



95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 29 August 2013
Sec. T Cell Biology
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00259
This article is part of the Research Topic The regulation of calcium homeostasis in T lymphocytes View all 10 articles
Calcium signaling is a universal signal transduction mechanism in animal and plant cells. In mammalian T-lymphocytes calcium signaling is essential for activation and re-activation and thus important for a functional immune response. Since many years it has been known that both calcium release from intracellular stores and calcium entry via plasma membrane calcium channels are involved in shaping spatio-temporal calcium signals. Second messengers derived from the adenine dinucleotides NAD and NADP have been implicated in T cell calcium signaling. Nicotinic acid adenine dinucleotide phosphate (NAADP) acts as a very early second messenger upon T cell receptor/CD3 engagement, while cyclic ADP-ribose (cADPR) is mainly involved in sustained partial depletion of the endoplasmic reticulum by stimulating calcium release via ryanodine receptors. Finally, adenosine diphosphoribose (ADPR) a breakdown product of both NAD and cADPR activates a plasma membrane cation channel termed TRPM2 thereby facilitating calcium (and sodium) entry into T cells. Receptor-mediated formation, metabolism, and mode of action of these novel second messengers in T-lymphocytes will be reviewed.
Adenine derived Ca2+ mobilizing second messengers comprise nicotinic acid adenine dinucleotide phosphate (NAADP), cyclic ADP-ribose (cADPR), and adenosine diphosphoribose (ADPR; Figure 1). They are all metabolites of nicotinamide adenine dinucleotide (NAD), a dinucleotide well known as coenzyme of oxidoreductases. NAD is converted by the multifunctional ectoenzyme NAD-glycohydrolase/ADP-ribosyl cyclase CD38 to ADPR and cADPR (Figure 1). The fact that the active site of CD38 faces the extracellular space while the targets for its products are located inside the cell, also known as topological paradox (1), has recently been investigated in detail. Importantly, in addition to the type II conformation with the active site facing the extracellular space, it was demonstrated that a smaller portion of CD38 is expressed in type III conformation thereby allowing for production of ADPR and cADPR within the cytosol (2). Another interesting feature of CD38 is the fact that it not only can make cADPR and ADPR, but also can synthesize NAADP, at least in vitro (Figure 1). However, this base-exchange mechanism needs nicotinamide adenine dinucleotide phosphate (NADP) and an excess of nicotinic acid as substrates and it works at acidic pH. Thus, it remains unclear whether this reaction is of physiological significance for second messenger formation in the cytosol. The substrate for the base-exchange reaction, NADP, is produced from NAD by NAD kinase (3). While mature, naïve T cells express only small amounts of CD38, it is upregulated as a consequence of mitogenic stimulation (4, 5). This is for instance seen after infection with HIV in activated antiviral CD8+ T cells (6). CD38 expression in the CD8 compartment is therefore used to monitor antiretroviral therapy (7). Whether CD38 upregulation in activated T cells affects Ca2+ signaling compared to naïve, mature T cells is not known, but it is easy to envision the production of Ca2+ mobilizing messengers in effector cells being facilitated by upregulation of CD38, allowing for faster Ca2+ responses necessary for secretion of cytokines or granzymes and perforin in contrast to activation of calcineurin and NFAT in naïve cells.
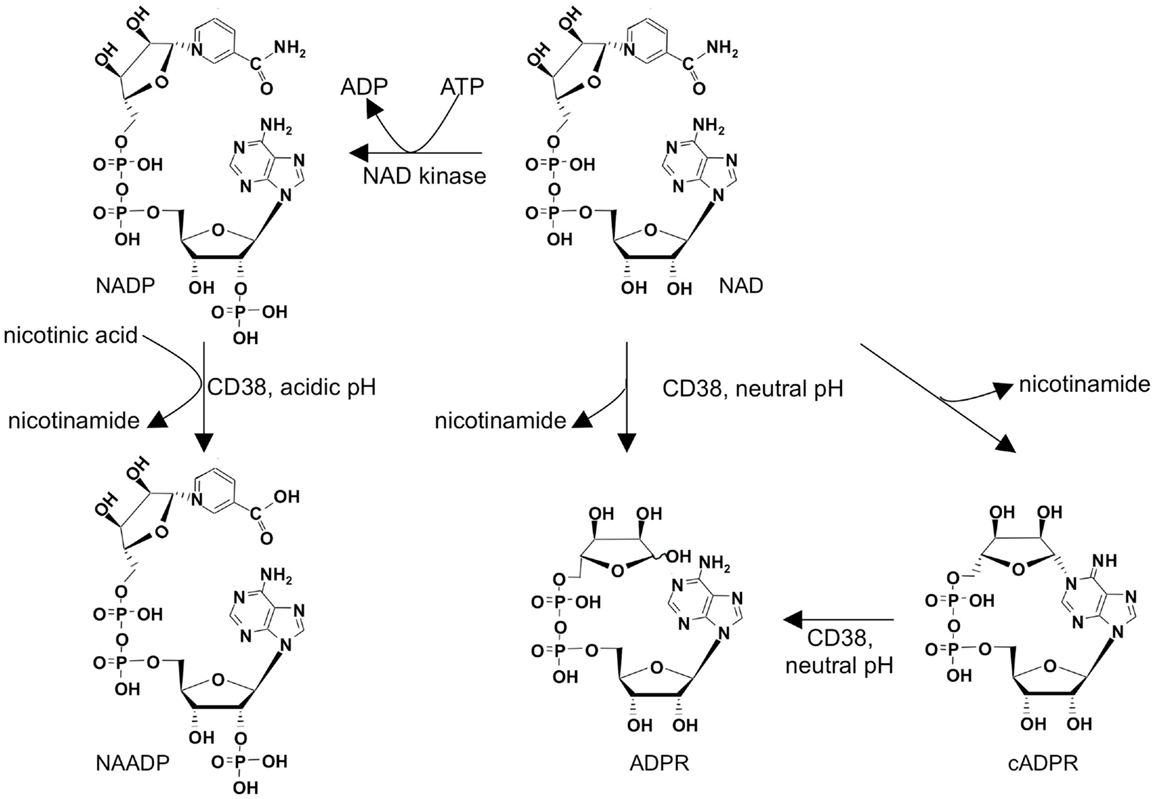
Figure 1. Metabolism of adenine derived second messengers cADPR, NAADP, and ADPR.
Ca2+ signaling is one of the essential intracellular signaling pathways involved in T cell activation. It has long been known that both Ca2+ release and Ca2+ entry contribute to global Ca2+ signaling in T cells. In addition to Ca2+ release and Ca2+ entry evoked by the adenine derived Ca2+ mobilizing second messengers introduced above, two “standard” Ca2+ signaling systems are involved: (i) Ca2+ release by d-myo-inositol 1,4,5-trisphosphate [IP3; (8)] and store-operated or capacitative Ca2+ entry (9). Since these systems have been thoroughly investigated and described in detail, they will not be reviewed in this article. However, due to their importance for T cell Ca2+ signaling, their roles will be mentioned and/or depicted, as for example in Figure 2.
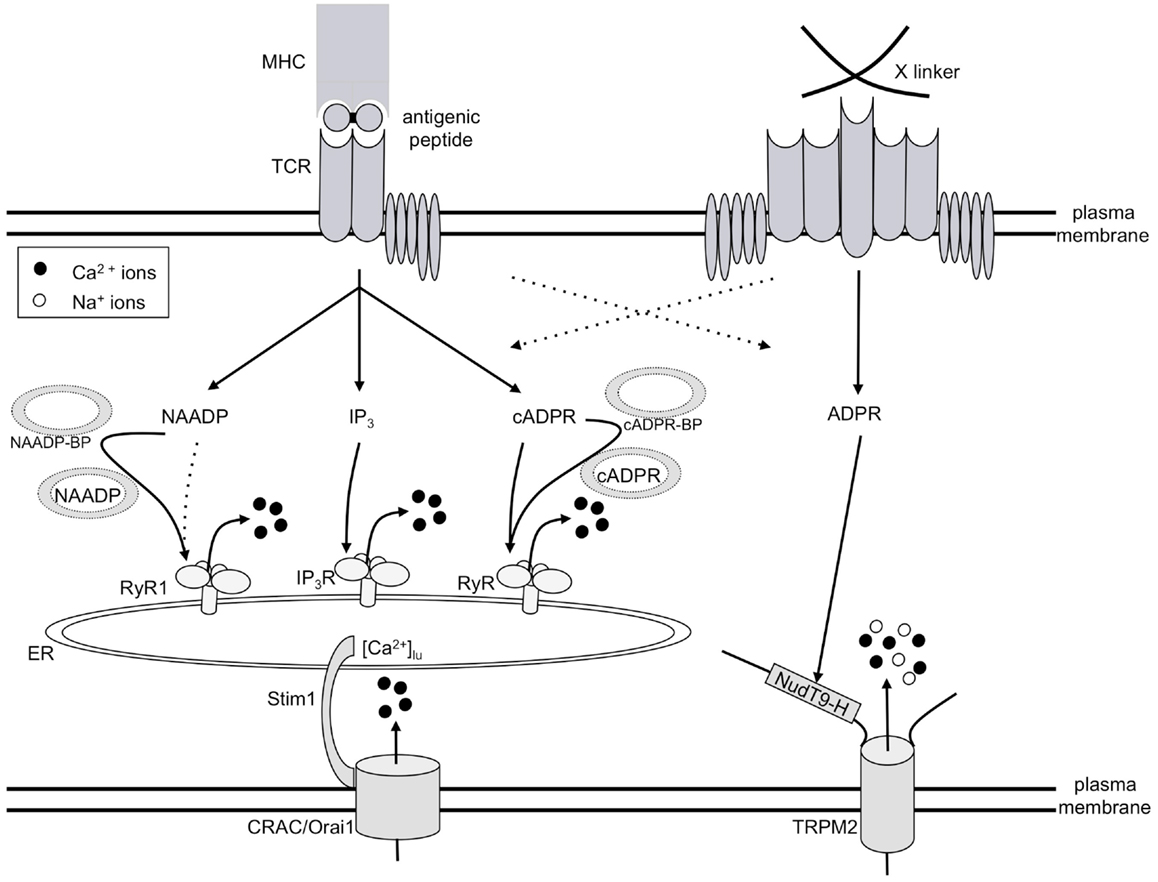
Figure 2. Model of T cell Ca2+ signaling. TCR/CD3 ligation by antigenic peptide presented by MHC molecules on antigen presenting cells results in consecutive formation of the second messengers NAADP, IP3, and cADPR, all of which release Ca2+ from the ER. Thus, a continuously decreased intraluminal free Ca2+ concentration in the ER ([Ca2+]lu) resulting from this constant Ca2+ release concomitantly activates CRAC/Orai1 channels in the plasma membrane. The mode of action of both NAADP and cADPR likely involves specific binding proteins for both second messengers (abbreviated here as NAADP-BP or cADPR-BP). A strong stimulus, e.g., cross-linking of receptors by concanavalin A (right side of Figure 2), triggers formation of ADPR and activation of TRPM2, in addition to the mechanisms described on the left side of the figure.
The initial player in our model of T cell Ca2+ signaling is NAADP (Figure 2) being formed within seconds upon TCR/CD3 ligation (10). However, NAADP is a rather short-lived second messenger, although after a rapid decrease to control levels, a second much smaller rise over several minutes was observed in Jurkat T cells (11). NAADP probably delivers the first local Ca2+ signals which then act as co-agonists at IP3 receptors (IP3R) and ryanodine receptors (RyR). IP3 is formed soon after the initial NAADP peak (12) and releases Ca2+ via IP3R (13). Finally, cADPR starts to increase and acts on RyR (14); likely, Ca2+ released by NAADP and/or IP3 facilitates the action of cADPR. Continuous Ca2+ release by these consecutively increased second messengers results in continuously decreased luminal Ca2+ concentration in the ER ([Ca2+]lu). Stromal interaction molecule-1 (Stim1) senses the decreased [Ca2+]lu and activates Ca2+ entry via orai/CRAC channels (15–17).
In addition to this Ca2+ signaling pathway involved in T cell activation or re-activation, high input signal strength, e.g., obtained by a high concentration of the cross-linking lectin Concanavalin A, activates another different Ca2+ entry system operated by ADPR and the transient receptor potential channel, subtype melastatin 2 (TRPM2; Figure 2).
Following we will review hallmarks of NAADP, cADPR, and ADPR as second messengers in T cell Ca2+ signaling.
Upon activation of the TCR/CD3 complex, formation of NAADP rapidly increases within 10–20 s in Jurkat T cells. Following a subsequent decrease within the first minute, a continuously elevated [NAADP] remains for 5–20 min (10). It has been proposed that NAADP may act as an early triggering messenger, mediating initial localized Ca2+ events which are subsequently amplified to a global signal, e.g., by recruitment of further channels, other second messengers like cADPR, IP3, and/or Ca2+ induced Ca2+ release (CICR). In T-lymphocytes a bell-shaped concentration-response curve following NAADP microinjection is observed. Compared to other second messengers such as IP3, already low concentrations in the nanomolar range (30–100 nM) induce Ca2+ signaling in T-lymphocytes (18).
The mechanism and very early kinetics of receptor-mediated formation of NAADP in vivo remains unclear and has been discussed previously [e.g., (19)]. In brief, NAADP is formed in vitro by a base-exchange of NADP in presence of nicotinic acid and a pH of 5 [Figure 1; (20)]. Further, at pH 5, but also at pH 7.4, 2′-phospho-cADPR may be converted to NAADP (21). Both reactions are catalyzed by the membrane bound, multifunctional enzyme CD38 and require presence of up to millimolar concentrations of nicotinic acid in vitro (20, 21). Furthermore, influx of extracellular NAADP may also induce Ca2+ signals as shown in a rat basophilic cell line (22). Interestingly, gene silencing of CD38 in Jurkat T-lymphocytes did not result in decreased NAADP levels. Rather, in thymus and spleen of CD38 knock-out mice increased NAADP levels were observed, thus indicating that CD38 may particularly drive degradation of NAADP (23, 24). Accordingly, in T cells to date CD38 may be primarily understood as degrading enzyme while its role in NAADP synthesis in vivo remains to be elucidated. In T-lymphocytes and other CD38+ cells, NAADP is degraded to 2′-phospho-ADPR at neutral and acidic pH by CD38, but degradation may also occur non-specifically via nucleotide pyrophosphatases (25).
The targeted receptor(s) and hence target organelle(s) of NAADP are still under debate (19, 26–29). In general, RyR have been implicated as NAADP targets in different cell types, e.g., skeletal muscle cell (30), or pancreatic acinar cells (31, 32). Nonetheless, in the majority of mammalian cells as well as in sea urchin eggs there is evidence that NAADP may primarily target acidic stores and that the RyR located on the ER may rather play a central role in the amplification of the Ca2+ signal (33–35).
However focusing particularly on data obtained in T-lymphocytes, NAADP Ca2+ signaling strongly depends on RyR activity in the ER (36–40). Following either knock-down or inhibition of RyR by ryanodine in Jurkat T cells, subcellular and global Ca2+ signals by NAADP microinjection were inhibited or almost completely abolished (36, 38). In primary effector T-lymphocytes the NAADP antagonist BZ194 inhibits Ca2+ signaling, e.g., during formation of the immunological synapse. Furthermore, BZ194 has been shown to selectively inhibit NAADP dependent binding of [3H]ryanodine to RyR1 (39). Interestingly, in primary effector T cells of an animal model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE), BZ194 leads to a decrease in cell motility and invasive capacity as well as a decrease in cytokine expression, all of which indicate the central role of NAADP-mediated Ca2+ signaling in T cells possibly via RyR (40). In contrast to these results obtained in CD4+ T cells, in cytotoxic T cells NAADP appears to target two-pore channels (TPC) on cytolytic granules (41). In general, overexpression or inhibition of the endolysosomal TPC1 and TPC2 suggest that NAADP initiates Ca2+ events via TPC [e.g., (42–44)]. Recently, the N-terminus of TPC1 has been identified as functional region for NAADP-mediated Ca2+ signaling (26). In contrast, it was shown that NAADP-mediated Ca2+ signaling in TPC1−/−/TPC2−/− mice does not differ from wild-type mice (27). Thus, whether NAADP primarily targets TPCs is controversial and particularly the effect of NAADP on TPCs in T-lymphocytes is not yet clear.
Further, in T-lymphocytes and neutrophils TRPM2 is activated by micromolar concentrations of NAADP per se, but particularly in synergism with cADPR (45, 46). The effect of cADPR on TRPM2 however, could not be confirmed in HEK293 cells overexpressing TRPM2 and contamination of commercial cADPR preparations with ADPR have been discussed (47, 48). NAADP has been shown to target the unspecific Ca2+ channel TRPML1 in smooth muscle myocytes (49). Whether TRPML1 and TRPM2 are of functional relevance within NAADP – mediated Ca2+ signaling in T-lymphocytes, remains to be elucidated.
Despite the questions which organelles are targeted by NAADP and which specific downstream mechanisms may underlie the initiated Ca2+ events, also the identity of the NAADP receptor remains unclear. Photoaffinity labeling in mammalian cells using a probe specific for NAADP binding proteins was not altered upon overexpression or knock-out of TPC1 or TPC2, but suggests that a yet not identified 22/23 kDa protein binds NAADP and may hence couple NAADP to its respective Ca2+ channels (50, 51), a mechanism recently introduced as unifying hypothesis of NAADP action (29).
Cyclic adenosine diphosphoribose (cADPR) was the first Ca2+ mobilizing second messenger discovered as derivative of an adenine dinucleotide (52, 53). Though first described in sea urchin egg homogenates, the Ca2+ mobilizing activity of cADPR was soon detected in many cells types. In 1995 we published the first report demonstrating specific Ca2+ release in human Jurkat T cells (54). Central aspects of the role of cADPR in T cell Ca2+ signaling were subsequently published by our laboratory: (i) formation of cADPR upon TCR/CD3 ligation (14, 55), and (ii) mode of action of cADPR by activation of Ca2+ release via RyR, as shown by gene silencing of RyR (56). Further, we demonstrated tyrosine phosphorylation of RyR upon TCR/CD3 ligation; in permeabilized T cells enhancement of cADPR evoked Ca2+ release by tyrosine kinase p59fyn was observed (57). Importantly, we demonstrated amplification and propagation of pacemaker Ca2+ signals by cADPR (58). A connection of cADPR signaling to Ca2+ entry was also observed: microinjection of cADPR in the absence of extracellular Ca2+ or in the presence of Ca2+ channel blockers resulted in much reduced Ca2+ signals (59). Finally, using a specific cADPR antagonist it was shown that downstream activation parameters of primary human T cells, such as activation antigen expression or proliferation, were concentration-dependently inhibited (14) suggesting a pivotal role of cADPR in T cell biology.
A detailed structure-activity analysis of cADPR in T cells has been conducted over the past couple of years. The main results from these studies were recently reviewed (60) and are (i) critical dependence of agonist vs. antagonist properties on the substituent at the C-atom 6 of the purine base, (ii) maintenance of biological activity, albeit at a lower level, when both southern and northern ribose were replaced by carbocyclic moieties or simplified ether/alkane bridges, and (iii) the possibility of radical simplification of the purine structure, e.g., the 1,2,3-triazole-4-amide mimic of adenine within cADPR retains biological activity.
A relatively new addition to the realm of adenine based Ca2+ mobilizing second messengers in T-lymphocytes is ADPR. Presence of ADPR in eukaryotic cells has been known for quite a while (61), but since ADPR is rather dangerous for the cell – its reactive ribose can non-enzymatically form Schiff-bases with amino groups of cellular proteins (62, 63) – it was mostly considered a toxic cellular waste product. This casually explained the presence of efficient mechanisms for the degradation of ADPR in form of cytosolic (64, 65) and mitochondrial ADPR pyrophosphatases (66, 67). These enzymes hydrolyze the pyrophosphate bridge of ADPR yielding AMP and ribose-5′-phosphate that are fed back into metabolism.
Two discoveries suggested that there might be more to ADPR: in 2001 it was reported that TRPM2 (formerly termed TRPC7 or LTRPC2), a Ca2+-permeable cation channel of the melastatin subfamily of TRP channels, can be activated by binding of ADPR to a cytoplasmic domain homologous to the mitochondrial ADPR pyrophosphatase NUDT9 (68, 69). This channel shows expression in a variety of tissues with highest levels being found in brain and cells of the immune system. A year later Bastide et al. showed that ADPR is also able to activate type I RyR isolated from rat skeletal muscle in the presence of micromolar concentrations of Ca2+ (70). Since there has been little news on the action of ADPR on RyR, we will focus on the role of ADPR for TRPM2 activation in T-lymphocytes.
Most of the work on ADPR and TRPM2 in T cells so far has been done in Jurkat cells that express TRPM2 on transcript and protein level and respond with a typical TRPM2 current to ADPR infusion (11, 45, 69). Microinjection of ADPR (11) and uncaging of photoactivatable ADPR (71) in these cells results in Ca2+ entry-dependent Ca2+ signals. By HPLC analysis the cellular ADPR concentration of roughly 40 μmol/L in resting Jurkat cells (72) was shown to nearly double after stimulation with high concentrations of concanavalin A (11).
There are different conceivable ways how this ADPR might be generated. CD38 expressed in Jurkat as well as primary T cells (73) can metabolize β-NAD+ and cADPR to yield ADPR (74). The topological paradox initially described for cADPR also holds true for ADPR (1). This paradox might be resolved by specific uptake mechanisms for ADPR as have been reported for erythrocytes (75, 76). Another possibility is the presence of CD38 in a type III orientation (2). While the contribution of CD38 to the increase in ADPR after stimulation is still unclear, the basal ADPR seems to be independent of CD38 as the murine T-lymphoma line BW5147 that lacks transcripts for CD38 (77) has even higher basal ADPR levels [73 μmol/L (72)] than Jurkat cells.
Another way that has been discussed for the production of ADPR is the consecutive action of poly-ADPR polymerase (PARP) and poly-ADPR glycohydrolase (PARG) (78). While PARP activity and poly-ADPR levels are quite low in non-stimulated cells, there is a constant turn-over due to the low KM of PARG [reviewed in (79)] that might contribute to the basal ADPR detected in Jurkat cells. Under DNA damaging conditions like strong oxidative stress the activity of PARP increases to such levels that a large part of cellular β-NAD+ can be metabolized as has been shown for DT-40 cells (80). Data for a range of cells suggest activation of TRPM2 by oxidative stress results in cell death by apoptosis (78, 81), most likely due to mitochondrial calcium overload and downstream activation of caspases [reviewed in (82)]. While murine CD4+ T cells also die after exposure to hydrogen peroxide, this apparently does not involve TRPM2 (83).
Interestingly there have been reports that in T cells PARP-1 activation can occur after TCR stimulation in a way independent of oxidative stress or DNA damage resulting in poly-ADP ribosylation of NFAT [(84, 85); reviewed in (86)]. It might be speculated that this increased pADPR turn-over will result in increased cellular ADPR and TRPM2 activation hinting to a possible role for ADPR/TRPM2 in TCR signaling. In accordance with this, naïve CD4+ T cells from the wild-type mice upregulated TRPM2 after stimulation with α-CD3/α-CD28-beads and CD4+ T-lymphocytes from TRPM2−/− mice showed not only reduced proliferation, but also reduced production of pro-inflammatory cytokines upon activation (83).
Most work on the role of TRPM2 in the immune response has been done using the TRPM2−/− mouse (87). In a model for ulcerative colitis the inflammation was suppressed, but this was shown to be due to a reduced production of the chemokine CXCL2 in monocytes whereas the infiltration of T cells in the colon was not affected by the knock-out of TRPM2 (87). Recent work has shown that TRPM2 knock-out does not affect airway inflammation either induced by oxidative stressors (88) or as a result of exposure to ovalbumin in a mouse model for acute asthma (89). On the other hand TRPM2−/− noticeably reduced inflammation and spinal cord lesions in EAE induced by a peptide from myelin oligodendrocyte glycoprotein (83). Since the effect of TRPM2−/− on EAE might also involve reduced neuronal cell death or microglia activation, it will be interesting to see whether proliferation and effector functions of T cells from such animals are affected.
Taken together, adenine derived Ca2+ mobilizing second messengers play essential roles in T cell Ca2+ signaling, both during activation/re-activation or apoptosis. While NAADP is important as rapid Ca2+ trigger, particularly in effector T cells, cADPR apparently holds a central role in maintenance of long-lasting Ca2+ signaling. Interestingly, also apoptosis induction via TRPM2 involves an adenine derived second messenger, the dinucleotide ADPR. The documented involvement of these adenine derived Ca2+ mobilizing second messengers in central aspects of immune regulation make the pathways described in this review suitable targets for therapeutic intervention. In fact, we have recently shown that the NAADP antagonist BZ194 ameliorated the clinical course of transfer EAE, an animal model of multiple sclerosis (40).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Work in the Calcium Signalling Group has been supported over the past couple of years by the Deutsche Forschungsgemeinschaft, the Gemeinnützige Hertie-Stiftung, the Wellcome Trust, and the Deutsche Akademische Austauschdienst. We would like to appreciate our long-standing collaborators in the field of cADPR research, Professor Potter (University of Bath, UK), Professor Zhang (Peking University, China), and Professor Shuto (Hokkaido University, Japan). Last but not least we would like to express sincere thanks to our hard working colleagues in the Calcium Signalling Group.
ADPR, adenosine diphosphoribose; cADPR, cyclic ADP-ribose; [Ca2+]i, free cytosolic Ca2+ concentration; [Ca2+]lu, free endoplasmic reticular luminal Ca2+ concentration; CICR, Ca2+-induced Ca2+ release; EAE, experimental autoimmune encephalomyelitis; IP3, D-myo inositol 1,4,5-trisphosphate; IP3R, D-myo inositol 1,4,5-trisphosphate receptor(s); NAADP, nicotinic acid adenine dinucleotide phosphate; NAD(P), nicotinamide adenine dinucleotide (phosphate); pADPr, poly-ADP-ribose; PARG, poly-ADP-ribose glycohydrolase; PARP, poly-ADP-ribose polymerase; RyR, ryanodine receptor(s); Stim1, stromal interaction molecule-1; TPC, two-pore channel(s); TRPM2, transient receptor potential channel, subtype melastatin 2.
1. De Flora A, Guida L, Franco L, Zocchi E. The CD38/cyclic ADP-ribose system: a topological paradox. Int J Biochem Cell Biol (1997) 29:1149–66. doi:10.1016/S1357-2725(97)00062-9
2. Zhao YJ, Lam CM, Lee HC. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci Signal (2012) 5:ra67. doi:10.1126/scisignal.2002700
3. Pollak N, Niere M, Ziegler M. NAD kinase levels control the NADPH concentration in human cells. J Biol Chem (2007) 282:33562–71. doi:10.1074/jbc.M704442200
4. Malavasi F, Funaro A, Alessio M, DeMonte LB, Ausiello CM, Dianzani U, et al. CD38: a multi-lineage cell activation molecule with a split personality. Int J Clin Lab Res (1992) 22:73–80. doi:10.1007/BF02591400
5. Deterre P, Berthelier V, Bauvois B, Dalloul A, Schuber F, Lund F. CD38 in T- and B-cell functions. Chem Immunol (2000) 75:146–68. doi:10.1159/000058767
6. Ho HN, Hultin LE, Mitsuyasu RT, Matud JL, Hausner MA, Bockstoce D, et al. Circulating HIV-specific CD8+ cytotoxic T cells express CD38 and HLA-DR antigens. J Immunol (1993) 150:3070–9.
7. Coetzee LM, Tay SS, Lawrie D, Janossy G, Glencross DK. From research tool to routine test: CD38 monitoring in HIV patients. Cytometry B Clin Cytom (2009) 76:375–84. doi:10.1002/cyto.b.20478
8. Mikoshiba K. IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem (2007) 102:1426–46. doi:10.1111/j.1471-4159.2007.04825.x
9. Putney JW. Capacitative calcium entry: from concept to molecules. Immunol Rev (2009) 231:10–22. doi:10.1111/j.1600-065X.2009.00810.x
10. Gasser A, Bruhn S, Guse AH. Second messenger function of nicotinic acid adenine dinucleotide phosphate revealed by an improved enzymatic cycling assay. J Biol Chem (2006) 281:16906–13. doi:10.1074/jbc.M601347200
11. Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, et al. Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem (2006) 281:2489–96. doi:10.1074/jbc.M506525200
12. Guse AH, Goldwich A, Weber K, Mayr GW. Non-radioactive, isomer-specific inositol phosphate mass determinations: high-performance liquid chromatography-micro-metal-dye detection strongly improves speed and sensitivity of analyses from cells and micro-enzyme assays. J Chromatogr B Biomed Appl (1995) 672:189–98. doi:10.1016/0378-4347(95)00219-9
13. Guse AH, Roth E, Emmrich F. D-myo-inositol 1,3,4,5-tetrakisphosphate releases Ca2+ from crude microsomes and enriched vesicular plasma membranes, but not from intracellular stores of permeabilized T-lymphocytes and monocytes. Biochem J (1992) 288(Pt 2):489–95.
14. Guse AH, da Silva CP, Berg I, Skapenko AL, Weber K, Heyer P, et al. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature (1999) 398:70–3. doi:10.1038/18024
15. Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel S-H, Tanasa B, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature (2006) 441:179–85. doi:10.1038/nature04702
16. Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature (2006) 443:230–3. doi:10.1038/nature05122
17. Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science (2006) 312:1220–3. doi:10.1126/science.1127883
18. Berg I, Potter BVL, Mayr GW, Guse AH. Nicotinic acid adenine dinucleotide phosphate (Naadp+) is an essential regulator of T-lymphocyte Ca2+-signaling. J Cell Biol (2000) 150:581–8. doi:10.1083/jcb.150.3.581
19. Guse AH, Lee HC. NAADP: a universal Ca2+ trigger. Sci Signal (2008) 1:re10. doi:10.1126/scisignal.144re10
20. Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J Biol Chem (1995) 270:30327–33. doi:10.1074/jbc.270.51.30327
21. Moreschi I, Bruzzone S, Melone L, De Flora A, Zocchi E. NAADP+ synthesis from cADPRP and nicotinic acid by ADP-ribosyl cyclases. Biochem Biophys Res Commun (2006) 345:573–80. doi:10.1016/j.bbrc.2006.04.096
22. Billington RA, Bellomo EA, Floriddia EM, Erriquez J, Distasi C, Genazzani AA. A transport mechanism for NAADP in a rat basophilic cell line. FASEB J (2006) 20:521–3.
23. Schmid F, Bruhn S, Weber K, Mittrücker H-W, Guse AH. CD38: a NAADP degrading enzyme. FEBS Lett (2011) 585:3544–8. doi:10.1016/j.febslet.2011.10.017
24. Soares S, Thompson M, White T, Isbell A, Yamasaki M, Prakash Y, et al. NAADP as a second messenger: neither CD38 nor base-exchange reaction are necessary for in vivo generation of NAADP in myometrial cells. Am J Physiol Cell Physiol (2007) 292:C227–39. doi:10.1152/ajpcell.00638.2005
25. Graeff R. Acidic residues at the active sites of CD38 and ADP-ribosyl cyclase determine nicotinic acid adenine dinucleotide phosphate (NAADP) synthesis and hydrolysis activities. J Biol Chem (2006) 281:28951–7. doi:10.1074/jbc.M604370200
26. Churamani D, Hooper R, Rahman T, Brailoiu E, Patel S. The N-terminal region of two-pore channel 1 regulates trafficking and activation by NAADP. Biochem J (2013) 453(1):147–51. doi:10.1042/BJ20130474
27. Wang X, Zhang X, Dong X-P, Samie M, Li X, Cheng X, et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell (2012) 151:372–83. doi:10.1016/j.cell.2012.08.036
28. Cang C, Zhou Y, Navarro B, Seo Y-J, Aranda K, Shi L, et al. mTOR regulates lysosomal ATP-sensitive two-pore Na(+) channels to adapt to metabolic state. Cell (2013) 152:778–90. doi:10.1016/j.cell.2013.01.023
29. Guse AH. Linking NAADP to ion channel activity: a unifying hypothesis. Sci Signal (2012) 5:e18. doi:10.1126/scisignal.2002890
30. Hohenegger M, Suko J, Gscheidlinger R, Drobny H, Zidar A. Nicotinic acid-adenine dinucleotide phosphate activates the skeletal muscle ryanodine receptor. Biochem J (2002) 367:423–31. doi:10.1042/BJ20020584
31. Gerasimenko JV, Maruyama Y, Yano K, Dolman NJ, Tepikin AV, Petersen OH, et al. NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J Cell Biol (2003) 163:271–82. doi:10.1083/jcb.200306134
32. Gerasimenko JV, Sherwood M, Tepikin AV, Petersen OH, Gerasimenko OV. NAADP, cADPR and IP3 all release Ca2+ from the endoplasmic reticulum and an acidic store in the secretory granule area. J Cell Sci (2006) 119:226–38. doi:10.1242/jcs.02721
33. Lee HC, Aarhus R. Functional visualization of the separate but interacting calcium stores sensitive to NAADP and cyclic ADP-ribose. J Cell Sci (2000) 113(Pt 24):4413–20.
34. Kinnear NP, Boittin F-X, Thomas JM, Galione A, Evans AM. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J Biol Chem (2004) 279:54319–26. doi:10.1074/jbc.M406132200
35. Cancela JM, Churchill GC, Galione A. Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature (1999) 398:74–6. doi:10.1038/18032
36. Dammermann W, Guse AH. Functional ryanodine receptor expression is required for NAADP-mediated local Ca2+ signaling in T-lymphocytes. J Biol Chem (2005) 280:21394–9. doi:10.1074/jbc.M413085200
37. Steen M, Kirchberger T, Guse AH. NAADP mobilizes calcium from the endoplasmic reticular Ca(2+) store in T-lymphocytes. J Biol Chem (2007) 282:18864–71. doi:10.1074/jbc.M610925200
38. Langhorst MF, Schwarzmann N, Guse AH. Ca2+ release via ryanodine receptors and Ca2+ entry: major mechanisms in NAADP-mediated Ca2+ signaling in T-lymphocytes. Cell Signal (2004) 16:1283–9. doi:10.1016/j.cellsig.2004.03.013
39. Dammermann W, Zhang B, Nebel M, Cordiglieri C, Odoardi F, Kirchberger T, et al. NAADP-mediated Ca2+ signaling via type 1 ryanodine receptor in T cells revealed by a synthetic NAADP antagonist. Proc Natl Acad Sci U S A (2009) 106:10678–83. doi:10.1073/pnas.0809997106
40. Cordiglieri C, Odoardi F, Zhang B, Nebel M, Kawakami N, Klinkert WEF, et al. Nicotinic acid adenine dinucleotide phosphate-mediated calcium signalling in effector T cells regulates autoimmunity of the central nervous system. Brain J Neurol (2010) 133:1930–43. doi:10.1093/brain/awq135
41. Davis LC, Morgan AJ, Chen JL, Snead CM, Bloor-Young D, Shenderov E, et al. NAADP activates two-pore channels on T cell cytolytic granules to stimulate exocytosis and killing. Curr Biol (2012) 22:2331–7. doi:10.1016/j.cub.2012.10.035
42. Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature (2009) 459:596–600. doi:10.1038/nature08030
43. Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol (2009) 186:201–9. doi:10.1083/jcb.200904073
44. Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rötzer K, et al. The two-pore channel TPCN2 mediates NAADP-dependent Ca(2+)-release from lysosomal stores. Pflügers Arch (2009) 458:891–9. doi:10.1007/s00424-009-0690-y
45. Beck A, Kolisek M, Bagley LA, Fleig A, Penner R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J (2006) 20:962–4. doi:10.1096/fj.05-5538fje
46. Lange I, Penner R, Fleig A, Beck A. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium (2008) 44:604–15. doi:10.1016/j.ceca.2008.05.001
47. Kirchberger T, Moreau C, Wagner GK, Fliegert R, Siebrands CC, Nebel M, et al. 8-Bromo-cyclic inosine diphosphoribose: towards a selective cyclic ADP-ribose agonist. Biochem J (2009) 422:139–49. doi:10.1042/BJ20082308
48. Tóth B, Csanády L. Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J Biol Chem (2010) 285:30091–102. doi:10.1074/jbc.M109.066464
49. Zhang F, Jin S, Yi F, Li P-L. TRP-ML1 functions as a lysosomal NAADP-sensitive Ca2+ release channel in coronary arterial myocytes. J Cell Mol Med (2009) 13:3174–85. doi:10.1111/j.1582-4934.2008.00486.x
50. Walseth TF, Lin-Moshier Y, Jain P, Ruas M, Parrington J, Galione A, et al. Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide phosphate (NAADP)-binding proteins in sea urchin egg. J Biol Chem (2012) 287:2308–15. doi:10.1074/jbc.M111.306563
51. Lin-Moshier Y, Walseth TF, Churamani D, Davidson SM, Slama JT, Hooper R, et al. Photoaffinity labeling of nicotinic acid adenine dinucleotide phosphate (NAADP) targets in mammalian cells. J Biol Chem (2012) 287:2296–307. doi:10.1074/jbc.M111.305813
52. Clapper DL, Walseth TF, Dargie PJ, Lee HC. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J Biol Chem (1987) 262:9561–8.
53. Lee HC, Walseth TF, Bratt GT, Hayes RN, Clapper DL. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J Biol Chem (1989) 264:1608–15.
54. Guse AH, da Silva CP, Emmrich F, Ashamu GA, Potter BV, Mayr GW. Characterization of cyclic adenosine diphosphate-ribose-induced Ca2+ release in T lymphocyte cell lines. J Immunol (1995) 1950(155):3353–9.
55. da Silva CP, Potter BV, Mayr GW, Guse AH. Quantification of intracellular levels of cyclic ADP-ribose by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl (1998) 707:43–50. doi:10.1016/S0378-4347(97)00622-1
56. Schwarzmann N, Kunerth S, Weber K, Mayr GW, Guse AH. Knock-down of the type 3 ryanodine receptor impairs sustained Ca2+ signaling via the T cell receptor/CD3 complex. J Biol Chem (2002) 277:50636–42. doi:10.1074/jbc.M209061200
57. Guse AH, Tsygankov AY, Weber K, Mayr GW. Transient tyrosine phosphorylation of human ryanodine receptor upon T cell stimulation. J Biol Chem (2001) 276:34722–7. doi:10.1074/jbc.M100715200
58. Kunerth S, Langhorst MF, Schwarzmann N, Gu X, Huang L, Yang Z, et al. Amplification and propagation of pacemaker Ca2+ signals by cyclic ADP-ribose and the type 3 ryanodine receptor in T cells. J Cell Sci (2004) 117:2141–9. doi:10.1242/jcs.01063
59. Guse AH, Berg I, da Silva CP, Potter BV, Mayr GW. Ca2+ entry induced by cyclic ADP-ribose in intact T-lymphocytes. J Biol Chem (1997) 272:8546–50. doi:10.1074/jbc.272.13.8546
60. Guse AH. Structure-activity relationship of cyclic ADP-ribose, an update. J Chin Pharm Sci (2013) 22:127–36. doi:10.5246/jcps.2013.02.017
61. Zocchi E, Guida L, Franco L, Silvestro L, Guerrini M, Benatti U, et al. Free ADP-ribose in human erythrocytes: pathways of intra-erythrocytic conversion and non-enzymic binding to membrane proteins. Biochem J (1993) 295(Pt 1):121–30.
62. Kun E, Chang AC, Sharma ML, Ferro AM, Nitecki D. Covalent modification of proteins by metabolites of NAD+. Proc Natl Acad Sci U S A (1976) 73:3131–5. doi:10.1073/pnas.73.9.3131
63. Jacobson EL, Cervantes-Laurean D, Jacobson MK. ADP-ribose in glycation and glycoxidation reactions. Adv Exp Med Biol (1997) 419:371–9. doi:10.1007/978-1-4419-8632-0_49
64. Canales J, Pinto RM, Costas MJ, Hernández MT, Miró A, Bernet D, et al. Rat liver nucleoside diphosphosugar or diphosphoalcohol pyrophosphatases different from nucleotide pyrophosphatase or phosphodiesterase I: substrate specificities of Mg(2+)-and/or Mn(2+)-dependent hydrolases acting on ADP-ribose. Biochim Biophys Acta (1995) 1246:167–77. doi:10.1016/0167-4838(94)00191-I
65. Yang H, Slupska MM, Wei YF, Tai JH, Luther WM, Xia YR, et al. Cloning and characterization of a new member of the Nudix hydrolases from human and mouse. J Biol Chem (2000) 275:8844–53. doi:10.1074/jbc.275.12.8844
66. Bernet D, Pinto RM, Costas MJ, Canales J, Cameselle JC. Rat liver mitochondrial ADP-ribose pyrophosphatase in the matrix space with low Km for free ADP-ribose. Biochem J (1994) 299(Pt 3):679–82.
67. Perraud A-L, Shen B, Dunn CA, Rippe K, Smith MK, Bessman MJ, et al. NUDT9, a member of the Nudix hydrolase family, is an evolutionarily conserved mitochondrial ADP-ribose pyrophosphatase. J Biol Chem (2003) 278:1794–801. doi:10.1074/jbc.M205601200
68. Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature (2001) 411:595–9. doi:10.1038/35079100
69. Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, et al. Immunocyte Ca2+ influx system mediated by LTRPC2. Science (2001) 293:1327–30. doi:10.1126/science.1062473
70. Bastide B, Snoeckx K, Mounier Y. ADP-ribose stimulates the calcium release channel RyR1 in skeletal muscle of rat. Biochem Biophys Res Commun (2002) 296:1267–71. doi:10.1016/S0006-291X(02)02073-9
71. Yu P, Wang Q, Zhang L-H, Lee H-C, Zhang L, Yue J. A cell permeable NPE caged ADP-ribose for studying TRPM2. PLoS ONE (2012) 7:e51028. doi:10.1371/journal.pone.0051028
72. Gasser A, Guse AH. Determination of intracellular concentrations of the TRPM2 agonist ADP-ribose by reversed-phase HPLC. J Chromatogr B Analyt Technol Biomed Life Sci (2005) 821:181–7. doi:10.1016/j.jchromb.2005.05.002
73. Gelman L, Deterre P, Gouy H, Boumsell L, Debré P, Bismuth G. The lymphocyte surface antigen CD38 acts as a nicotinamide adenine dinucleotide glycohydrolase in human T lymphocytes. Eur J Immunol (1993) 23:3361–4. doi:10.1002/eji.1830231245
74. Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, et al. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science (1993) 262:1056–9. doi:10.1126/science.8235624
75. Kim UH, Han MK, Park BH, Kim HR, An NH. Function of NAD glycohydrolase in ADP-ribose uptake from NAD by human erythrocytes. Biochim Biophys Acta (1993) 1178:121–6. doi:10.1016/0167-4889(93)90001-6
76. Albeniz I, Demir O, Nurten R, Bermek E. NAD glycohydrolase activities and ADP-ribose uptake in erythrocytes from normal subjects and cancer patients. Biosci Rep (2004) 24:41–53. doi:10.1023/B:BIRE.0000037755.42767.a4
77. Ferrero E, Saccucci F, Malavasi F. The making of a leukocyte receptor: origin, genes and regulation of human CD38 and related molecules. Chem Immunol (2000) 75:1–19. doi:10.1159/000058763
78. Fonfria E, Marshall ICB, Benham CD, Boyfield I, Brown JD, Hill K, et al. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol (2004) 143:186–92. doi:10.1038/sj.bjp.0705914
79. Bonicalzi M-E, Haince J-F, Droit A, Poirier GG. Regulation of poly(ADP-ribose) metabolism by poly(ADP-ribose) glycohydrolase: where and when? Cell Mol Life Sci (2005) 62:739–50. doi:10.1007/s00018-004-4505-1
80. Buelow B, Song Y, Scharenberg AM. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J Biol Chem (2008) 283:24571–83. doi:10.1074/jbc.M802673200
81. Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell (2002) 9:163–73. doi:10.1016/S1097-2765(01)00438-5
82. Miller BA. The role of TRP channels in oxidative stress-induced cell death. J Membr Biol (2006) 209:31–41. doi:10.1007/s00232-005-0839-3
83. Melzer N, Hicking G, Göbel K, Wiendl H. TRPM2 cation channels modulate T cell effector functions and contribute to autoimmune CNS inflammation. PLoS ONE (2012) 7:e47617. doi:10.1371/journal.pone.0047617
84. Olabisi OA, Soto-Nieves N, Nieves E, Yang TTC, Yang X, Yu RYL, et al. Regulation of transcription factor NFAT by ADP-ribosylation. Mol Cell Biol (2008) 28:2860–71. doi:10.1128/MCB.01746-07
85. Valdor R, Schreiber V, Saenz L, Martínez T, Muñoz-Suano A, Dominguez-Villar M, et al. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol Immunol (2008) 45:1863–71. doi:10.1016/j.molimm.2007.10.044
86. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13:907–15. doi:10.1038/ni.2386
87. Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med (2008) 14:738–47. doi:10.1038/nm1758
88. Hardaker L, Bahra P, de Billy BC, Freeman M, Kupfer N, Wyss D, et al. The ion channel transient receptor potential melastatin-2 does not play a role in inflammatory mouse models of chronic obstructive pulmonary diseases. Respir Res (2012) 13:30. doi:10.1186/1465-9921-13-30
Keywords: calcium signaling, T-lymphocyte, calcium release, nicotinic acid adenine dinucleotide phosphate, cyclic ADP-ribose, adenosine diphosphoribose, TRPM2 cation channels, calcium entry
Citation: Ernst IMA, Fliegert R and Guse AH (2013) Adenine dinucleotide second messengers and T-lymphocyte calcium signaling. Front. Immunol. 4:259. doi: 10.3389/fimmu.2013.00259
Received: 06 June 2013; Accepted: 15 August 2013;
Published online: 29 August 2013.
Edited by:
Gergely Toldi, Semmelweis University, HungaryCopyright: © 2013 Ernst, Fliegert and Guse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas H. Guse, The Calcium Signalling Group, Department of Biochemistry and Molecular Cell Biology, University Medical Center Hamburg-Eppendorf, Martinistrasse 52, 20246 Hamburg, Germany e-mail:Z3VzZUB1a2UuZGU=
†Insa M. A. Ernst and Ralf Fliegert have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.