
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 August 2013
Sec. Inflammation
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00237
This article is part of the Research Topic The role of arginase in endothelial dysfunction View all 13 articles
 Lih Kuo1,2*
Lih Kuo1,2* Travis W. Hein2
Travis W. Hein2
Overproduction of reactive oxygen species, i.e., oxidative stress, is associated with the activation of redox signaling pathways linking to inflammatory insults and cardiovascular diseases by impairing endothelial function and consequently blood flow dysregulation due to microvascular dysfunction. This review focuses on the regulation of vasomotor function in the coronary microcirculation by endothelial nitric oxide (NO) during oxidative stress and inflammation related to the activation of l-arginine consuming enzyme arginase. Superoxide produced in the vascular wall compromises vasomotor function by not only scavenging endothelium-derived NO but also inhibiting prostacyclin synthesis due to formation of peroxynitrite. The upregulation of arginase contributes to the deficiency of endothelial NO and microvascular dysfunction in various vascular diseases by initiating or following oxidative stress and inflammation. Hydrogen peroxide, a diffusible and stable oxidizing agent, exerts vasodilator function and plays important roles in the physiological regulation of coronary blood flow. In occlusive coronary ischemia, the release of hydrogen peroxide from the microvasculature helps to restore vasomotor function of coronary collateral microvessels with exercise training. However, excessive production and prolonged exposure of microvessels to hydrogen peroxide impairs NO-mediated endothelial function by reducing l-arginine availability through hydroxyl radical-dependent upregulation of arginase. The redox signaling can be a double-edged sword in the microcirculation, which helps tissue survival in one way by improving vasomotor regulation and elicits oxidative stress and tissue injury in the other way by causing vascular dysfunction. The impact of vascular arginase on the development of vasomotor dysfunction associated with angiotensin II receptor activation, hypertension, ischemia-reperfusion, hypercholesterolemia, and inflammatory insults is discussed.
A normal function of the vascular endothelium involving responses to physical (1), chemical (2, 3), and electrical (4, 5) stimuli is essential to maintain microvascular homeostasis and regulate local blood flow by changing vasomotor activity via release of endothelium-derived vasodilators, e.g., nitric oxide (NO), prostacyclin (PGI2), C-type natriuretic peptide, and hyperpolarizing factors (EDHF). The endothelium also releases vasoconstrictors such as endothelin-1, prostaglandin H/F, thromboxane, and angiotensin. Endothelial dysfunction is one of the earliest markers of vascular abnormalities observed in many cardiovascular diseases associated with oxidative stress due to excessive production of reactive oxygen species (ROS). Redox regulation of proteins by moderate levels of ROS is indispensable for signaling pathways underlying the regulation of subcellular and cellular activity as well as cardiovascular function (6–8). Notably, superoxide and hydrogen peroxide (H2O2) are the most common and important ROS involved in the physiological and pathophysiological events (6–8).
Superoxide is produced by several enzyme systems in the cell and it is converted to H2O2 by superoxide dismutase. H2O2 itself is a potent oxidizing agent that can be converted to hydroxyl radical in the presence of ferric compounds. H2O2 can be degraded by catalase to form H2O and an oxygen molecule. Compared with superoxide, H2O2 is stable, lacks charge, has longer half-life, is cell permeable, and can diffuse across longer distances. Therefore, its physical properties are suitable for second-messenger signaling (7, 8). Because a proper delivery of oxygen and nutrients to the tissue is essential for the normal function of an organ, in this review we will discuss the roles of superoxide and H2O2 in the physiological and pathophysiological regulation of vasomotor activity of resistance arterioles where blood flow is primarily controlled, with special focus on the coronary microcirculation. The deficiency of endothelium-derived vasodilators such as NO and PGI2 in relation to oxidative stress and the l-arginine consuming enzyme arginase is discussed.
l-Arginine is the precursor for NO synthesis from three different isoforms of NO synthase (NOS). The endothelial NOS (eNOS) is the main isoform contributing directly to the regulation of vasomotor activity. In healthy human adults, it was estimated that 1.2% of arginine flux in the plasma contributes to the formation of NO and about 54% of whole body NO formation is derived from plasma arginine (9), although the fraction of l-arginine flux for NO production in the vasculature is unclear. Experimental data demonstrate that acute exogenous arginine provision can increase NO production (10, 11) and NO-mediated vasodilation (11, 12) despite the fact that the intracellular arginine level far exceeds the Km of eNOS (13). It appears that the extracellular l-arginine exerts a significant impact on the synthesis of NO from the membrane-bound eNOS.
Besides NOS, arginase is another major l-arginine consuming enzyme, which converts l-arginine to ornithine and urea. Arginase is expressed most abundantly in the liver for ammonia detoxification via the urea cycle (14). Studies in the cardiovascular system have shown that endothelia (12, 15–18), vascular smooth muscle cells (12, 17, 19), macrophages (20, 21), and red blood cells (22), which do not possess the complete urea cycle enzymes, also express arginase. In humans, about 15% of plasma arginine flux is associated with extrahepatic arginase activity (9). There are two isoforms of arginase. Type 1 arginase (Arg-I) is cytosolic and predominately expressed in the liver. In extrahepatic tissues and cells, a low level of Arg-I expression has also been detected. Type 2, or mitochondrial, arginase (Arg-II) is expressed with low levels in brain, kidney, intestine, red blood cells, and immune cells. Arg-I and -II are derived from distinct genes located on different chromosomes (14) and can be induced or regulated independently by a wide array of agents/factors (23, 24). Although these two arginase isoforms are expressed in a variety of cells, their distribution varies with tissue/organ and cell types (25). In the vasculature, both isoforms of arginase have been identified and their expression is highly regulated for physiological and pathophysiological processes (17) but the relative level of expression may be species dependent (19, 26–28).
Synthesis and release of the vasodilator NO from eNOS, in response to various physiological or pharmacological stimulations, can be related to the substrate bioavailability (10, 11) and thus influence vasomotor activity (11, 12). In this regard, change of protein expression and activity of arginase is expected to have an impact on NO synthesis by affecting the l-arginine level. From the biochemical standpoint, the Km of arginase for l-arginine in mammals, including humans, is reported to be around 0.5–29 mM (14). Although the Km of NOS (1–20 μM) (29) is much lower than that of arginase, taking into consideration their Vmax enzyme activities (1400 μmol⋅min−1⋅mg−1 for arginase vs. 900 μmol⋅min−1⋅mg−1 for NOS), the arginase is capable of competing with NOS for their substrate arginine (24). Based on the kinetic analysis of these two enzymes, the relative activity of NOS to arginase, in terms of consuming arginine, is diminished with either increasing arginine concentration or decreasing NOS to arginase molar ratio (24). Therefore, arginase activity can exceed NOS activity at higher levels of arginine or at higher arginase to NOS molar concentrations. Interestingly, the competition between NOS and arginase for arginine is more pronounced at lower levels of arginine (24). In terms of functional interpretation, the competitiveness (or importance) of arginase against NOS becomes apparent under conditions with upregulated arginase protein and limited supply of l-arginine.
Interestingly, intravenous administration of arginase causes constriction of cerebral arterioles and enhances platelet aggregation in mice (30), implicating that exogenous arginase may influence endothelial function through attenuation of NO production. However, the direct role of arginase in vasomotor regulation is unclear since the confounding effects from the changes in systemic hemodynamics and neuro-humoral factors cannot be excluded in this in vivo preparation. Using an isolated vessel approach, the role of endogenous arginase in vasomotor regulation of NO-mediated vasodilation was demonstrated for the first time in pressurized coronary arterioles (12). It was found that coronary arterioles express Arg-I in both endothelial and smooth muscle cells, and the NO production, as well as NO-mediated vasodilation, is enhanced by inhibiting arginase activity (12). It appears that endogenous arginase plays a counteracting role in the regulation of NO production and thus its associated vasomotor activity. The l-arginine-dependent NO-mediated vasodilation was also observed in various microvascular beds (11, 30–32) including human coronary arterioles (33), suggesting that l-arginine can be a limiting factor for the stimulated NO synthesis in the microcirculation. On the other hand, recent studies on cardiovascular diseases have implicated that upregulation of a specific arginase isoform in the vasculature may contribute to the development of vascular disease linked to l-arginine deficiency and reduced NO production (34, 35), especially under conditions with elevated level of angiotensin II (Ang II), hypertension, and inflammation, all of which are closely associated with oxidative stress (36).
In animal models of hypertension and myocardial hypertrophy, the excessive ROS release associated with renin-angiotensin system activation has been well documented (37, 38). However, the vasomotor action of Ang II in the intact heart is controversial. For example, a decrease (39, 40), an increase (41, 42), or a transient decrease followed by an increase (43, 44) in coronary blood flow by Ang II was reported. Although this inconsistency may be a result of using different animal models or experimental approaches, the complexity of flow regulation in the intact heart may be largely responsible for these diverse findings. Moreover, coronary vasomotor responses are influenced by the neural activity and by the changes in local hemodynamics and cardiac metabolism (44–46). The precise action of Ang II in the coronary microvasculature is difficult to assess in the intact heart because this peptide has direct and indirect actions on these biological factors (43, 44). It is also unclear whether the ROS generated by Ang II can modulate coronary microvascular reactivity in view that enhanced superoxide production by Ang II in endothelial cells is well recognized (47, 48).
Using isolated vessel approaches to eliminate the confounding influences from systemic and local effects inherited in in vivo preparations, it was found that Ang II, via activation of its type 1 (AT1) receptors, evokes a moderate vasoconstriction of porcine coronary arterioles (50–80 μm in diameter) at low concentrations (∼1 nM) but a marked vasodilation at higher concentrations (>10 nM) via AT2 receptor activation (49). This vasodilator effect is likely mediated by the released endothelial NO via bradykinin receptor signaling (50). Interestingly, in the human coronary circulation AT2 receptors were found expressed in the microvasculature only (50). Depending upon the concentration used, Ang II appears to exert different vasomotor activities in the coronary microvessels, and thus may explain the inconsistent observations on coronary flow changes in vivo. Moreover, pre-treating the isolated coronary arterioles with a sub-vasomotor concentration of Ang II (0.1 nM) for 60 min caused an elevation of superoxide production in the vessel wall and inhibited NO production and endothelium-dependent, NO-mediated dilation in response to adenosine, a potent metabolic vasodilator in the heart. This inhibitory effect was prevented by AT1 receptor blocker losartan, superoxide scavenger TEMPOL, or NAD(P)H oxidase inhibitor apocynin (49). These microvascular findings indicate that Ang II, at the level without causing vasomotor activity, exerts an adverse effect on NO-mediated vasodilator function via superoxide generated by AT1 receptor-dependent activation of NAD(P)H oxidase. Because acute myocardial ischemia (<60 min) upregulates the cardiac renin-angiotensin system and impairs coronary flow regulation (51, 52), it is speculated that the small elevation of local Ang II at sub-vasomotor levels in the heart during disease states may cause oxidative stress at the local microvascular domain and elicit focal vasoconstriction and myocardial ischemia secondary to the reduced NO bioavailability. In addition to the local vascular spasm, the deficiency of basal NO release from the endothelium, which is subjected to continuous shear stress stimulation, is expected to aggravate ischemic insult by promoting platelet aggregation and thrombosis formation (53, 54) in the microvasculature.
The blunted endothelium-dependent vasorelaxation in aging animals was recently reported to be associated with excessive vascular formation of ROS and upregulation of NAD(P)H oxidase subunits (e.g., Nox-1 and p22-phox), Arg-I, and AT1 and AT2 receptor expression in a manner sensitive to NAD(P)H oxidase inhibition and antioxidants (55). These findings suggest the initiation of vascular dysfunction by oxidative stress linking to Ang II receptors and arginase. However, the role and signaling pathway for Ang II receptor activation leading to NO deficiency in relation to arginase activity and vasomotor regulation is incompletely understood. In cultured bovine aortic endothelial cells, Ang II (0.1 μM, 24 h incubation) was recently shown to increase arginase activity and Arg-I expression through Gα12/13 protein-coupled AT1 receptor activation (56). The upregulated Arg-I appears to reduce l-arginine bioavailability and hamper NO production. The adverse effect of Arg-I is mediated by the activation of p38 mitogen-activated protein kinase (MAPK) pathways through RhoA/Rho kinase signaling (56). Although the threshold concentration of Ang II necessary for evoking NO deficiency and endothelial dysfunction in the above cell-culture study is unclear, chronic administration of Ang II (42 μg/kg/h, 2 weeks) in the mice was recently shown to impair endothelium-dependent NO-mediated relaxation of a tissue strip from corpus cavernosum (57). In agreement with the findings in cell culture (56), the Ang II-evoked endothelial dysfunction is mediated by the p38 MAPK-dependent upregulation of arginase (57). However, the responsible isozyme is Arg-II rather than Arg-I. Interestingly, inhibition of p38 MAPK not only prevents the effects of Ang II on endothelial function and arginase activity/expression, it also attenuates the increased systemic blood pressure by Ang II.
Hypertension is a major risk factor for coronary artery disease by impairing endothelium-dependent NO-mediated vasodilation (58) in the form of diminished bioavailability of NO, increased Ang II-dependent production of superoxide (59), and decreased endothelial levels of eNOS co-factor tetrahydrobiopterin (BH4) (60) or substrate l-arginine (61). In some studies, administration of l-arginine has been shown to restore endothelium-dependent vasodilator function in patients with essential hypertension (61) and to normalize coronary hemodynamics (62) and systemic blood pressure with enhanced NO production in hypertensive rats (63, 64). In deoxycorticosterone acetate (DOCA)-salt hypertensive rats, expression and activity of Arg-I protein in the aorta are elevated and correlate positively with blood pressure, suggesting the participation of this enzyme in pathophysiology of arterial hypertension (65). The upregulation of Arg-I in the coronary arteriolar wall was reported to contribute in part to the impairment of endothelial NO production and vasodilation by reducing l-arginine availability in hypertensive pigs (66). In the animal model of genetic (67, 68) or metabolic (69) form of hypertension, chronic inhibition of arginase was recently shown to improve endothelium-dependent vascular function (67–69), reduce cardiac fibrosis (68), prevent vascular remodeling and Arg-I overexpression (68), inhibit insulin-resistance (69), reduce oxidative stress (69), and alleviate hypertension (67–69). Although the evidence for the link of oxidative stress and inflammation to the pathogenesis of hypertension, and vice versa, is well supported in both experimental and clinical studies (70), it is unclear whether the direct elevation of mechanical stress on the vascular wall or the associated oxidative stress and inflammation contribute to the upregulation of vascular arginase during hypertension. Moreover, oxidative stress can probably promote inflammation and, conversely, inflammation per se may induce tissue damage and promote oxidative stress. Their individual contributions to the vasomotor dysfunction related to NO deficiency are difficult to define in vivo due to the complex and intertwined biological events and multifactorial processes involved in the development of vascular pathophysiology. However, recent studies using cell culture (71–75) and isolated vessel (49, 73, 76–80) approaches suggest that pro-inflammatory factors such as C-reactive protein (CRP), tumor necrosis factor-α (TNF-α), and oxidized low-density lipoprotein (Ox-LDL) are capable of causing endothelial NO deficiency and vasomotor dysfunction through elevated arginase and oxidative stress.
The dysfunction of coronary microvascular endothelial cells is closely associated with the development of various inflammatory diseases in the heart (81, 82). The inflammatory marker CRP, which has recently been established as a cardiovascular risk factor, also exhibits adverse effects on endothelium-dependent NO-mediated vasodilator function and NO production in isolated coronary (78) and retinal (83) arterioles by enhancing NAD(P)H oxidase-mediated superoxide production via p38 MAPK activation. Since activation of the endothelial p38 MAPK pathway by oxidative stress also has been documented (84, 85), the positive feedback between p38 MAPK and superoxide production is expected to exacerbate the oxidative insult on the vascular wall. In rats, treatment with human CRP at concentrations achievable in patients with cardiovascular disease impairs endothelium-dependent vasomotor function linked with uncoupling of eNOS due to reduction in dimerization of the enzyme, as well as inhibition of GTP cyclohydrolase I (GTPCH1), the rate-limiting enzyme in BH4 biosynthesis, and decrease in BH4 levels (86). Human CRP also causes the activation of NAD(P)H oxidase resulting in eNOS uncoupling directly or via inhibition of GTPCH1 or oxidation of BH4 (86). These studies provide the first evidence for the adverse action of human CRP in vivo manifested by impairing eNOS-dependent vasodilation and uncoupling of eNOS. Thus, given the importance of CRP-induced pro-oxidative effects and resultant eNOS inhibition, CRP appears to be a key molecule to accentuate endothelial dysfunction and contribute to blood flow dysregulation.
The CRP also displays an adverse effect on arachidonic acid-prostanoid pathways in the endothelium (79). The formation of peroxynitrite from NO (basal release) and superoxide (CRP-stimulated release) in the endothelium appears to compromise PGI2 production, and thus PGI2-mediated vasodilation, by inhibiting PGI2 synthase activity through tyrosine nitration (79). Peroxynitrite also contributes to eNOS uncoupling by oxidizing the co-factor BH4 (87) and thus reduces NO production. Although there is no evidence at the present time to suggest a direct impact of CRP on vascular arginase expression or activity, the elevation of serum Arg-I has been shown to correlate positively with CRP in asthmatic patients (88). Interestingly, the imbalance in l-arginine metabolism via arginase and NOS has been considered as a unifying element of asthma pathophysiology (89). The upregulation of arginase in the vasculature is expected to compromise endothelial NO with enhanced oxidative stress promoting peroxynitrite formation and hypertension during inflammatory insults in a manner similar to the development of allergic asthma in chronic inflammatory airway diseases (90) with primary and secondary forms of pulmonary hypertension (91–93). The recent report on the close relation between asthma and metabolic syndrome (94), a major risk of cardiovascular disease with dysregulation of l-arginine metabolism (69), supports the emerging role of arginase in the general regulation of NO production and oxidative stress in inflammatory diseases.
Tumor necrosis factor-α is a pro-inflammatory cytokine and an important mediator of cardiovascular complications such as acute myocardial infarction, ischemia-reperfusion injury, atherosclerosis, chronic heart failure, and coronary arterial disease in association with diminished coronary blood flow. Treating the isolated coronary arterioles with a pathological concentration of TNF-α (1 ng/ml, 90 min) caused a significant reduction of NO release, enhanced superoxide production, and c-Jun N-terminal kinase (JNK) phosphorylation in arteriolar endothelial cells and impaired endothelium-dependent dilation to adenosine (77). TNF-α participates in the pathogenesis and progression of myocardial injury induced by ischemia-reperfusion (95). In the model of ischemia-reperfusion injury of porcine coronary arterioles, the upregulation of Arg-I, via de novo protein synthesis pathway, causes endothelial dysfunction and NO deficiency (96). Using genetic tools to manipulate TNF-α expression in the mouse, it was found that myocardial ischemia-reperfusion evokes superoxide-dependent endothelial dysfunction and NO deficiency via upregulation of Arg-I, in a manner correlating with TNF-α expression (97). In contrast with the insults elicited by Ang II and CRP, the TNF-α induced oxidative stress and endothelial dysfunction are associated with the activation of ceramide-induced activation of JNK and subsequent production of superoxide via xanthine oxidase (77, 98) rather than the signaling via p38 MAPK-activated NAD(P)H oxidase (49, 78). Recent clinical evidence has shown that arginase blockade improves endothelium-dependent NO-mediated vasodilation in patients with coronary artery disease (99) and increases NO-dependent microvascular perfusion in patients with heart failure (100). Interestingly, the systemic level of Arg-I correlates with the severity of heart failure (100) and Arg-I polymorphisms are associated with myocardial infarction and vascular remodeling (101). The elevated level of Arg-I appears to be a major risk and/or pathogenic factor in developing coronary ischemic disease and vascular pathophysiology.
Experimental studies have shown that the expression of arginase is elevated in a variety of vascular and immune cells with inflammation and oxidative stress (20, 21, 102–104), the conditions that are known to be associated with atherogenesis. Interestingly, l-arginine deficiency coupled to impaired NO-mediated vascular function has been reported in animals (105–108) and humans (109–112) with hypercholesterolemia or atherosclerosis, possibly due to upregulation of arginase in the disease state (34). Furthermore, transgenic mice with overexpression of endothelial Arg-II exhibit increased aortic atherosclerotic lesions (113). In apolipoprotein E deficient mice, the arginase activity of atherosclerotic aorta is significantly elevated (28, 108). In the same mouse model, inhibition of arginase activity or deletion of Arg-II gene alleviates oxidative stress in the endothelium, prevents NO deficiency, and restores endothelial function, suggesting the critical role of Arg-II in triggering ROS-dependent endothelial dysfunction in hypercholesterolemia (114). Since Arg-II blockade reduces superoxide formation via a pathway sensitive to NOS inhibition (114), the uncoupling of eNOS, due to l-arginine deficiency, appears to be involved in the arginase-dependent oxidative stress. It was found that Arg-II activity positively correlates with RhoA protein level in atherosclerotic aortas and that manipulation of RhoA/Rho kinase activity and expression directly affects enzymatic activity of Arg-II (28). In this regard, RhoA/Rho kinase activation is likely responsible for the increased Arg-II activity leading to vascular dysfunction and atheroma formation. Rho kinase activation also contributes to Arg-I-mediated coronary vascular dysfunction in diabetic rats and to NO deficiency induced by hyperglycemia in bovine aortic endothelial cells (115). In the rabbit model of hypercholesterolemia, the expression of both arginase isozymes is elevated in atherosclerotic aortas (27). However, the regulation and role of specific arginase isoforms in disease development remains to be determined.
In the coronary microcirculation, the endothelium-dependent NO-mediated dilation, compared to that mediated by the EDHF and the endothelial prostanoids, is more susceptible to the insult of Ox-LDL (116) than that of native LDL (3). The enhanced superoxide production and reduced l-arginine bioavailability are responsible for the observed endothelial dysfunction of coronary arterioles (3). In cultured human aortic endothelial cells, Ox-LDL activates lectin-like Ox-LDL receptor-1 (LOX-1) and subsequently increases Arg-II activity/expression and reciprocally inhibits NO production via RhoA/Rho kinase activation (117). Interestingly, the NO deficiency, as well as the increased arginase activity and ROS production, evoked by hypercholesterolemia or Ox-LDL are not observed in endothelial cells absent of LOX-1, suggesting the critical role of LOX-1 in mediating arginase-dependent NO deficiency and oxidative stress (117). The accumulation of superoxide is likely derived from the uncoupled eNOS and NAD(P)H oxidase because blockade of these enzymes attenuates oxidative stress (117). In the intact porcine coronary arterioles, the upregulated Arg-I contributes, in part, to the reduced NO production and impaired endothelium-dependent dilation evoked by Ox-LDL (118). However, it is unclear whether LOX-1 plays a role in this experimental model.
The elevated level of H2O2 has been detected under various pathophysiological conditions, including ischemia-reperfusion, inflammation, hypertension, diabetes, and atherosclerosis. The H2O2 can be released from various types of cells, including vascular cells (119, 120) and has been implicated, in some tissues, as an endothelium-derived hyperpolarizing factor exhibiting vasodilator activity (119). Extraluminal administration of H2O2 (1–100 μM) elicits concentration-dependent dilation of isolated coronary arterioles in part via an endothelium-dependent mechanism through cyclooxygenase (COX)-1-mediated release of PGE2 (121). H2O2 can also cause smooth muscle hyperpolarization and lead to vasodilation through the opening of calcium-activated potassium channels (121, 122). This vasodilator response plays a role in regulating coronary perfusion by recruiting blood flow to the heart during pressure reduction (i.e., autoregulation) (123) or metabolic activation (i.e., functional hyperemia) (124). Interestingly, in disease states, the vasodilator action of H2O2 appears to compensate for the impaired NO-mediated dilation linking to the uncoupling of eNOS with its co-factor BH4 (125) and to protect ischemia-reperfusion injury in the coronary microcirculation (126). In the pig model of coronary ischemia, the impaired NO-mediated vasodilation in collateral-dependent arterioles distal to chronic coronary occlusion was restored by exercise training (127). The beneficial effect of exercise on coronary arteriolar function was abolished by catalase, suggesting the contribution of H2O2 in compensating and restoring endothelium-dependent vasomotor function in the phase of collateral microvessel adaption to myocardial ischemia (127).
On the other hand, H2O2 can exert an adverse effect by reducing endothelial release of NO for vasodilation when the endothelium is exposed to a prolonged (e.g., 60 min) elevation of excessive H2O2 (e.g., 100 μM) (128). Interestingly, the dilation mechanisms involving the activation of COX, guanylyl cyclase, cytochrome-P450 monooxygenase, and potassium channels are not affected by H2O2 (128). Moreover, supplementation of l-arginine or inhibition of arginase restores H2O2-impaired vasomotor function, and the adverse effect of H2O2 can be prevented by inhibiting hydroxyl radical production (128). It appears that a high intravascular level of H2O2 selectively impairs endothelium-dependent NO-mediated dilation of coronary microvessels by reducing l-arginine availability. The formation of hydroxyl radicals leading to Arg-I overexpression is responsible for the adverse effect of H2O2 (128). Interestingly, it was recently shown that the oxidative stress elicited by peroxynitrite or H2O2 increases Arg-I activity/expression through protein kinase C-mediated activation of RhoA/Rho kinase in bovine aortic endothelial cells (129). It remains unclear whether hydroxyl radicals and protein kinase C contribute to the activation of Rho kinase in intact microvessels.
Although COX activation is known to mediate tissue inflammation and participate in vasomotor regulation (130), its linkage to arginase, another important enzyme related to the inflammation process (89), remains unclear. A recent study has shown that inhibition of arginase improves endothelial function and attenuates vascular COX-2, thromboxane synthase, and PGI2 synthase activities in the rat model of adjuvant-induced arthritis (131). Thus, arginase activation contributes to the augmentation of inflammatory enzyme activity related to prostanoid synthesis. Interestingly, arginase inhibition improved endothelial function, but it had no effect on the arthritis severity of the animal (131). It appears that this type of inflammatory insult targets vascular arginase and consequently leads to vascular disorder. While COX-2 inhibitors have been shown to reduce tumor growth through arginase inhibition (132, 133), administration of diclofenac, a non-steroidal anti-inflammatory drug against COX-2 (134) and phospholipase A2 (135), was found to cause tumor suppression via a mechanism related to the inhibition of tumor vascularization (136). Although the expression and activity of arginase in the vasculature was not evaluated in this study, it is speculated that the observed tumor suppression is attributable to the inhibition of vascular arginase since this enzyme has been shown to play an important role in the growth of vascular cells (35, 137–139). Although the direct link between COX and arginase in vasomotor regulation remains to be determined, the finding of the close association between these two enzymes in tumor-promoted angiogenesis (140) and in alleviating chronic hypertension and improving vascular endothelial function and vasomotor activity (68) may provide new direction and insights into this underdeveloped research area.
The arginase inhibitors currently available are not isoform selective and their specificity may be species dependent (17). Therefore, it is difficult to identify the role and function of a specific arginase isoform using pharmacological tools. With above limitations, genetic manipulation of an arginase isoform becomes an important strategy for more precise study of arginase function in a living system. Homozygous deletion of Arg-I is lethal to the animal in the perinatal period (141). In contrast, homozygous deletion of Arg-II in the mice does not cause significant changes in phenotype, except an elevation of plasma level of arginine (142). The observed increase in endothelial NO production and NO-mediated vasorelaxation, in conjunction with reduced vasoconstrictor response, in carotid arteries from Arg-II knockout mice (143) supports the idea that endothelial Arg-II plays a counteracting role in NO production and the associated vasomotor dysfunction. Deletion of Arg-II gene attenuates vascular disorder (i.e., impaired NO-mediated endothelial function and enhanced sympathetic vasoconstriction) in corpora cavernosal tissue of mice with type 1 diabetes, suggesting the detrimental role of Arg-II in this disease model (144). Arg-II appears to modulate not only vasomotor reactivity but also the physical property of the vascular wall by influencing NOS activity because Arg-II deficient mice exhibit decreased vascular stiffness in a manner sensitive to NOS inhibition (143). On the other hand, selective overexpression of human Arg-II gene in the endothelium causes systemic hypertension, impairs endothelium-dependent NO-mediated vasorelaxation, and promotes atherosclerotic lesions (113). These in vivo findings are in agreement with the observed adverse effect of Arg-II on NO-mediated endothelial function in cell culture. Moreover, the experimental data from an Arg-II knockout study indicate that the renal injuries observed in spontaneous or streptozotocin-induced diabetes animals are also mediated by Arg-II (145). However, genetic manipulation of Arg-I (partial deletion) in Arg-II deficient mice shows that upregulation of vascular Arg-I, rather than Arg-II, contributes to the diabetes (type I)-induced endothelial dysfunction, vascular stiffness, and coronary fibrosis (146), in which Rho kinase activation can be responsible for the observed pathophysiology (147). Using the same genetic approach, the detrimental role of Arg-I in mediating blood pressure elevation and vascular endothelial dysfunction was recently reported in the mice subjected to systemic hypertension induced by DOCA-salt (148). In diabetic human patients, Arg-I upregulation appears to be responsible for the impairment of coronary arteriolar dilation to an endothelium-dependent NO-mediated agonist (149).
Surprisingly, a recent study by Huynh et al. showed that Arg-II knockout mice start to display hypertension at 8 weeks old, despite the reduction in vasoconstrictor responsiveness (150). The observed changes in systemic hemodynamics are associated with left ventricular hypertrophy, diastolic dysfunction, and increased sympathetic activity (150). In contrast to the previous report in carotid arteries with Arg-II deletion by Lim et al. (143), the aortic relaxation to an NO-dependent agonist was not significantly enhanced in the Arg-II knockout mice (150), suggesting that the observed reduction in the vasoconstrictor response was not attributable to alterations in NO production. There is no clear explanation to the apparent discrepancies between these two Arg-II knockout studies, especially in the observed global changes in cardiovascular function and vasomotor regulation related to endothelial NO. Nevertheless, the study of Huynh et al. demonstrated a correlation between Arg-II and Rho kinase, suggesting a contribution of downregulation of Rho kinase to the observed reduction in the vasoconstrictor response in Arg-II deficiency (150). This is in agreement with the context that upregulation of arginase in the disease state may enhance Rho kinase activity/expression and consequently alter vasomotor activity because numerous studies have implicated a close association between Rho kinase and arginase in the development of vascular dysfunction (28, 56, 115, 117, 129).
Although recent studies using genetic approaches have provided significant insights into the contribution of specific arginase isozymes in vasomotor regulation in health and disease, the inconsistent results are often reported as discussed above. In view that arginase gene deletion might also alter expression of other genes or activate alternate signaling pathways to confound the consequences of initial gene deletion (151, 152), the interpretation of these results should be cautious. The gene–gene interaction and the development of compensatory and/or decompensatory biological responses, at local or systemic levels, with gene manipulation may contribute to the observed discrepancies, in addition to the variation of involved signaling molecules, age, gender, tissue/organ, species/strain, and experimental conditions.
At the protein level, although Arg-I and -II carry out the same catalytic function, they have different physicochemical characteristics, including immunological cross-reactivity, charge, and subcellular location (14). Because the expression pattern of specific arginase isoforms can be cell/tissue and animal species dependent (17), it is unclear at the present time why and how Arg-I and -II can be targeted differently. Interestingly, recent studies suggest that the catalytic efficiency of arginase can be modulated without altering protein expression (153). It appears that cysteine residues 303 in Arg-I can undergo S-nitrosylation and subsequently increase stability of the arginase trimer and reduce its Km for arginine (153). This increase in arginase activity can contribute to the endothelial dysfunction and reduced NO bioavailability (153). On the other hand, there is no cysteine in mammalian Arg-II that corresponds to cysteine 303 in Arg-I, suggesting that the post-translational modulation via S-nitrosylation might not occur in Arg-II (17). It is likely that S-nitrosylation elicited by the excessive production of NO during iNOS induction (e.g., inflammation) or by the formation of peroxynitrite during oxidative stress may contribute to a selective activation of Arg-I leading to endothelium-dependent vasomotor dysfunction. Moreover, uric acid has been demonstrated to increase arginase activity by increasing the affinity for arginine (154). This phenomenon is unlikely isoform selective because it is observed in the pulmonary arterial endothelial cell lysates (Arg-II) and rat kidney (Arg-II) and liver (Arg-I) homogenates (154). However, it was recently found that uric acid, at the concentrations reported to affect arginase activity (154), does not alter Arg-II activity in cultured human umbilical vein endothelial cells (155). The explanation for these inconsistent findings on uric acid-arginase interaction remains unclear. Hydroxyl radicals derived from H2O2 appear to specifically induce Arg-I expression and lead to endothelial dysfunction in coronary microvessels (128). Interestingly, biochemical studies in vitro indicate that Arg-I enzyme activity can be enhanced by hydroxyl radicals (156). Although it has not been demonstrated whether hydroxyl radicals also alter Arg-II activity, the activation of Arg-I, both in protein expression and activity, by oxidative stress (i.e., peroxynitrite and H2O2) in cultured endothelial cells also has been reported recently (129). In view that the increase of Arg-I activity (50%) is more than that of protein expression (35%) (129), the direct impact of these insults on arginase enzyme activity per se is apparent. Collectively, the above studies suggest the differential activation of arginase isozymes, depending upon the environment and the nature of the stimulation, in addition to the selective regulation of its protein expression in the vasculature. These differential regulation mechanisms may also contribute to the observed diversity and heterogeneity in involved arginase isoforms in vascular cell, as well as the exerted function, in different tissues, species, and diseases. Further studies on the differential activation of specific arginase isoforms are required.
Collectively, the NO bioavailability, determined by the synthesis/release and utilization/scavenging at the level of the endothelium, plays an important role in maintaining vascular homeostasis and function, as well as disease development linking to oxidative stress and inflammation. Redox signaling with a low level of ROS released from cardiomyocytes and/or vascular cells displays an indispensable role in maintaining microcirculatory homeostasis by regulating vasomotor activity in response to physiological challenges. The release of H2O2 from the vasculature helps to restore vasomotor function by compensating for NO deficiency in coronary collateral microvessels adapted to chronic myocardial ischemia with exercise training. Depending upon the disease model and the pathophysiological insult, the excessive and prolonged production of superoxide, via stress kinase-activated NAD(P)H oxidase or xanthine oxidase, and the subsequent exorbitant formation of H2O2, appear to generate oxidative stress and inflammation, which outweighs the benefits of vasoregulation by impairing endothelial function and possibly exhausting vasodilator reserve (Figure 1). The status and the balance of redox signaling in the vascular cells and their surrounding parenchymal tissues appear to modulate the vasomotor function of microvessels in health and disease.
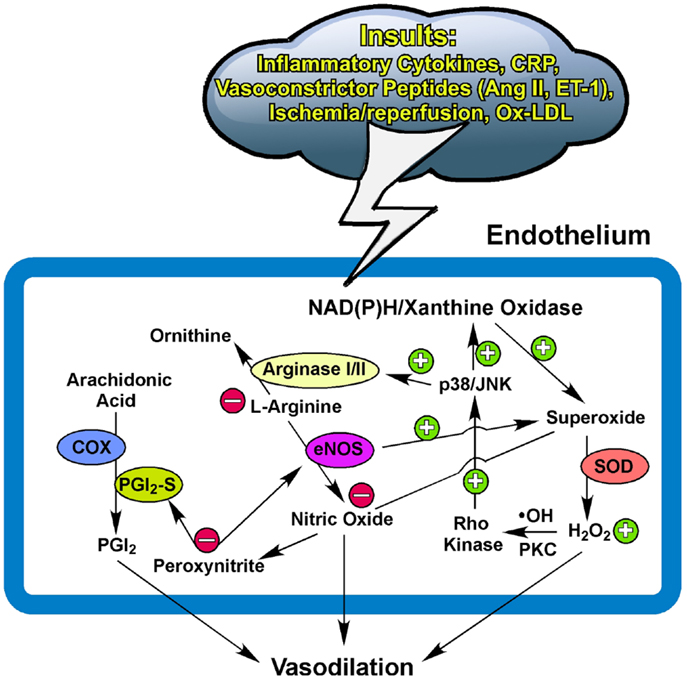
Figure 1. Potential pathways for redox and arginase modulation of vasomotor function. The low level of superoxide and hydrogen peroxide (H2O2) is essential for maintaining normal homeostasis of the endothelium to exert vasodilation in response to physiological stimulation. The excessive production of superoxide from the activated NAD(P)H oxidase and/or xanthine oxidase by insults from inflammatory cytokines, C-reactive protein (CRP), ischemia/reperfusion, oxidized low-density lipoprotein (Ox-LDL), or vasoconstrictor peptides such as angiotensin II (Ang II) and endothelin-1 (ET-1) (162– 165) scavenges the released nitric oxide and subsequently forms peroxynitrite. The prolonged and elevated production of H2O2 from superoxide dismutase (SOD) suppresses NO production by up-regulating vascular arginase via p38 mitogen-activated protein kinase or c-Jun-N-terminal kinase (JNK) signaling following the hydroxyl radical (∙OH)- or protein kinase C (PKC)-mediated activation of Rho kinase. The upregulated arginase limits substrate L-arginine availability to nitric oxide synthase (eNOS) for nitric oxide synthesis and also uncouples eNOS to promote superoxide production. The production of nitric oxide from eNOS and prostacyclin (PGI2) from cyclooxygenase (COX) and PGI2 synthase (PGI2-S) is inhibited by peroxynitrite due to uncoupling of eNOS and nitration of PGI2-S (see text for details). Peroxynitrite also activates arginase and consequently reduces nitric oxide production. These redox events, in company with arginase upregulation, compromise endothelial function and thus augment vascular tone and reduce nitric oxide-mediated arteriolar dilation for blood flow recruitment and regulation. +, enhance/increase/upregulate; −, inhibit/reduce/downregulate.
The converging evidence suggests that NO-mediated vascular function, including vasomotor activity, can be influenced by the arginase activity in the endothelium and/or its surroundings. The upregulation of arginase, in either protein or activity, contributes to vascular dysfunction in various vascular diseases by initiating or following oxidative stress and inflammation (Figure 1). Therefore, therapeutic inhibition of arginase may be useful for disease treatment. However, a global Arg-II deletion can develop hypertension, ventricular hypertrophy, and cardiac dysfunction with age (150). Because these cardiovascular disorders are not present at young age with Arg-II ablation, chronic Arg-II deficiency appears to elicit a series of cardiovascular remodeling (e.g., compensation and decompensation). Moreover, biochemical studies indicate that Arg-I and -II can exhibit different enzyme kinetics for substrate binding and products, as well as different sensitivities and responsiveness toward inhibitors (17, 157). These isozyme-dependent characteristics, in combination with the use of different experimental models and animal species, may complicate the experimental results, interpretations, and conclusions on the effect of arginase inhibition on endothelial function and vasomotor regulation under physiological and pathophysiological conditions. In this regard, the clinical benefits of inhibition of specific arginase isoforms for cardiovascular disease treatment are uncertain and deserve further investigation. It is worth noting that the systemic supplementation of antioxidants showed no benefit but instead promoted possible harmful effects in cardiovascular disease prevention or therapy (158–161). Oxidative stress and inflammation are two sides of the same coin and can be the cause or result of arginase upregulation in the vasculature via diverse signaling mechanisms. Localized manipulation of the redox system and arginase activity in a diseased vessel might be a useful strategy to improve flow regulation and thus enhance oxygen and nutrient delivery for tissue survival and recovery.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by National Institutes of Health grant HL-48179 (to Lih Kuo), Retina Research Foundation (to Lih Kuo and Travis W. Hein), and Scott & White Research Foundation (to Lih Kuo).
1. Kuo L, Davis MJ, Chilian WM. Endothelial modulation of arteriolar tone. News Physiol Sci (1992) 7:5–9.
2. Hein TW, Kuo L. cAMP-independent dilation of coronary arterioles to adenosine: role of nitric oxide, G proteins, and KATP channels. Circ Res (1999) 85:634–42. doi:10.1161/01.RES.85.7.634
3. Hein TW, Kuo L. LDLs impair vasomotor function of the coronary microcirculation: role of superoxide anions. Circ Res (1998) 83:404–14. doi:10.1161/01.RES.83.4.404
4. Kotecha N, Neild TO. Effects of endothelium-derived relaxing factor on the smooth muscle of the rat tail artery. Clin Exp Pharmacol Physiol (1986) 13:249–57. doi:10.1111/j.1440-1681.1986.tb00343.x
5. Jiang F, Li CG, Rand MJ. Mechanisms of electrical field stimulation-induced vasodilatation in the guinea-pig basilar artery: the role of endothelium. J Auton Pharmacol (1997) 17:71–6. doi:10.1046/j.1365-2680.1997.00444.x
6. Queisser N, Fazeli G, Schupp N. Superoxide anion and hydrogen peroxide-induced signaling and damage in angiotensin II and aldosterone action. Biol Chem (2010) 391:1265–79. doi:10.1515/BC.2010.136
7. Schroder E, Eaton P. Hydrogen peroxide as an endogenous mediator and exogenous tool in cardiovascular research: issues and considerations. Curr Opin Pharmacol (2008) 8:153–9. doi:10.1016/j.coph.2007.12.012
8. Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res (2005) 68:26–36. doi:10.1016/j.cardiores.2005.06.021
9. Castillo L, Beaumier L, Ajami AM, Young VR. Whole body nitric oxide synthesis in healthy men determined from [15N] arginine-to-[15N]citrulline labeling. Proc Natl Acad Sci U S A (1996) 93:11460–5. doi:10.1073/pnas.93.21.11460
10. Shin S, Mohan S, Fung HL. Intracellular L-arginine concentration does not determine NO production in endothelial cells: implications on the “L-arginine paradox.” Biochem Biophys Res Commun (2011) 414:660–3. doi:10.1016/j.bbrc.2011.09.112
11. Vukosavljevic N, Jaron D, Barbee KA, Buerk DG. Quantifying the L-arginine paradox in vivo. Microvasc Res (2006) 71:48–54. doi:10.1016/j.mvr.2005.10.006
12. Zhang C, Hein TW, Wang W, Chang CI, Kuo L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J (2001) 15:1264–6.
13. Flam BR, Eichler DC, Solomonson LP. Endothelial nitric oxide production is tightly coupled to the citrulline-NO cycle. Nitric Oxide (2007) 17:115–21. doi:10.1016/j.niox.2007.07.001
14. Jenkinson CP, Grody WW, Cederbaum SD. Comparative properties of arginases. Comp Biochem Physiol B Biochem Mol Biol (1996) 114:107–32. doi:10.1016/0305-0491(95)02138-8
15. Wu G, Meininger CJ. Impaired arginine metabolism and NO synthesis in coronary endothelial cells of the spontaneously diabetic BB rat. Am J Physiol (1995) 269:H1312–8.
16. Krause BJ, Carrasco-Wong I, Caniuguir A, Carvajal J, Farias M, Casanello P. Endothelial eNOS/arginase imbalance contributes to vascular dysfunction in IUGR umbilical and placental vessels. Placenta (2013) 34:20–8. doi:10.1016/j.placenta.2012.09.015
17. Morris SM Jr. Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol (2009) 157:922–30. doi:10.1111/j.1476-5381.2009.00278.x
18. Buga GM, Singh R, Pervin S, Rogers NE, Schmitz DA, Jenkinson CP, et al. Arginase activity in endothelial cells: inhibition by NG-hydroxy-L-arginine during high-output NO production. Am J Physiol (1996) 271:H1988–98.
19. Wei LH, Jacobs AT, Morris SMJ, Ignarro LJ. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol (2000) 279:C248–56.
20. Chang C-I, Liao JC, Kuo L. Arginase modulates nitric oxide production in activated macrophages. Am J Physiol (1998) 274:H342–8.
21. Chang CI, Zoghi B, Liao JC, Kuo L. The involvement of tyrosine kinases, cyclic AMP/protein kinase A, and p38 mitogen-activated protein kinase in IL-13-mediated arginase I induction in macrophages: its implications in IL-13-inhibited nitric oxide production. J Immunol (2000) 165:2134–41.
22. Jiang M, Ding Y, Su Y, Hu X, Li J, Zhang Z. Arginase-flotillin interaction brings arginase to red blood cell membrane. FEBS Lett (2006) 580:6561–4. doi:10.1016/j.febslet.2006.11.003
23. Mori M. Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. J Nutr (2007) 137:1616S–20.
24. Santhanam L, Christianson DW, Nyhan D, Berkowitz DE. Arginase and vascular aging. J Appl Physiol (2008) 105:1632–42. doi:10.1152/japplphysiol.90627.2008
25. Choi S, Park C, Ahn M, Lee JH, Shin T. Immunohistochemical study of arginase 1 and 2 in various tissues of rats. Acta Histochem (2012) 114:487–94. doi:10.1016/j.acthis.2011.09.002
26. Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation (2003) 108:2000–6. doi:10.1161/01.CIR.0000092948.04444.C7
27. Hayashi T, Esaki T, Sumi D, Mukherjee T, Iguchi A, Chaudhuri G. Modulating role of estradiol on arginase II expression in hyperlipidemic rabbits as an atheroprotective mechanism. Proc Natl Acad Sci U S A (2006) 103:10485–90. doi:10.1073/pnas.0603918103
28. Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, et al. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation (2004) 110:3708–14. doi:10.1161/01.CIR.0000142867.26182.32
29. Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol (1995) 57:707–36. doi:10.1146/annurev.ph.57.030195.003423
30. Nishimura H, Rosenblum WI, Nelson GH, Boynton S. Agents that modify EDRF formation alter antiplatelet properties of brain arteriolar endothelium in vivo. Am J Physiol Heart Circ Physiol (1991) 261:H15–21.
31. Sun D, Messina EJ, Koller A, Wolin MS, Kaley G. Endothelium-dependent dilation to L-arginine in isolated rat skeletal muscle arterioles. Am J Physiol Heart Circ Physiol (1992) 262:H1211–6.
32. Frame MD, Sarelius IH. L-arginine-induced conducted signals alter upstream arteriolar responsivity to L-arginine. Circ Res (1995) 77:695–701. doi:10.1161/01.RES.77.4.695
33. Quyyumi AA, Dakak N, Diodati JG, Gilligan DM, Panza JA, Cannon RI. Effect of L-arginine on human coronary endothelium-dependent and physiologic vasodilation. J Am Coll Cardiol (1997) 30:1220–7. doi:10.1016/S0735-1097(97)00279-9
34. Yang Z, Ming XF. Endothelial arginase: a new target in atherosclerosis. Curr Hypertens Rep (2006) 8:54–9. doi:10.1007/s11906-006-0041-8
35. Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol (2007) 34:906–11. doi:10.1111/j.1440-1681.2007.04638.x
36. Caldwell RB, Zhang W, Romero MJ, Caldwell RW. Vascular dysfunction in retinopathy – an emerging role for arginase. Brain Res Bull (2010) 81:303–9. doi:10.1016/j.brainresbull.2009.08.025
37. Ito N, Ohishi M, Yamamoto K, Tatara Y, Shiota A, Hayashi N, et al. Renin-angiotensin inhibition reverses advanced cardiac remodeling in aging spontaneously hypertensive rats. Am J Hypertens (2007) 20:792–9. doi:10.1016/j.amjhyper.2007.02.004
38. Xu J, Carretero OA, Liao TD, Peng H, Shesely EG, Liu TS, et al. Local angiotensin II aggravates cardiac remodeling in hypertension. Am J Physiol Heart Circ Physiol (2010) 299:H1328–38. doi:10.1152/ajpheart.00538.2010
39. Downing SE, Sonnenblick EH. Effects of continuous administration of angiotensin II on ventricular performance. J Appl Physiol (1963) 18:585–92.
40. Heyndrickx GR, Boettcher DH, Vatner SF. Effects of angiotensin, vasopressin, and methoxamine on cardiac function and blood flow distribution in conscious dogs. Am J Physiol (1976) 231:1579–87.
41. Forte IE, Potgieter L, Schmitthenner JE. Effects of hypertension on arterial pressure, heart work and cardiac oxygen utilization. Circ Res (1960) 8:1235–41. doi:10.1161/01.RES.8.6.1235
42. Needleman P, Marshall GR, Sobel BE. Hormone interactions in the isolated rabbit heart. Synthesis and coronary vasomotor effects of prostaglandins, angiotensin, and bradykinin. Circ Res (1975) 37:802–8. doi:10.1161/01.RES.37.6.802
43. Fowler NO, Holmes JC. Coronary and myocardial actions of angiotensin. Circ Res (1964) 14:191–201. doi:10.1161/01.RES.14.3.191
44. Pörsti I, Hecker M, Bassenge E, Busse R. Dual action of angiotensin II on coronary resistance in the isolated perfused rabbit heart. Naunyn Schmiedebergs Arch Pharmacol (1993) 348:650–8. doi:10.1007/BF00167243
45. Kuo L, Chilian WM, Davis MJ. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am J Physiol Heart Circ Physiol (1991) 261:H1706–15.
46. Jones CJH, Kuo L, Davis MJ, Chilian WM. Regulation of coronary blood flow: coordination of heterogeneous control mechanisms in vascular microdomains. Cardiovasc Res (1995) 29:585–9. doi:10.1016/S0008-6363(96)88626-3
47. Li JM, Mullen AM, Shah AM. Phenotypic properties and characteristics of superoxide production by mouse coronary microvascular endothelial cells. J Mol Cell Cardiol (2001) 33:1119–31. doi:10.1006/jmcc.2001.1372
48. Sohn HY, Raff U, Hoffmann A, Gloe T, Heermeier K, Galle J, et al. Differential role of angiotensin II receptor subtypes on endothelial superoxide formation. Br J Pharmacol (2000) 131:667–72. doi:10.1038/sj.bjp.0703566
49. Zhang C, Hein TH, Wang W, Kuo L. Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ Res (2003) 92:322–9. doi:10.1161/01.RES.0000056759.53828.2C
50. Batenburg WW, Garrelds IM, Bernasconi CC, Juillerat-Jeanneret L, van Kats JP, Saxena PR, et al. Angiotensin II type 2 receptor-mediated vasodilation in human coronary microarteries. Circulation (2004) 109:2296–301. doi:10.1161/01.CIR.0000128696.12245.57
51. Yang BC, Phillips MI, Ambuehl PE, Shen LP, Mehta P, Mehta JL. Increase in angiotensin II type 1 receptor expression immediately after ischemia-reperfusion in isolated rat hearts. Circulation (1997) 96:922–6. doi:10.1161/01.CIR.96.3.922
52. Werrmann JG, Cohen SM. Use of losartan to examine the role of the cardiac renin-angiotensin system in myocardial dysfunction during ischemia and reperfusion. J Cardiovasc Pharmacol (1996) 27:177–82. doi:10.1097/00005344-199602000-00001
53. Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res (2001) 88:756–62. doi:10.1161/hh0801.089861
54. Moore C, Tymvios C, Emerson M. Functional regulation of vascular and platelet activity during thrombosis by nitric oxide and endothelial nitric oxide synthase. Thromb Haemost (2010) 104:342–9. doi:10.1160/TH09-11-0764
55. Dal-Ros S, Bronner C, Auger C, Schini-Kerth VB. Red wine polyphenols improve an established aging-related endothelial dysfunction in the mesenteric artery of middle-aged rats: role of oxidative stress. Biochem Biophys Res Commun (2012) 419:381–7. doi:10.1016/j.bbrc.2012.02.031
56. Shatanawi A, Romero MJ, Iddings JA, Chandra S, Umapathy NS, Verin AD, et al. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol (2011) 300:C1181–92. doi:10.1152/ajpcell.00328.2010
57. Toque HA, Romero MJ, Tostes RC, Shatanawi A, Chandra S, Carneiro ZN, et al. p38 Mitogen-activated protein kinase (MAPK) increases arginase activity and contributes to endothelial dysfunction in corpora cavernosa from angiotensin-II-treated mice. J Sex Med (2010) 7:3857–67. doi:10.1111/j.1743-6109.2010.01996.x
58. Luscher TF, Richard V, Tschudi M, Yang ZH, Boulanger C. Endothelial control of vascular tone in large and small coronary arteries. J Am Coll Cardiol (1990) 15:519–27. doi:10.1016/0735-1097(90)90619-Z
59. Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens (2001) 19:1245–54. doi:10.1097/00004872-200107000-00009
60. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest (2003) 111:1201–9. doi:10.1172/JCI200314172
61. Lekakis JP, Papathanassiou S, Papaioannou TG, Papamichael CM, Zakopoulos N, Kotsis V, et al. Oral L-arginine improves endothelial dysfunction in patients with essential hypertension. Int J Cardiol (2002) 86:317–23. doi:10.1016/S0167-5273(02)00413-8
62. Susic D, Francischetti A, Frohlich ED. Prolonged L-arginine on cardiovascular mass and myocardial hemodynamics and collagen in aged spontaneously hypertensive rats and normal rats. Hypertension (1999) 33:451–5. doi:10.1161/01.HYP.33.1.451
63. Asab I, Peer G, Blum M, Wollman Y, Chernihovsky T, Hassner A, et al. Oral administration of L-arginine and captopril in rats prevents chronic renal failure by nitric oxide production. Kidney Int (1995) 47:1515–21. doi:10.1038/ki.1995.214
64. Tousoulis D, Antoniades C, Tentolouris C, Goumas G, Stefanadis C, Toutouzas P. L-arginine in cardiovascular disease: dream or reality? Vasc Med (2002) 7:203–11. doi:10.1191/1358863x02vm434ra
65. Rodriguez S, Richert L, Berthelot A. Increased arginase activity in aorta of mineralocorticoid-salt hypertensive rats. Clin Exp Hypertens (2000) 22:75–85. doi:10.1081/CEH-100100063
66. Zhang C, Hein TW, Wang W, Miller MW, Fossum TW, McDonald MM, et al. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension (2004) 44:935–43. doi:10.1161/01.HYP.0000146907.82869.f2
67. Bagnost T, Berthelot A, Bouhaddi M, Laurant P, Andre C, Guillaume Y, et al. Treatment with the arginase inhibitor Nw-hydroxy-nor-L-arginine improves vascular function and lowers blood pressure in adult spontaneously hypertensive rat. J Hypertens (2008) 26:1110–8. doi:10.1097/HJH.0b013e3282fcc357
68. Bagnost T, Ma L, da Silva RF, Rezakhaniha R, Houdayer C, Stergiopulos N, et al. Cardiovascular effects of arginase inhibition in spontaneously hypertensive rats with fully developed hypertension. Cardiovasc Res (2010) 87:569–77. doi:10.1093/cvr/cvq081
69. El-Bassossy HM, El-Fawal R, Fahmy A, Watson ML. Arginase inhibition alleviates hypertension in the metabolic syndrome. Br J Pharmacol (2013) 169(3):693–703. doi:10.1111/bph.12144
70. Vaziri ND. Causal link between oxidative stress, inflammation, and hypertension. Iran J Kidney Dis (2008) 2:1–10.
71. Horowitz S, Binion DG, Nelson VM, Kanaa Y, Javadi P, Lazarova Z, et al. Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol (2007) 292:G1323–36. doi:10.1152/ajpgi.00499.2006
72. Nelin LD, Nash HE, Chicoine LG. Cytokine treatment increases arginine metabolism and uptake in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol (2001) 281:L1232–9.
73. Joe Y, Zheng M, Kim HJ, Kim S, Uddin MJ, Park C, et al. Salvianolic acid B exerts vasoprotective effects through the modulation of heme oxygenase-1 and arginase activities. J Pharmacol Exp Ther (2012) 341:850–8. doi:10.1124/jpet.111.190736
74. Wilke CM, Wei S, Wang L, Kryczek I, Kao J, Zou W. Dual biological effects of the cytokines interleukin-10 and interferon-gamma. Cancer Immunol Immunother (2011) 60:1529–41. doi:10.1007/s00262-011-1104-5
75. Chang R, Chicoine LG, Cui H, Kanagy NL, Walker BR, Liu Y, et al. Cytokine-induced arginase activity in pulmonary endothelial cells is dependent on Src family tyrosine kinase activity. Am J Physiol Lung Cell Mol Physiol (2008) 295:L688–97. doi:10.1152/ajplung.00504.2007
76. Zhang DX, Yi FX, Zou AP, Li PL. Role of ceramide in TNF-α-induced impairment of endothelium-dependent vasorelaxation in coronary arteries. Am J Physiol Heart Circ Physiol (2002) 283:H1785–94.
77. Zhang C, Hein TW, Wang W, Ren Y, Shipley RD, Kuo L. Activation of JNK and xanthine oxidase by TNF-α impairs nitric oxide-mediated dilation of coronary arterioles. J Mol Cell Cardiol (2006) 40:247–57. doi:10.1016/j.yjmcc.2005.11.010
78. Qamirani E, Ren Y, Kuo L, Hein TW. C-reactive protein inhibits endothelium-dependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD(P)H oxidase. Arterioscler Thromb Vasc Biol (2005) 25:995–1001. doi:10.1161/01.ATV.0000159890.10526.1e
79. Hein TW, Qamirani E, Ren Y, Kuo L. C-reactive protein impairs coronary arteriolar dilation to prostacyclin synthase activation: role of peroxynitrite. J Mol Cell Cardiol (2009) 47:196–202. doi:10.1016/j.yjmcc.2009.04.015
80. Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, et al. TNF-α contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol (2007) 27:1269–75. doi:10.1161/ATVBAHA.107.142521
81. Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol (2008) 103:398–406. doi:10.1007/s00395-008-0733-0
82. Hansen PR. Inflammatory alterations in the myocardial microcirculation. J Mol Cell Cardiol (1998) 30:2555–9. doi:10.1006/jmcc.1998.0827
83. Nagaoka T, Kuo L, Ren Y, Yoshida A, Hein TW. C-reactive protein inhibits endothelium-dependent nitric oxide-mediated dilation of retinal arterioles via enhanced superoxide production. Invest Ophthalmol Vis Sci (2008) 49:2053–60. doi:10.1167/iovs.07-1387
84. Yamada T, Egashira N, Bando A, Nishime Y, Tonogai Y, Imuta M, et al. Activation of p38 MAPK by oxidative stress underlying epirubicin-induced vascular endothelial cell injury. Free Radic Biol Med (2012) 52:1285–93. doi:10.1016/j.freeradbiomed.2012.02.003
85. Schweitzer KS, Hatoum H, Brown MB, Gupta M, Justice MJ, Beteck B, et al. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: role of oxidative stress and ceramides. Am J Physiol Lung Cell Mol Physiol (2011) 301:L836–46. doi:10.1152/ajplung.00385.2010
86. Hein TW, Singh U, Vasquez-Vivar J, Devaraj S, Kuo L, Jialal I. Human C-reactive protein induces endothelial dysfunction and uncoupling of eNOS in vivo. Atherosclerosis (2009) 206:61–8. doi:10.1016/j.atherosclerosis.2009.02.002
87. Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest (2002) 109:817–26. doi:10.1172/JCI0214442
88. Ogino K, Obase Y, Takahashi N, Shimizu H, Takigawa T, Wang DH, et al. High serum arginase I levels in asthma: its correlation with high-sensitivity C-reactive protein. J Asthma (2011) 48:1–7. doi:10.3109/02770903.2010.528496
89. Munder M. Role of arginase in asthma: potential clinical applications. Expert Rev Clin Pharmacol (2010) 3:17–23. doi:10.1586/ecp.09.53
90. Maarsingh H, Zaagsma J, Meurs H. Arginase: a key enzyme in the pathophysiology of allergic asthma opening novel therapeutic perspectives. Br J Pharmacol (2009) 158:652–64. doi:10.1111/j.1476-5381.2009.00374.x
91. Maarsingh H, Pera T, Meurs H. Arginase and pulmonary diseases. Naunyn Schmiedebergs Arch Pharmacol (2008) 378:171–84. doi:10.1007/s00210-008-0286-7
92. Sasaki A, Doi S, Mizutani S, Azuma H. Roles of accumulated endogenous nitric oxide synthase inhibitors, enhanced arginase activity, and attenuated nitric oxide synthase activity in endothelial cells for pulmonary hypertension in rats. Am J Physiol Lung Cell Mol Physiol (2007) 292:L1480–7. doi:10.1152/ajplung.00360.2006
93. Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J (2004) 18:1746–8.
94. Agrawal A, Mabalirajan U, Ahmad T, Ghosh B. Emerging interface between metabolic syndrome and asthma. Am J Respir Cell Mol Biol (2011) 44:270–5. doi:10.1165/rcmb.2010-0141TR
95. Kleinbongard P, Schulz R, Heusch G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail Rev (2011) 16:49–69. doi:10.1007/s10741-010-9180-8
96. Hein TH, Zhang C, Wang W, Chang CI, Thengchaisri N, Kuo L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. FASEB J (2003) 17:2328–30.
97. Zhang C, Wu J, Xu X, Potter BJ, Gao X. Direct relationship between levels of TNF-α expression and endothelial dysfunction in reperfusion injury. Basic Res Cardiol (2010) 105:453–64. doi:10.1007/s00395-010-0083-6
98. Zhang C, Xu X, Potter BJ, Wang W, Kuo L, Michael L, et al. TNF-α contributes to endothelial dysfunction in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol (2006) 26:475–80. doi:10.1161/01.ATV.0000201932.32678.7e
99. Shemyakin A, Kovamees O, Rafnsson A, Bohm F, Svenarud P, Settergren M, et al. Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation (2012) 126:2943–50. doi:10.1161/CIRCULATIONAHA.112.140335
100. Quitter F, Figulla HR, Ferrari M, Pernow J, Jung C. Increased arginase levels in heart failure represent a therapeutic target to rescue microvascular perfusion. Clin Hemorheol Microcirc (2012) 54:75–85. doi:10.3233/CH-2012-1617
101. Dumont J, Zureik M, Cottel D, Montaye M, Ducimetiere P, Amouyel P, et al. Association of arginase 1 gene polymorphisms with the risk of myocardial infarction and common carotid intima media thickness. J Med Genet (2007) 44:526–31. doi:10.1136/jmg.2006.047449
102. Bachetti T, Comini L, Francolini G, Bastianon D, Valetti B, Cadei M, et al. Arginase pathway in human endothelial cells in pathophysiological conditions. J Mol Cell Cardiol (2004) 37:515–23. doi:10.1016/j.yjmcc.2004.05.004
103. Sankaralingam S, Xu H, Davidge ST. Arginase contributes to endothelial cell oxidative stress in response to plasma from women with preeclampsia. Cardiovasc Res (2010) 85:194–203. doi:10.1093/cvr/cvp277
104. Waddington SN, Mosley K, Cook HT, Tam FW, Cattell V. Arginase AI is upregulated in acute immune complex-induced inflammation. Biochem Biophys Res Commun (1998) 247:84–7. doi:10.1006/bbrc.1998.8755
105. Cooke JP, Andon NA, Girerd XJ, Hirsch AT, Creager MA. Arginine restores cholinergic relaxation of hypercholesterolemic rabbit thoracic aorta. Circulation (1991) 83:1057–62. doi:10.1161/01.CIR.83.3.1057
106. Girerd XJ, Hirsch AT, Cooke JP, Dzau VJ, Creager MA. L-arginine augments endothelium-dependent vasodilation in cholesterol-fed rabbits. Circ Res (1990) 67:1301–8. doi:10.1161/01.RES.67.6.1301
107. Kuo L, Davis MJ, Cannon MS, Chilian WM. Pathophysiological consequences of atherosclerosis extend into the coronary microcirculation. Restoration of endothelium-dependent responses by L-arginine. Circ Res (1992) 70:465–76. doi:10.1161/01.RES.70.3.465
108. Woo A, Shin W, Cuong TD, Min B, Lee JH, Jeon BH, et al. Arginase inhibition by piceatannol-3’-O-beta-D-glucopyranoside improves endothelial dysfunction via activation of endothelial nitric oxide synthase in ApoE-null mice fed a high-cholesterol diet. Int J Mol Med (2013) 31:803–10. doi:10.3892/ijmm.2013.1261
109. Creager MA, Gallagher SJ, Girerd XJ, Coleman SM, Dzau VJ, Cooke JP. L-arginine improves endothelium-dependent vasodilation in hypercholesterolemic humans. J Clin Invest (1992) 90:1248–53. doi:10.1172/JCI115987
110. Clarkson P, Adams MR, Powe AJ, Donald AE, McCredie R, Robinson J, et al. Oral L-arginine improves endothelium-dependent dilation in hypercholesterolemic young adults. J Clin Invest (1996) 97:1989–94. doi:10.1172/JCI118632
111. Tousoulis D, Boger RH, Antoniades C, Siasos G, Stefanadi E, Stefanadis C. Mechanisms of disease: L-arginine in coronary atherosclerosis – a clinical perspective. Nat Clin Pract Cardiovasc Med (2007) 4:274–83. doi:10.1038/ncpcardio0878
112. Lauer T, Kleinbongard P, Rath J, Schulz R, Kelm M, Rassaf T. L-arginine preferentially dilates stenotic segments of coronary arteries thereby increasing coronary flow. J Intern Med (2008) 264:237–44. doi:10.1111/j.1365-2796.2008.01943.x
113. Vaisman BL, Andrews KL, Khong SM, Wood KC, Moore XL, Fu Y, et al. Selective endothelial overexpression of arginase II induces endothelial dysfunction and hypertension and enhances atherosclerosis in mice. PLoS ONE (2012) 7:e39487. doi:10.1371/journal.pone.0039487
114. Ryoo S, Gupta G, Benjo A, Lim HK, Camara A, Sikka G, et al. Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ Res (2008) 102:923–32. doi:10.1161/CIRCRESAHA.107.169573
115. Romero MJ, Platt DH, Tawfik HE, Labazi M, El-Remessy AB, Bartoli M, et al. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res (2008) 102:95–102. doi:10.1161/CIRCRESAHA.107.155028
116. Hein TW, Liao JC, Kuo L. oxLDL specifically impairs endothelium-dependent, NO-mediated dilation of coronary arterioles. Am J Physiol Heart Circ Physiol (2000) 278:H175–83.
117. Ryoo S, Bhunia A, Chang F, Shoukas A, Berkowitz DE, Romer LH. OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling. Atherosclerosis (2011) 214:279–87. doi:10.1016/j.atherosclerosis.2010.10.044
118. Wang W, Hein TW, Zhang C, Zawieja DC, Liao JC, Kuo L. Oxidized low-density lipoprotein inhibits nitric oxide-mediated coronary arteriolar dilation by up-regulating endothelial arginase I. Microcirculation (2011) 18:36–45. doi:10.1111/j.1549-8719.2010.00066.x
119. Matoba T, Shimokawa H, Morikawa K, Kubota H, Kunihiro I, Urakami-Harasawa L, et al. Electron spin resonance detection of hydrogen peroxide as an endothelium-derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler Thromb Vasc Biol (2003) 23:1224–30. doi:10.1161/01.ATV.0000078601.79536.6C
120. Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med (2008) 45:1340–51. doi:10.1016/j.freeradbiomed.2008.08.013
121. Thengchaisri N, Kuo L. Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: role of cyclooxygenase and potassium channels. Am J Physiol Heart Circ Physiol (2003) 285:H2255–63.
122. Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, et al. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res (2012) 110:471–80. doi:10.1161/CIRCRESAHA.111.258871
123. Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M, et al. Hydrogen peroxide, an endogenous endothelium-derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation (2003) 107:1040–5. doi:10.1161/01.CIR.0000050145.25589.65
124. Saitoh S, Kiyooka T, Rocic P, Rogers PA, Zhang C, Swafford A, et al. Redox-dependent coronary metabolic dilation. Am J Physiol Heart Circ Physiol (2007) 293:H3720–5. doi:10.1152/ajpheart.00436.2007
125. Katusic ZS. Vascular endothelial dysfunction: does tetrahydrobiopterin play a role? Am J Physiol Heart Circ Physiol (2001) 281:H981–6.
126. Yada T, Shimokawa H, Hiramatsu O, Haruna Y, Morita Y, Kashihara N, et al. Cardioprotective role of endogenous hydrogen peroxide during ischemia-reperfusion injury in canine coronary microcirculation in vivo. Am J Physiol Heart Circ Physiol (2006) 291:H1138–46. doi:10.1152/ajpheart.00187.2006
127. Thengchaisri N, Shipley R, Ren Y, Parker J, Kuo L. Exercise training restores coronary arteriolar dilation to NOS activation distal to coronary artery occlusion: role of hydrogen peroxide. Arterioscler Thromb Vasc Biol (2007) 27:791–8. doi:10.1161/01.ATV.0000258416.47953.9a
128. Thengchaisri N, Hein TW, Wang W, Xu X, Li Z, Fossum TW, et al. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol (2006) 26:2035–42. doi:10.1161/01.ATV.0000233334.24805.62
129. Chandra S, Romero MJ, Shatanawi A, Alkilany AM, Caldwell RB, Caldwell RW. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br J Pharmacol (2012) 165:506–19. doi:10.1111/j.1476-5381.2011.01584.x
130. Feletou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol (2011) 164:894–912. doi:10.1111/j.1476-5381.2011.01276.x
131. Prati C, Berthelot A, Kantelip B, Wendling D, Demougeot C. Treatment with the arginase inhibitor Nw-hydroxy-nor-L-arginine restores endothelial function in rat adjuvant-induced arthritis. Arthritis Res Ther (2012) 14:R130. doi:10.1186/ar3860
132. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202:931–9. doi:10.1084/jem.20050715
133. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res (2007) 67:4507–13. doi:10.1158/0008-5472.CAN-06-4174
134. Van Hecken A, Schwartz JI, Depre M, De Lepeleire I, Dallob A, Tanaka W, et al. Comparative inhibitory activity of rofecoxib, meloxicam, diclofenac, ibuprofen, and naproxen on COX-2 versus COX-1 in healthy volunteers. J Clin Pharmacol (2000) 40:1109–20.
135. Makela A, Kuusi T, Schroder T. Inhibition of serum phospholipase-A2 in acute pancreatitis by pharmacological agents in vitro. Scand J Clin Lab Invest (1997) 57:401–7. doi:10.3109/00365519709084587
136. Mayorek N, Naftali-Shani N, Grunewald M. Diclofenac inhibits tumor growth in a murine model of pancreatic cancer by modulation of VEGF levels and arginase activity. PLoS ONE (2010) 5:e12715. doi:10.1371/journal.pone.0012715
137. Li H, Meininger CJ, Hawker JR Jr, Haynes TE, Kepka-Lenhart D, Mistry SK, et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab (2001) 280:E75–82.
138. Yoon J, Ryoo S. Arginase inhibition reduces interleukin-1β-stimulated vascular smooth muscle cell proliferation by increasing nitric oxide synthase-dependent nitric oxide production. Biochem Biophys Res Commun (2013) 435:428–33. doi:10.1016/j.bbrc.2013.05.002
139. Chen B, Calvert AE, Cui H, Nelin LD. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol (2009) 297:L1151–9. doi:10.1152/ajplung.00183.2009
140. Davel LE, Rimmaudo L, Espanol A, de la Torre E, Jasnis MA, Ribeiro ML, et al. Different mechanisms lead to the angiogenic process induced by three adenocarcinoma cell lines. Angiogenesis (2004) 7:45–51. doi:10.1023/B:AGEN.0000037329.45326.a8
141. Iyer RK, Yoo PK, Kern RM, Rozengurt N, Tsoa R, O’Brien WE, et al. Mouse model for human arginase deficiency. Mol Cell Biol (2002) 22:4491–8. doi:10.1128/MCB.22.13.4491-4498.2002
142. Shi O, Morris SM Jr, Zoghbi H, Porter CW, O’Brien WE. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Mol Cell Biol (2001) 21:811–3. doi:10.1128/MCB.21.3.811-813.2001
143. Lim HK, Lim HK, Ryoo S, Benjo A, Shuleri K, Miriel V, et al. Mitochondrial arginase II constrains endothelial NOS-3 activity. Am J Physiol Heart Circ Physiol (2007) 293:H3317–24. doi:10.1152/ajpheart.00700.2007
144. Toque HA, Tostes RC, Yao L, Xu Z, Webb RC, Caldwell RB, et al. Arginase II deletion increases corpora cavernosa relaxation in diabetic mice. J Sex Med (2011) 8:722–33. doi:10.1111/j.1743-6109.2010.02098.x
145. Morris SM Jr, Gao T, Cooper TK, Kepka-Lenhart D, Awad AS. Arginase-2 mediates diabetic renal injury. Diabetes (2011) 60:3015–22. doi:10.2337/db11-0901
146. Romero MJ, Iddings JA, Platt DH, Ali MI, Cederbaum SD, Stepp DW, et al. Diabetes-induced vascular dysfunction involves arginase I. Am J Physiol Heart Circ Physiol (2012) 302:H159–66. doi:10.1152/ajpheart.00774.2011
147. Yao L, Chandra S, Toque HA, Bhatta A, Rojas M, Caldwell RB, et al. Prevention of diabetes-induced arginase activation and vascular dysfunction by Rho kinase (ROCK) knockout. Cardiovasc Res (2013) 97:509–19. doi:10.1093/cvr/cvs371
148. Toque HA, Nunes KP, Rojas M, Bhatta A, Yao L, Xu Z, et al. Arginase 1 mediates increased blood pressure and contributes to vascular endothelial dysfunction in deoxycorticosterone acetate (DOCA)-salt hypertension. Front Immunol (2013) 4:219. doi:10.3389/fimmu.2013.00219
149. Beleznai T, Feher A, Spielvogel D, Lansman SL, Bagi Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol (2011) 300:H777–83. doi:10.1152/ajpheart.00831.2010
150. Huynh NN, Andrews KL, Head GA, Khong SM, Mayorov DN, Murphy AJ, et al. Arginase II knockout mouse displays a hypertensive phenotype despite a decreased vasoconstrictory profile. Hypertension (2009) 54:294–301. doi:10.1161/HYPERTENSIONAHA.108.121731
151. Becker-Catania SG, Gregory TL, Yang Y, Gau CL, de Vellis J, Cederbaum SD, et al. Loss of arginase I results in increased proliferation of neural stem cells. J Neurosci Res (2006) 84:735–46. doi:10.1002/jnr.20964
152. Mumenthaler SM, Rozengurt N, Livesay JC, Sabaghian A, Cederbaum SD, Grody WW. Disruption of arginase II alters prostate tumor formation in TRAMP mice. Prostate (2008) 68:1561–9. doi:10.1002/pros.20816
153. Santhanam L, Lim HK, Lim HK, Miriel V, Brown T, Patel M, et al. Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res (2007) 101:692–702. doi:10.1161/CIRCRESAHA.107.157727
154. Zharikov S, Krotova K, Hu H, Baylis C, Johnson RJ, Block ER, et al. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am J Physiol Cell Physiol (2008) 295:C1183–90. doi:10.1152/ajpcell.00075.2008
155. Park JH, Jin YM, Hwang S, Cho DH, Kang DH, Jo I. Uric acid attenuates nitric oxide production by decreasing the interaction between endothelial nitric oxide synthase and calmodulin in human umbilical vein endothelial cells: a mechanism for uric acid-induced cardiovascular disease development. Nitric Oxide (2013) 32:36–42. doi:10.1016/j.niox.2013.04.003
156. Iyamu EW, Perdew H, Woods GM. Cysteine-iron promotes arginase activity by driving the Fenton reaction. Biochem Biophys Res Commun (2008) 376:116–20. doi:10.1016/j.bbrc.2008.08.102
157. Colleluori DM, Morris SM Jr, Ash DE. Expression, purification, and characterization of human type II arginase. Arch Biochem Biophys (2001) 389:135–43. doi:10.1006/abbi.2001.2324
158. Miller ER III, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med (2005) 142:37–46. doi:10.7326/0003-4819-142-1-200501040-00110
159. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev (2012) 3:CD007176.
160. Tinkel J, Hassanain H, Khouri SJ. Cardiovascular antioxidant therapy: a review of supplements, pharmacotherapies, and mechanisms. Cardiol Rev (2012) 20:77–83. doi:10.1097/CRD.0b013e31823dbbad
161. Lee IM, Cook NR, Gaziano JM, Gordon D, Ridker PM, Manson JE, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized controlled trial. JAMA (2005) 294:56–65. doi:10.1001/jama.294.19.2432-b
162. Cerrato R, Cunnington C, Crabtree MJ, Antoniades C, Pernow J, Channon KM, et al. Endothelin-1 increases superoxide production in human coronary artery bypass grafts. Life Sci (2012) 91:723–8. doi:10.1016/j.lfs.2012.03.024
163. Romero M, Jimenez R, Sanchez M, Lopez-Sepulveda R, Zarzuelo A, Tamargo J, et al. Vascular superoxide production by endothelin-1 requires Src non-receptor protein tyrosine kinase and MAPK activation. Atherosclerosis (2010) 212:78–85. doi:10.1016/j.atherosclerosis.2010.04.031
164. Matsuo J, Oku H, Kanbara Y, Kobayashi T, Sugiyama T, Ikeda T. Involvement of NADPH oxidase and protein kinase C in endothelin-1-induced superoxide production in retinal microvessels. Exp Eye Res (2009) 89:693–9. doi:10.1016/j.exer.2009.06.012
Keywords: endothelium, superoxide, nitric oxide, inflammation, arterioles, vasodilation
Citation: Kuo L and Hein TW (2013) Vasomotor regulation of coronary microcirculation by oxidative stress: role of arginase. Front. Immunol. 4:237. doi: 10.3389/fimmu.2013.00237
Received: 16 May 2013; Paper pending published: 13 June 2013;
Accepted: 01 August 2013; Published online: 19 August 2013.
Edited by:
Rudolf Lucas, Medical College of Georgia, USAReviewed by:
Kottarappat Dileepan, University of Kansas Medical Center, USACopyright: © 2013 Kuo and Hein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lih Kuo, Department of Medical Physiology, Texas A&M Health Science Center, 702 Southwest H.K. Dodgen Loop, Temple, TX 76504, USA e-mail:bGt1b0B0YW1oc2MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.