

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 11 July 2013
Sec. Cancer Immunity and Immunotherapy
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00192
This article is part of the Research Topic Tumor Cell/Dendritic Cell Interactions and the Influence of Tumors on Dendritic Cell-mediated Anti-Tumor Immune Responses and Dendritic Cell-Based Tumor Immunotherapies View all 18 articles
Dendritic cells (DC) are key regulators of both innate and adaptive immunity, and the array of immunoregulatory functions exhibited by these cells is dictated by their differentiation, maturation, and activation status. Although a major role for these cells in the induction of immunity to pathogens has long been appreciated, data accumulated over the last several years has demonstrated that DC are also critical regulators of anti-tumor immune responses. However, despite the potential for stimulation of robust anti-tumor immunity by DC, tumor-altered DC function has been observed in many cancer patients and tumor-bearing animals and is often associated with tumor immune escape. Such dysfunction has significant implications for both the induction of natural anti-tumor immune responses as well as the efficacy of immunotherapeutic strategies that target endogenous DC in situ or that employ exogenous DC as part of anti-cancer immunization maneuvers. In this review, the major types of tumor-altered DC function will be described, with emphasis on recent insights into the mechanistic bases for the inhibition of DC differentiation from hematopoietic precursors, the altered programing of DC precursors to differentiate into myeloid-derived suppressor cells or tumor-associated macrophages, the suppression of DC maturation and activation, and the induction of immunoregulatory DC by tumors, tumor-derived factors, and tumor-associated cells within the milieu of the tumor microenvironment. The impact of these tumor-altered cells on the quality of the overall anti-tumor immune response will also be discussed. Finally, this review will also highlight questions concerning tumor-altered DC function that remain unanswered, and it will address factors that have limited advances in the study of this phenomenon in order to focus future research efforts in the field on identifying strategies for interfering with tumor-associated DC dysfunction and improving DC-mediated anti-tumor immunity.
Dendritic cells (DC) are critical regulators of host immune responses that serve as a bridge between innate and adaptive immunity. Following their differentiation from either myeloid or lymphoid bone marrow-derived progenitors, DC populate both lymphoid and peripheral tissues, where they are involved in immune surveillance and control of immune reactivity in the host. DC precursors may differentiate into a variety of specialized subsets that exhibit numerous immunoregulatory activities, and the diverse functions of these cells are tightly linked to their maturation and activation status (1). Immature DC are highly phagocytic and function to sample both soluble and cell-associated antigens (Ag) in host tissues. In the steady state, such immature DC either fail to elicit immune responsiveness to Ag they have acquired (2), or they actively induce immune tolerance to these Ag (3–6). On the other hand, stimulation of immature DC by a variety of factors [including pathogen-associated molecular patterns, danger-associated molecular patterns (DAMPs), inflammatory mediators, CD40L, etc.] induces the maturation and activation of these cells, thereby converting DC into potent stimulators of immune activation. Such DC upregulate expression of costimulatory molecules, cytokines, and chemokines necessary for the activation and recruitment of T lymphocytes and other immune effectors into a response to eliminate the source of Ag representing danger to the host (7–9).
In addition to their role in activation of immunity against foreign pathogens, DC have also been shown to be critical players in the induction of anti-tumor immune responses (10–12). The role of DC in eliciting such responses is highlighted by studies demonstrating immunologic ignorance of tumors under conditions in which cross-presentation of tumor Ag by DC is precluded (13–17). However, despite the ability of DC to elicit tumor Ag-specific T lymphocyte responses, in many cases these responses are dysfunctional and ineffective in clearing the tumor (18–24). While such immune dysfunction might result from direct suppression of T cells by tumors or tumor-derived factors, it may also arise indirectly from suboptimal stimulation or suppression of T cells by tumor-altered DC. Tumor-altered DC function has now been documented in many cancer patients and tumor-bearing animals and ranges from influences of tumors on the differentiation of DC from hematopoietic precursors to effects on the behavior of fully differentiated DC. These effects on DC and their precursors can lead to accumulation in the tumor microenvironment of a variety of cells that include myeloid-derived suppressor cells (MDSC), tumor-associated macrophages (TAM), immature DC, and immunoregulatory myeloid DC (mDC) and plasmacytoid DC (pDC), each of which exhibit distinct phenotypic characteristics (Table 1). The identification of such cells in cancer patients not only has important prognostic value, but it also has significant implications for (1) the induction of natural anti-tumor immune responses and (2) the efficacy of immunotherapeutic strategies that target endogenous DC in situ or that employ exogenous DC as part of immunization maneuvers. Therefore, because of the importance of DC differentiation, maturation, and activation in dictating the immune stimulatory versus inhibitory activities of these cells, interference with any of these processes by factors or cells within the tumor microenvironment may greatly influence the induction and maintenance of anti-tumor immune responses in the host. This review will summarize the current knowledge regarding tumor-altered DC function and its impact on anti-tumor immunity, and it will highlight both recent advances in the field as well as important questions that will need to be answered as efforts are made to improve the quality of DC-mediated anti-tumor immune responses and DC-based cancer immunotherapies in the future.
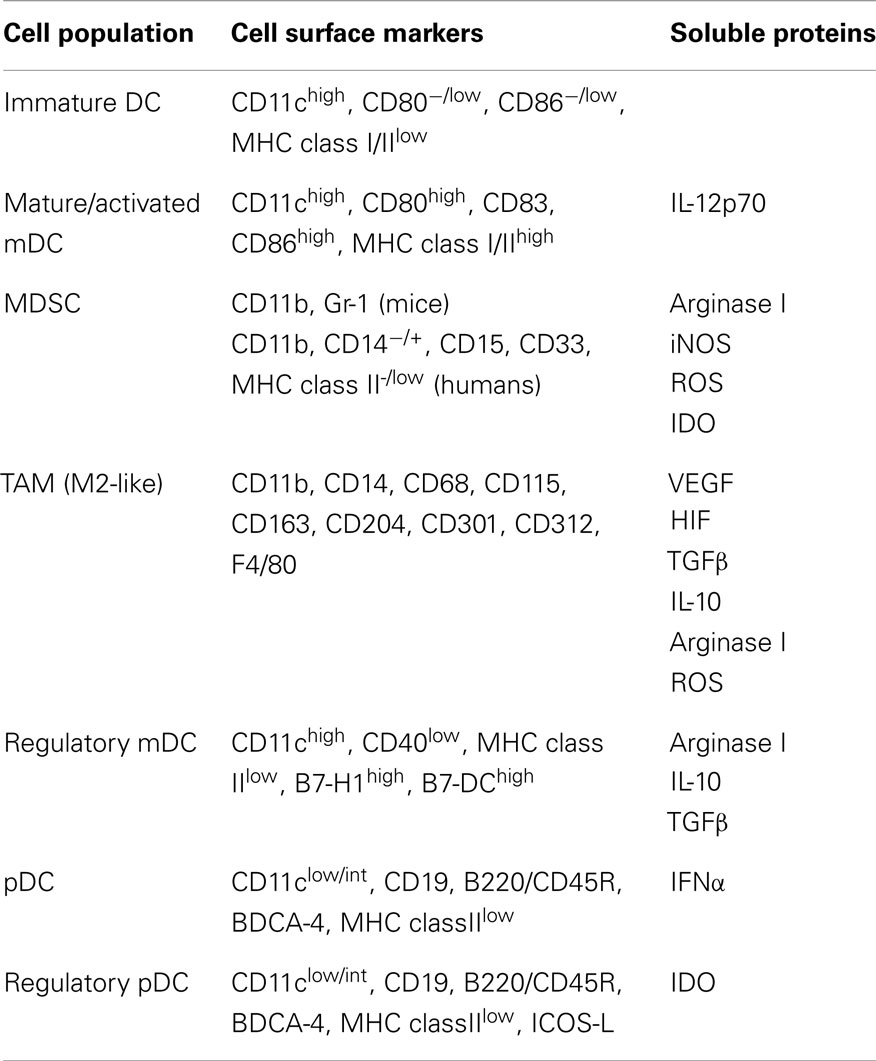
Table 1. Phenotypic characteristics of tumor-associated DC and populations derived from DC precursors.
Dendritic cells are specialized cells that differentiate from both myeloid and lymphoid progenitors before acquiring their unique functions as Ag presenting cells (APC), and a number of studies have described factors derived from both tumors and associated cells within the tumor microenvironment that interfere with DC differentiation from precursors, thereby contributing to a loss of stimulatory APC activity in tumor-bearing hosts. Soluble factors secreted by human renal cell carcinomas and pancreatic cancers, including IL-6 and M-CSF, have been shown to block DC differentiation from CD34+ progenitors and promote lineage commitment toward CD14+ monocytes that express little to no MHC and costimulatory molecules and that fail to induce allogeneic T cell proliferation in mixed leukocyte reaction (MLR) assays (25, 26). Similar inhibition of CD34+ precursor cell differentiation into DC has been attributed to tumor-derived VEGF (27), and this blockade of DC differentiation is associated with suppression of NF-κB activity in these cells (28). VEGF has also been implicated in suppressing the differentiation of skin-resident Langerhans cells in a murine fibrosarcoma model (29). In addition to secreting cytokines that inhibit DC differentiation, tumors may also secrete other factors that interfere with the development of different subsets of DC. The gangliosides GD2 and GM3 secreted by human and murine neuroblastoma cell lines have been shown to inhibit differentiation of DC from CD34+ progenitors (30), and human melanomas secrete GM3 and GD3 gangliosides that not only inhibit DC differentiation from monocytic precursors but also induce apoptosis of monocyte-derived DC (31, 32). Likewise, cyclooxygenase-1 (COX-1)- and COX-2-derived prostanoids present in primary tumor-derived supernatants from several freshly isolated human tumor types block DC differentiation as well (33), and the source of these suppressive mediators may be not only tumor cells themselves but also stromal cells within the tumor microenvironment, as stromal cell-derived prostaglandin-E2 (PGE2) was recently shown to inhibit the differentiation of both bone marrow- and monocyte-derived DC (34). Regardless of the mechanism of inhibition, the loss of APC function associated with suppressing DC differentiation may significantly limit the induction of anti-tumor immune responses and contribute greatly to tumor immune escape.
In addition to the inhibitory effects of tumor-derived and tumor-associated factors on DC differentiation that preclude the development of cells with APC function, there is an abundance of data documenting how these factors can also alter the differentiation program of DC precursors and promote the accumulation of immature myeloid cells with immunosuppressive function (Table 2). These MDSC, characterized by expression of CD11b and Gr-1 in mice and a number of cell surface markers in humans (Table 1), are associated with various cancer types and have been recovered at high levels from both tumors and tumor-draining lymph nodes (35–38). Their induction may be driven by a number of factors released by tumors and tumor-associated cells, including VEGF (39), TGFβ (40), IL-1β (41), IL-13 (42), GM-CSF (43), prostaglandins (44), reactive oxygen species (ROS) (45), and components of the complement system (46). Differentiation into MDSC is associated with hyperactivation of STAT3 (39, 47, 48) and is accompanied by acquisition of a number of immunosuppressive properties. In a murine sarcoma model, MDSC suppression of Ag-specific CD8+ T cell responses required direct cell–cell contact via TCR/MHC class I and was mediated by release of ROS (49–51). Similarly, MDSC lines generated from mice bearing adenocarcinomas exhibit nitric oxide-mediated suppression of IL-2 signaling in activated T cells. In this model, nitric oxide production by MDSC required direct contact with, and IFNγ secretion by, the activated T cell, and this nitric oxide inhibited T cell proliferation and induced T cell apoptosis (52, 53). More recently, the increased production of ROS by MDSC was shown to be the result of upregulated NADPH oxidase activity in these cells in several different murine models and in head and neck cancer patients (54). MDSC have also been shown to induce T cell tolerance through release of indoleamine 2,3-dioxygenase (IDO) and arginase I, enzymes involved in degradation of tryptophan and arginine, respectively (55, 56). In regard to the latter, tumor-derived COX-2 can mediate PGE2 signaling in MDSC, thereby triggering overexpression of arginase I in these cells (57). In both murine models and cancer patients, tumors are enriched in arginase I-producing MDSC, and arginine metabolism in the tumor microenvironment leads to downregulation of CD3ζ chain and suppression of proliferative capacity in both CD4+ and CD8+ T cells (58, 59). Interestingly, recent studies evaluating the abundance and activity of MDSC in tumors, tumor-draining lymph nodes, and peripheral blood of cancer patients have shown that both the frequency and arginase I activity of these cells correlates with the clinical stage of the tumor, thus suggesting a critical role for these immunosuppressive cells in disease progression (60, 61).

Table 2. Mechanisms of immune suppression by MDSC.
In addition to the direct tolerization of anti-tumor T lymphocyte responses by MDSC, these cells are also known to induce the development of regulatory T cells (Tregs) that can also suppress T cell activation. In this light, MDSC-derived TGFβ not only suppresses cytolytic activity of T lymphocytes (42), but it has also been demonstrated in the B16 murine melanoma model to promote expansion of CD4+ CD25+ FOXP3+ Tregs in both tumors and tumor-draining lymph nodes (62). Others have reported that tumor-infiltrating MDSC isolated from B16 melanomas also express high levels of the chemokines CCL3, CCL4, and CCL5, the ligands for the CCR5 chemokine receptor that is preferentially expressed on Tregs (63). These results indicate that MDSC likely also play a critical role in recruitment of expanded Tregs into the tumor microenvironment. Additionally, TGFβ-independent MDSC induction of Tregs has been reported in a B cell lymphoma model, where expansion of preexisting natural Tregs required Ag presentation and arginase I activity by MDSC (64). A link between MDSC and Tregs has also recently been reported in a study of breast cancer patients, where the presence of IDO-expressing MDSC correlated with increased infiltration of Tregs into primary tumors and lymph node metastases (56).
The immunosuppressive activities of MDSC extend beyond regulatory effects on T lymphocytes as well. In a murine model of gestation-enhanced metastasis of B16 melanoma, MDSC diminished the number and activity of NK cells (65). Likewise, in both mammary carcinoma and hepatic tumor models, MDSC suppressed NKG2D expression, IFNγ secretion, and cytotoxic activity by NK cells (66, 67). In the hepatic tumor model, suppression required direct cell–cell contact and was mediated by membrane-bound TGFβ on MDSC, and this interaction caused NK cells to be hyporesponsive to activating stimuli, indicating that they had acquired an anergic phenotype. Similar findings have been reported in patients with hepatocellular carcinoma, where suppression of NK cell activity was dependent on MDSC engagement of the NKp30 receptor on NK cells (68). MDSC have also been shown both to impede the maturation and T cell stimulatory capacity of DC (69) and to engage in cross-talk with macrophages, leading to diminished IL-12 secretion by macrophages and increased IL-10 production by MDSC (70). Such alteration of cytokine secretion patterns has the potential to polarize helper and cytotoxic T cells toward a type 2 response that is less robust in its anti-tumor efficacy.
In addition to shifting the differentiation of myeloid precursors away from DC lineage commitment and promoting development of MDSC, tumors can also drive the differentiation of DC precursors into other immunosuppressive cells of myeloid origin, most notably TAM. Recently, these cells have been shown to descend from both bone marrow-derived and splenic precursors, and some populations are believed to reflect the culmination of MDSC differentiation (71). Importantly, accumulation of TAM, particularly those with an anti-inflammatory M2-like phenotype, correlates with poor prognosis in patients with a variety of cancers (72–76). TH2 cytokines, glucocorticoids, and growth factors present in the tumor milieu are all known to induce M2-like macrophages (77), and tumor-derived IL-10 has specifically been shown to inhibit DC differentiation from monocytic precursors and to promote the development of TAM from these cells (78). Much like MDSC, these TAM can suppress a variety of immune effectors and promote Treg suppressive functions through production of TGFβ, IL-10, and arginase I (38, 58, 79, 80). They have also been shown to induce T cell apoptosis by upregulating expression of B7-H1 (81), the ligand for the PD-1 receptor on T cells. Taken together, the diverse effects exerted by TAM and MDSC on cells of both the innate and adaptive immune systems contribute greatly to the immunosuppressive nature of the tumor microenvironment, and these phenomena highlight the role that tumor-altered differentiation of DC progenitors into MDSC and TAM plays in promoting tumor immune escape.
In addition to subverting anti-tumor immunity by altering the differentiation of DC precursors and either preventing acquisition of APC function by these cells or inducing their development into immunosuppressive MDSC or TAM, tumors may also interfere with the maturation and activation of fully differentiated DC. While in vitro studies have shown that the release of heat-shock proteins by necrotic tumors triggers DAMP-mediated DC maturation (82, 83), and the presence of mature tumor-infiltrating DC correlates with the magnitude of anti-tumor T cell responses and disease prognosis in cancer patients (84, 85), a number of studies have described the accumulation of fully differentiated, yet immature, DC in tumors as well (86–88). Although a lack of mature DC in tumor tissue might reflect tumor-induced death of these cells (31, 32, 89), this phenomenon does not explain the accumulation of immature DC often seen in tumors. In cases where immature DC are recovered from tumors, it is often unclear whether the immature phenotype of these cells reflects a simple failure of tumors to support DC maturation and activation or, alternatively, an active suppression of DC maturation by tumors. One study demonstrated that administration of anti-CD40 Ab to tumor-bearing animals leads to maturation of DC capable of stimulating T cell activation (90), suggesting that the tumor either fails to support DC maturation or that suppression of DC maturation by the tumor is a reversible process. In support of the latter possibility, it has been shown that the maturation of tumor-infiltrating DC is enhanced following dissociation of DC from the tumor and overnight culture ex vivo, demonstrating that the tumor had actively limited DC maturation in vivo (91). Other studies have revealed that tumor-infiltrating DC are refractory to some maturation stimuli but not others, indicating that tumors can actively suppress DC maturation but that in some cases this suppression can be reversed under appropriate stimulatory conditions (92–94). Interestingly, in a comparative study of melanoma patients exhibiting either progressing or regressing metastases, DC isolated from patients with progressive disease expressed significantly lower levels of costimulatory molecules than those taken from patients with regressing tumors. Furthermore, DC from patients with regressing metastases induced robust T cell proliferation, while DC from patients with progressing metastases induced T cell anergy (95). Collectively, these data suggest that the context in which the tumor is encountered by DC is likely to impact the quality of their maturation, activation, and immunostimulatory capacity, and they emphasize the need to understand the role of tumor-derived factors and the tumor microenvironment in regulating the function of tumor-associated DC.
The limiting number of DC that can be isolated from tumor-bearing animals and cancer patients and the complex nature of the cell types and soluble proteins present within the tumor microenvironment have made it difficult to gain mechanistic insights into tumor-associated suppression of DC maturation in vivo. Ex vivo experiments with monocyte-derived and bone marrow-derived DC (BMDC) have been used as an alternative to in vivo studies for evaluating the suppression of DC maturation by tumor cells or tumor-conditioned media (96–98). Recent studies using these and similar ex vivo models have shown that interference with the HIF-1-induced COX-2/PGE2 and VEGF pathways in colon cancer cells and knockdown of TGFβ expression in hepatocellular carcinoma both restore DC maturation that is otherwise suppressed by these tumors (99, 100). In another system involving a multicellular tumor spheroid three-dimensional model of melanoma, tumor-derived lactic acid was shown to suppress the production of several proinflammatory cytokines, including IL-12, by monocyte-derived DC and to limit the ability of these cells to induce T cell proliferation (101). Importantly, though, because these ex vivo systems often require differentiation of DC from progenitors in culture, it is often unclear from these studies whether the effects observed stem from a direct influence of tumors on DC or instead from an indirect action mediated by an influence of tumors on other cells in the culture that have not differentiated into DC. Therefore, to overcome the limitations inherent with studying the influence of tumors on DC function in both in vivo and ex vivo settings, DC lines that can be maintained as highly pure populations in culture have been generated and are a useful tool for in vitro studies aimed at understanding the basic biology of these cells (102–105). Such lines have enabled direct analyses of tumor/DC interactions, and it has recently been shown that melanoma-derived factors suppress the LPS-induced maturation of both the DC2.4 and JAWSII DC lines (106). In a related study, a comparative analysis of multiple murine melanoma cell lines demonstrated that the suppression of DC2.4 costimulatory molecule and proinflammatory cytokine/chemokine expression correlates with the tumorigenicity of the melanoma under study (107), with the highly tumorigenic B16 melanoma exhibiting significantly greater suppression than its poorly tumorigenic, chemically mutated variant D5.1G4. These findings again point to a potentially vital role for tumor/DC interactions in the regulation of overall anti-tumor immunity and tumor outgrowth. It will be interesting to evaluate differences in the profile of immunosuppressive mediators released by these particular melanoma cell lines, as this analysis will identify potential candidate molecules involved in the suppression of DC maturation and activation by this cancer. While concerns have been raised that maneuvers employed to immortalize DC lines may alter the maturation state of these cells and their responsiveness to regulatory factors, many of these lines do exhibit the characteristics of immature DC and are responsive to traditional maturation stimuli (108–110). Therefore, additional studies using these DC lines and other tumor systems can offer proof-of-principle data that tumors interfere with DC maturation in a straightforward, cost-effective model, and such investigations will provide further mechanistic insight into tumor-associated suppression of DC maturation and activation. Furthermore, observations made in such in vitro systems are likely to inform the design of experiments evaluating the role of tumor-derived factors in the suppression of DC maturation and activation in more physiologically relevant ex vivo and in vivo settings. Collectively, use of these different models will increase our understanding of tumor-induced suppression of DC function, and these insights will suggest immunotherapeutic strategies designed to reverse or prevent this suppression and enhance the immunostimulatory capacity of tumor-associated DC.
The suppression of DC maturation and activation by tumor cells or factors within the tumor microenvironment has significant implications for the induction of T cell immunity to tumors, as immature DC are poor APC and do not efficiently stimulate T cell activation. There is also now substantial evidence that tumors not only suppress DC maturation but that they can also induce the development of regulatory DC that actively display immunosuppressive activity themselves. In fact, recent studies have demonstrated that progression of ovarian cancer from an immunologically controlled state to metastatic disease is accompanied by a switch in the phenotype and function of tumor-associated DC. Whereas DC isolated from ascites or draining lymph nodes of early-stage tumor-bearing mice elicited robust T cell responses, those isolated from mice with advanced disease induced minimal T cell proliferation and suppressed T cell activation by immunocompetent DC (111, 112). Immunosuppressive DC isolated from late-stage tumor-bearing animals downregulated MHC class II and CD40 expression but significantly upregulated the co-inhibitory molecule B7-H1 and exhibited arginase I activity comparable to that seen in MDSC. These immunosuppressive activities were driven by tumor-derived PGE2 and TGFβ (112). Other studies have also demonstrated tumor-induced upregulation of DC co-inhibitory molecules, including both B7-H1 and B7-DC (10, 96), as well as tumor-enhanced secretion of arginase I (113, 114) and TGFβ (115) by DC that inhibit T cell effector function and promote Treg development, respectively. In both tumor-bearing mice and prostate cancer patients, the expression of these and other immunoregulatory molecules by tumor-associated DC resulted from elevated expression of FOXO3 (116), a transcription factor recently shown to mitigate DC stimulatory capacity (117). Additionally, inhibition of T cell effector activity by tumor-associated regulatory DC has also been associated with increased IL-10 secretion by these cells. A variety of soluble factors present in colorectal tumor explant cultures, including VEGF and the chemokines CCL2, CXCL1, and CXCL5, were shown to enhance IL-10 production by DC (118, 119). Non-soluble mediators expressed on colorectal carcinoma cells can contribute to this process as well, as IL-10 production by DC was increased following engagement of DC-SIGN by tumor-associated cell surface glycans (120). Likewise, recombinant MUC1 mucins glycosylated in a manner equivalent to those expressed on breast carcinoma cells and natural MUC1 mucins in supernatants of human pancreatic carcinoma cell lines both suppress IL-12 production and promote IL-10 production by monocyte-derived DC, and these regulatory DC are poor stimulators of T cell proliferation and CTL activity but potent inducers of T cell anergy and CD4+ Tregs (121, 122). IL-10 production by tumor-associated DC that inhibit anti-tumor T cell responses and promote tumor outgrowth has also been reported to be induced by COX-2/PGE2 (123, 124). Similarly, in a murine myeloma model, tumor-derived IL-6, IL-10, and TGFβ were all shown to contribute to p38 MAPK signaling-mediated effects on BMDC maturation that led to decreased production of IL-12 and increased production of IL-10 by DC, and these cells elicited poor tumor-specific TH1, CTL, and antibody responses (125). Hyperactivation of MAPK signaling similarly inhibited IL-12 production and TH1 stimulation by melanoma-altered DC, though these effects were independent of IL-10, TGFβ, VEGF, and PGE2 in tumor lysates (97). In addition to suppressing the development of TH1-type immunity, other studies have shown that melanoma, as well as breast cancer, triggers DC-mediated induction of TH2-like responses that promote tumor development (126, 127). Identification of factors produced by these tumors and their role in MAPK hyperactivation in DC will be crucial to developing strategies for skewing anti-tumor T cells toward type 1 responses that are more efficient in mediating tumor rejection.
In addition to the regulatory DC activities described above, which are largely associated with conventional mDC, a specialized subset of DC that develop immunosuppressive activity in the context of many tumors is the pDC. IDO-expressing pDC can be induced by tumor-derived PGE2 (128) and have been recovered from tumor-draining lymph nodes of both melanoma-bearing animals and cancer patients (129). These cells suppress CD8+ T cell responses to Ag presented by the pDC themselves as well as to those presented by third-party, non-suppressive APC. In addition to inducing CD8+ T cell anergy, IDO production by pDC also promotes the differentiation of CD4+ CD25+ FOXP3+ Tregs (130). Interestingly, pharmacologic blockade of IDO leads to enhanced IL-6 production by pDC that converts tolerogenic CD4+ Tregs into TH17-like cells, and this conversion correlates with enhanced CD8+ T cell activation and anti-tumor immunity (131). CD4+ Treg induction by pDC can also be mediated by engagement of ICOS on T cells with ICOS-L on pDC, and ICOS-L+ pDC infiltration of tumors is associated with poor prognosis and disease progression in both breast and ovarian cancer patients (132–134). Tumors can also subvert immunity by regulating pDC production of IFN-α, a type I IFN that functions as a “signal 3” cytokine for CD8+ T cell activation (135) and that promotes the survival and Ag retention of CD8α+ DC that cross-prime tumor-specific CD8+ T cells (11). In clinical studies, tumor-associated pDC have been isolated by magnetic activated cell sorting via BDCA-4 positive selection of lineage-negative enriched mononuclear cells obtained from patient biopsies. In patients with aggressive breast cancers, these pDC exhibit suppressed IFN-α secretion and are able to sustain CD4+ Treg expansion (136), and the suppression of IFN-α production by pDC has been attributed to tumor-derived TGFβ and TNFα mediated-signaling in these cells (137). Finally, pDC isolated from ascites of ovarian carcinoma patients have also been shown to induce CD8+ Tregs that secrete high levels of IL-10 and suppress T cell proliferation (138). Altogether, these findings demonstrate the complexity of the tumor microenvironment and its ability to induce a variety of immunoregulatory activities in DC that impact the function of multiple cell types involved in anti-tumor immune responses (Table 3). Tumor-associated conversion of these potentially immunostimulatory APC into suppressive cells is therefore a significant hurdle to the induction of effective anti-tumor immunity that contributes greatly to tumor immune evasion.

Table 3. Induction and suppressive activity of tumor-associated regulatory DC.
The induction of DC dysfunction is a major impediment to the activation and maintenance of successful anti-tumor immunity (Figure 1). Indeed, in addition to its documented impacts on anti-tumor T cell responses summarized herein, this phenomenon may also explain the unaccounted for presence of dysfunctional T cells associated with naturally generated immune responses in other experimental animal models and cancer patients (18–24). Additionally, tumor-associated DC dysfunction may limit the efficacy of immunotherapeutic strategies that rely on the activity of DC in situ to stimulate anti-tumor immunity, and it may therefore explain the lack of success observed thus far with many DNA-, peptide-, and protein-based immunization maneuvers that require endogenous DC to process and present tumor Ag to specific T cells (139–143). Even the quality of responses elicited following immunization with previously activated, exogenous DC may be compromised by an influence of tumor-associated factors on DC function. Importantly, though, insights into the mechanistic bases for tumor-associated DC dysfunction have informed the design of novel DC-based cancer immunotherapies, and many of these strategies have enhanced the T cell stimulatory capacity of DC and led to induction of more robust and efficacious anti-tumor immune responses.
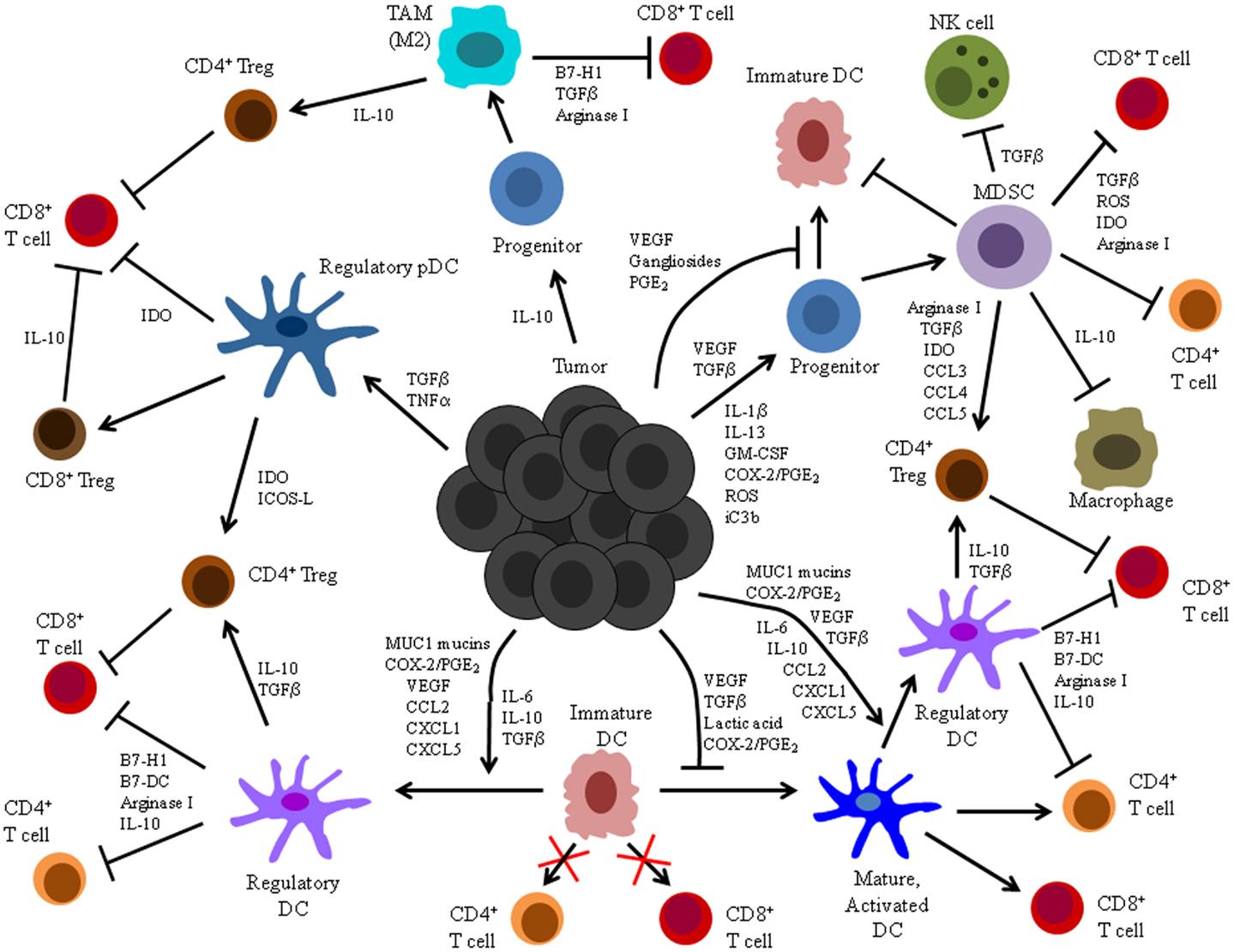
Figure 1. Summary of tumor-altered DC function. Illustrated here are the mechanisms by which tumors alter DC function and the processes by which these altered cells impact host anti-tumor immunity. Tumors secrete a variety of factors that can: (1) inhibit differentiation of DC from precursors, (2) induce differentiation of DC precursors into immunosuppressive MDSC or TAM, (3) suppress maturation, activation, and stimulatory APC function of already differentiated DC, and (4) induce development of immunosuppressive regulatory DC.
Several strategies have been employed to promote DC differentiation from hematopoietic precursors and prevent the accumulation and suppressive activities of tumor-associated cells of myeloid origin. For instance, both IL-4 and IL-13 were shown to prevent renal cell carcinoma-induced blockade of DC differentiation (144). Similarly, administration of the anti-VEGF Ab bevacizumab to patients with lung, breast, and colorectal carcinoma led to a decrease in the frequency of MDSC and enhanced the T cell stimulatory capacity of DC (145). Abrogation of MDSC immunosuppression can also be achieved by exposure of these cells to all-trans retinoic acid, which induces the differentiation of MDSC isolated from a number of murine tumors and renal cell carcinoma patients into mature immunostimulatory DC (146, 147). Others have demonstrated that interference with STAT3-mediated-signaling reverses immune suppression by MDSC and enables differentiation of these cells into mature DC (39, 61). One study also showed that interference with both STAT3 and p38 MAPK signaling pathways in monocyte progenitors further improved the quality of tumor-associated DC, blocking the inhibitory effects of tumor-derived factors on DC differentiation from these progenitors and skewing the IL-12/IL-10 cytokine profile of the resulting DC toward a TH1-promoting phenotype (148). Based on these data, it is not surprising that vaccination with exogenous, STAT3-depleted DC was shown to enhance anti-tumor CD8+ T cell responses and improve control of tumor outgrowth (149).
In addition to strategies that interfere with the development and suppressive activities of tumor-associated myeloid cells, several approaches are being explored for improving the quality of fully differentiated DC in the context of tumors as well. In vivo administration of nanoparticles carrying immunostimulatory miRNA converts endogenous immunosuppressive DC into cells capable of activating robust anti-tumor responses that inhibit progression of established ovarian cancers (150). Moreover, supplementation of stimulatory cytokines whose expression is often suppressed in tumor-associated DC, such as IL-12 and IFNα, can enhance T cell effector function elicited by endogenous DC (151, 152). Significant efforts have also been made to optimize exogenous DC-based cancer immunotherapies. Several studies have investigated various maturation protocols for exogenous DC in order to best promote the immunostimulatory capacity and vaccine efficacy of these cells (153–156). One group has reported that treatment of PBMC-derived immature DC with various combinations of cytokines and inflammatory stimuli, namely LPS + IFNγ, LPS + IFNγ + IL-1β, and LPS + IFNγ + IL-1β + TNFα, results in no discernible difference in DC expression of costimulatory molecules or IL-12 (153). On the other hand, substantial differences in DC maturation have been observed following exposure of immature DC to a mixture of various other inflammatory mediator/cytokine cocktails. Stimulation with lipid A and IFNγ resulted in significantly higher DC expression of costimulatory molecules and IL-12 than stimulation with a combination of TNF-α, IFN-α, IFN-γ, IL-1β, and poly(I:C) or a combination of TNF-α, IFN-γ, IL-1β, and CL097 (156). Still others have evaluated DC maturation following exposure to tumor lysates. PBMC-derived DC treated with lysates from heat-shocked melanoma cells exhibited robust maturation and immunostimulatory capacity, as these cells were capable of cross-presenting melanoma-associated Ag and inducing anti-tumor CD8+ T cell responses (154). Importantly, heat-shocking of melanoma cells induced membrane translocation of CRT and expression of HMGB1, and the maturation of immunostimulatory DC in this study was dependent on their recognition of these tumor-derived “danger signals.” It has also recently been shown that TNFα can augment tumor lysate-induced DC maturation (155). In addition to investigating strategies for optimal induction of DC maturation, many researchers have employed strategies to block the suppressive effects of tumor-derived factors on exogenous DC. In this light, DC genetically engineered to secrete a VEGF/vascular permeability factor decoy receptor that neutralizes soluble VEGF and precludes signaling in DC resulted in increased expression of costimulatory molecules and proinflammatory cytokines/chemokines by DC and improved CTL activity and anti-tumor immune control in a murine colon cancer model (157). Similar improvements in the efficacy of an exogenous DC vaccine were observed following neutralization of tumor-derived TGFβ (158). Alternatively, other approaches for enhancing exogenous DC-induced anti-tumor immune responses aim at blocking either the immunosuppressive mediators expressed by tumor-altered DC or the targets of these mediators expressed on other immune cells. In a murine model of breast cancer, siRNA-mediated silencing of IDO in vaccinating DC enhanced the ability of these cells to stimulate T cell proliferation and CTL effector function, decreased the induction of CD4+ CD25+ FOXP3+ Tregs, and led to enhanced control of tumor outgrowth (159). Similarly, immunization with IL-10-deficient DC conferred enhanced protective and therapeutic immunity against a murine hepatocellular carcinoma (160). Furthermore, DC genetically engineered to interfere with immunomodulatory receptors expressed on endogenous immune cells, such as CTLA-4 on effector T cells and GITR on Tregs, can enhance the overall immunogenicity of these cells as well (161, 162). Improved anti-tumor immunity has also been observed for a DC-based vaccine administered in combination with anti-CTLA-4 Ab and Treg-depleting anti-CD25 Ab (163). Likewise, administration of neutralizing Ab that interferes with the B7-H1/PD-1 pathway improved the efficacy of a DC/tumor fusion vaccine in multiple myeloma patients (164). Finally, it is also possible to improve the efficacy of both exogenous and endogenous DC-based vaccines by transducing DC either ex vivo or in vivo with viral vectors that encode immunostimulatory molecules. A number of studies have reported improved anti-tumor immunity when this approach was used to drive expression of CD80/CD86 costimulatory molecules (165, 166) or IL-12 (167) by DC. Collectively, these strategies highlight the advances made in tumor immunotherapy as our understanding of tumor immune suppression and evasion has evolved over the last several years. As additional insights into tumor-altered DC function are gained, optimization of these current immunotherapies and development of novel strategies for enhancing anti-tumor immune responses will further improve the efficacy of DC-based cancer vaccines.
Tumor immunosurveillance is now a well-documented phenomenon whereby host immune cells and effector molecules function to recognize and eradicate developing tumors in the body. At the heart of this process are DC, innate immune cells that function to acquire tumor Ag through phagocytosis, activate adaptive immunity against these specific tumor Ag, and recruit immune effectors to the site of the tumor for immunologic destruction of these transformed cells. However, one of the hallmarks of cancer growth is immune evasion, and tumor cells may evolve a number of escape mechanisms during their progression that subvert immunosurveillance. A significant contributor to tumor immune evasion is the alteration of DC function by tumors and associated factors present in the tumor microenvironment. As discussed, such alteration of DC function may include effects on the differentiation of DC from bone marrow-derived precursors, suppression of the maturation and activation of already differentiated DC, and the induction of immunosuppressive regulatory DC that inhibit anti-tumor immune responses. Over the last several years, significant efforts have been made to gain mechanistic insights into these processes of tumor-altered DC function. These findings have in turn led to the development of several immunotherapeutic strategies for improving the function of tumor-associated DC. Still, much remains to be learned about the processes by which tumors impact the function of DC and how such altered DC influence the quality of other immune effectors. As this field moves forward, it will be important to increase our understanding of factors that contribute to tumor recognition by DC and to identify additional tumor-associated DAMPs and inflammatory stimuli that promote optimal maturation and activation of immunostimulatory DC. Additionally, a better understanding of how tumor microenvironmental factors impact the quality of DC differentiation, maturation, and activation will suggest new possibilities for interfering with the suppression of these processes by tumors. Such knowledge will enable the optimization of current, and the development of novel, DC-based immunotherapies that aim to improve the quality and outcome of host anti-tumor immune responses.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research in Kristian M. Hargadon’s laboratory is funded by a Virginia Academy of Science Jeffress Research Grant from the Thomas F. Jeffress and Kate Miller Jeffress Memorial Trust, a Virginia Foundation for Independent Colleges Mednick Memorial Fellowship, and a Hampden-Sydney College Research Grant from the Arthur Vining Davis endowment.
1. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi:10.1038/32588
2. Albert ML, Jegathesan M, Darnell RB. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat Immunol (2001) 2:1010–7. doi:10.1038/ni722
3. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med (2001) 194:769–80. doi:10.1084/jem.194.6.769
4. Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med (2001) 193:233–8. doi:10.1084/jem.193.2.233
5. Probst HC, Lagnel J, Kollias G, Van den Broek M. Inducible transgenic mice reveal resting dendritic cells as potent inducers of CD8+ T cell tolerance. Immunity (2003) 18:713–20. doi:10.1016/S1074-7613(03)00120-1
6. Probst HC, McCoy K, Okazaki T, Honjo T, Van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol (2005) 6:280–6. doi:10.1038/ni1165
7. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi:10.1146/annurev.immunol.12.1.991
8. Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
9. Joffre O, Nolte MA, Spörri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev (2009) 227:234–47. doi:10.1111/j.1600-065X.2008.00718.x
10. McDonnell AM, Prosser AC, Van Bruggen I, Robinson BWS, Currie AJ. CD8alpha+ DC are not the sole subset cross-presenting cell-associated tumor antigens from a solid tumor. Eur J Immunol (2010) 40:1617–27. doi:10.1002/eji.200940153
11. Lorenzi S, Mattei F, Sistigu A, Bracci L, Spadaro F, Sanchez M, et al. Type I IFNs control antigen retention and survival of CD8α(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J Immunol (2011) 186:5142–50. doi:10.4049/jimmunol.1004163
12. Tel J, Schreibelt G, Sittig SP, Mathan TSM, Buschow SI, Cruz LJ, et al. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood (2013) 121:459–67. doi:10.1182/blood-2012-06-435644
13. Speiser DE, Miranda R, Zakarian A, Bachmann MF, McKall-Faienza K, Odermatt B, et al. Self antigens expressed by solid tumors do not efficiently stimulate naive or activated T cells: implications for immunotherapy. J Exp Med (1997) 186:645–53. doi:10.1084/jem.186.5.645
14. Hermans IF, Daish A, Yang J, Ritchie DS, Ronchese F. Antigen expressed on tumor cells fails to elicit an immune response, even in the presence of increased numbers of tumor-specific cytotoxic T lymphocyte precursors. Cancer Res (1998) 58:3909–17.
15. Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M, Hengartner H, et al. Immune surveillance against a solid tumor fails because of immunological ignorance. Proc Natl Acad Sci U S A (1999) 96:2233–8. doi:10.1073/pnas.96.5.2233
16. Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, et al. Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature (2001) 411:1058–64. doi:10.1038/35082583
17. Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, et al. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity (2002) 17:737–47. doi:10.1016/S1074-7613(02)00480-6
18. Radoja S, Saio M, Schaer D, Koneru M, Vukmanovic S, Frey AB. CD8(+) tumor-infiltrating T cells are deficient in perforin-mediated cytolytic activity due to defective microtubule-organizing center mobilization and lytic granule exocytosis. J Immunol (2001) 167:5042–51.
19. Blohm U, Roth E, Brommer K, Dumrese T, Rosenthal FM, Pircher H. Lack of effector cell function and altered tetramer binding of tumor-infiltrating lymphocytes. J Immunol (2002) 169:5522–30.
20. Anichini A, Scarito A, Molla A, Parmiani G, Mortarini R. Differentiation of CD8+ T cells from tumor-invaded and tumor-free lymph nodes of melanoma patients: role of common gamma-chain cytokines. J Immunol (2003) 171:2134–41.
21. Mortarini R, Piris A, Maurichi A, Molla A, Bersani I, Bono A, et al. Lack of terminally differentiated tumor-specific CD8+ T cells at tumor site in spite of antitumor immunity to self-antigens in human metastatic melanoma. Cancer Res (2003) 63:2535–45.
22. Zippelius A, Batard P, Rubio-godoy V, Bioley G, Lie D, Lejeune F, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res (2004) 64:2865–73. doi:10.1158/0008-5472.CAN-03-3066
23. Whiteside TL. Down-regulation of zeta-chain expression in T cells: a biomarker of prognosis in cancer? Cancer Immunol Immunother (2004) 53:865–78. doi:10.1007/s00262-004-0521-0
24. Hargadon KM, Brinkman CC, Sheasley-O’Neill SL, Nichols LA, Bullock TNJ, Engelhard VH. Incomplete differentiation of antigen-specific CD8 T cells in tumor-draining lymph nodes. J Immunol (2006) 177:6081–90.
25. Menetrier-Caux C, Montmain G, Dieu MC, Bain C, Favrot MC, Caux C, et al. Inhibition of the differentiation of dendritic cells from CD34(+) progenitors by tumor cells: role of interleukin-6 and macrophage colony-stimulating factor. Blood (1998) 92:4778–91.
26. Bharadwaj U, Li M, Zhang R, Chen C, Yao Q. Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res (2007) 67:5479–88. doi:10.1158/0008-5472.CAN-06-3963
27. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2:1096–103. doi:10.1038/nm1096-1096
28. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol (1998) 160:1224–32.
29. Ishida T, Oyama T, Carbone DP, Gabrilovich DI. Defective function of Langerhans cells in tumor-bearing animals is the result of defective maturation from hemopoietic progenitors. J Immunol (1998) 161:4842–51.
30. Shurin GV, Shurin MR, Bykovskaia S, Shogan J, Lotze MT, Barksdale EM. Neuroblastoma-derived gangliosides inhibit dendritic cell generation and function. Cancer Res (2001) 61:363–9.
31. Péguet-Navarro J, Sportouch M, Popa I, Berthier O, Schmitt D, Portoukalian J. Gangliosides from human melanoma tumors impair dendritic cell differentiation from monocytes and induce their apoptosis. J Immunol (2003) 170:3488–94.
32. Bennaceur K, Popa I, Chapman JA, Migdal C, Péguet-Navarro J, Touraine J-L, et al. Different mechanisms are involved in apoptosis induced by melanoma gangliosides on human monocyte-derived dendritic cells. Glycobiology (2009) 19:576–82. doi:10.1093/glycob/cwp015
33. Sombroek CC, Stam AGM, Masterson AJ, Lougheed SM, Schakel MJAG, Meijer CJLM, et al. Prostanoids play a major role in the primary tumor-induced inhibition of dendritic cell differentiation. J Immunol (2002) 168:4333–43.
34. Stock A, Booth S, Cerundolo V. Prostaglandin E2 suppresses the differentiation of retinoic acid-producing dendritic cells in mice and humans. J Exp Med (2011) 208:761–73. doi:10.1084/jem.20101967
35. Almand B, Clark JI, Nikitina E, Van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol (2001) 166:678–89.
36. Watanabe S, Deguchi K, Zheng R, Tamai H, Wang L-X, Cohen PA, et al. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J Immunol (2008) 181:3291–300.
37. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182:4499–506. doi:10.4049/jimmunol.0802740
38. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi:10.1038/nri3175
39. Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol (2004) 172:464–74.
40. Xiang X, Poliakov A, Liu C, Liu Y, Deng Z, Wang J, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer (2009) 124:2621–33. doi:10.1002/ijc.24249
41. Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, et al. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol (2005) 175:8200–8.
42. Terabe M, Matsui S, Park J-M, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med (2003) 198:1741–52. doi:10.1084/jem.20022227
43. Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol (1999) 162:5728–37.
44. Obermajer N, Wong JL, Edwards RP, Odunsi K, Moysich K, Kalinski P. PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunol Invest (2012) 41:635–57. doi:10.3109/08820139.2012.695417
45. Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J Immunol (2012) 188:5365–76. doi:10.4049/jimmunol.1103553
46. Hsieh C-C, Chou H-S, Yang H-R, Lin F, Bhatt S, Qin J, et al. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood (2013) 121:1760–8. doi:10.1182/blood-2012-06-440214
47. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med (2008) 205:2235–49. doi:10.1084/jem.20080132
48. Mace TA, Ameen Z, Collins A, Wojcik SE, Mair M, Young GS, et al. Pancreatic cancer associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res (2013) 73:3007–18. doi:10.1158/0008-5472.CAN-12-4601
49. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol (2001) 166:5398–406.
50. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol (2004) 172:989–99.
51. Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol (2005) 175:4583–92.
52. Apolloni E, Bronte V, Mazzoni A, Serafini P, Cabrelle A, Segal DM, et al. Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated T lymphocytes. J Immunol (2000) 165:6723–30.
53. Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol (2002) 168:689–95.
54. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182:5693–701. doi:10.4049/jimmunol.0900092
55. Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222:180–91. doi:10.1111/j.1600-065X.2008.00608.x
56. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (2013) 190:3783–97. doi:10.4049/jimmunol.1201449
57. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202:931–9. doi:10.1084/jem.20050715
58. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64:5839–49. doi:10.1158/0008-5472.CAN-04-0465
59. Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res (2007) 13:721s–6. doi:10.1158/1078-0432.CCR-06-2197
60. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother (2009) 58:49–59. doi:10.1007/s00262-008-0523-4
61. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest (2013) 123:1580–9. doi:10.1172/JCI60083
62. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med (2005) 202:919–29. doi:10.1084/jem.20050463
63. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189:5602–11. doi:10.4049/jimmunol.1201018
64. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res (2008) 68:5439–49. doi:10.1158/0008-5472.CAN-07-6621
65. Mauti LA, Le Bitoux M-A, Baumer K, Stehle J-C, Golshayan D, Provero P, et al. Myeloid-derived suppressor cells are implicated in regulating permissiveness for tumor metastasis during mouse gestation. J Clin Invest (2011) 121:2794–807. doi:10.1172/JCI41936
66. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182:240–9.
67. Elkabets M, Ribeiro VSG, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, et al. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol (2010) 40:3347–57. doi:10.1002/eji.201041037
68. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (2009) 50:799–807. doi:10.1002/hep.23054
69. Poschke I, Mao Y, Adamson L, Salazar-Onfray F, Masucci G, Kiessling R. Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol Immunother (2012) 61:827–38. doi:10.1007/s00262-011-1143-y
70. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol (2007) 179:977–83.
71. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A (2012) 109:2491–6. doi:10.1073/pnas.1113744109
72. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi:10.1126/science.1129139
73. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362:875–85. doi:10.1056/NEJMoa0905680
74. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffel B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov (2011) 1:54–67. doi:10.1158/2159-8274.CD-10-0028
75. Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, et al. Significance of M2-polarized tumor-associated macrophages in pancreatic cancer. J Surg Res (2011) 167:e211–9. doi:10.1016/j.jss.2009.05.026
76. Zhang BC, Gao J, Wang J, Rao ZG, Wang BC, Gao JF. Tumor-associated macrophages infiltration is associated with peritumoral lymphangiogenesis and poor prognosis in lung adenocarcinoma. Med Oncol (2011) 28:1447–52. doi:10.1007/s12032-010-9638-5
77. Schouppe E, De Baetselier P, Van Ginderachter JA, Sarukhan A. Instruction of myeloid cells by the tumor microenvironment: open questions on the dynamics and plasticity of different tumor-associated myeloid cell populations. Oncoimmunology (2012) 1:1135–45. doi:10.4161/onci.21566
78. Allavena P, Piemonti L, Longoni D, Bernasconi S, Stoppacciaro A, Ruco L, et al. IL-10 prevents the differentiation of monocytes to dendritic cells but promotes their maturation to macrophages. Eur J Immunol (1998) 28:359–69. doi:10.1002/(SICI)1521-4141(199801)28:01<359::AID-IMMU359>3.0.CO;2-4
79. Chong H, Vodovotz Y, Cox GW, Barcellos-Hoff MH. Immunocytochemical localization of latent transforming growth factor-beta1 activation by stimulated macrophages. J Cell Physiol (1999) 178:275–83. doi:10.1002/(SICI)1097-4652(199903)178:3<275::AID-JCP1>3.0.CO;2-Q
80. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10:942–9. doi:10.1038/nm1093
81. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206:1327–37. doi:10.1084/jem.20082173
82. Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med (2000) 191:423–34. doi:10.1084/jem.191.3.423
83. Somersan S, Larsson M, Fonteneau JF, Basu S, Srivastava P, Bhardwaj N. Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J Immunol (2001) 167:4844–52.
84. Movassagh M, Spatz A, Davoust J, Lebecque S, Romero P, Pittet M, et al. Selective accumulation of mature DC-Lamp+ dendritic cells in tumor sites is associated with efficient T-cell-mediated antitumor response and control of metastatic dissemination in melanoma. Cancer Res (2004) 64:2192–8. doi:10.1158/0008-5472.CAN-03-2969
85. Elliott B, Scolyer RA, Suciu S, Lebecque S, Rimoldi D, Gugerli O, et al. Long-term protective effect of mature DC-LAMP+ dendritic cell accumulation in sentinel lymph nodes containing micrometastatic melanoma. Clin Cancer Res (2007) 13:3825–30. doi:10.1158/1078-0432.CCR-07-0358
86. Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med (1999) 190:1417–26. doi:10.1084/jem.190.10.1417
87. Treilleux I, Blay J-Y, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla J-P, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res (2004) 10:7466–74. doi:10.1158/1078-0432.CCR-04-0684
88. Bergeron A, El-Hage F, Kambouchner M, Lecossier D, Tazi A. Characterisation of dendritic cell subsets in lung cancer micro-environments. Eur Respir J (2006) 28:1170–7.
89. Manabu I, Minamiya Y, Kawai H, Saito S, Saito H, Nakagawa T, et al. Tumor-derived TGFβ-1 induces dendritic cell apoptosis in the sentinel lymph node. J Immunol (2006) 176:5637–43.
90. Van Mierlo GJD, Boonman ZFHM, Dumortier HMH, Den Boer AT, Fransen MF, Nouta J, et al. Activation of dendritic cells that cross-present tumor-derived antigen licenses CD8+ CTL to cause tumor eradication. J Immunol (2004) 173:6753–9.
91. Preynat-Seauve O, Schuler P, Contassot E, Beermann F, Huard B, French LE. Tumor-infiltrating dendritic cells are potent antigen-presenting cells able to activate T cells and mediate tumor rejection. J Immunol (2006) 176:61–7.
92. Chaux P, Favre N, Martin M, Martin F. Tumor-infiltrating dendritic cells are defective in their antigen-presenting function and inducible B7 expression in rats. Int J Cancer (1997) 72:619–24. doi:10.1002/(SICI)1097-0215(19970807)72:4<619::AID-IJC12>3.0.CO;2-6
93. Vicari AP, Chiodoni C, Vaure C, Ait-Yahia S, Dercamp C, Matsos F, et al. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med (2002) 196:541–9. doi:10.1084/jem.20020732
94. Perrot I, Blanchard D, Freymond N, Isaac S, Guibert B, Pacheco Y, et al. Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J Immunol (2007) 178:2763–9.
95. Enk AH, Jonuleit H, Saloga J, Knop J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int J Cancer (1997) 73:309–16. doi:10.1002/(SICI)1097-0215(19971104)73:3<309::AID-IJC1>3.3.CO;2-B
96. Idoyaga J, Moreno J, Bonifaz L. Tumor cells prevent mouse dendritic cell maturation induced by TLR ligands. Cancer Immunol Immunother (2007) 56:1237–50. doi:10.1007/s00262-006-0275-y
97. Jackson AM, Mulcahy LA, Zhu XW, O’Donnell D, Patel PM. Tumour-mediated disruption of dendritic cell function: inhibiting the MEK1/2-p44/42 axis restores IL-12 production and Th1-generation. Int J Cancer (2008) 123:623–32. doi:10.1002/ijc.23530
98. Alshamsan A, Hamdy S, Das S, Lavasanifar A, Samuel J, El-Kadi AOS. Validation of bone marrow derived dendritic cells as an appropriate model to study tumor-mediated suppression of DC maturation through STAT3 hyperactivation. J Pharm Pharm Sci (2010) 13:21–6.
99. Garufi A, Pistritto G, Ceci C, Di Renzo L, Santarelli R, Faggioni A, et al. Targeting COX-2/PGE(2) pathway in HIPK2 knockdown cancer cells: impact on dendritic cell maturation. PLoS ONE (2012) 7:e48342. doi:10.1371/journal.pone.0048342
100. Wang Y, Deng B, Tang W, Liu T, Shen X. TGF-β1 secreted by hepatocellular carcinoma induces the expression of the Foxp3 gene and suppresses antitumor immunity in the tumor microenvironment. Dig Dis Sci (2013) 58:1644–52. doi:10.1007/s10620-012-2550-4. [Epub ahead of print].
101. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood (2006) 107:2013–21. doi:10.1182/blood-2005-05-1795
102. Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol (1997) 158:2723–30.
103. Winzler C, Rovere P, Rescigno M, Granucci F, Penna G, Adorini L, et al. Maturation stages of mouse dendritic cells in growth factor-dependent long-term cultures. J Exp Med (1997) 185:317–28. doi:10.1084/jem.185.2.317
104. van Helden SFG, Van Leeuwen FN, Figdor CG. Human and murine model cell lines for dendritic cell biology evaluated. Immunol Lett (2008) 117:191–7. doi:10.1016/j.imlet.2008.02.003
105. Fuertes Marraco SA, Grosjean F, Duval A, Rosa M, Lavanchy C, Ashok D, et al. Novel murine dendritic cell lines: a powerful auxiliary tool for dendritic cell research. Front Immunol (2012) 3:331. doi:10.3389/fimmu.2012.00331
106. Hargadon KM, Ararso YT, Forrest OA, Harte CM. Melanoma-associated suppression of the dendritic cell lines DC2.4 and JAWSII. Am J Immunol (2012) 8:179–90.
107. Hargadon KM, Forrest OA, Reddy PR. Suppression of the maturation and activation of the dendritic cell line DC2.4 by melanoma-derived factors. Cell Immunol (2012) 272:275–82. doi:10.1016/j.cellimm.2011.10.003
108. He T, Tang C, Xu S, Moyana T, Xiang J. Interferon gamma stimulates cellular maturation of dendritic cell line DC2.4 leading to induction of efficient cytotoxic T cell responses and antitumor immunity. Cell Mol Immunol (2007) 4:105–11.
109. Rhule A, Rase B, Smith JR, Shepherd DM. Toll-like receptor ligand-induced activation of murine DC2.4 cells is attenuated by Panax notoginseng. J Ethnopharmacol (2008) 116:179–86. doi:10.1016/j.jep.2007.11.019
110. Jiang X, Shen C, Rey-Ladino J, Yu H, Brunham RC. Characterization of murine dendritic cell line JAWS II and primary bone marrow-derived dendritic cells in Chlamydia muridarum antigen presentation and induction of protective immunity. Infect Immun (2008) 76:2392–401. doi:10.1128/IAI.01584-07
111. Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, et al. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol (2011) 186:6905–13. doi:10.4049/jimmunol.1100274
112. Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med (2012) 209:495–506. doi:10.1084/jem.20111413
113. Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J Immunol (2009) 182:6207–16. doi:10.4049/jimmunol.0803926
114. Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res (2009) 69:3086–94. doi:10.1158/0008-5472.CAN-08-2826
115. Dumitriu IE, Dunbar DR, Howie SE, Sethi T, Gregory CD. Human dendritic cells produce TGF-beta 1 under the influence of lung carcinoma cells and prime the differentiation of CD4+CD25+Foxp3+ regulatory T cells. J Immunol (2009) 182:2795–807. doi:10.4049/jimmunol.0712671
116. Watkins SK, Zhu Z, Riboldi E, Shafer-Weaver KA, Stagliano KER, Sklavos MM, et al. FOXO3 programs tumor-associated DCs to become tolerogenic in human and murine prostate cancer. J Clin Invest (2011) 121:1361–72. doi:10.1172/JCI44325
117. Dejean AS, Beisner DR, Chen IL, Kerdiles YM, Babour A, Arden KC, et al. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat Immunol (2009) 10:504–13. doi:10.1038/ni.1729
118. Michielsen AJ, Hogan AE, Marry J, Tosetto M, Cox F, Hyland JM, et al. Tumour tissue microenvironment can inhibit dendritic cell maturation in colorectal cancer. PLoS ONE (2011) 6:e27944. doi:10.1371/journal.pone.0027944
119. Michielsen AJ, O’Sullivan JN, Ryan EJ. Tumor conditioned media from colorectal cancer patients inhibits dendritic cell maturation. Oncoimmunology (2012) 1:751–3. doi:10.4161/onci.19570
120. Nonaka M, Ma BY, Murai R, Nakamura N, Baba M, Kawasaki N, et al. Glycosylation-dependent interactions of C-type lectin DC-SIGN with colorectal tumor-associated Lewis glycans impair the function and differentiation of monocyte-derived dendritic cells. J Immunol (2008) 180:3347–56.
121. Monti P, Leone BE, Zerbi A, Balzano G, Cainarca S, Sordi V, et al. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. J Immunol (2004) 172:7341–9.
122. Rughetti A, Pellicciotta I, Biffoni M, Backstrom M, Link T, Bennet EP, et al. Recombinant tumor-associated MUC1 glycoprotein impairs the differentiation and function of dendritic cells. J Immunol (2005) 174:7764–72.
123. Sharma S, Stolina M, Yang S-C, Baratelli F, Lin JF, Atianzar K, et al. Tumor cyclooxygenase 2-dependent suppression of dendritic cell function. Clin Cancer Res (2003) 9:961–8.
124. Ahmadi M, Emery DC, Morgan DJ. Prevention of both direct and cross-priming of antitumor CD8+ T-cell responses following overproduction of prostaglandin E2 by tumor cells in vivo. Cancer Res (2008) 68:7520–9. doi:10.1158/0008-5472.CAN-08-1060
125. Wang S, Yang J, Qian J, Wezeman M, Kwak LW, Yi Q. Tumor evasion of the immune system: inhibiting p38 MAPK signaling restores the function of dendritic cells in multiple myeloma. Blood (2006) 107:2432–9. doi:10.1182/blood-2005-06-2486
126. McCarter M, Clarke J, Richter D, Wilson C. Melanoma skews dendritic cells to facilitate a T helper 2 profile. Surgery (2005) 138:321–8. doi:10.1016/j.surg.2005.06.011
127. Aspord C, Pedroza-Gonzalez A, Gallegos M, Tindle S, Burton EC, Su D, et al. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med (2007) 204:1037–47. doi:10.1084/jem.20061120
128. Von Bergwelt-Baildon MS, Popov A, Saric T, Chemnitz J, Classen S, Stoffel MS, et al. CD25 and indoleamine 2,3-dioxygenase are up-regulated by prostaglandin E2 and expressed by tumor-associated dendritic cells in vivo: additional mechanisms of T-cell inhibition. Blood (2006) 108:228–37. doi:10.1182/blood-2005-08-3507
129. Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest (2004) 114:280–90. doi:10.1172/JCI21583E1
130. Chen W, Liang X, Peterson AJ, Munn DH, Blazar BR. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J Immunol (2008) 181:5396–404.
131. Sharma MD, Hou D-Y, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood (2009) 113:6102–11. doi:10.1182/blood-2008-12-195354
132. Conrad C, Gregorio J, Wang Y-H, Ito T, Meller S, Hanabuchi S, et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res (2012) 72:5240–9. doi:10.1158/0008-5472.CAN-12-2271
133. Faget J, Bendriss-Vermare N, Gobert M, Durand I, Olive D, Biota C, et al. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells. Cancer Res (2012) 72:6130–41. doi:10.1158/0008-5472.CAN-12-2409
134. Labidi-Galy SI, Treilleux I, Goddard-Leon S, Combes J-D, Blay J-Y, Ray-Coquard I, et al. Plasmacytoid dendritic cells infiltrating ovarian cancer are associated with poor prognosis. Oncoimmunology (2012) 1:380–2. doi:10.4161/onci.18801
135. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol (2005) 174:4465–9.
136. Sisirak V, Faget J, Gobert M, Goutagny N, Vey N, Treilleux I, et al. Impaired IFN-α production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res (2012) 72:5188–97. doi:10.1158/0008-5472.CAN-11-3468
137. Sisirak V, Vey N, Goutagny N, Renaudineau S, Malfroy M, Thys S, et al. Breast cancer-derived transforming growth factor-β and tumor necrosis factor-α compromise interferon-α production by tumor-associated plasmacytoid dendritic cells. Int J Cancer (2013) 133:771–8. doi:10.1002/ijc.28072
138. Wei S, Kryczek I, Zou L, Daniel B, Cheng P, Mottram P, et al. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res (2005) 65:5020–6. doi:10.1158/0008-5472.CAN-04-4043
139. Marchand M, Weynants P, Rankin E, Arienti F, Belli F, Parmiani G, et al. Tumor regression responses in melanoma patients treated with a peptide encoded by gene MAGE-3. Int J Cancer (1995) 63:883–5. doi:10.1002/ijc.2910630622
140. Cormier JN, Salgaller ML, Prevette T, Barracchini KC, Rivoltini L, Restifo NP, et al. Enhancement of cellular immunity in melanoma patients immunized with a peptide from MART-1/Melan A. Cancer J Sci Am (1997) 3:37–44.
141. Tagawa ST, Lee P, Snively J, Boswell W, Ounpraseuth S, Lee S, et al. Phase I study of intranodal delivery of a plasmid DNA vaccine for patients with Stage IV melanoma. Cancer (2003) 98:144–54. doi:10.1002/cncr.11462
142. Nicholaou T, Ebert LM, Davis ID, McArthur GA, Jackson H, Dimopoulos N, et al. Regulatory T-cell-mediated attenuation of T-cell responses to the NY-ESO-1 ISCOMATRIX vaccine in patients with advanced malignant melanoma. Clin Cancer Res (2009) 15:2166–73. doi:10.1158/1078-0432.CCR-08-2484
143. Ebert LM, MacRaild SE, Zanker D, Davis ID, Cebon J, Chen W. A cancer vaccine induces expansion of NY-ESO-1-specific regulatory T cells in patients with advanced melanoma. PLoS ONE (2012) 7:e48424. doi:10.1371/journal.pone.0048424
144. Menetrier-Caux C, Thomachot MC, Alberti L, Montmain G, Blay JY. IL-4 prevents the blockade of dendritic cell differentiation induced by tumor cells. Cancer Res (2001) 61:3096–104.
145. Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother (2008) 57:1115–24. doi:10.1007/s00262-007-0441-x
146. Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res (2003) 63:4441–9.
147. Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kübler H, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res (2008) 14:8270–8. doi:10.1158/1078-0432.CCR-08-0165
148. Oosterhoff D, Lougheed S, Van de Ven R, Lindenberg J, Van Cruijsen H, Hiddingh L, et al. Tumor-mediated inhibition of human dendritic cell differentiation and function is consistently counteracted by combined p38 MAPK and STAT3 inhibition. Oncoimmunology (2012) 1:649–58. doi:10.4161/onci.20365
149. Iwata-Kajihara T, Sumimoto H, Kawamura N, Ueda R, Takahashi T, Mizuguchi H, et al. Enhanced cancer immunotherapy using STAT3-depleted dendritic cells with high Th1-inducing ability and resistance to cancer cell-derived inhibitory factors. J Immunol (2011) 187:27–36. doi:10.4049/jimmunol.1002067
150. Cubillos-Ruiz JR, Baird JR, Tesone AJ, Rutkowski MR, Scarlett UK, Camposeco-Jacobs AL, et al. Reprogramming tumor-associated dendritic cells in vivo using miRNA mimetics triggers protective immunity against ovarian cancer. Cancer Res (2012) 72:1683–93. doi:10.1158/0008-5472.CAN-11-3160
151. Ferguson AR, Nichols LA, Zarling AL, Thompson ED, Brinkman CC, Hargadon KM, et al. Strategies and challenges in eliciting immunity to melanoma. Immunol Rev (2008) 222:28–42. doi:10.1111/j.1600-065X.2008.00620.x
152. Hong S, Qian J, Li H, Yang J, Lu Y, Zheng Y, et al. CpG or IFN-α are more potent adjuvants than GM-CSF to promote anti-tumor immunity following idiotype vaccine in multiple myeloma. Cancer Immunol Immunother (2012) 61:561–71. doi:10.1007/s00262-011-1123-2
153. Han TH, Jin P, Ren J, Slezak S, Marincola FM, Stroncek DF. Evaluation of 3 clinical dendritic cell maturation protocols containing lipopolysaccharide and interferon-gamma. J Immunother (1991) (2009) 32:399–407. doi:10.1097/CJI.0b013e31819e1773
154. Aguilera R, Saffie C, Tittarelli A, González FE, Ramírez M, Reyes D, et al. Heat-shock induction of tumor-derived danger signals mediates rapid monocyte differentiation into clinically effective dendritic cells. Clin Cancer Res (2011) 17:2474–83. doi:10.1158/1078-0432.CCR-10-2384
155. Miwa S, Nishida H, Tanzawa Y, Takata M, Takeuchi A, Yamamoto N, et al. TNF-α and tumor lysate promote the maturation of dendritic cells for immunotherapy for advanced malignant bone and soft tissue tumors. PLoS ONE (2012) 7:e52926. doi:10.1371/journal.pone.0052926
156. Massa C, Seliger B. Fast dendritic cells stimulated with alternative maturation mixtures induce polyfunctional and long-lasting activation of innate and adaptive effector cells with tumor-killing capabilities. J Immunol (2013) 190:3328–37. doi:10.4049/jimmunol.1202024
157. Sugiyama M, Kakeji Y, Tsujitani S, Harada Y, Onimaru M, Yoshida K, et al. Antagonism of VEGF by genetically engineered dendritic cells is essential to induce antitumor immunity against malignant ascites. Mol Cancer Ther (2011) 10:540–9. doi:10.1158/1535-7163.MCT-10-0479
158. Zhang M, Berndt BE, Chen J-J, Kao JY. Expression of a soluble TGF-beta receptor by tumor cells enhances dendritic cell/tumor fusion vaccine efficacy. J Immunol (2008) 181:3690–7.
159. Zheng X, Koropatnick J, Chen D, Velenosi T, Ling H, Zhang X, et al. Silencing IDO in dendritic cells: a novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int J Cancer (2013) 132:967–77. doi:10.1002/ijc.27710
160. Chen Y-X, Man K, Ling GS, Chen Y, Sun B-S, Cheng Q, et al. A crucial role for dendritic cell (DC) IL-10 in inhibiting successful DC-based immunotherapy: superior antitumor immunity against hepatocellular carcinoma evoked by DC devoid of IL-10. J Immunol (2007) 179:6009–15.
161. Boczkowski D, Lee J, Pruitt S, Nair S. Dendritic cells engineered to secrete anti-GITR antibodies are effective adjuvants to dendritic cell-based immunotherapy. Cancer Gene Ther (2009) 16:900–11. doi:10.1038/cgt.2009.39
162. Pruitt SK, Boczkowski D, De Rosa N, Haley NR, Morse MA, Tyler DS, et al. Enhancement of anti-tumor immunity through local modulation of CTLA-4 and GITR by dendritic cells. Eur J Immunol (2011) 41:3553–63. doi:10.1002/eji.201141383
163. Saha A, Chatterjee SK. Combination of CTL-associated antigen-4 blockade and depletion of CD25 regulatory T cells enhance tumour immunity of dendritic cell-based vaccine in a mouse model of colon cancer. Scand J Immunol (2010) 71:70–82. doi:10.1111/j.1365-3083.2009.02355.x
164. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother (1991) (2011) 34:409–18. doi:10.1097/CJI.0b013e31821ca6ce
165. Oertli D, Marti WR, Zajac P, Noppen C, Kocher T, Padovan E, et al. Rapid induction of specific cytotoxic T lymphocytes against melanoma-associated antigens by a recombinant vaccinia virus vector expressing multiple immunodominant epitopes and costimulatory molecules in vivo. Hum Gene Ther (2002) 13:569–75. doi:10.1089/10430340252809856
166. Adamina M, Rosenthal R, Weber WP, Frey DM, Viehl CT, Bolli M, et al. Intranodal immunization with a vaccinia virus encoding multiple antigenic epitopes and costimulatory molecules in metastatic melanoma. Mol Ther (2010) 18:651–9. doi:10.1038/mt.2009.275
Keywords: dendritic cell, tumor, differentiation, maturation, activation, immunosuppression, tumor microenvironment, immunotherapy
Citation: Hargadon KM (2013) Tumor-altered dendritic cell function: implications for anti-tumor immunity. Front. Immunol. 4:192. doi: 10.3389/fimmu.2013.00192
Received: 07 May 2013; Accepted: 27 June 2013;
Published online: 11 July 2013.
Edited by:
Timothy Bullock, University of Virginia, USAReviewed by:
Janet V. Cross, University of Virginia, USACopyright: © 2013 Hargadon. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Kristian M. Hargadon, Department of Biology, Hampden-Sydney College, Box 837, Hampden-Sydney, VA 23943, USA e-mail:a2hhcmdhZG9uQGhzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.