



95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 March 2013
Sec. Alloimmunity and Transplantation
Volume 4 - 2013 | https://doi.org/10.3389/fimmu.2013.00076
This article is part of the Research Topic How to improve immune reconstitution in allogeneic hematopoietic stem cell transplantation ? View all 10 articles
In the last decade several therapeutic antibodies have been Federal Drug Administration (FDA) and European Medicines Agency (EMEA) approved. Although their mechanisms of action in vivo is not fully elucidated, antibody-dependent cellular cytotoxicity (ADCC) mediated by natural killer (NK) cells is presumed to be a key effector function. A substantial role of ADCC has been demonstrated in vitro and in mouse tumor models. However, a direct in vivo effect of ADCC in tumor reactivity in humans remains to be shown. Several studies revealed a predictive value of FcγRIIIa-V158F polymorphism in monoclonal antibody treatment, indicating a potential effect of ADCC on outcome for certain indications. Furthermore, the use of therapeutic antibodies after allogeneic hematopoietic stem cell transplantation is an interesting option. Studying the role of the FcγRIIIa-V158F polymorphism and the influence of Killer-cell Immunoglobuline-like Receptor (KIR) receptor ligand incompatibility on ADCC in this approach may contribute to future transplantation strategies. Despite the success of approved second-generation antibodies in the treatment of several malignancies, efforts are made to further augment ADCC in vivo by antibody engineering. Here, we review currently used therapeutic antibodies for which ADCC has been suggested as effector function.
Naturally cytotoxic cells against tumor cells were first described in humans and mice in the 1970s of the last century (Rosenberg et al., 1972; Oldham and Herberman, 1973; Herberman et al., 1975a,b; Kiessling et al., 1975a,b). Initially regarded as artifacts, these cells were recognized eventually as a novel lymphocyte population and named natural killer (NK) cells after their natural occurrence and spontaneous capacity to kill lymphomas and leukemic cells in non-immunized animals (Kiessling et al., 1975a,b). Nowadays, NK cells are recognized as a subset of cytotoxic innate lymphoid cells (ILCs) which are able to directly kill virus-infected cells and tumor cells and participate in shaping the adaptive immunity by secretion of cytokines (e.g., IFN-γ) (Vivier et al., 2011). The role of NK cells in human cancer is highlighted by a study linking low peripheral blood NK cell activity with increased cancer risk (Imai et al., 2000). Furthermore, an association of NK cell infiltration into the tumor site with better disease prognosis has been shown for several malignancies (Carrega et al., 2008; Halama et al., 2011; Platonova et al., 2011; Eckl et al., 2012).
Human NK cells are defined by the phenotype CD3− CD56+; additionally they are CD19 and CD14 negative. The only marker that is specific for NK cells is NKp46. NK cells comprise 5–15% of all circulating lymphocytes (Lanier et al., 1986; Walzer et al., 2007). They are commonly divided into two major subpopulations, CD56dim CD16+ and CD56bright CD16−, with each of those possessing distinct effector functions. The CD56dim CD16+ subset comprises 90% of all peripheral blood NK cells and mediates an early response via direct cellular cytotoxicity induced by perforin and granzyme, FasL, and TRAIL interactions as well as cytokine production (De Maria et al., 2011). One major characteristic of NK cells is their constant state of readiness to respond immediately. In contrast to T cells, NK cells constitutively express perforin. This facilitates the instant polarized delivery of apoptosis-inducing granzymes after formation of a lytic synapse between the NK cell and a target cell (Shresta et al., 1995). The CD56bright CD16− subset mediates a late but sustained effector function via potent pro-inflammatory cytokine and chemokine release of mainly IFN-γ, but is poorly cytotoxic (De Maria et al., 2011). NK cell activation and cytotoxicity is controlled by a complex balance between activating receptors, inhibitory receptors and co-receptors (Lanier, 2003; Leung, 2011). Positive and negative downstream signals of these receptors are integrated and decisive for NK cell activation (Figure 1). Hence, absence of inhibitory signals on target cells together with engagement of activating receptors as NKG2D, DNAM-1, and 2B4 as well as the natural cytotoxicity receptors (NCRs), including NKp46, NKp44, and NKp30 mediate triggering of resting NK cells (Moretta et al., 2001; Bryceson and Long, 2008; Lanier, 2008). Whereas for NKG2D, DNAM-1, and 2B4 multiple ligands are known (Bottino et al., 2005), despite their involvement in tumor cell lysis the NCR ligands have remained rather elusive (Moretta et al., 2006). Nevertheless, NKp30 has been shown to recognize a tumor cell ligand of the B7 family, B7-H6 (Brandt et al., 2009) and NKp44 is suggested to recognize proliferating cell nuclear antigen (PCNA), which surprisingly triggers inhibition of NK cells (Rosental et al., 2011). However, several of those receptors need to be triggered by target cells for activating downstream signals to prevail over inhibitory signals and NK cells to be activated and to mediate target cell lysis (Moretta et al., 2001; Bryceson et al., 2006).
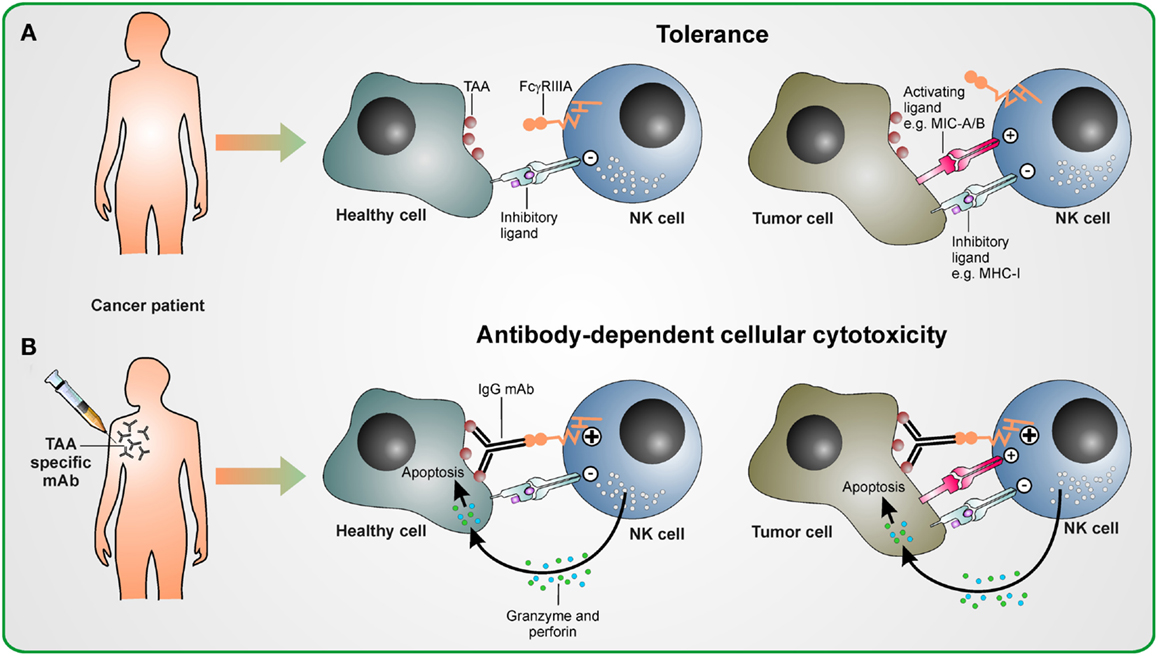
Figure 1. Antibody-dependent cellular cytotoxicity in therapeutic antibody treatment. (A) Without antibody therapy, NK cells are tolerant to healthy cells and tumor cells, if the strength of activating signal they receive upon encountering activating ligands on these malignant cells does not overcome the inhibitory signaling delivered by inhibitory ligands as, e.g., MHC class I molecules. (B) Upon treatment with tumor antigen-associated (TAA) specifc antibody, the activating stimulus from FcγRIIIa induced by antibodies cross-linking NK cells with TAA-expressing healthy and malignant cells overcomes inhibitory signals. This leads to the activation of NK cells and ADCC is mediated by releasing cytotoxic granules containing perforin and granzyme.
Most hematopoietic cells, except most T cell subsets, express Fcγ receptors (FcγRs) (Nimmerjahn and Ravetch, 2008). There are three types of FcγRs which recognize the Fc part of IgG antibody subclasses with different affinities. The activating FcγRI (CD64) binds to human IgG1 and IgG3 with high affinity, is expressed on macrophages and neutrophils, and mediates phagocytosis of target cells. The FcγRII (CD32) class comprises activating low affinity FcγRIIa (which binds human IgG1, IgG2, and IgG3) and inhibitory FcγRIIb (which recognizes human IgG1 and IgG3 with low affinity) and may attenuate signaling from activating receptors as FcγRI if engaged in phagocytes. FcγRIIIb is a protein expressed by neutrophils and may play a role in neutrophil activation. Activating low affinity FcγRIIIa (type III receptor for IgG; CD16) mediates antibody-dependent cellular cytotoxicity (ADCC) and is highly expressed on the cytotoxic CD56dim CD16+ NK cell subset as well as on other hematopoietic cells. Nevertheless, NK cells are thought to be the key mediators of ADCC, since only NK cells do not co-express the inhibitory FcγRIIb. In contrast, all other FcγR-expressing cells are tightly controlled by the balance between activating and inhibitory FcγRs. Efficient FcγRIIIa signaling depends on high avidity for specific binding to antibodies which ensures that NK cells are activated when antibodies have bound to a multimeric or multivalent cognate antigen only (Banks et al., 2002). These antibodies of the subclasses IgG1 and IgG3 binding to FcγRIIIa induce a potent activating signal which overcomes inhibitory signals and results in both cytotoxicity and a cytokine response (Chan et al., 2012) (Figure 1). Co-engagement of other activating receptors has a synergistic effect and may enhance NK cell activation (Bryceson et al., 2006).
Several mouse model studies postulated the impact of activating FcγRs on anti-tumor effects in antibody therapy indicating that ADCC has a substantial effect on tumor rejection. A study using a xenograft breast carcinoma model has shown that an antibody engineered to prevent Fc binding to FcγRIIIa and specific to Her2/neu [Human Epidermal Growth Factor Receptor (EGFR) 2, CD340] which is overexpressed in certain aggressive types of breast cancer, was, in contrary to its murine parental antibody targeting Her2/neu, unable to arrest tumor growth in vivo (Clynes et al., 2000). Additionally, this study in mice showed in a murine lymphoma model that treatment with CD20-specific antibody rituximab was capable of preventing tumor growth in FcR wild-type mice whereas FcR knock-out mice failed to mediate antibody-dependent tumor protection. Another study demonstrated ADCC in mice in vivo by showing formation of ADCC synapses in immunocompetent mice bearing a murine breast tumor treated with an antibody specific to Tn, a glycopeptidic antigen which is expressed by breast cancer and a variety of other epithelial tumors in mice and humans (Hubert et al., 2011). Additionally, tumor rejection was abolished in mice deficient for FcγRs – emphasizing the potential role of ADCC (Hubert et al., 2011).
Another study investigated the relative contributions of complement-dependent cytotoxicity (CDC) and ADCC in a murine GD2-expressing metastatic lymphoma model treated with an antibody specific to the disialoganglioside GD2 utilizing wild-type, complement-deficient, complement-receptor-deficient, and FcγRI/III-deficient mice. Outcome after treatment with the ADCC and CDC mediating GD2 antibody was unaffected in mice incapable for CDC but was almost completely abrogated in FcγRI/III-deficient mice that were disqualified for ADCC (Imai et al., 2005). These results further highlight the key role of ADCC in anti-tumor effects in mice in vivo. In men it has been shown, that breast cancer patients who responded with partial or complete remission to Her2/neu-specific antibody trastuzumab have a higher capacity to mediate ADCC in vitro than patients which failed to respond to antibody therapy (Musolino et al., 2008).
These studies indicate that, at least in murine models, ADCC is a considerable component of the in vivo activity of therapeutic antibodies against tumors. However, studies with therapeutic antibodies in humans which demonstrate the direct influence of ADCC, as, e.g., ADCC as the single effector mechanism, remain to be conducted. Therefore, the direct in vivo effect of ADCC in tumor reactivity in humans remains to be shown.
In the past 15 years, 12 therapeutic antibodies have reached FDA approval for hematological malignancies as well as solid tumors (Scott et al., 2012). Their mechanisms of action include direct anti-tumor effects as induction of apoptosis, blocking receptor signaling or acting as an agonist, delivery of a cytotoxic agent, immune-mediated effects as CDC and ADCC as well as effects on the tumor microenvironment. At least five of these antibodies mediate efficient ADCC and a large number of new constructs are currently under investigation in early and late phase clinical trials (Table 1). In this review we will use anti-CD20-specific monoclonal antibody (mAb) rituximab, CD52-specific mAb alemtuzumab, Her2/neu-specific mAb trastuzumab, EGFR-specific mAb cetuximab, and anti-GD2 antibodies to discuss the influence of ADCC in treatment with therapeutic antibodies.

Table 1. ADCC-mediating therapeutic antibodies currently FDA approved for cancer therapy.
The CD16 encoding gene FCGR3A bears a single nucleotide polymorphism (SNP) at nucleotide 526 [thymidine (T) → guanine (G)] resulting in an amino acid (aa) exchange at aa position 158 of phenylalanine (F) to valine (V). It has been demonstrated that human IgG1 binds more efficiently to NK cells expressing the FcγRIIIa-158V allotype than to the FcγRIIIa-158F allotype (Koene et al., 1997; Wu et al., 1997). In vitro studies have shown that the increased binding is caused by a significantly higher affinity of IgG antibodies to FcγRIIIa-158V receptors, whereas expression levels of FcγRIIIa on NK cells are not influenced by the FcγRIIIa polymorphism (Dall’Ozzo et al., 2004; Congy-Jolivet et al., 2008). Furthermore, the adequate rituximab concentration exciting 50% lysis of a CD20+ tumor cell line has been shown to be significantly lower in FcγRIIIa-158V/V donors compared to FcγRIIIa-158F/F donors (Dall’Ozzo et al., 2004).
Several clinical studies investigating antibodies which utilize an ADCC-mediating therapeutic antibody suggest that patients with the FcγRIIIa-158V allotype have a better clinical outcome. However, conflicting data have been published. First, we will discuss the chimeric IgG1 CD20-specific antibody rituximab which is approved for treatment of CD20+ B-cell non-Hodgkin’s lymphoma (NHL), CD20+ follicular NHL, and chronic lymphocytic leukemia (CLL). Rituximab is the most extensively studied antibody mediating ADCC as its key effector mechanism (Cheson and Leonard, 2008; Alduaij and Illidge, 2011). The first study by Cartron et al. (2002) suggested that the homozygous FcγRIIIa-158V/V genotype is beneficial in patients with follicular NHL regarding clinical and molecular responses to single agent rituximab. These results were confirmed in a larger follicular lymphoma patient cohort treated with rituximab as monotherapy and an association of the FcγRIIIa-158V/V genotype with progression-free survival could be shown (Weng and Levy, 2003). Another study analyzing the effect of the FcγRIIIa genotype on the outcome of patients to single agent rituximab promoted some of the above mentioned reports by suggesting a significant improvement in event-free survival in FcγRIIIa-158V/V patients (Ghielmini et al., 2005). Furthermore, it has been postulated that NK cells from lymphoma patients with the FcγRIIIa-158V allotype but not from patients that are homozygous for FcγRIIIa-158F/F were activated after rituximab application (Veeramani et al., 2011).
On the contrary, no influence of the FcγRIIIa-F158V polymorphism on the outcome of follicular lymphoma patients treated with risk-adapted chemotherapy and rituximab has been shown (Prochazka et al., 2011). Similarly, in relapsed or refractory CLL patients treated with chemotherapy and rituximab, no significant influence of the FcγRIIIa-F158V polymorphism was demonstrated (Dornan et al., 2010). Another study evaluated the impact of the FcγRIIIa-F158V polymorphism on the response to rituximab in combination with a chemotherapy regimen (rituximab with cyclophosphamide, hydroxydaunorubicin, oncovin, and predniso(lo)ne, R-CHOP) in diffuse large B cell lymphoma (DLBCL) patients. The FcγRIIIa-158V allotype was shown to be significantly correlated with a higher complete response rate compared with the FcγRIIIa-158F allotype (Kim et al., 2006). However, overall survival did not correlate with the FcγRIIIa-158V/V genotype and several other studies investigating the impact of the FcγRIIIa-F158V polymorphism in DLBCL found no association with the response to R-CHOP (Carlotti et al., 2007; Mitrovic et al., 2007; Varoczy et al., 2012). Interestingly, all studies conducted with rituximab as monotherapy show an impact of the FcγRIIIa-F158V polymorphism and therefore indicate an important influence of ADCC on clinical outcome. However, the picture is less clear when rituximab is combined with chemotherapy. Since combined chemotherapy may hamper ADCC due to impaired NK cell function the combination of rituximab with chemotherapy might mask the influence of the FcγRIIIa genotype and ADCC. Nevertheless, a combination of rituximab with chemotherapy generally improves clinical results.
The humanized IgG1 mAb alemtuzumab was approved for treatment of B-CLL and is directed against the glycoprotein CD52 (Hillmen et al., 2007). The therapeutic antibody was withdrawn from the market in 2012 in order to optimize the pending launch of the agent as a treatment for multiple sclerosis. The predominant mechanism of action of alemtuzumab in vivo has not been clearly defined but involvement of ADCC mediated by NK cells and neutrophils has been suggested by studies employing a human CD52 transgenic mouse model and a xenograft model (Hu et al., 2009; Siders et al., 2010). In addition, CDC and direct induction of apoptosis have been identified as other potential mechanisms of action in in vitro studies (Table 1) (Crowe et al., 1992; Mone et al., 2006). A study exploiting the predictive value of FcγRIIIa polymorphism in a small CLL patient cohort (n = 33) treated with alemtuzumab found no association of responsiveness to the mAb with a FcγRIIIa-F158V allotype (Lin et al., 2005). However, further studies with larger patient cohorts are required in order to determine the potential influence of FcγRIIIa-F158V polymorphism and ADCC in treatment with alemtuzumab.
The humanized IgG1 mAb trastuzumab is applicable in HER2/neu-positive breast cancer patients and mediates abrogation of tumor cell signaling and ADCC (Table 1) (Hudis, 2007). A study investigating breast cancer patients treated with trastuzumab and a chemotherapeutic agent postulated a higher response rate and longer progression-free survival in homozygous FcγRIIIa-158V/V patients compared to patients with the FcγRIIIa-158F allotype (Musolino et al., 2008). In contrast, another study suggested a correlation of the FcγRIIa-131H/H genotype with objective response and progression-free survival in HER2/neu-positive non-metastatic and metastatic breast cancer patients treated with chemotherapy and trastuzumab but no significant correlation was observed for the FcγRIIIa-F158V/V genotype (Tamura et al., 2011). Furthermore, another recent study postulated a lack of correlation between FcγRIIIa genotypes and trastuzumab efficacy in HER2/neu-positive non-metastatic breast cancer patients treated with a combined trastuzumab-chemotherapy (Hurvitz et al., 2012). Since no studies with trastuzumab as single agent have been published and besides ADCC abrogation of tumor cell signaling being a major effector mechanism of trastuzumab the influence of FcγRIIIa-F158V polymorphism and ADCC on clinical outcome is hard to substantiate.
The chimeric IgG1 therapeutic antibody cetuximab targets EGFR in colorectal cancer (CRC) and has like trastuzumab ADCC and abrogation of tumor cell signaling as the modes of action (Table 1) (Van Cutsem et al., 2009). A study suggested a significantly higher clinical benefit after treatment with cetuximab and chemotherapy in homozygous FcγRIIIa-158V/V and or FcγRIIa-131H/H patients. These results were independent from the GTPase KRas (KRAS) mutation status in metastatic CRC (mCRC) (Bibeau et al., 2009). Furthermore, a higher disease control rate in KRAS-mutated mCRC patients harboring the FcγRIIa-131H/H genotype and treated with combined cetuximab-chemotherapy has been shown (Rodriguez et al., 2012). It has been suggested that the predictive value of FcγRIIa-H131R polymorphism may result from linkage disequilibrium between FcγRIIIa-V158F and FcγRIIa-H131R and therefore nevertheless indicates an effect of ADCC (Lejeune et al., 2008). Similarly, the association of the FcγRIIIa-F158V polymorphism with progression-free survival has been suggested for single agent cetuximab treated mCRC patients (Zhang et al., 2007). However, in this study and in another study from Dahan et al. investigating combined cetuximab-chemotherapy treatment, the FcγRIIIa-158F allele rather than the FcγRIIIa-158V allele had a favorable influence on overall and progression-free survival (Zhang et al., 2007; Dahan et al., 2011). Beyond that, a recent study found no association of clinical outcome in a patient cohort of 107 mCRC patients treated with cetuximab and chemotherapy and FcγRIIIa-F158V and FcγRIIa-H131R polymorphisms (Park et al., 2012). These discrepancies related to the predictive value of the FcγRIIIa-F158V polymorphism on cetuximab treatment are difficult to explain and indicate a demand for further elaborated investigations in larger patient cohorts. Interestingly, mutated KRAS induces a ligand-independent activation of EGFR downstream signaling pathways. Hence, the key effector mechanism of cetuximab besides ADCC, namely abrogation of tumor signaling, is invalidated by KRAS mutations. Clinical benefit of cetuximab yet seen in these patients and the predictive value of FcγRIIIa-V158F genotype emphasizes the impact of ADCC.
Disialoganglioside GD2 is a sialic acid containing glycosphingolipid which is uniformly expressed in neuroblastoma and most melanomas but also to a variable degree in some other tumors (Navid et al., 2010). Due to its tumor-associated expression, several anti-GD2 antibodies such as the chimeric IgG1 antibody ch14.18 and its humanized counterpart hu14.18 as well as 3F8 have been developed and deployed in the clinic. The mechanisms of actions of these antibodies are ADCC and CDC. Several years ago, a phase I study with pediatric neuroblastoma patients treated with anti-GD2 antibody ch14.18 was conducted at our institute and elicited some complete and partial tumor responses in neuroblastoma patients (Handgretinger et al., 1995). Currently, we are conducting an ongoing phase I/II-trial for relapsed metastatic neuroblastoma with subsequent immunotherapy with ch14.18/Chinese hamster ovary (CHO) after HLA mismatched, haploidentical stem cell transplantation (SCT) at our institution. Preliminary results show effective ADCC and complement-mediated anti-tumor effects against neuroblastoma cells with donor-derived NK cells in vitro as well as significant anti-tumor effects in vivo (unpublished data). Due to yet small patient cohorts, the role of FcγRIIIa-F158V polymorphism in treatment with GD2 antibodies has not been studied up to now. However, there are a variety of further studies ongoing which investigate the in vivo efficacy of GD2 antibodies (Navid et al., 2010; Shusterman et al., 2010; Yu et al., 2010; Alderson and Sondel, 2011; Simon et al., 2011). Yu et al. demonstrated in a randomized trial a significantly better event-free survival for patients who received a combination of ch14.18, GM-CSF, and interleukin 2 than patients without ch14.18. Additionally, the third-generation antibody hu14.18K322A, which bears an aa substitution in the CH2 part, has been suggested to induce increased dose-dependent ADCC compared to ch14.18 and hu14.18, whereas dose-limiting CDC is intercepted by the Fc modification (Navid et al., 2010). In fact, results from studies conducted with this antibody in vivo will be interesting regarding anti-tumor effects caused by ADCC as single mode of action without involvement of any other effector mechanisms.
In adult acute myeloid leukemia (AML) and pediatric acute lymphoblastic leukemia (ALL) donor versus recipient NK cell alloreactivity is a key mechanism after HLA mismatched, haploidentical SCT, and has been reviewed elsewhere (Velardi et al., 2012). These “unlicensed” NK cells are characterized in the autologous setting by lacking self-KIRs and are thought to be beneficial in patients with neuroblastoma as well (Venstrom et al., 2009; Delgado et al., 2010). Interestingly, a recent study investigating the differential potential for ADCC of “licensed” and “unlicensed” NK cells in neuroblastoma, showed that “unlicensed” NK cells mediate ADCC most effectively against neuroblastoma cell lines under inflammatory conditions (Tarek et al., 2012). Based on these studies and our ongoing trial with HLA mismatched, haploidentical stem cell transplanted neuroblastoma patients treated with ch14.18/CHO we suggest that in the allogeneic, HLA mismatched SCT setting, KIR receptor ligand incompatibility may have synergistic effects with subsequent antibody therapy and propose further exploration of this hypothesis in larger clinical trials.
Presuming that ADCC is a key mediator of anti-tumor effects in vivo, enhancing ADCC by engineering mAbs is expected to markedly improve clinical efficacy of therapeutic antibodies. Two main approaches of optimizing FcγRIIIa binding by enhancing the affinity of mAbs have been suggested and shown to induce 5- to 100-fold increased ADCC in vitro in recent years: molecular modifications in the Fc portion leading to aa substitutions (Shields et al., 2001; Lazar et al., 2006; Stavenhagen et al., 2007) and modifying Fc-linked glycosylation (Umana et al., 1999; Davies et al., 2001; Shinkawa et al., 2003).
The approach of optimizing the Fc portion of a therapeutic antibody via aa substitutions predominantly intends to enhance ADCC by increasing the affinity to activating FcγRIIIa and reducing the affinity to inhibitory FcγRIIb. A considerable number of these third-generation monoclonal antibodies are currently under early clinical investigation. These therapeutic antibodies target among others CD19, CD20, CD30, CD40, and FLT3 in hematological malignancies and all of them aim at achieving highly augmented ADCC in vivo (Awan et al., 2010; Foyil and Bartlett, 2010; Horton et al., 2010; Le Garff-Tavernier et al., 2011; Hofmann et al., 2012; Kellner et al., 2013). Furthermore, a CD19-specific Fc-optimized therapeutic antibody is currently evaluated in pediatric patients with refractory acute B-lineage leukemia after allogeneic SCT within the scope of a compassionate use program at our institution. In four out of six patients at very high risk of relapse, minimal residual disease could be significantly reduced or completely eradicated with a longest follow up of 15 months (unpublished data). Other attempts aim at reduction of CDC related toxicity as applied in GD2 targeting hu14.18K322A in neuroblastoma (Lazar et al., 2006; Sorkin et al., 2010) or enhancing affinity to low affinity allele FcγRIIIa-158F (Bowles et al., 2006; Nordstrom et al., 2011). Recently, a phase I trial investigating a Fc-engineered antibody targeting CD20 in follicular lymphoma patients has suggested encouraging results even in patients with the less favorable FcγRIIIa-F allotype (Forero-Torres et al., 2012).
Glyco-engineered therapeutic antibodies either bear oligosaccharides modified by bisecting GlcNAc, a β1,4-GlcnAc residue attached to a core β-mannose residue, or lack the core fucose of the Fc oligosaccharides and have been found to exhibit significantly higher ADCC than their unmodified counterparts (Lifely et al., 1995; Shinkawa et al., 2003). These modifications of oligosaccharides are mainly obtained by either transfecting CHO cell line with N-acetylglucosaminyltransferase III (GnTIII) or eliminating its intrinsic α-1,6-fucosyltransferase (FUT8) activity. Alternative expression systems such as Sf21 insect cells, resulting in fucosylated paucimannosidic N-linked glycosylation, have been suggested to elicit an enhanced ADCC and have been investigated with a chimeric CD19 antibody at our institution (Lang et al., 2004; Barbin et al., 2006). Several other glyco-engineered antibodies targeting a variety of different tumor associated antigens like CD19, CD20, EGFR, and GD2 are currently investigated in early clinical studies (Robak, 2009; Navid et al., 2010; Paz-Ares et al., 2011; Ward et al., 2011). All glyco-engineered antibodies in clinical trials have been reviewed in more detail elsewhere (Beck and Reichert, 2012). Recently, the C–C chemokine receptor 4 (CCR4) targeting glyco-engineered antibody mogamulizumab has been approved in Japan for use in patients with relapsed and refractory CCR4-positive adult T cell leukemia/lymphoma (ATL) (Beck and Reichert, 2012). Clinical superiority of these third-generation mAbs over clinically established therapeutic antibodies remains to be shown and may further portend the key role of ADCC for clinical use.
The studies discussed here clearly demonstrate a substantial role of ADCC in vitro and in mouse tumor models. However, the direct in vivo effect of ADCC in tumor reactivity in humans remains to be shown since no therapeutic antibody with ADCC as single mechanism of action has been investigated in clinical trials to date. Conflicting results have been obtained about the role of FcγRIIIa-V158F polymorphism in mAb treatment. Several studies have shown a predictive value, which underlines the influence of ADCC in vivo and suggests that ADCC is one of the key mechanisms for clinical efficacy of therapeutic antibodies. On the other hand, some studies could not confirm these results. Further studies investigating the clinical relevance of FcγRIIIa-V158F polymorphism may be required for each therapeutic antibody and its indications. Although additional chemotherapy can mask the influence of the FcγRIIIa genotype and may hamper ADCC due to impaired NK cell function, a combination of both is able to improve clinical results. In particular, the use of therapeutic antibodies after hematopoietic SCT is an interesting treatment option, as the new donor-derived immune system usually faces minimal tumor burden. Further exploration of the role of the FcγRIIIa-V158F polymorphism and the influence of KIR-receptor-ligand incompatibility on ADCC in allogeneic hematopoietic SCT may contribute to future transplantation strategies in this setting. Furthermore, clinical superiority of ADCC improved third-generation mAbs over clinically established second-generation antibodies remains to be shown and may further portend the key role of ADCC in cancer therapy with monoclonal antibodies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge support by Deutsche Forschungsgemeinschaft (DFG), CRC685 Immunotherapy, Open Access Publishing Fund of Tübingen University, Bundesministerium für Bildung und Forschung (BMBF iVac ALL and Reinhold Beitlich Stiftung. The authors thank David Martin and Christina Kyzirakos for help with editing this manuscript.
Alderson, K. L., and Sondel, P. M. (2011). Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J. Biomed. Biotechnol. 2011, 379123.
Alduaij, W., and Illidge, T. M. (2011). The future of anti-CD20 monoclonal antibodies: are we making progress? Blood 117, 2993–3001.
Awan, F. T., Lapalombella, R., Trotta, R., Butchar, J. P., Yu, B., Benson, D. M. Jr., et al. (2010). CD19 targeting of chronic lymphocytic leukemia with a novel Fc-domain-engineered monoclonal antibody. Blood 115, 1204–1213.
Banks, N. D., Kinsey, N., Clements, J., and Hildreth, J. E. (2002). Sustained antibody-dependent cell-mediated cytotoxicity (ADCC) in SIV-infected macaques correlates with delayed progression to AIDS. AIDS Res. Hum. Retroviruses 18, 1197–1205.
Barbin, K., Stieglmaier, J., Saul, D., Stieglmaier, K., Stockmeyer, B., Pfeiffer, M., et al. (2006). Influence of variable N-glycosylation on the cytolytic potential of chimeric CD19 antibodies. J. Immunother. 29, 122–133.
Beck, A., and Reichert, J. M. (2012). Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs 4, 419–425.
Bibeau, F., Lopez-Crapez, E., Di Fiore, F., Thezenas, S., Ychou, M., Blanchard, F., et al. (2009). Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 27, 1122–1129.
Bottino, C., Castriconi, R., Moretta, L., and Moretta, A. (2005). Cellular ligands of activating NK receptors. Trends Immunol. 26, 221–226.
Bowles, J. A., Wang, S. Y., Link, B. K., Allan, B., Beuerlein, G., Campbell, M. A., et al. (2006). Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood 108, 2648–2654.
Brandt, C. S., Baratin, M., Yi, E. C., Kennedy, J., Gao, Z., Fox, B., et al. (2009). The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 206, 1495–1503.
Bryceson, Y. T., and Long, E. O. (2008). Line of attack: NK cell specificity and integration of signals. Curr. Opin. Immunol. 20, 344–352.
Bryceson, Y. T., March, M. E., Ljunggren, H. G., and Long, E. O. (2006). Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107, 159–166.
Carlotti, E., Palumbo, G. A., Oldani, E., Tibullo, D., Salmoiraghi, S., Rossi, A., et al. (2007). FcgammaRIIIA and FcgammaRIIA polymorphisms do not predict clinical outcome of follicular non-Hodgkin’s lymphoma patients treated with sequential CHOP and rituximab. Haematologica 92, 1127–1130.
Carrega, P., Morandi, B., Costa, R., Frumento, G., Forte, G., Altavilla, G., et al. (2008). Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer 112, 863–875.
Cartron, G., Dacheux, L., Salles, G., Solal-Celigny, P., Bardos, P., Colombat, P., et al. (2002). Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99, 754–758.
Chan, W. K., Kung Sutherland, M., Li, Y., Zalevsky, J., Schell, S., and Leung, W. (2012). Antibody-dependent cell-mediated cytotoxicity overcomes NK cell resistance in MLL-rearranged leukemia expressing inhibitory KIR ligands but not activating ligands. Clin. Cancer Res. 18, 6296–6305.
Cheson, B. D., and Leonard, J. P. (2008). Monoclonal antibody therapy for B-cell non-Hodgkin’s lymphoma. N. Engl. J. Med. 359, 613–626.
Clynes, R. A., Towers, T. L., Presta, L. G., and Ravetch, J. V. (2000). Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 6, 443–446.
Congy-Jolivet, N., Bolzec, A., Ternant, D., Ohresser, M., Watier, H., and Thibault, G. (2008). Fc gamma RIIIa expression is not increased on natural killer cells expressing the Fc gamma RIIIa-158V allotype. Cancer Res. 68, 976–980.
Crowe, J. S., Hall, V. S., Smith, M. A., Cooper, H. J., and Tite, J. P. (1992). Humanized monoclonal antibody CAMPATH-1H: myeloma cell expression of genomic constructs, nucleotide sequence of cDNA constructs and comparison of effector mechanisms of myeloma and Chinese hamster ovary cell-derived material. Clin. Exp. Immunol. 87, 105–110.
Dahan, L., Norguet, E., Etienne-Grimaldi, M. C., Formento, J. L., Gasmi, M., Nanni, I., et al. (2011). Pharmacogenetic profiling and cetuximab outcome in patients with advanced colorectal cancer. BMC Cancer 11:496. doi:10.1186/1471-2407-11-496
Dall’Ozzo, S., Tartas, S., Paintaud, G., Cartron, G., Colombat, P., Bardos, P., et al. (2004). Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 64, 4664–4669.
Davies, J., Jiang, L., Pan, L. Z., LaBarre, M. J., Anderson, D., and Reff, M. (2001). Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. Biotechnol. Bioeng. 74, 288–294.
De Maria, A., Bozzano, F., Cantoni, C., and Moretta, L. (2011). Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc. Natl. Acad. Sci. U.S.A. 108, 728–732.
Delgado, D. C., Hank, J. A., Kolesar, J., Lorentzen, D., Gan, J., Seo, S., et al. (2010). Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res. 70, 9554–9561.
Dornan, D., Spleiss, O., Yeh, R. F., Duchateau-Nguyen, G., Dufour, A., Zhi, J., et al. (2010). Effect of FCGR2A and FCGR3A variants on CLL outcome. Blood 116, 4212–4222.
Eckl, J., Buchner, A., Prinz, P. U., Riesenberg, R., Siegert, S. I., Kammerer, R., et al. (2012). Transcript signature predicts tissue NK cell content and defines renal cell carcinoma subgroups independent of TNM staging. J. Mol. Med. 90, 55–66.
Forero-Torres, A., de Vos, S., Pohlman, B. L., Pashkevich, M., Cronier, D. M., Dang, N. H., et al. (2012). Results of a phase 1 study of AME-133v (LY2469298), an Fc-engineered humanized monoclonal anti-CD20 antibody, in FcgammaRIIIa-genotyped patients with previously treated follicular lymphoma. Clin. Cancer Res. 18, 1395–1403.
Foyil, K. V., and Bartlett, N. L. (2010). Anti-CD30 antibodies for Hodgkin lymphoma. Curr. Hematol. Malig. Rep. 5, 140–147.
Ghielmini, M., Rufibach, K., Salles, G., Leoncini-Franscini, L., Leger-Falandry, C., Cogliatti, S., et al. (2005). Single agent rituximab in patients with follicular or mantle cell lymphoma: clinical and biological factors that are predictive of response and event-free survival as well as the effect of rituximab on the immune system: a study of the Swiss Group for Clinical Cancer Research (SAKK). Ann. Oncol. 16, 1675–1682.
Halama, N., Braun, M., Kahlert, C., Spille, A., Quack, C., Rahbari, N., et al. (2011). Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin. Cancer Res. 17, 678–689.
Handgretinger, R., Anderson, K., Lang, P., Dopfer, R., Klingebiel, T., Schrappe, M., et al. (1995). A phase I study of human/mouse chimeric antiganglioside GD2 antibody ch14.18 in patients with neuroblastoma. Eur. J. Cancer 31A, 261–267.
Herberman, R. B., Nunn, M. E., Holden, H. T., and Lavrin, D. H. (1975a). Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 16, 230–239.
Herberman, R. B., Nunn, M. E., and Lavrin, D. H. (1975b). Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 16, 216–229.
Hillmen, P., Skotnicki, A. B., Robak, T., Jaksic, B., Dmoszynska, A., Wu, J., et al. (2007). Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J. Clin. Oncol. 25, 5616–5623.
Hofmann, M., Grosse-Hovest, L., Nubling, T., Pyz, E., Bamberg, M. L., Aulwurm, S., et al. (2012). Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 26, 1228–1237.
Horton, H. M., Bernett, M. J., Peipp, M., Pong, E., Karki, S., Chu, S. Y., et al. (2010). Fc-engineered anti-CD40 antibody enhances multiple effector functions and exhibits potent in vitro and in vivo antitumor activity against hematologic malignancies. Blood 116, 3004–3012.
Hu, Y., Turner, M. J., Shields, J., Gale, M. S., Hutto, E., Roberts, B. L., et al. (2009). Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology 128, 260–270.
Hubert, P., Heitzmann, A., Viel, S., Nicolas, A., Sastre-Garau, X., Oppezzo, P., et al. (2011). Antibody-dependent cell cytotoxicity synapses form in mice during tumor-specific antibody immunotherapy. Cancer Res. 71, 5134–5143.
Hudis, C. A. (2007). Trastuzumab – mechanism of action and use in clinical practice. N. Engl. J. Med. 357, 39–51.
Hurvitz, S. A., Betting, D. J., Stern, H. M., Quinaux, E., Stinson, J., Seshagiri, S., et al. (2012). Analysis of Fcgamma receptor IIIa and IIa polymorphisms: lack of correlation with outcome in trastuzumab-treated breast cancer patients. Clin. Cancer Res. 18, 3478–3486.
Imai, K., Matsuyama, S., Miyake, S., Suga, K., and Nakachi, K. (2000). Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet 356, 1795–1799.
Imai, M., Landen, C., Ohta, R., Cheung, N. K., and Tomlinson, S. (2005). Complement-mediated mechanisms in anti-GD2 monoclonal antibody therapy of murine metastatic cancer. Cancer Res. 65, 10562–10568.
Kellner, C., Zhukovsky, E. A., Pötzke, A., Brüggemann, M., Schrauder, A., Schrappe, M., et al. (2013). The Fc-engineered CD19 antibody MOR208 (XmAb5574) induces natural killer cell-mediated lysis of acute lymphoblastic leukemia cells from pediatric and adult patients. Leukemia. doi:10.1038/leu.2012.373. [Epub ahead of print].
Kiessling, R., Klein, E., Pross, H., and Wigzell, H. (1975a). “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 5, 117–121.
Kiessling, R., Klein, E., and Wigzell, H. (1975b). “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 5, 112–117.
Kim, D. H., Jung, H. D., Kim, J. G., Lee, J. J., Yang, D. H., Park, Y. H., et al. (2006). FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood 108, 2720–2725.
Koene, H. R., Kleijer, M., Algra, J., Roos, D., von dem Borne, A. E., and de Haas, M. (1997). Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood 90, 1109–1114.
Lang, P., Barbin, K., Feuchtinger, T., Greil, J., Peipp, M., Zunino, S. J., et al. (2004). Chimeric CD19 antibody mediates cytotoxic activity against leukemic blasts with effector cells from pediatric patients who received T-cell-depleted allografts. Blood 103, 3982–3985.
Lanier, L. L. (2008). Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 9, 495–502.
Lanier, L. L., Le, A. M., Civin, C. I., Loken, M. R., and Phillips, J. H. (1986). The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J. Immunol. 136, 4480–4486.
Lazar, G. A., Dang, W., Karki, S., Vafa, O., Peng, J. S., Hyun, L., et al. (2006). Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. U.S.A. 103, 4005–4010.
Le Garff-Tavernier, M., Decocq, J., de Romeuf, C., Parizot, C., Dutertre, C. A., Chapiro, E., et al. (2011). Analysis of CD16+CD56dim NK cells from CLL patients: evidence supporting a therapeutic strategy with optimized anti-CD20 monoclonal antibodies. Leukemia 25, 101–109.
Lejeune, J., Thibault, G., Ternant, D., Cartron, G., Watier, H., and Ohresser, M. (2008). Evidence for linkage disequilibrium between Fcgamma RIIIa-V158F and Fcgamma RIIa-H131R polymorphisms in white patients, and for an Fcgamma RIIIa-restricted influence on the response to therapeutic antibodies. J. Clin. Oncol. 26, 5489–5491; author reply 5491–5482.
Lifely, M. R., Hale, C., Boyce, S., Keen, M. J., and Phillips, J. (1995). Glycosylation and biological activity of CAMPATH-1H expressed in different cell lines and grown under different culture conditions. Glycobiology 5, 813–822.
Lin, T. S., Flinn, I. W., Modali, R., Lehman, T. A., Webb, J., Waymer, S., et al. (2005). FCGR3A and FCGR2A polymorphisms may not correlate with response to alemtuzumab in chronic lymphocytic leukemia. Blood 105, 289–291.
Mitrovic, Z., Aurer, I., Radman, I., Ajdukovic, R., Sertic, J., and Labar, B. (2007). FCgammaRIIIA and FCgammaRIIA polymorphisms are not associated with response to rituximab and CHOP in patients with diffuse large B-cell lymphoma. Haematologica 92, 998–999.
Mone, A. P., Cheney, C., Banks, A. L., Tridandapani, S., Mehter, N., Guster, S., et al. (2006). Alemtuzumab induces caspase-independent cell death in human chronic lymphocytic leukemia cells through a lipid raft-dependent mechanism. Leukemia 20, 272–279.
Moretta, A., Bottino, C., Vitale, M., Pende, D., Cantoni, C., Mingari, M. C., et al. (2001). Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 19, 197–223.
Moretta, L., Bottino, C., Pende, D., Castriconi, R., Mingari, M. C., and Moretta, A. (2006). Surface NK receptors and their ligands on tumor cells. Semin. Immunol. 18, 151–158.
Musolino, A., Naldi, N., Bortesi, B., Pezzuolo, D., Capelletti, M., Missale, G., et al. (2008). Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 26, 1789–1796.
Navid, F., Santana, V. M., and Barfield, R. C. (2010). Anti-GD2 antibody therapy for GD2-expressing tumors. Curr. Cancer Drug Targets 10, 200–209.
Nimmerjahn, F., and Ravetch, J. V. (2008). Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47.
Nordstrom, J. L., Gorlatov, S., Zhang, W., Yang, Y., Huang, L., Burke, S., et al. (2011). Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcgamma receptor binding properties. Breast Cancer Res. 13, R123.
Oldham, R. K., and Herberman, R. B. (1973). Evaluation of cell-mediated cytotoxic reactivity against tumor associated antigens with 125I-iododeoxyuridine labeled target cells. J. Immunol. 111, 862–871.
Park, S. J., Hong, Y. S., Lee, J. L., Ryu, M. H., Chang, H. M., Kim, K. P., et al. (2012). Genetic polymorphisms of FcgammaRIIa and FcgammaRIIIa are not predictive of clinical outcomes after cetuximab plus irinotecan chemotherapy in patients with metastatic colorectal cancer. Oncology 82, 83–89.
Paz-Ares, L. G., Gomez-Roca, C., Delord, J. P., Cervantes, A., Markman, B., Corral, J., et al. (2011). Phase I pharmacokinetic and pharmacodynamic dose-escalation study of RG7160 (GA201), the first glycoengineered monoclonal antibody against the epidermal growth factor receptor, in patients with advanced solid tumors. J. Clin. Oncol. 29, 3783–3790.
Platonova, S., Cherfils-Vicini, J., Damotte, D., Crozet, L., Vieillard, V., Validire, P., et al. (2011). Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 71, 5412–5422.
Prochazka, V., Papajik, T., Gazdova, J., Divoka, M., Rozmanova, S., Faber, E., et al. (2011). FcgammaRIIIA receptor genotype does not influence an outcome in patients with follicular lymphoma treated with risk-adapted immunochemotherapy. Neoplasma 58, 263–270.
Robak, T. (2009). GA-101, a third-generation, humanized and glyco-engineered anti-CD20 mAb for the treatment of B-cell lymphoid malignancies. Curr. Opin. Investig. Drugs 10, 588–596.
Rodriguez, J., Zarate, R., Bandres, E., Boni, V., Hernandez, A., Sola, J. J., et al. (2012). Fc gamma receptor polymorphisms as predictive markers of Cetuximab efficacy in epidermal growth factor receptor downstream-mutated metastatic colorectal cancer. Eur. J. Cancer 48, 1774–1780.
Rosenberg, E. B., Herberman, R. B., Levine, P. H., Halterman, R. H., McCoy, J. L., and Wunderlich, J. R. (1972). Lymphocyte cytotoxicity reactions to leukemia-associated antigens in identical twins. Int. J. Cancer 9, 648–658.
Rosental, B., Brusilovsky, M., Hadad, U., Oz, D., Appel, M. Y., Afergan, F., et al. (2011). Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J. Immunol. 187, 5693–5702.
Scott, A. M., Wolchok, J. D., and Old, L. J. (2012). Antibody therapy of cancer. Nat. Rev. Cancer 12, 278–287.
Shields, R. L., Namenuk, A. K., Hong, K., Meng, Y. G., Rae, J., Briggs, J., et al. (2001). High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem. 276, 6591–6604.
Shinkawa, T., Nakamura, K., Yamane, N., Shoji-Hosaka, E., Kanda, Y., Sakurada, M., et al. (2003). The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 278, 3466–3473.
Shresta, S., Heusel, J. W., Macivor, D. M., Wesselschmidt, R. L., Russell, J. H., and Ley, T. J. (1995). Granzyme B plays a critical role in cytotoxic lymphocyte-induced apoptosis. Immunol. Rev. 146, 211–221.
Shusterman, S., London, W. B., Gillies, S. D., Hank, J. A., Voss, S. D., Seeger, R. C., et al. (2010). Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children’s Oncology Group (COG) phase II study. J. Clin. Oncol. 28, 4969–4975.
Siders, W. M., Shields, J., Garron, C., Hu, Y., Boutin, P., Shankara, S., et al. (2010). Involvement of neutrophils and natural killer cells in the anti-tumor activity of alemtuzumab in xenograft tumor models. Leuk. Lymphoma 51, 1293–1304.
Simon, T., Hero, B., Faldum, A., Handgretinger, R., Schrappe, M., Klingebiel, T., et al. (2011). Long term outcome of high-risk neuroblastoma patients after immunotherapy with antibody ch14.18 or oral metronomic chemotherapy. BMC Cancer 11:21. doi:10.1186/1471-2407-11-21
Sorkin, L. S., Otto, M., Baldwin, W. M. III, Vail, E., Gillies, S. D., Handgretinger, R., et al. (2010). Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain 149, 135–142.
Stavenhagen, J. B., Gorlatov, S., Tuaillon, N., Rankin, C. T., Li, H., Burke, S., et al. (2007). Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res. 67, 8882–8890.
Tamura, K., Shimizu, C., Hojo, T., Akashi-Tanaka, S., Kinoshita, T., Yonemori, K., et al. (2011). FcgammaR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann. Oncol. 22, 1302–1307.
Tarek, N., Le Luduec, J. B., Gallagher, M. M., Zheng, J., Venstrom, J. M., Chamberlain, E., et al. (2012). Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J. Clin. Invest. 122, 3260–3270.
Umana, P., Jean-Mairet, J., Moudry, R., Amstutz, H., and Bailey, J. E. (1999). Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 17, 176–180.
Van Cutsem, E., Kohne, C. H., Hitre, E., Zaluski, J., Chang Chien, C. R., Makhson, A., et al. (2009). Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 360, 1408–1417.
Varoczy, L., Zilahi, E., Gyetvai, A., Kajtar, B., Gergely, L., Sipka, S., et al. (2012). Fc-gamma-receptor IIIa polymorphism and gene expression profile do not predict the prognosis in diffuse large B-cell lymphoma treated with R-CHOP protocol. Pathol. Oncol. Res. 18, 43–48.
Veeramani, S., Wang, S. Y., Dahle, C., Blackwell, S., Jacobus, L., Knutson, T., et al. (2011). Rituximab infusion induces NK activation in lymphoma patients with the high-affinity CD16 polymorphism. Blood 118, 3347–3349.
Velardi, A., Ruggeri, L., and Mancusi, A. (2012). Killer-cell immunoglobulin-like receptors reactivity and outcome of stem cell transplant. Curr. Opin. Hematol. 19, 319–323.
Venstrom, J. M., Zheng, J., Noor, N., Danis, K. E., Yeh, A. W., Cheung, I. Y., et al. (2009). KIR and HLA genotypes are associated with disease progression and survival following autologous hematopoietic stem cell transplantation for high-risk neuroblastoma. Clin. Cancer Res. 15, 7330–7334.
Vivier, E., Raulet, D. H., Moretta, A., Caligiuri, M. A., Zitvogel, L., Lanier, L. L., et al. (2011). Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–49.
Walzer, T., Jaeger, S., Chaix, J., and Vivier, E. (2007). Natural killer cells: from CD3(-)NKp46(+) to post-genomics meta-analyses. Curr. Opin. Immunol. 19, 365–372.
Ward, E., Mittereder, N., Kuta, E., Sims, G. P., Bowen, M. A., Dall’Acqua, W., et al. (2011). A glycoengineered anti-CD19 antibody with potent antibody-dependent cellular cytotoxicity activity in vitro and lymphoma growth inhibition in vivo. Br. J. Haematol. 155, 426–437.
Weng, W. K., and Levy, R. (2003). Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 21, 3940–3947.
Wu, J., Edberg, J. C., Redecha, P. B., Bansal, V., Guyre, P. M., Coleman, K., et al. (1997). A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J. Clin. Invest. 100, 1059–1070.
Yu, A. L., Gilman, A. L., Ozkaynak, M. F., London, W. B., Kreissman, S. G., Chen, H. X., et al. (2010). Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 363, 1324–1334.
Zhang, W., Gordon, M., Schultheis, A. M., Yang, D. Y., Nagashima, F., Azuma, M., et al. (2007). FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J. Clin. Oncol. 25, 3712–3718.
Keywords: natural killer cells, ADCC, tumor immunotherapy, therapeutic antibodies, allogeneic stem cell transplantation
Citation: Seidel UJE, Schlegel P and Lang P (2013) Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 4:76. doi: 10.3389/fimmu.2013.00076
Received: 16 October 2012; Accepted: 08 March 2013;
Published online: 27 March 2013.
Edited by:
Hermann Einsele, University Hospital Würzburg, GermanyReviewed by:
Rupali Das, Children’s Hospital of Philadelphia, USACopyright: © 2013 Seidel, Schlegel and Lang. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Peter Lang, Department of General Paediatrics, Oncology/Haematology, University Children’s Hospital Tübingen, Hoppe-Seyler-Str. 1, 72076 Tübingen, Germany. e-mail:cGV0ZXIubGFuZ0BtZWQudW5pLXR1ZWJpbmdlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.