin C, Marsh S, et al. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res (1997) 25:206–11. doi:10.1093/nar/25.1.206
69. Lefranc MP, Pommié C, Ruiz M, Giudicelli V, Foulquier E, Truong L, et al. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol (2003) 27:55–77. doi:10.1016/S0145-305X(02)00039-3
70. Honegger A, Pluckthun A. Yet another numbering scheme for immunoglobulin variable domains: an automatic modeling and analysis tool. J Mol Biol (2001) 309:657–70. doi:10.1006/jmbi.2001.4662
71. Wang F, Ekiert DC, Ahmad I, Yu W, Zhang Y, Bazirgan O, et al. Reshaping antibody diversity. Cell (2013) 153:1379–93. doi:10.1016/j.cell.2013.04.049
72. Padlan EA. Anatomy of the antibody molecule. Mol Immunol (1994) 31:169–217. doi:10.1016/0161-5890(94)90001-9
73. MacCallum RM, Martin AC, Thornton JM. Antibody-antigen interactions: contact analysis and binding site topography. J Mol Biol (1996) 262:732–45. doi:10.1006/jmbi.1996.0548
74. Kunik V, Ashkenazi S, Ofran Y. Paratome: an online tool for systematic identification of antigen binding regions in antibodies based on sequence or structure. Nucleic Acids Res (2012) 40(Web Server issue):W521–4. doi:10.1093/nar/gks480
75. Jones S, Thornton JM. Prediction of protein-protein interaction sites using patch analysis. J Mol Biol (1997) 272:133–43. doi:10.1006/jmbi.1997.1234
76. Cohen GH, Silverton EW, Padlan EA, Dyda F, Wibbenmeyer JA, Willson RC, et al. Water molecules in the antibody-antigen interface of the structure of the Fab HyHEL-5-lysozyme complex at 1.7 A resolution: comparison with results from isothermal titration calorimetry. Acta Crystallogr D Biol Crystallogr (2005) 61:628–33. doi:10.1107/S0907444905007870
77. Collis AV, Brouwer AP, Martin AC. Analysis of the antigen combining site: correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J Mol Biol (2003) 325:337–54. doi:10.1016/S0022-2836(02)01222-6
78. Raghunathan G, Smart J, Williams J, Almagro J. Antigen-binding site anatomy and somatic mutations in antibodies that recognize different types of antigens. J Mol Recognit (2012) 25:103–13. doi:10.1002/jmr.2158
79. Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nat Biotechnol (2009) 27:331–7. doi:10.1038/nbt0409-331
80. Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol (2005) 23:1126–36. doi:10.1038/nbt1142
82. Jain M, Kamal N, Batra SK. Engineering antibodies for clinical applications. Trends Biotechnol (2007) 25:307–16. doi:10.1016/j.tibtech.2007.05.001
83. Ward ES, Güssow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature (1989) 341:544–6. doi:10.1038/341544a0
84. Rinfret A, Horne C, Dorrington KJ, Klein M. Noncovalent association of heavy and light chains of human immunoglobulins. IV. The roles of the CH1 and CL domains in idiotypic expression. J Immunol (1985) 135:2574–81.
85. Berry MJ, Davies J. Use of antibody fragments in immunoaffinity chromatography. Comparison of FV fragments, VH fragments and paralog peptides. J Chromatogr (1992) 597:239–45. doi:10.1016/0021-9673(92)80116-C
86. Pereira B, Benedict CR, Le A, Shapiro SS, Thiagarajan P. Cardiolipin binding a light chain from lupus-prone mice. Biochemistry (1998) 37:1430–7. doi:10.1021/bi972277q
87. Dubnovitsky AP, Kravchuk ZI, Chumanevich AA, Cozzi A, Arosio P, Martsev SP. Expression, refolding, and ferritin-binding activity of the isolated VL-domain of monoclonal antibody F11. Biochemistry (Mosc) (2000) 65:1011–8.
88. Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science (1995) 267:383–6. doi:10.1126/science.7529940
89. Sheinerman FB, Norel R, Honig B. Electrostatic aspects of protein-protein interactions. Curr Opin Struct Biol (2000) 10:153–9. doi:10.1016/S0959-440X(00)00065-8
90. Ofran Y. Prediction of Protein Interaction Sites. Computational Protein-Protein Interactions R Nussinov and G Schreiber. Boca Raton: CRC Press (2009). p. 167–84.
91. Park BW, Zhang HT, Wu C, Berezov A, Zhang X, Dua R, et al. Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo. Nat Biotechnol (2000) 18:194–8. doi:10.1038/72651
92. Polonelli L, Pontón J, Elguezabal N, Moragues MD, Casoli C, Pilotti E, et al. Antibody complementarity-determining regions (CDRs) can display differential antimicrobial, antiviral and antitumor activities. PLoS One (2008) 3(6):e2371. doi:10.1371/journal.pone.0002371
93. Kang CY, Brunck TK, Kieber-Emmons T, Blalock JE, Kohler H. Inhibition of self-binding antibodies (autobodies) by a VH-derived peptide. Science (1988) 240:1034–6. doi:10.1126/science.3368787
94. Taub R, Hsu JC, Garsky VM, Hill BL, Erlanger BF, Kohn LD. Peptide sequences from the hypervariable regions of two monoclonal anti-idiotypic antibodies against the thyrotropin (TSH) receptor are similar to TSH and inhibit TSH-increased cAMP production in FRTL-5 thyroid cells. J Biol Chem (1992) 267:5977–84.
95. Saragovi HU, Fitzpatrick D, Raktabutr A, Nakanishi H, Kahn M, Greene MI. Design and synthesis of a mimetic from an antibody complementarity-determining region. Science (1991) 253:792–5. doi:10.1126/science.1876837
96. Levi M, Sällberg M, Rudén U, Herlyn D, Maruyama H, Wigzell H, et al. A complementarity-determining region synthetic peptide acts as a miniantibody and neutralizes human immunodeficiency virus type 1 in vitro. Proc Natl Acad Sci U S A (1993) 90:4374–8. doi:10.1073/pnas.90.10.4374
97. Tsumoto K, Misawa S, Ohba Y, Ueno T, Hayashi H, Kasai N, et al. Inhibition of hepatitis C virus NS3 protease by peptides derived from complementarity-determining regions (CDRs) of the monoclonal antibody 8D4: tolerance of a CDR peptide to conformational changes of a target. FEBS Lett (2002) 525:77–82. doi:10.1016/S0014-5793(02)03090-9
98. Feng Y, Chung D, Garrard L, McEnroe G, Lim D, Scardina J, et al. Peptides derived from the complementarity-determining regions of anti-Mac-1 antibodies block intercellular adhesion molecule-1 interaction with Mac-1. J Biol Chem (1998) 273:5625–30. doi:10.1074/jbc.273.10.5625
99. Feng J, Li Y, Zhang W, Shen B. Rational design of potent mimic peptide derived from monoclonal antibody: antibody mimic design. Immunol Lett (2005) 98:311–6. doi:10.1016/j.imlet.2004.12.006
100. Lasonder E, Bloemhoff W, Welling GW. Interaction of lysozyme with synthetic anti-lysozyme D1.3 antibody fragments studied by affinity chromatography and surface plasmon resonance. J Chromatogr A (1994) 676:91–8. doi:10.1016/0021-9673(94)00125-1
101. Schellekens GA. Molecular Aspects of Antibody-Antigen Interactions: Size Reduction of a Herpes Simplex Virus Neutralizing Antibody and its Antigen. Groningen: University of Groningen (1996).
102. Burkovitz A, Leiderman O, Sela-Culang I, Byk G, Ofran Y. Computational identification of antigen-binding antibody fragments. J Immunol (2013) 190:2327–34. doi:10.4049/jimmunol.1200757
103. Bourgeois C, Bour JB, Aho LS, Pothier P. Prophylactic administration of a complementarity-determining region derived from a neutralizing monoclonal antibody is effective against respiratory syncytial virus infection in BALB/c mice. J Virol (1998) 72:807–10.
104. Williams WV, Kieber-Emmons T, VonFeldt J, Greene MI, Weiner DB. Design of bioactive peptides based on antibody hypervariable region structures. Development of conformationally constrained and dimeric peptides with enhanced affinity. J Biol Chem (1991) 266:5182–90.
105. Barrios Y, Jirholt P, Ohlin M. Length of the antibody heavy chain complementarity determining region 3 as a specificity-determining factor. J Mol Recognit (2004) 17:332–8. doi:10.1002/jmr.679
106. Kuroda D, Shirai H, Kobori M, Nakamura H. Structural classification of CDR-H3 revisited: a lesson in antibody modeling. Proteins (2008) 73:608–20. doi:10.1002/prot.22087
107. Sela-Culang I, Alon S, Ofran Y. A systematic comparison of free and bound antibodies reveals binding-related conformational changes. J Immunol (2012) 189:4890–9. doi:10.4049/jimmunol.1201493
108. Sedrak P, Hsu K, Mohan C. Molecular signatures of anti-nuclear antibodies – contribution of heavy chain framework residues. Mol Immunol (2003) 40:491–9. doi:10.1016/S0161-5890(03)00223-2
109. Xiang J, Sha Y, Jia Z, Prasad L, Delbaere L. Framework residue-71 and residue-93 of the chimeric B72.3 antibody are major determinants of the conformation of heavy-chain hypervariable loops. J Mol Biol (1995) 253:385–90. doi:10.1006/jmbi.1995.0560
110. Verhoeyen M, Milstein C, Winter G. Reshaping human antibodies: grafting an antilysozyme activity. Science (1988) 239:1534–6. doi:10.1126/science.2451287
111. Kettleborough CA, Saldanha J, Heath VJ, Morrison CJ, Bendig MM. Humanization of a mouse monoclonal antibody by CDR-grafting: the importance of framework residues on loop conformation. Protein Eng (1991) 4:773–83. doi:10.1093/protein/4.7.773
112. Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A (1992) 89:4285–9. doi:10.1073/pnas.89.10.4285
113. Baca M, Presta L, O’Connor S, Wells J. Antibody humanization using monovalent phage display. J Biol Chem (1997) 272:10678–84. doi:10.1074/jbc.272.16.10678
114. Rodríguez-Rodríguez ER, Ledezma-Candanoza LM, Contreras-Ferrat LG, Olamendi-Portugal T, Possani LD, Becerril B, et al. A single mutation in framework 2 of the heavy variable domain improves the properties of a diabody and a related single-chain antibody. J Mol Biol (2012) 423:337–50. doi:10.1016/j.jmb.2012.07.007
115. Chiu WC, Lai YP, Chou MY. Humanization and characterization of an anti-human TNF-alpha murine monoclonal antibody. PLoS One (2011) 6:e16373. doi:10.1371/journal.pone.0016373
117. Schroeder H, Hillson J, Perlmutter R. Structure and evolution of mammalian VH families. Int Immunol (1990) 2:41–50. doi:10.1093/intimm/2.1.41
118. Amit A, Mariuzza R, Phillips S, Poljak R. 3-Dimensional structure of an antigen-antibody complex at 2.8 a resolution. Science (1986) 233:747–53. doi:10.1126/science.2426778
119. Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol (1992) 224:487–99. doi:10.1016/0022-2836(92)91010-M
120. Tramontano A, Chothia C, Lesk A. Framework residue-71 is a major determinant of the position and conformation of the 2nd hypervariable region in the VH domains of immunoglobulins. J Mol Biol (1990) 215:175–82. doi:10.1016/S0022-2836(05)80102-0
121. Holmes M, Buss T, Foote J. Structural effects of framework mutations on a humanized anti-lysozyme antibody. J Immunol (2001) 167:296–301.
122. Potter K, Hobby P, Klijn S, Stevenson F, Sutton B. Evidence for involvement of a hydrophobic patch in framework region 1 of human v4-34-encoded igs in recognition of the red blood cell I antigen. J Immunol (2002) 169:3777–82.
123. Pospisil R, Youngcooper G, Mage R. Preferential expansion and survival of B-lymphocytes based on VH framework-1 and framework-3 expression: “positive” selection in appendix of normal and VH-mutant rabbits. Proc Natl Acad Sci USA (1995) 92:6961–5. doi:10.1073/pnas.92.15.6961
124. Capra J, Kehoe J. Variable region sequences of 5 human immunoglobulin heavy chains of VH3 subgroup: definitive identification of four heavy chain hypervariable regions. Proc Natl Acad Sci USA (1974) 71:845–8. doi:10.1073/pnas.71.3.845
125. Banfield M, King D, Mountain A, Brady R. V-L:V-H domain rotations in engineered antibodies: crystal structures of the Fab fragments from two murine antitumor antibodies and their engineered human constructs. Proteins (1997) 29:161–71. doi:10.1002/(SICI)1097-0134(199710)29:2<161::AID-PROT4>3.0.CO;2-G
126. Nakanishi T, Tsumoto K, Yokota A, Kondo H, Kumagai I. Critical contribution of VH-VL interaction to reshaping of an antibody: the case of humanization of anti-lysozyme antibody, HyHEL-10. Protein Sci (2008) 17:261–70. doi:10.1110/ps.073156708
127. Stanfield R, Takimotokamimura M, Rini J, Profy A, Wilson I. Major antigen-induced domain rearrangements in an antibody. Structure (1993) 1:83–93. doi:10.1016/0969-2126(93)90024-B
128. Tan P, Sandmaier B, Stayton P. Contributions of a highly conserved VHNL hydrogen bonding interaction to scFv folding stability and refolding efficiency. Biophys J (1998) 75:1473–82. doi:10.1016/S0006-3495(98)74066-4
129. Essen L, Skerra A. The de-novo design of an antibody combining site – crystallographic analysis of the V-L domain confirms the structural model. J Mol Biol (1994) 238:226–44. doi:10.1006/jmbi.1994.1284
130. Masuda K, Sakamoto K, Kojima M, Aburatani T, Ueda T, Ueda H. The role of interface framework residues in determining antibody V-H/V-L interaction strength and antigen-binding affinity. Febs J (2006) 273:2184–94. doi:10.1111/j.1742-4658.2006.05232.x
131. Torres M, Casadevall A. The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol (2008) 29: 91–7. doi:10.1016/j.it.2007.11.004
132. Casadevall A, Janda A. Immunoglobulin isotype influences affinity and specificity. Proc Natl Acad Sci U S A (2012) 109:12272–3. doi:10.1073/pnas.1209750109
133. Janda A, Casadevall A. Circular dichroism reveals evidence of coupling between immunoglobulin constant and variable region secondary structure. Mol Immunol (2010) 47:1421–5. doi:10.1016/j.molimm.2010.02.018
134. Dam TK, Torres M, Brewer CF, Casadevall A. Isothermal titration calorimetry reveals differential binding thermodynamics of variable region-identical antibodies differing in constant region for a univalent ligand. J Biol Chem (2008) 283:31366–70. doi:10.1074/jbc.M806473200
135. Pritsch O, Hudry-Clergeon G, Buckle M, Petillot Y, Bouvet JP, Gagnon J, et al. Can immunoglobulin C(H)1 constant region domain modulate antigen binding affinity of antibodies? J Clin Invest (1996) 98:2235–43. doi:10.1172/JCI119033
136. Pritsch O, Magnac C, Dumas G, Bouvet JP, Alzari P, Dighiero G. Can isotype switch modulate antigen-binding affinity and influence clonal selection? Eur J Immunol (2000) 30(12):3387–95. doi:10.1002/1521-4141(2000012)30:12<3387::AID-IMMU3387>3.0.CO;2-K
137. Torres M, May R, Scharff MD, Casadevall A. Variable-region- identical antibodies differing in isotype demonstrate differences in fine specificity and idiotype. J Immunol (2005) 174:2132–42.
138. Torres M, Fernández-Fuentes N, Fiser A, Casadevall A. The immunoglobulin heavy chain constant region affects kinetic and thermodynamic parameters of antibody variable region interactions with antigen. J Biol Chem (2007) 282:13917–27. doi:10.1074/jbc.M700661200
139. McLean GR, Torres M, Elguezabal N, Nakouzi A, Casadevall A. Isotype can affect the fine specificity of an antibody for a polysaccharide antigen. J Immunol (2002) 169:1379–86.
140. Cooper LJ, Shikhman AR, Glass DD, Kangisser D, Cunningham MW, Greenspan NS. Role of heavy chain constant domains in antibody-antigen interaction. Apparent specificity differences among streptococcal IgG antibodies expressing identical variable domains. J Immunol (1993) 150:2231–42.
141. McCloskey N, Turner MW, Steffner P, Owens R, Goldblatt D. Human constant regions influence the antibody binding characteristics of mouse-human chimeric IgG subclasses. Immunology (1996) 88:169–73. doi:10.1111/j.1365-2567.1996.tb00001.x
142. Michaelsen TE, Ihle Ø, Beckstrøm KJ, Herstad TK, Sandin RH, Kolberg J, et al. Binding properties and anti-bacterial activities of V-region identical, human IgG and IgM antibodies, against group B Neisseria meningitidis. Biochem Soc Trans (2003) 31:1032–5. doi:10.1042/BST0311032
143. Liu F, Bergami PL, Duval M, Kuhrt D, Posner M, Cavacini L. Expression and functional activity of isotype and subclass switched human monoclonal antibody reactive with the base of the V3 loop of HIV-1 gp120. AIDS Res Hum Retroviruses (2003) 19:597–607. doi:10.1089/088922203322230969
144. Tudor D, Yu H, Maupetit J, Drillet AS, Bouceba T, Schwartz-Cornil I, et al. Isotype modulates epitope specificity, affinity, and antiviral activities of anti-HIV-1 human broadly neutralizing 2F5 antibody. Proc Natl Acad Sci U S A (2012) 109:12680–5. doi:10.1073/pnas.1200024109
145. Torosantucci A, Chiani P, Bromuro C, De Bernardis F, Palma AS, Liu Y, et al. Protection by anti-beta-glucan antibodies is associated with restricted beta-1,3 glucan binding specificity and inhibition of fungal growth and adherence. PLoS One (2009) 4:e5392. doi:10.1371/journal.pone.0005392
146. Xia Y, Janda A, Eryilmaz E, Casadevall A, Putterman C. The constant region affects antigen binding of antibodies to DNA by altering secondary structure. Mol Immunol (2013) 56:28–37. doi:10.1016/j.molimm.2013.04.004
147. Adachi M, Kurihara Y, Nojima H, Takeda-Shitaka M, Kamiya K, Umeyama H. Interaction between the antigen and antibody is controlled by the constant domains: normal mode dynamics of the HEL-HyHEL-10 complex. Protein Sci (2003) 12:2125–31. doi:10.1110/ps.03100803
148. Xia Y, Pawar RD, Nakouzi AS, Herlitz L, Broder A, Liu K, et al. The constant region contributes to the antigenic specificity and renal pathogenicity of murine anti-DNA antibodies. J Autoimmun (2012) 39:398–411. doi:10.1016/j.jaut.2012.06.005
149. Oda M, Kozono H, Morii H, Azuma T. Evidence of allosteric conformational changes in the antibody constant region upon antigen binding. Int Immunol (2003) 15:417–26. doi:10.1093/intimm/dxg036
150. Piekarska B, Drozd A, Konieczny L, Król M, Jurkowski W, Roterman I, et al. The indirect generation of long-distance structural changes in antibodies upon their binding to antigen. Chem Biol Drug Des (2006) 68:276–83. doi:10.1111/j.1747-0285.2006.00448.x
151. Horgan C, Brown K, Pincus SH. Effect of H-chain V-region on complement activation by immobilized immune-complexes. J Immunol (1992) 149:127–35.
152. Vidarte L, Pastor C, Mas S, Blazquez AB, de los Rios V, Guerrero R, et al. Serine 132 is the C3 covalent attachment point on the CH1 domain of human IgG1. J Biol Chem (2001) 276(41): 38217–23.
153. Braden BC, Poljak RJ. Structural features of the reactions – between antibodies and protein antigens. FASEB J (1995) 9:9–16.
154. Pellequer JL, Chen SW, Roberts VA, Tainer JA, Getzoff ED. Unraveling the effect of changes in conformation and compactness at the antibody V-L-V-H interface upon antigen binding. J Mol Recog (1999) 12:267–75. doi:10.1002/(SICI)1099-1352(199907/08)12:4<267::AID-JMR465>3.3.CO;2-0
Keywords: antibody, CDRs, antigen, paratope, epitope, framework, constant domain
Citation: Sela-Culang I, Kunik V and Ofran Y (2013) The structural basis of antibody-antigen recognition. Front. Immunol. 4:302. doi: 10.3389/fimmu.2013.00302
Received: 04 August 2013; Accepted: 12 September 2013;
Published online: 08 October 2013.
Edited by:
Michal Or-Guil, Humboldt University Berlin, GermanyReviewed by:
Gur Yaari, Yale University, USACopyright: © 2013 Sela-Culang, Kunik and Ofran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanay Ofran, The Goodman Faculty of Life Sciences, Bar Ilan University, Ramat-Gan 52900, Israel e-mail:eWFuYXlAb2ZyYW5sYWIub3Jn
†Inbal Sela-Culang and Vered Kunik have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.