

- 1Faculté de Médecine, Sorbonne Paris Cité, Université Paris Descartes, Paris, France
- 2Unité 1013, Institut National de la Santé et de le Recherche Médicale, Hôpital Necker, Paris, France
- 3Immunoregulation and Immunopathology Team, INEM, Paris, France
- 4Breast Cancer Biology Research Unit, Institut de Recherches Cliniques de Montreal (IRCM), McGill University, Université de Montreal, Montreal, QC, Canada
The influence of CD4+CD25+Foxp3+ regulatory T-cells (Tregs) on cancer progression has been demonstrated in a large number of preclinical models and confirmed in several types of malignancies. Neoplastic processes trigger an increase of Treg numbers in draining lymph nodes, spleen, blood, and tumors, leading to the suppression of anti-tumor responses. Treg-depletion before or early in tumor development may lead to complete tumor eradication and extends survival of mice and humans. However this strategy is ineffective in established tumors, highlighting the critical role of the early Treg-tumor encounters. In this review, after discussing old and new concepts of immunological tumor tolerance, we focus on the nature (thymus-derived vs. peripherally derived) and status (naïve or activated/memory) of the regulatory T-cells at tumor emergence. The recent discoveries in this field suggest that the activation status of Tregs and effector T-cells (Teffs) at the first encounter with the tumor are essential to shape the fate and speed of the immune response across a variety of tumor models. The relative timing of activation/recruitment of anti-tumor cells vs. tolerogenic cells at tumor emergence appears to be crucial in the identification of tumor cells as friend or foe, which has broad implications for the design of cancer immunotherapies.
Tumor Recognition by the Immune System: Ignorance, Surveillance, and Tolerance
A now receding branch of tumor immunology literature favors the view that antigens expressed by many tumors would be ignored by the immune system due to inadequate antigen presentation (1–3). Hewitt, after examining the immunogenicity of many tumor cell lines, concluded that only virus-induced tumors are likely to induce an immune response against them (4). Beyond this founding observation, and the reports that many tumor cells do not express MHC proteins, several groups have found functional alterations of the proteasome (5, 6) and TAP (7) in tumor cells and APCs (8), reducing tumor visibility to the immune system. In contrast, many studies have proven that the immune system is activated in the presence of spontaneous tumors (9–12), revitalizing the concept of immunosurveillance.
This concept, first proposed in 1909 (13), was formally defined in 1957 when – based on the findings that the immune system can specifically recognize and reject tumor cells in a chemically induced murine sarcoma model (14) – Burnet proposed that the immune system may prevent tumor development by recognizing antigens absent in normal tissues (15). According to his theory, the immune reactions against tumor antigens expressed by neoplastic cells generally eliminate them at an early stage before any clinical hint of their existence, and frank tumors can grow only after escaping the immune system by diminishing their immunogenicity. The existence of tumor-specific antigens was indeed confirmed in the 1960s by Klein (16). Later, tumors developing in immunodeficient mice were proven to be more immunogenic than tumors developing in immunoproficient mice (17), suggesting that tumors undergo selection by the immune system.
However, in other mouse models, immunodeficiency did not promote tumor development (18–20), and the ability of tumor cells to diminish the expression of their most immunogenic epitopes by adaptation or selection, also called cancer immunoediting, has been questioned (21). The involvement of innate immunity in tumor surveillance was further explored in studies on natural killer cells (NKs), and has produced similar arguments pro (22–24) and con (20, 25, 26). The conflicting accounts on the role of ongoing anti-tumor surveillance produced in animal models are mirrored by findings in clinical studies that measured the risk of tumor development in patients with immunodeficiencies. Numerous publications have observed a significant increase of cancer occurrence in immunodeficient patients, but at the same time there is no study reporting an “explosion” of cancer cases in these patients. For example, in a study of 2005 on a very large number of immunosuppressed renal transplantation recipients, Hollenbeak et al. observed that of the 89,786 patients who underwent transplantation, 246 patients developed melanoma, with an age-adjusted incidence rate of 55.9 diagnoses per 100,000 individuals. This represented an increase in age-adjusted, standardized risk that was 3.6 times greater than the general population (27). Thus, while such studies support a real role of immunosuppression in promoting cancer susceptibility, the risk of developing a melanoma in the absence of a functional immune system, if increased and non-negligible, is still only 0.056%. One interpretation is that tumor development in the absence of immune system is still a rare event, another interpretation is that the immunodeficient state in patients mostly increases risk of cancers of viral etiology, and that the impact of immunosurveillance on preventing non-viral human cancer may actually be relatively minor (28–31). On the other hand, in already established tumors, presence of tumor-infiltrating lymphocytes is correlated with improved survival (32), and intra-tumoral CD8 T-cells infiltration is associated with delayed recurrence and extended survival in oncologic patients (33). A consensus is that the immune surveillance may guard against cancer under certain conditions, but the precise nature of these conditions is unclear.
The first clue that immune tolerance might be a part of the equation came from the works of Nishizuka and Sakakura. While investigating the role of the thymus in tumor immunity in mice susceptible to mouse mammary tumor virus (MMTV)-induced cancer, they observed that neonatal thymectomy at 3 days of age (day 3 nTx) resulted in reduced frequency of breast cancer in tumor-prone (C3H/HeMs × 129/J)F1 females (34), suggesting that cells produced by the thymus after day 3 may protect the tumor. In the following studies, they also looked at tumor development in extra-mammary tissues. There was no increase in the lung and liver tumors after neonatal thymectomy, but the authors reported increased ovarian, lymphoreticular, and pituitary tumor development (35). A notable point of these studies was the discovery that mammary gland development in day 3 nTx female mice was delayed (34) and that mice became infertile secondary to the development of oophoritis (36). At the time, Sakakura and Nishizuka attributed these features to an endocrine role of the thymus, although it is now known to be the manifestation of T-cell-mediated autoimmunity, which paved the road to the discovery of thymic-derived suppressor T-cells, and active tolerance to the tumors.
Treg-Mediated Tumor Surveillance: Expand to Reign
T-cells capable of suppressing the rejection of implanted tumors were first observed in the late 1970s (37–40). These reports remained underappreciated as were most findings pointing to the existence of suppressor cells, caused, in part, by lack of suppressor-specific cellular markers. The doubts have disappeared only in the 1990s, when Sakaguchi, a former student of Nishizuka demonstrated that CD4+CD25+ T-cells, baptized “regulatory,” were responsible for the induction of dominant immune tolerance to tumors. First, the transplantable tumors grew in immunodeficient hosts transferred with whole splenocytes, but were rejected in hosts that have received splenocytes depleted of CD25+ cells (41). Second, the tumors were rejected following preventive treatments with anti-CD25 antibody (42). In both cases, the presence of CD25+ cells inhibited the anti-tumor immune response and their removal led to the complete elimination of the tumor.
In a short time, an impressive number of reports confirmed the association between malignant tumors and the regulatory T-cells (Tregs). Clinical studies have shown that CD4+CD25+ cells are often present within the tumor mass, and have reported a link between a presence of a tumor and an increase in the proportion and/or the number of CD4+CD25+ Tregs in the blood (43–47). However some results were more heterogeneous depending of the cancer type, and in some studies, no Treg increase was observed (48). Moreover, sometimes the observed proportion of Tregs seems falsely increased by the reduction of the absolute number of CD4+CD25− effector cells (Teffs) (49). Regardless of its causes, an important question was whether the observed increase in Tregs is informative for prognosis. Animal models have argued that Tregs have pro-tumorigenic effects (see above), and tumor volume appears directly correlated to the number of Tregs present in the secondary lymphoid organs in several models (50–52).
Starting with the report correlating presence of Tregs within the tumor infiltrate and a poor survival prognosis in patients with ovarian cancer (Curiel, 200,456), the majority of studies have agreed that an increase in Tregs/Teffs ratio or in an absolute Treg number confers a poor prognosis in cancer patients [see below, and in these recent reviews (48, 53, 54)]. Yet there are instances in which Treg increase is actually linked to a good prognosis, for example in lymphomas (55, 56) and in colorectal cancer (57–59). The reasons for this discrepancy appear to depend on the special nature of these cancers, in which inflammation may promote tumor growth if not regulated by Tregs, but may also be related to a difference in the origin of cells with Treg characteristics observed in individual malignancies.
Concerning the causes of the tumor-induced increase in Tregs, the literature describes several mechanisms: (i) Preferential recruitment of existing thymic-derived Tregs (tTregs), which may be mediated, in part, by chemokines produced by tumors, such as CCL22, that attracts regulatory T-cells, which predominantly express the cognate agonist receptor (60, 61). However, as effector lymphocytes express chemokine receptors as well, chemokine secretion alone cannot explain the preferential recruitment of Tregs to tumor sites (62, 63). The two alternative explanations are (ii) fate conversion – de novo induction of peripheral Treg (pTregs) out of effector T-cells; and (iii) clonal expansion – cytokine and/or antigen-induced proliferation in the periphery of tTregs. Given the vast variety of tumor systems in which all these scenarios have been explored, it is conceivable that the nature of the transforming event, or the tissue of origin of the tumor may determine the specific biological mechanism leading to an increase in Tregs.
Peripherally and Thymic-Derived Tregs in Cancer
Discovered in the early 2000s in mice (64) and in humans (65), pTregs quickly became the subject of active investigation in tumor immunology, generating evidence both for and against their role in tumor tolerance. Adoptive transfer of CD4+CD25− T-cells in mice challenged with either colon cancer or B cell lymphoma resulted in induction of CD25 expression in a significant proportion of donor Teffs, as well as appearance of Foxp3 transcript (66, 67).
A major line of research pursued a possible instructive role of TGF-ß, a signaling molecule with pleiotropic functions in both immunity and cancer, and in the conversion of CD4+CD25− T-cells to pTreg cells (65). TGF-ß acts by binding to the type II TGF-ß receptor (TGF-ßRII), which is constitutively active as a serine/threonine kinase (68, 69). A CD4+ cell-restricted blockade of TGF-ß signaling in mice expressing a dominant negative version of the receptor resulted in eradication of TGF-ß expressing lymphoma or metastatic B16F10 melanoma (70) and has established a firm link between TGF-ß and tumor immune tolerance. In part, such a blockade may impair the pro-tumorigenic conversion to pTregs. Indeed, an in vitro study has implied that TGF-ß expressing kidney or prostate tumor cells can stimulate the pro-tumorigenic conversion to pTregs (71). Accordingly, the anti-TGF-ß treatment of mice injected with these tumor cells resulted in fewer tumor nodules; but the in vivo experiments did not exclude a possibility of a direct effect of TGF-ß-blockade on RENCA and TRAMP-C2 cell growth. Moreover, pancreatic tumor-derived TGF-ß was shown to activate Foxp3 expression in tumor cells themselves (72). The functional significance of this upregulation is unclear, as in the tumor cells the Foxp3 transcription factor remains restricted to the cytoplasm, contrary to nuclear localization in Tregs, but it may result in a lower immunogenicity of the tumor, as siRNA-mediated inhibition of Foxp3 expression in tumor cells may shift their cytokine expression pattern toward IL-6 and IL-9 secretion (72).
The effect of TGF-ß on the conversion in vivo in tumor-bearing mice was addressed more recently using adoptive transfer of CD4+25−Foxp3−T-cells into Rag−/− mice. In the presence of a TGF-ß-producing pancreatic Pan02 tumor, the transferred T-cells converted into Foxp3+ pTregs, but few FoxP3+-converted cells were found when mice were transplanted with a TGF-ß-negative esophageal Eso2 tumors (73). As predicted, the induction of cells with Tregs characteristics in Pan02-bearing mice was blocked by systemic injection of an anti-TGF-ß-antibody. This finding mirrors the clinical situation, when increase of Foxp3+ Tregs is observed in patients with a TGF-ß-producing pancreatic adenocarcinoma but not in those with a TGF-ß-negative esophageal tumor (74). Similarly, in non-small cell lung cancer patients, TGF-ß plasma concentrations directly correlated with the frequency of circulating Tregs (75).
As stated above, the spectrum of biological effects of TGF-ß is wide, and is spread beyond the pTreg induction to regulate other Teff responses. For example, anti-TGF-ß treatment significantly and synergistically improved vaccine efficacy as measured by a reduction in growth of the TC1 lung tumor allografts, but anti-TGF-ß alone without vaccine had no impact (76). Moreover, anti-TGF-ß treatment did not affect Treg numbers in lymph nodes and tumors, or their function (76). The resultant synergistic protection induced by anti-TGF-ß plus vaccine combined treatment was likely mediated by CD8+ T-cells since anti-CD8 treatment completely abrogated this effect (76). These results, of course, do not exclude a role for peripherally derived-CD4+ pTregs, but greatly diminish the chances that CD4+ pTregs are the sole culprit behind the TGF-ß effects on tumor tolerance.
Overall, the role of TGF-ß in Treg maintenance is mixed, as it inhibits Teffs and Treg cell proliferation, but is important for tTreg and pTreg survival in the periphery (77). In fact, the nature of TGF-ß/Treg interactions may be more complex than a direct conversion scenario would suggest. For example, a mammary tumor cell line, 4T1, can induce recruitment of TGF-ß-producing Gr-1+CD11b+ monocytes (78), and a mouse melanoma and a rat colon tumor were shown to convert dendritic cells (DCs) into the TGF-ß-producing cells, which then led to Treg proliferation (79), possibly through a GILZ-dependent mechanism (80). A similar hierarchy of APC/Treg exchange has been clearly demonstrated in colitis. There, DC-produced TGF-ß was shown to be critical to avoid colitis due to its Treg inducing power (81). In this paper, the Sheppard team showed that DCs lacking the TGF-β-activating integrin αvβ8 failed to induce Tregs in vitro, and that mice with conditional deletion of αvβ8 in DCs presented reduced proportions of Treg cells in colonic tissue. If It should not be excluded that effector cell expansion may contributes to this observed reduction in the fractional number of Treg cells in the colon, these in vitro and in vivo results reinforce observations that DCs are essential in the maintenance of both pTreg and tTreg cells in the periphery (82–85).
A major complication that weakens the accounts of de novo pTreg induction after adoptive transfer of Teffs in tumor-bearing mice is that the CD4+CD25− Teffs subset purified in the majority of the conversion experiments of the pioneer articles, contains around 2% of CD4+CD25−Foxp3+ T-cells that exhibit suppressive functions (86) and can gain CD25 expression and expand after stimulation (87, 88). The experimental approaches based on the sorting of CD25−T-cells do not provide supportive evidence for a de novo induction of pTregs, and do not exclude a possibility of tumor-driven activation and expansion of CD4+CD25−Foxp3+ thymus-derived tTregs. Accordingly, experiments using Teffs transfer from donor mice expressing a Foxp3-reporter indicate that generation of peripherally derived FoxP3+ pTregs out of GFP− Teffs within tumors is inefficient and that tumor-infiltrating GFP+FoxP3+ tTregs are highly stable and do not readily convert back to FoxP3− T-cells contrary to pTregs (89). Some authors suggest that proliferating Helios+ Treg cells are a major population in tumors (90), which may be interpreted against pTregs conversion in tumors, Helios being a tTreg marker (91, 92). But Helios may be upregulated in peripherally derived pTregs after activation by DCs (93).
Another line of evidence questioning the primary role of pTregs in tumor tolerance comes from a recent paper describing the tolerogenic response against the prostate-associated MJ23 self-antigen expressed by prostate tumors induced by an SV40 TAg transgene. On an immunoproficient background, these tumors are infiltrated with MJ23 tumor-specific Tregs, but no MJ23 tumor-specific Tregs were found in tumors that have developed in Aire−/−mice. As Aire is important for tTreg development but dispensable for pTreg induction, these findings indicate that the tumor-infiltrating Treg cells specific for the highly immunogenic MJ23 are principally of the thymic origin (94).
Overall, there is little doubt that pTregs may appear from CD25− subsets, probably from recent tumor emigrant cells (95), in the presence of tumors under certain experimental conditions. Whether this subset plays a substantial role during spontaneous tumor development, is less clear. The difficulty is best illustrated by recalling the original report of Sakaguchi, showing that the immunodeficient mice reconstituted with CD25 depleted splenocytes acquired efficient anti-tumor responses in various cancer models (41). Stated otherwise, any spontaneous pTreg conversion that may occur in this experimental setup does not prevent clearance of the transplantable tumors.
Activated/Memory Tregs in the Early Immune Response to Cancer
When we were studying the kinetic of early immune responses in various models of cancer by adoptive transfer of CFSE-labeled T-cells, we were struck by the fact that Treg response was not a late event, secondary to the activation of IL-2-releasing anti-TAA effector T-cells (Tumor-associated antigen-specific Teffs), but was actually a very early event, preceding any Teffs activation (52). Such a rapid tumor-specific response of the immune system was counterintuitive in a model of primary tumor exposure, but it bore well with the earlier reports that tumor growth can activate immune cells very quickly.
In 1975, Bhatnagar and colleagues have measured ex vivo thymidine incorporation by splenocytes and detected substantial cellular immune responses as early as 1–2 days after i.p. injection of methylcholanthrene-induced fibrosarcoma cells (96). The intensity and rapidity of the cellular response was dependent of the number of cells injected and was always followed by a gradual loss of cellular reactivity against the tumor cells. The progressive loss of immune recognition for tumor cells correlated with progression of tumor growth (96). These observations were confirmed by Berendt and North, who provided evidence supporting the hypothesis that immunity to tumors declines with time as a result of T-cell-mediated immunosuppression (40). More recently, in several injected tumor models (B16 melanoma, 4T1 carcinoma, AB1 mesothelioma, and more) and in an inducible-oncogene-driven breast tumor model, an increase in T-cell division was detected as soon as 2 days after the emergence of the tumor by measure of CFSE dilution as well as BrdU incorporation (52). But this early response was restricted to CD4+CD25+Foxp3+ regulatory T-cells, and it appeared to precede the response of conventional T-cells (Figure 1). The responding Treg cells were specific to the antigens, which, although expressed by tumors, were already present in mice before tumor appearance (Figure 1). In other words, the tumor-derived antigens able to stimulate Tregs were self-antigens. Indeed, no Treg expansion was observed against tumors that were not bearing a cognate self-antigen recognized by the transferred tTregs. These observations confirmed previous observations that the self-specific Tregs suppress anti-tumor responses (97, 98), although it did not exclude a possibility that Tregs specific for tumor neoantigens may also participate to the induction of tolerance to the tumor (99, 100). Recently, it was demonstrated that Aire-mediated expression of peripheral tissue antigens drives thymic development of a subset of organ-specific tTregs, which are likely recruited by tumors developing within the associated organ (94).
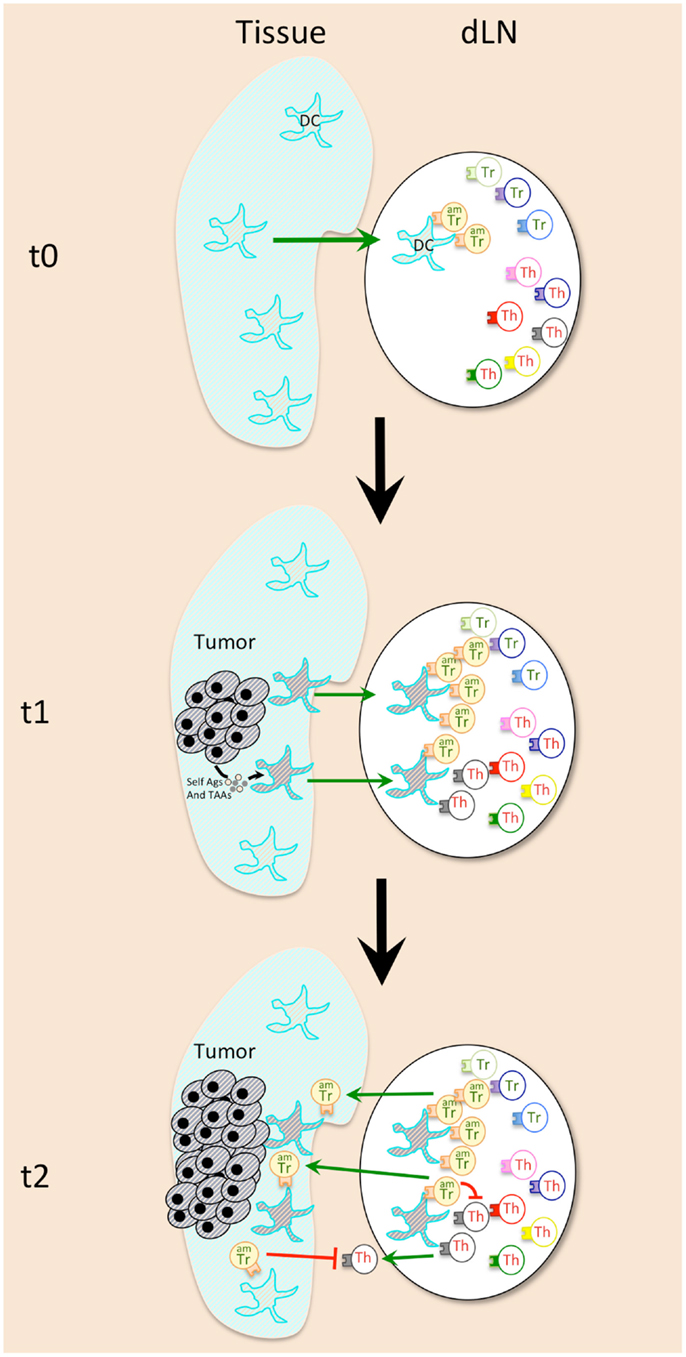
Figure 1. Early events during cancer emergence lead to immune tolerance against tumor. Activated memory Tregs (AmTregs or amTr, beige lymphocytes) are the first to be stimulated by the presence of the tumor (gray round-shaped cells) via recognition of self-Ag presented by dendritic cells (DCs, star-shaped cells) coming from the tumor site (t1). AmTreg will then proliferate faster than TAA-specific Teffs (Th, gray lymphocytes) that are naïve (or have already been suppressed at the steady state). AmTreg will then inhibit either Teff activation, proliferation, migration, and function either/or DCs presentation and costimulation (t2).
Concerning the APCs that may be responsible for presentation of the tumor self-Ags to Tregs, the good candidates are tissue DCs, which are known to be especially potent in stimulating and maintaining the actively dividing Treg pool (83). Indeed, DCs from tumor-bearing mice were shown to recruit Tregs and to favor their proliferation in the draining lymph nodes (79) (Figure 1). These DCs may present antigens derived from proteins secreted by the live tumor cells, or those derived from tumor cells that die during transformation-induced apoptosis. Of note, microvesicles that are released by tumors and may be captured by DCs for tumor antigen presentation (101) appear to have a role in Treg expansion and activation (102) (Figure 1). Moreover, Treg subset expands after adoptive transfer in MHCII+/+ but not in MHCII−/− tumor-bearing mice, which proves that cytokines released in the tumor-bearing mice are not sufficient by themselves to favor Treg recruitment, and that antigen-driven proliferation is mandatory (83).
Isolation of Tregs with activated/memory vs. naïve phenotype from tumor-free mice followed by adoptive transfer to tumor-bearing mice showed that the initial proliferation of Tregs in tumor-draining lymph nodes was confined to the pool of activated/memory Tregs (amTregs) present in naive mice, (52). These cells were previously characterized as an activated/memory subtype of Tregs, constantly stimulated by self-antigens at the steady state (103). These amTregs are phenotypically and functionally distinct from naïve Tregs (103, 104), and are highly potent at suppressing autoimmune responses (105, 106). The intensity of the early anti-tumor Treg response is thus explained by their self-specificity and activated/memory status.
The early dividing cells described in tumor-bearing mice since 1975 are thus the tolerogenic amTregs cells, a conclusion that is further confirmed by observing tumor rejection following short-term depletion of proliferating immune cells via early administration of anti-mitotic hydroxyurea (HU) or cyclophosphamide (CY) in mice bearing HU/Cy-resistant tumors (50, 52, 107, 108). The early administration of these drugs has a much stronger effect than the late administration, once again suggesting that the immune cells that divide early in the presence of an emerging tumor favor tolerance. Accordingly, a recent analysis of Treg subsets in Her2/Neu-expressing mammary tumor-bearing mice revealed the existence of a Cy-sensitive CD4+Foxp3+CD25+ subset with tumor-seeking migratory phenotype, characteristic of amTregs, and capable of high avidity T-cell suppression (109). In addition, the tumor-infiltrating Foxp3+ T-cells express high levels of memory/tumor-associated CCR8 and CXCR4 receptors, and antigen priming is required for the induction of this trafficking receptor phenotype. Thus only antigen-primed, but not antigen-inexperienced naive, FoxP3+ T-cells can efficiently migrate into tumors (89). Of course, the effector T-cells also start to proliferate after an adoptive transfer into tumor-bearing mice, but with a primary kinetics that is much slower (9–12 days) than that observed in Treg subset (2–4 days) (83). This delay appears to be sufficient for the establishment of a stable immunosuppressive environment.
To test if tolerance to tumors was due to the Treg/Teffs imbalance induced by the delays between their respective activation/expansion, we adoptively transferred high numbers of HA-specific Teffs in mice bearing HA-expressing tumor cells. We observed complete remission in mice adoptively transferred with antigen-experienced HA-specific Teffs (52). Complete regression was also found (i) in secondary-challenged mice cured from first tumor challenge by temporary Treg-depletion (42, 52) and (ii) in tumor-pre-immunized mice (52, 110–112). The activated/memory Teffs, are able to eradicate very efficiently even poorly immunogenic tumors like B16 melanoma (110, 111), regardless of the number of Tregs present in the mice (52). Even highly suppressive adoptively transferred tumor-specific Tregs are not able to reverse the anti-tumor memory response (52). The resistance of activated/memory Teffs (amTeffs) to Treg-mediated suppression demonstrated was also observed in other conditions like allograft rejection (113) and autoimmune inflammation (114). Nishikawa and colleagues also observed that CD45RO+ but not CD45RA+ tumor-specific CD4 T-cells from cancer patients were resistant to Treg suppression (115). This resistance could be due to the fact that activated Tregs can downregulate expression of costimulation molecules by DC (116), but activation/function of amTeffs is much less dependent on costimulation than that of naive T-cells (117). Together, these observations suggest that anti-tumor amTeffs could be inherently more resistant to Tregs, and explain why detection of amTeffs correlates with good prognosis in cancer patients (118, 119).
The memory status of Treg and Teffs in early tolerance induction might be important in other settings than just cancer. Several analogies between pregnancy and cancer [reviewed in (120)] point to similarities between the early Treg responses to embryo implantation and tumor emergence. In a just-released study, we observed that early Treg responses to embryo implantation obey to the same rules as those in cancer setting: Tregs expressing markers of the amTreg subset are rapidly recruited to para-aortic conceptus-draining lymph nodes and are activated in the first days after embryo implantation in both syngeneic and allogeneic matings (121). They are also at least in part self-Ag specific, as seen in tumor emergence. Finally, pre-immunization against paternal tissue Ags results in the increase of aborted fetus frequency, and additional Treg-depletion (by anti-CD25) at the time of pre-immunization against paternal tissue Ags, leads to very high frequencies of fetus loss (121). Thus, thymic-derived amTregs appear as a driving force of tolerance to self-ambiguous tissues in the absence of infectious danger signals or pre-immunization.
One can then wonder how an immune system that protects deadly tumor cells may survive evolution. We speculate that the AmTreg tolerant response has been actually positively selected to protect allogeneic fetuses against immune rejection. Indeed, Foxp3-expressing Treg-like cells appeared in the first live-bearing animals like Tetraodon (2400 million years) (122) and zebrafish (123), both histotrophic viviparous species. Tregs were thus probably selected in part to protect allogeneic fetuses against immune rejection (121, 124), but the pro-tumorigenic activity of Tregs was not counter-selected because cancers mostly develop late in life (125) without affecting reproductive life span.
Implications for the Design of Anti-Cancer Immunotherapies
Activation kinetics and memory status of different T-cell subsets at tumor emergence are pivotal in the outcome of cancer (Figure 2) and explains why preventive immunization is more effective than therapeutic immunization and suggests (i) that preventive vaccination against cancer should be considered seriously and (ii) that therapeutic vaccination could actually worsen host tolerance to tumor antigens (126, 127). Development of vaccination strategies must include treatments aimed at Treg-depletion (128–130) or at inhibition of their function (131–133), with mandatory validation of the effect of therapeutic vaccination on the level/function of Tregs. Preventive vaccination with tumor-specific antigens presented in a context that would not stimulate amTregs will improve development of efficient amTeffs, which may mount efficient effector responses when a tumor emerges.
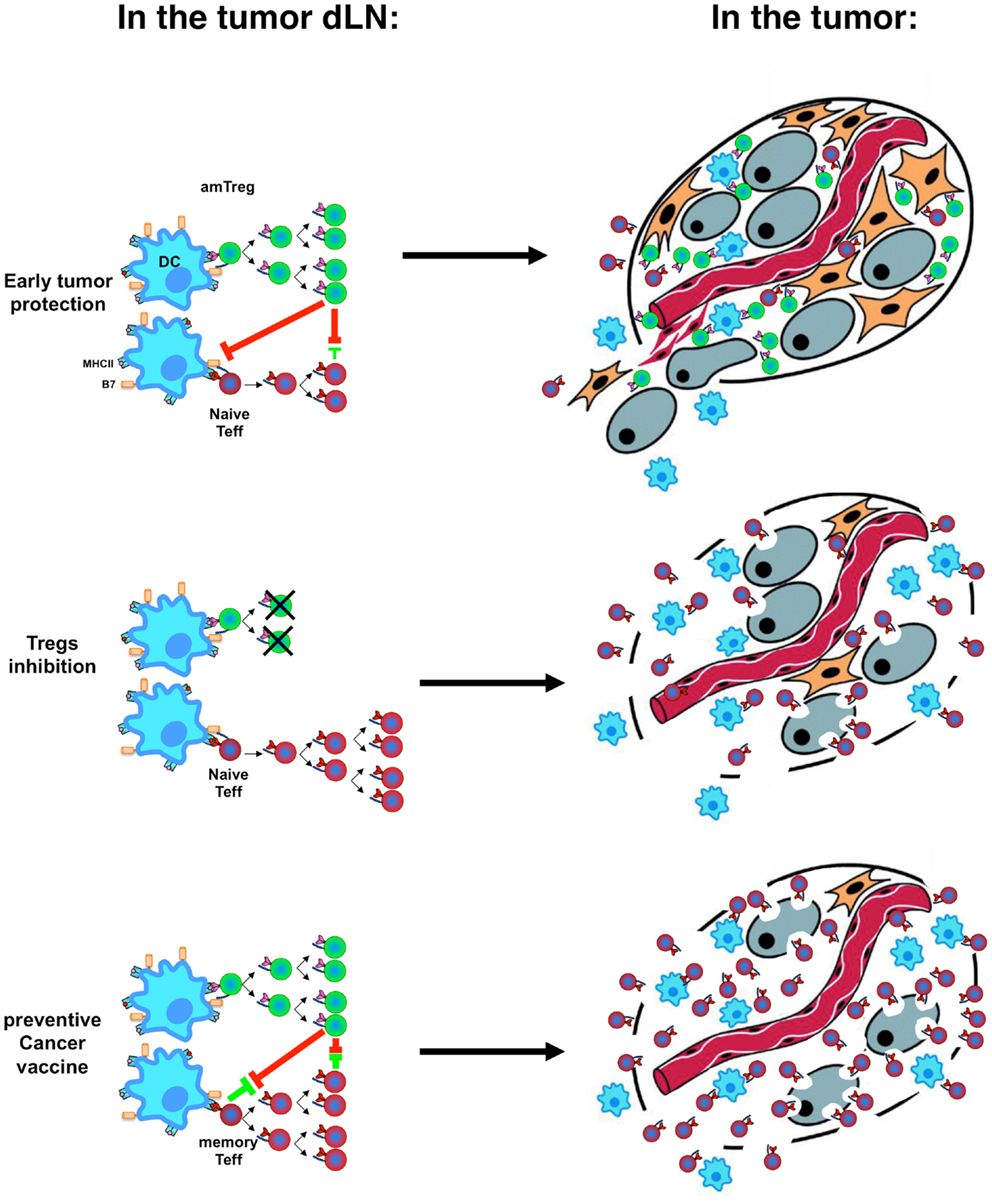
Figure 2. Immune tolerance vs. immune rejection decision process. Activation kinetics and memory status of Tregs (green) and Teffs (red) in the tumor-draining lymph nodes (dLNs, left) after stimulation by dendritic cells (DC, blue) result in the infiltration of the tumor by different cell subsets with different speed and different tumor fate (right, with tumor cells in gray).
Noteworthy, although amTeffs are resistant to Tregs, and can cure mice if provided at the time of tumor implantation, the global immunosuppressive environment established by Tregs in draining lymph modes and at the tumor site (134) can develop to a point where later therapeutic administration of amTeffs would no longer be effective (52).
Together with vaccination and beyond, ablation of Tregs in cancer patients appears to be a promising direction, especially if performed early in the course of the disease (129, 135). Nonetheless, we need to remember that the efficiency of anti-tumor responses after Treg ablation is certainly tumor- and genetic background- dependent: Treg ablation results in minimal rejection and delayed growth of B16 tumors in B6 mice, 60% rejection of 4T1 tumors in BALB/c mice (83), and close to a 100% rejection of RL♂1, MOPC-70A, and Meth A tumors in BALB/c mice (41, 42). These diverse outcomes may depend upon (a) the percentage of Treg cells in a given strain of mice in the steady state, (b) the natural ability of some mouse backgrounds to favor strong Th1 responses, and (c) the tumor-specific expression of immunodominant antigens able to trigger strong anti-tumor effector responses (136). These observations from tumor-bearing mice must be kept in mind while designing new immunotherapies strategies in cancer patients.
Altogether, these recent discoveries on the events taking place during the early tumor immune response highlight the importance of the timing and kinetic of Treg and Teff engagement, which depends on their memory status (Figures 1 and 2). In theory, this may disqualify tumor-induced pTregs from playing a substantial role during the early tumor development as they arise preferentially from naïve recent thymic emigrants (95). This does not exclude their eventual involvement in some later events that may sustain the ongoing tolerance. But the fate of the tumor is being decided early, pTregs are unlikely to have much impact in most cancers. Their late arrival in the battle and the absence of memory status puts pTregs at disadvantage during the early tumor development. In tumor immunology and beyond, the timing of engagement dictates the final outcome of an immune response.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dan Avi Landau for stimulating discussions and critical reading of the manuscript, and David Klatzmann for his concepts discussed in this review, and notably the role of Tregs in the early immune response to cancer and embryo.
References
1. Melero I, Bach N, Chen L. Costimulation, tolerance and ignorance of cytolytic T lymphocytes in immune responses to tumor antigens. Life Sci (1997) 60:2035–41. doi: 10.1016/S0024-3205(96)00686-8
2. Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M, Hengartner H, et al. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc Natl Acad Sci USA (1999) 96:2233–8. doi:10.1073/pnas.96.5.2233
3. Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature (2001) 411:1058–64. doi:10.1038/35082583
4. Hewitt HB, Blake ER, Walder AS. A critique of the evidence for active host defence against cancer, based on personal studies of 27 murine tumours of spontaneous origin. Br J Cancer (1976) 33:241–59. doi:10.1038/bjc.1976.37
5. Restifo NP, Esquivel F, Kawakami Y, Yewdell JW, Mule JJ, Rosenberg SA, et al. Identification of human cancers deficient in antigen processing. J Exp Med (1993) 177:265–72. doi:10.1084/jem.177.2.265
6. Spataro V, Norbury C, Harris AL. The ubiquitin-proteasome pathway in cancer. Br J Cancer (1998) 77:448–55. doi:10.1038/bjc.1998.71
7. Cromme FV, Airey J, Heemels MT, Ploegh HL, Keating PJ, Stern PL, et al. Loss of transporter protein, encoded by the TAP-1 gene, is highly correlated with loss of HLA expression in cervical carcinomas. J Exp Med (1994) 179:335–40. doi:10.1084/jem.179.1.335
8. Whiteside TL, Stanson J, Shurin MR, Ferrone S. Antigen-processing machinery in human dendritic cells: up-regulation by maturation and down-regulation by tumor cells. J Immunol (2004) 173:1526–34.
9. Marzo AL, Lake RA, Lo D, Sherman L, Mcwilliam A, Nelson D, et al. Tumor antigens are constitutively presented in the draining lymph nodes. J Immunol (1999) 162:5838–45.
10. Robinson BW, Scott BM, Lake RA, Stumbles PA, Nelson DJ, Fisher S, et al. Lack of ignorance to tumor antigens: evaluation using nominal antigen transfection and T-cell receptor transgenic lymphocytes in Lyons-Parish analysis – implications for tumor tolerance. Clin Cancer Res (2001) 7:811s–7s.
11. Cuenca A, Cheng F, Wang H, Brayer J, Horna P, Gu L, et al. Extra-lymphatic solid tumor growth is not immunologically ignored and results in early induction of antigen-specific T-cell anergy: dominant role of cross-tolerance to tumor antigens. Cancer Res (2003) 63:9007–15.
12. Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol (2005) 175:6169–76.
13. Ehrlich P. Ueber den jetzigen Stand der Karzinomforschung. Ned Tijdschr Geneeskd (1909) 5:273–90.
14. Foley EJ. Antigenic properties of methylcholanthrene-induced tumors in mice of the strain of origin. Cancer Res (1953) 13:835–7.
15. Burnet FM. Cancer: a biological approach. III. Viruses associated with neoplastic conditions. IV. Practical applications. Br Med J (1957) 1(5023):841–7. doi:10.1136/bmj.1.5023.841
16. Klein G. Tumor antigens. Annu Rev Microbiol (1966) 20:223–52. doi:10.1146/annurev.mi.20.100166.001255
17. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410:1107–11. doi:10.1038/35074122
18. Stutman O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science (1974) 183:534–6. doi:10.1126/science.183.4124.534
19. Stutman O. Chemical carcinogenesis in nude mice: comparison between nude mice from homozygous matings and heterozygous matings and effect of age and carcinogen dose. J Natl Cancer Inst (1979) 62:353–8.
20. Gallo-Hendrikx E, Percy D, Copps J, Mckeown B, Quinton M, McMillan I, et al. Evaluation of three lines of immunodeficient mice for the study of spontaneous metastatic tumors. APMIS (1999) 107:245–56. doi:10.1111/j.1699-0463.1999.tb01551.x
21. Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature (2005) 437:141–6. doi:10.1038/nature03954
22. Gorelik E, Wiltrout RH, Okumura K, Habu S, Herberman RB. Role of NK cells in the control of metastatic spread and growth of tumor cells in mice. Int J Cancer (1982) 30:107–12. doi:10.1002/ijc.2910300118
23. Haliotis T, Ball JK, Dexter D, Roder JC. Spontaneous and induced primary oncogenesis in natural killer (NK)-cell-deficient beige mutant mice. Int J Cancer (1985) 35:505–13. doi:10.1002/ijc.2910350414
24. Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM. In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proc Natl Acad Sci USA (2000) 97:2731–6. doi:10.1073/pnas.050588297
25. Fodstad O, Hansen CT, Cannon GB, Boyd MR. Immune characteristics of the beige-nude mouse. A model for studying immune surveillance. Scand J Immunol (1984) 20:267–72. doi:10.1111/j.1365-3083.1984.tb01002.x
26. Fodstad O, Hansen CT, Cannon GB, Statham CN, Lichtenstein GR, Boyd MR. Lack of correlation between natural killer activity and tumor growth control in nude mice with different immune defects. Cancer Res (1984) 44:4403–8.
27. Hollenbeak CS, Todd MM, Billingsley EM, Harper G, Dyer AM, Lengerich EJ. Increased incidence of melanoma in renal transplantation recipients. Cancer (2005) 104:1962–7. doi:10.1002/cncr.21404
28. Beral V, Newton R. Overview of the epidemiology of immunodeficiency-associated cancers. J Natl Cancer Inst Monogr (1998) 23:1–6. doi:10.1093/oxfordjournals.jncimonographs.a024164
29. Mueller N. Overview of the epidemiology of malignancy in immune deficiency. J Acquir Immune Defic Syndr (1999) 21(Suppl 1):S5–10.
30. Dal Maso L, Serraino D, Franceschi S. Epidemiology of AIDS-related tumours in developed and developing countries. Eur J Cancer (2001) 37:1188–201. doi:10.1016/S0959-8049(01)00120-4
31. Oluwole SF, Ali AO, Shafaee Z, Depaz HA. Breast cancer in women with HIV/AIDS: report of five cases with a review of the literature. J Surg Oncol (2005) 89:23–7. doi:10.1002/jso.20171
32. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA (2005) 102:18538–43. doi:10.1073/pnas.0509182102
33. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348:203–13. doi:10.1056/NEJMoa020177
34. Sakakura T, Nishizuka Y. Effect of thymectomy on mammary tumorigenesis, noduligenesis, and mammogenesis in the mouse. Gann (1967) 58:441–50.
35. Kojima A, Taguchi O, Sakakura T, Nishizuka Y. Prevalent types of tumors developing in neonatally thymectomized mice. Gann (1979) 70:839–43.
36. Nishizuka Y, Sakakura T. Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science (1969) 166:753–5. doi:10.1126/science.166.3906.753
37. Fujimoto S, Greene M, Sehon AH. Immunosuppressor T cells in tumor bearing host. Immunol Commun (1975) 4:201–17.
38. Fujimoto S, Greene MI, Sehon AH. Regualtion of the immune response to tumor antigens. I. Immunosuppressor cells in tumor-bearing hosts. J Immunol (1976) 116:791–9.
39. Fujimoto S, Greene MI, Sehon AH. Regulation of the immune response to tumor antigens. II. The nature of immunosuppressor cells in tumor-bearing hosts. J Immunol (1976) 116:800–6.
40. Berendt MJ, North RJ. T-cell-mediated suppression of anti-tumor immunity. An explanation for progressive growth of an immunogenic tumor. J Exp Med (1980) 151:69–80. doi:10.1084/jem.151.1.69
41. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol (1999) 163:5211–8.
42. Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res (1999) 59:3128–33.
43. Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early- stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res (2001) 61:4766–72.
44. Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol (2002) 169:2756–61.
45. Somasundaram R, Jacob L, Swoboda R, Caputo L, Song H, Basak S, et al. Inhibition of cytolytic T lymphocyte proliferation by autologous CD4+/CD25+ regulatory T cells in a colorectal carcinoma patient is mediated by transforming growth factor-beta. Cancer Res (2002) 62:5267–72.
46. Woo EY, Yeh H, Chu CS, Schlienger K, Carroll RG, Riley JL, et al. Cutting edge: regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol (2002) 168:4272–6.
47. Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res (2003) 9:606–12.
48. deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res (2012) 18:3022–9. doi:10.1158/1078-0432.CCR-11-3216
49. Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer (2003) 98:1089–99. doi:10.1002/cncr.11618
50. Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol (2004) 34:336–44. doi:10.1002/eji.200324181
51. Liu JY, Zhang XS, Ding Y, Peng RQ, Cheng X, Zhang NH, et al. The changes of CD4+CD25+/CD4+ proportion in spleen of tumor-bearing BALB/c mice. J Transl Med (2005) 3:5. doi:10.1186/1479-5876-3-5
52. Darrasse-Jeze G, Bergot AS, Durgeau A, Billiard F, Salomon BL, Cohen JL, et al. Tumor emergence is sensed by self-specific CD44hi memory Tregs that create a dominant tolerogenic environment for tumors in mice. J Clin Invest (2009) 119:2648–62. doi:10.1172/JCI36628
53. Martin F, Ladoire S, Mignot G, Apetoh L, Ghiringhelli F. Human FOXP3 and cancer. Oncogene (2010) 29:4121–9. doi:10.1038/onc.2010.174
54. Tanchot C, Terme M, Pere H, Tran T, Benhamouda N, Strioga M, et al. Tumor-infiltrating regulatory T cells: phenotype, role, mechanism of expansion in situ and clinical significance. Cancer Microenviron (2012) 6(2):147–57. doi:10.1007/s12307-012-0122-y
55. Alvaro T, Lejeune M, Salvado MT, Bosch R, Garcia JF, Jaen J, et al. Outcome in Hodgkin’s lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res (2005) 11:1467–73. doi:10.1158/1078-0432.CCR-04-1869
56. Carreras J, Lopez-Guillermo A, Fox BC, Colomo L, Martinez A, Roncador G, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood (2006) 108:2957–64. doi:10.1182/blood-2006-04-018218
57. Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother (2009) 60:909–18. doi:10.1007/s00262-011-1046-y
58. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol (2009) 27:186–92. doi:10.1200/JCO.2008.18.7229
59. Correale P, Rotundo MS, Del Vecchio MT, Remondo C, Migali C, Ginanneschi C, et al. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother (2010) 33:435–41. doi:10.1097/CJI.0b013e3181d32f01
60. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10:942–9. doi:10.1038/nm1093
61. Yang ZZ, Novak AJ, Stenson MJ, Witzig TE, Ansell SM. Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood (2006) 107:3639–46. doi:10.1182/blood-2005-08-3376
62. Gutierrez-Ramos JC, Lloyd C, Kapsenberg ML, Gonzalo JA, Coyle AJ. Non-redundant functional groups of chemokines operate in a coordinate manner during the inflammatory response in the lung. Immunol Rev (2000) 177:31–42. doi:10.1034/j.1600-065X.2000.17713.x
63. Ghoreschi K, Laurence A, Yang XP, Tato CM, Mcgeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature (2010) 467:967–71. doi:10.1038/nature09447
64. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi:10.1084/jem.20030152
65. Zheng SG, Gray JD, Ohtsuka K, Yamagiwa S, Horwitz DA. Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25− precursors. J Immunol (2002) 169:4183–9.
66. Valzasina B, Piconese S, Guiducci C, Colombo MP. Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25− lymphocytes is thymus and proliferation independent. Cancer Res (2006) 66:4488–95. doi:10.1158/0008-5472.CAN-05-4217
67. Zhou G, Levitsky HI. Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J Immunol (2007) 178:2155–62.
68. Heldin CH, Miyazono K, Ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature (1997) 390:465–71. doi:10.1038/37284
69. Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev (2000) 14:627–44. doi:10.1101/gad.14.6.627
70. Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med (2001) 7:1118–22. doi:10.1038/nm1001-1118
71. Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor evasion of the immune system by converting CD4+CD25− T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol (2007) 178:2883–92.
72. Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B, et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res (2007) 67:8344–50. doi:10.1158/0008-5472.CAN-06-3304
73. Moo-Young TA, Larson JW, Belt BA, Tan MC, Hawkins WG, Eberlein TJ, et al. Tumor-derived TGF-beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J Immunother (2009) 32:12–21. doi:10.1097/CJI.0b013e318189f13c
74. Liyanage UK, Goedegebuure PS, Moore TT, Viehl CT, Moo-Young TA, Larson JW, et al. Increased prevalence of regulatory T cells (Treg) is induced by pancreas adenocarcinoma. J Immunother (2006) 29:416–24. doi:10.1097/01.cji.0000205644.43735.4e
75. Baratelli F, Lee JM, Hazra S, Lin Y, Walser TC, Schaue D, et al. PGE(2) contributes to TGF-beta induced T regulatory cell function in human non-small cell lung cancer. Am J Trans Res (2010) 2:356–67.
76. Terabe M, Ambrosino E, Takaku S, O’Konek JJ, Venzon D, Lonning S, et al. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res (2009) 15:6560–9. doi:10.1158/1078-0432.CCR-09-1066
77. Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity (2006) 25:455–71. doi:10.1016/j.immuni.2006.07.011
78. Li Z, Pang Y, Gara SK, Achyut BR, Heger C, Goldsmith PK, et al. Gr-1+CD11b+ cells are responsible for tumor promoting effect of TGF-beta in breast cancer progression. Int J Cancer (2012) 131:2584–95. doi:10.1002/ijc.27572
79. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med (2005) 202:919–29. doi:10.1084/jem.20050463
80. Lebson L, Wang T, Jiang Q, Whartenby KA. Induction of the glucocorticoid-induced leucine zipper gene limits the efficacy of dendritic cell vaccines. Cancer Gene Ther (2011) 18:563–70. doi:10.1038/cgt.2011.23
81. Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature (2007) 449:361–5. doi:10.1038/nature06110
82. Birnberg T, Bar-On L, Sapoznikov A, Caton ML, Cervantes-Barragan L, Makia D, et al. Lack of conventional dendritic cells is compatible with normal development and T cell homeostasis, but causes myeloid proliferative syndrome. Immunity (2008) 29:986–97. doi:10.1016/j.immuni.2008.10.012
83. Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med (2009) 206:1853–62. doi:10.1084/jem.20090746
84. Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med (2009) 206:549–59. doi:10.1084/jem.20082394
85. Billiard F, Lobry C, Darrasse-Jeze G, Waite J, Liu X, Mouquet H, et al. Dll4-Notch signaling in Flt3-independent dendritic cell development and autoimmunity in mice. J Exp Med (2012) 209:1011–28. doi:10.1084/jem.20111615
86. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity (2005) 22:329–41. doi:10.1016/j.immuni.2005.01.016
87. Zelenay S, Lopes-Carvalho T, Caramalho I, Moraes-Fontes MF, Rebelo M, Demengeot J. Foxp3+ CD25− CD4 T cells constitute a reservoir of committed regulatory cells that regain CD25 expression upon homeostatic expansion. Proc Natl Acad Sci USA (2005) 102:4091–6. doi:10.1073/pnas.0408679102
88. Schallenberg S, Tsai PY, Riewaldt J, Kretschmer K. Identification of an immediate Foxp3(−) precursor to Foxp3(+) regulatory T cells in peripheral lymphoid organs of nonmanipulated mice. J Exp Med (2010) 207:1393–407. doi:10.1084/jem.20100045
89. Wang C, Lee JH, Kim CH. Optimal population of FoxP3+ T cells in tumors requires an antigen priming-dependent trafficking receptor switch. PLoS ONE (2012) 7:e30793. doi:10.1371/journal.pone.0030793
90. Zabransky DJ, Nirschl CJ, Durham NM, Park BV, Ceccato CM, Bruno TC, et al. Phenotypic and functional properties of Helios+ regulatory T cells. PLoS ONE (2012) 7:e34547. doi:10.1371/journal.pone.0034547
91. Getnet D, Grosso JF, Goldberg MV, Harris TJ, Yen HR, Bruno TC, et al. A role for the transcription factor Helios in human CD4(+)CD25(+) regulatory T cells. Mol Immunol (2010) 47:1595–600. doi:10.1016/j.molimm.2010.02.001
92. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol (2010) 184:3433–41. doi:10.4049/jimmunol.0904028
93. Verhagen J, Wraith DC. Comment on “Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells”. J Immunol (2010) 185:7129. doi:10.4049/jimmunol.1090105 author reply 7130,
94. Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science (2013) 339:1219–24. doi:10.1126/science.1233913
95. Paiva RS, Lino AC, Bergman ML, Caramalho I, Sousa AE, Zelenay S, et al. Recent thymic emigrants are the preferential precursors of regulatory T cells differentiated in the periphery. Proc Natl Acad Sci USA (2013) 110:6494–9. doi:10.1073/pnas.1221955110
96. Bhatnagar RM, Zabriskie JB, Rausen AR. Cellular immune responses to methylcholanthrene-induced fibrosarcoma in BALB/c mice. J Exp Med (1975) 142:839–55. doi:10.1084/jem.142.4.839
97. Nishikawa H, Kato T, Tanida K, Hiasa A, Tawara I, Ikeda H, et al. CD4+ CD25+ T cells responding to serologically defined autoantigens suppress antitumor immune responses. Proc Natl Acad Sci USA (2003) 100:10902–6. doi:10.1073/pnas.1834479100
98. Vence L, Palucka AK, Fay JW, Ito T, Liu YJ, Banchereau J, et al. Circulating tumor antigen-specific regulatory T cells in patients with metastatic melanoma. Proc Natl Acad Sci USA (2007) 104:20884–9. doi:10.1073/pnas.0710557105
99. Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, et al. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity (2004) 20:107–18. doi:10.1016/S1074-7613(03)00359-5
100. Wang HY, Peng G, Guo Z, Shevach EM, Wang RF. Recognition of a new ARTC1 peptide ligand uniquely expressed in tumor cells by antigen-specific CD4+ regulatory T cells. J Immunol (2005) 174:2661–70.
101. Valenti R, Huber V, Filipazzi P, Pilla L, Sovena G, Villa A, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res (2006) 66:9290–8. doi:10.1158/0008-5472.CAN-06-1819
102. Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE (2010) 5:e11469. doi:10.1371/journal.pone.0011469
103. Fisson S, Darrasse-Jèze G, Litvinova E, Septier F, Klatzmann D, Liblau R, et al. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med (2003) 198:737–46. doi:10.1084/jem.20030686
104. Fritzsching B, Oberle N, Pauly E, Geffers R, Buer J, Poschl J, et al. Naive regulatory T cells: a novel subpopulation defined by resistance toward CD95L-mediated cell death. Blood (2006) 108:3371–8. doi:10.1182/blood-2006-02-005660
105. Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med (2004) 199:303–13. doi:10.1084/jem.20031562
106. Setiady YY, Ohno K, Samy ET, Bagavant H, Qiao H, Sharp C, et al. Physiologic self antigens rapidly capacitate autoimmune disease-specific polyclonal CD4+ CD25+ regulatory T cells. Blood (2006) 107:1056–62. doi:10.1182/blood-2005-08-3088
107. North RJ. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med (1982) 155:1063–74. doi:10.1084/jem.155.4.1063
108. Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood (2005) 105:2862–8. doi:10.1182/blood-2004-06-2410
109. Weiss VL, Lee TH, Song H, Kouo TS, Black CM, Sgouros G, et al. Trafficking of high avidity HER-2/neu-specific T cells into HER-2/neu-expressing tumors after depletion of effector/memory-like regulatory T cells. PLoS ONE (2012) 7:e31962. doi:10.1371/journal.pone.0031962
110. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med (1998) 188:2357–68. doi:10.1084/jem.188.12.2357
111. Overwijk WW, Lee DS, Surman DR, Irvine KR, Touloukian CE, Chan CC, et al. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc Natl Acad Sci USA (1999) 96:2982–7. doi:10.1073/pnas.96.6.2982
112. Lollini PL, Cavallo F, Nanni P, Forni G. Vaccines for tumour prevention. Nat Rev Cancer (2006) 6:204–16. doi:10.1038/nrc1815
113. Yang J, Brook MO, Carvalho-Gaspar M, Zhang J, Ramon HE, Sayegh MH, et al. Allograft rejection mediated by memory T cells is resistant to regulation. Proc Natl Acad Sci USA (2007) 104:19954–9. doi:10.1073/pnas.0704397104
114. Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, Van Loosdregt J, et al. Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood (2011) 118:3538–48. doi:10.1182/blood-2010-12-328187
115. Nishikawa H, Jager E, Ritter G, Old LJ, Gnjatic S. CD4+ CD25+ regulatory T cells control the induction of antigen-specific CD4+ helper T cell responses in cancer patients. Blood (2005) 106:1008–11. doi:10.1182/blood-2005-02-0607
116. Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur J Immunol (2000) 30:1538–43. doi:10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X
117. Dubey C, Croft M, Swain SL. Naive and effector CD4 T cells differ in their requirements for T cell receptor versus costimulatory signals. J Immunol (1996) 157:3280–9.
118. Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med (2005) 353:2654–66. doi:10.1056/NEJMoa051424
119. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi:10.1126/science.1129139
120. Holtan SG, Creedon DJ, Haluska P, Markovic SN. Cancer and pregnancy: parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clin Proc (2009) 84:985–1000. doi:10.1016/S0025-6196(11)60669-1
121. Chen T, Darrasse-Jeze G, Bergot AS, Courau T, Churlaud G, Valdivia K, et al. Self-specific memory regulatory T cells protect embryos at implantation in mice. J Immunol (2013) 191:2273–81. doi:10.4049/jimmunol.1202413
122. Wen Y, Fang W, Xiang LX, Pan RL, Shao JZ. Identification of Treg-like cells in Tetraodon: insight into the origin of regulatory T subsets during early vertebrate evolution. Cell Mol Life Sci (2011) 68:2615–26. doi:10.1007/s00018-010-0574-5
123. Quintana FJ, Iglesias AH, Farez MF, Caccamo M, Burns EJ, Kassam N, et al. Adaptive autoimmunity and Foxp3-based immunoregulation in zebrafish. PLoS ONE (2010) 5:e9478. doi:10.1371/journal.pone.0009478
124. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell (2012) 150:29–38. doi:10.1016/j.cell.2012.05.031
126. Zhou G, Drake CG, Levitsky HI. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood (2006) 107:628–36. doi:10.1182/blood-2005-07-2737
127. Ebert LM, Macraild SE, Zanker D, Davis ID, Cebon J, Chen W. A cancer vaccine induces expansion of NY-ESO-1-specific regulatory T cells in patients with advanced melanoma. PLoS ONE (2012) 7:e48424. doi:10.1371/journal.pone.0048424
128. Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest (2005) 115:3623–33. doi:10.1172/JCI25947
129. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr., et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Trans Med (2012) 4:134ra162. doi:10.1126/scitranslmed.3003330
130. Tan C, Reddy V, Dannull J, Ding E, Nair SK, Tyler DS, et al. Impact of anti-CD25 monoclonal antibody on dendritic cell-tumor fusion vaccine efficacy in a murine melanoma model. J Transl Med (2013) 11:148. doi:10.1186/1479-5876-11-148
131. Taieb J, Chaput N, Schartz N, Roux S, Novault S, Menard C, et al. Chemoimmunotherapy of tumors: cyclophosphamide synergizes with exosome based vaccines. J Immunol (2006) 176:2722–9.
132. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res (2013) 73:3591–603. doi:10.1158/0008-5472.CAN-12-4100
133. Hirayama M, Nishikawa H, Nagata Y, Tsuji T, Kato T, Kageyama S, et al. Overcoming regulatory T-cell suppression by a lyophilized preparation of Streptococcus pyogenes. Eur J Immunol (2013) 43:989–1000. doi:10.1002/eji.201242800
134. Chaput N, Darrasse-Jeze G, Bergot AS, Cordier C, Ngo-Abdalla S, Klatzmann D, et al. Regulatory T cells prevent CD8 T cell maturation by inhibiting CD4 Th cells at tumor sites. J Immunol (2007) 179:4969–78.
135. Maury S, Lemoine FM, Hicheri Y, Rosenzwajg M, Badoual C, Cherai M, et al. CD4+CD25+ regulatory T cell depletion improves the graft-versus-tumor effect of donor lymphocytes after allogeneic hematopoietic stem cell transplantation. Sci Trans Med (2010) 2:41ra52. doi:10.1126/scitranslmed.3001302
Keywords: Treg, Foxp3, memory, cancer, tolerance, tumor cells, vaccination, early immune response
Citation: Darrasse-Jèze G and Podsypanina K (2013) How numbers, nature, and immune status of Foxp3+ regulatory T-cells shape the early immunological events in tumor development. Front. Immunol. 4:292. doi: 10.3389/fimmu.2013.00292
Received: 14 June 2013; Paper pending published: 14 July 2013;
Accepted: 05 September 2013; Published online: 26 September 2013.
Edited by:
Eyad Elkord, United Arab Emirates University, UAE; University of Salford, UK; University of Manchester, UKReviewed by:
Ye Zheng, Salk Institute for Biological Studies, USALionel Apetoh, Institut National de la Santé et de la Recherche Médicale, France
Copyright: © 2013 Darrasse-Jèze and Podsypanina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillaume Darrasse-Jèze, Institut National de la Santé et de la Recherche Médicale, Université Paris Descartes, Sorbonne Paris Cité, Hôpital Necker, 149 rue de Sevres, F-75015 Paris, France e-mail:Z3VpbGxhdW1lLmRhcnJhc3NlLWplemVAaW5zZXJtLmZy