

- 1The University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
- 2Department of Surgery, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
- 3Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
- 4Department of Bioengineering, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
High-mobility group box 1 (HMGB1) is a leaderless cytokine, like the IL-1 and FGF family members, that has primary roles within the nucleus and the cytosol. Within the nucleus, it serves as another guardian of the genome, protecting it from oxidant injury and promoting access to transcriptional complexes such as nuclear hormone/nuclear hormone receptors and p53/p73 complexes. Within the cytosol it promotes autophagy and recruitment of the myddosome to Toll-like receptor (TLR) 9 vesicular compartments. Outside of the cell, it can either bind to specific receptors itself, or with high affinity to DNA, nucleosomes, IL-1β, lipopolysaccharide, and lipoteichoic acid to mediate responses in specific physiological or pathological conditions. Currently identified receptors include TLR2, TLR4, the receptor for advanced glycation end products, CD24-Siglec G/10, chemokine CXC receptor 4, and TIM-3. In terms of its effects or functions within lymphoid cells, HMGB1 is principally secreted from mature dendritic cells (DCs) to promote T-cell and B-cell reactivity and expansion and from activated natural killer cells to promote DC maturation during the afferent immune response. Some studies suggest that its primary role in the setting of chronic inflammation is to promote immunosuppression. As such, HMGB1 is a central cytokine for all lymphoid cells playing a role complementary to its better studied role in myeloid cells.
Introduction
Damage-associated molecular pattern (DAMPs) molecules, are endogenous danger signals that elicit inflammation and subsequent immune responses once released from dead or stressed cells following injury or infection (Rubartelli and Lotze, 2007; Sims et al., 2010; Tang et al., 2012). Although various DAMPs have been identified, the best characterized is the prototypic nuclear protein high-mobility group box 1 (HMGB1). HMGB1 is an evolutionarily ancient protein that was first discovered as a chromatin-associated protein more than three decades ago (Goodwin et al., 1977). We now know that it displays many other functions depending on its location and its synergizing partners. Present within the nuclei of almost all eukaryotic cells, HMGB1 functions as a DNA chaperone that stabilizes nucleosome formation and promotes access to transcriptional factors that target specific genes (Müller et al., 2001; Thomas, 2001), although HMGB1 itself is not sequence specific. Our group demonstrated that cytosolic HMGB1 also promotes autophagy, a conserved programed survival pathway evoked following environmental and intracellular stress (Tang et al., 2010a, 2012). Apart from its nuclear and cytosolic roles, HMGB1 possesses a previously unexpected multifaceted role in immunity when released or secreted into the extracellular milieu. This occurs in two principal ways: either (1) passively released from necrotic cells (Scaffidi et al., 2002) or (2) actively secreted by inflammatory cells, such as monocytes or macrophages (Gardella et al., 2002; Bonaldi et al., 2003; Tang et al., 2007) and natural killer (NK) cells (Semino et al., 2005; Gougeon and Bras, 2011). It this way, HMGB1 evokes innate immune response via its interaction with cell surface receptors.
Previous studies highlight the importance of HMGB1 at the core of inflammation-associated events, acting as an irreplaceable modulator of immune responses and the “universal” biosensor for nucleic acids (Yanai et al., 2012). In spite of its well-established divergent functions in myeloid cells which predominantly participate in innate immune response, its roles in adaptive immunity involving T-cells and B-cells is so far not fully understood and surprisingly, one which needs substantially more study. Here, we describe the cytokine-like biology of HMGB1 protein, with a focus on lymphoid cells, including NK cells, T-cells, and B-cells.
Lessons from HMGB1 Knockouts
High-mobility group box 1 is vital for ex utero growth, as shown by inborn defects and rapid death (within 24 h following birth) in hmgb1−/− mice, as early as E15 in inbred species, because of hypoglycemia. This was initially postulated to be the result from deficient glucocorticoids receptor function (Calogero et al., 1999), but we would now attribute this to reduced autophagy, critically important for survival in the neonatal period (Kuma et al., 2004). Necrotic HMGB1−/− cells only weakly activate dendritic cells (DCs) (Rovere-Querini et al., 2004), and HMGB1-deficient DCs display sharply impaired capacity to trigger inflammation (Scaffidi et al., 2002). We now know that floxed HMGB1 deleted in a tissue- or cell type-specific fashion within the pancreas, liver, small bowel, DCs, and NK cells, is associated with prolonged viability of animals compared with complete knockout of HMGB1 (unpublished observations) in the whole animal, suggesting that these are not the target tissues associated with lethality.
HMGB1 as the Cytokine for Lymphoid Cells
High-mobility group box 1 was identified as a delayed mediator of inflammation released from macrophages (Wang et al., 1999), found in the serum 24–48 h later than secretion IL-1β and tumor necrosis factor (TNF)-α, the classical early pro-inflammatory cytokines which are dissipated by 24 h. Afterwards, it was demonstrated to be liberated from cells undergoing necrosis, followed by production of TNF-α from monocytes (Scaffidi et al., 2002). Subsequent investigations uncovered an amazingly profligate role in mediating local or systemic immune responses through its interaction with several receptors. As a cytokine, it transduces signals and coordinates cellular activities through several pattern-recognition receptors including the receptor for advanced glycation end products (RAGE), Toll-like receptor (TLR)2, TLR4, TIM-3, chemokine CXC receptor (CXCR)4, CD24-Siglec G/10 (Park et al., 2004, 2006; Dumitriu et al., 2005; Lotze and Tracey, 2005; Bianchi, 2009; Chen et al., 2009; Tang and Lotze, 2012; Tang et al., 2012; Yanai et al., 2012), and TLR-9 when combined with DNA (Tian et al., 2007). Extracellular HMGB1 thus functions as a modulator, modifying the immunogenic potentials of DNA and potentially other PAMPs and DAMPs and cytokines. Indeed, given the differences in the all thiol form of HMGB1, promoting primarily chemokine activity and the dithiol form which promotes TNF/IL-6 production (cytokine activity), it is quite likely that the molecule secreted by activated cells, endowed with autocrine and paracrine actions, differs biochemically and functionally from the molecule released as a consequence of cell and tissue necrosis (Venereau et al., 2012). Given this difference with the all thiol form promoting release of the chemokine CXCL12, and the dithiol not, environmental conditions likely dictate the eventual outcome of HMGB1 interactions with lymphoid cells in the tissues. For example, well perfused and non-hypoxic environments may promote different T-cell responses that hypoxic, reducing conditions (Venereau et al., 2012). TLRs, the best-studied pattern-recognition receptors (PRRs), are highly conserved proteins initiating immune responses following recognition of various molecules derived from pathogens (PAMPs) as well as endogenous danger signals (DAMPs) sharing similar structures (Medzhitov and Janeway, 1997; Aderem and Ulevitch, 2000; Medzhitov, 2001). The intracellular signaling cascades after recognition principally involve two specific adaptors, the Toll/IL-1R domain-containing adaptor TRIF and myeloid differentiation primary response protein (MyD88), which is primarily involved in HMGB1-mediated signaling pathway and acts as a component of myddosome with IRAK2 and IRAK4 assembled in response to primary stimulation (Motshwene et al., 2009; Lin et al., 2010; George et al., 2011). RAGE is a PRR with a wide variety of ligands including advanced glycation end products (AGEs) and DAMPs (Sparvero et al., 2009; Sims et al., 2010). The list of receptors that interact with HMGB1 continues to grow, as does interest in understanding the signaling pathways and their cooperative functions in specific cell types. Current insights on these receptors, based on experimental observations, is that TLRs principally are involved in the activation of myeloid cells, whereas RAGE is primarily activated in endothelial and somatic cells (Yanai et al., 2012).
High-mobility group box 1 signaling has been studied in many cell types following interaction with individual receptors, with most studies centering on myeloid cells – the maturation of conventional DCs, their role in plasmacytoid DCs, activation of monocytes or macrophages, and the production of pro-inflammatory cytokines (Lotze and Tracey, 2005; Yang et al., 2007; Bianchi, 2009; Yanai et al., 2012). Their effects on lymphoid cells, however, are surprisingly not well characterized. We have extraordinarily limited information about the expression of receptors RAGE and, TLR2/4 and TIM-3 on both helper and regulatory T-cells (Wild et al., 2012), RAGE and TLR2/4/9 on B-cells (Tian et al., 2007; Avalos et al., 2010), and TLR2/4 and TIM-3 on NK cells (Tang and Lotze, 2012), shown in Figure 1. Beyond that there is quite little information. In this review, we summarize the critical roles of HMGB1 in lymphoid cells (Table 1), with a focus on its extracellular role acting as a cytokine.
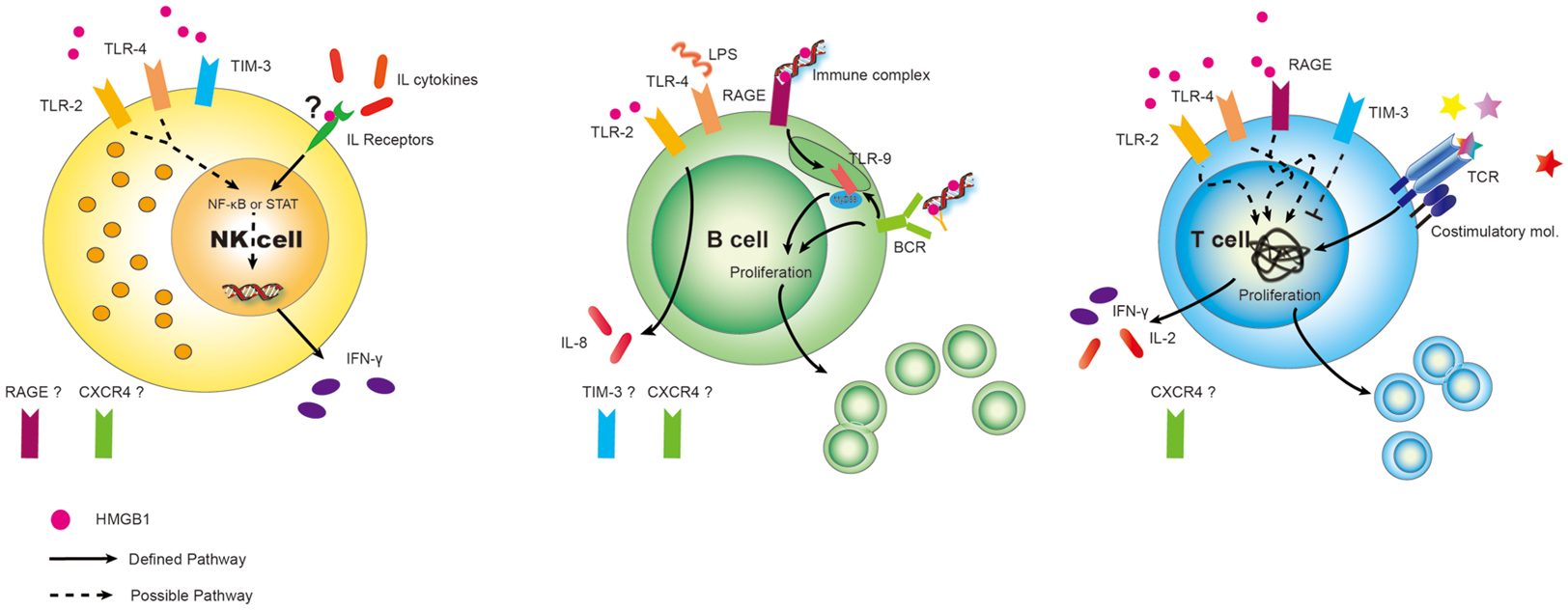
Figure 1. Differential expression of DAMP receptors for HMGB1 on lymphoid cells. Surprisingly, given the surfeit of receptors now shown to be expressed on various cell types, relatively little has been done to fully characterize the panoply of receptors on lymphoid cells. Best defined are the expression of RAGE on Ba and T-cells (but not to our knowledge on NK cells). Similarly TLR2, TLR4, CXCR4, and possible TIM-3 could play critical roles on subserving cell functions in response to HMGB1 on immune cells and this needs to be more carefully studied. Possible and defined roles are shown. “?” represents we hypothesize that HMGB1 binds to IL receptors, assisting in interleukin (cytokine) interaction. Also, expression of other HMGB1 receptors are unknown at present.

Table 1. Lymphoid cells respond to HMGB1.
Most cytokines function distinctly in synergy or antagonism with other cytokines acting collectively. This is also true for HMGB1. Moreover, HMGB1 shares pleiotropic and redundant characteristics with other cytokines (Lotze and Tracey, 2005), sometimes binding them to enhance immunologic function, thereby endowing them with a more potent capacity to elicit biological and immunological responses, consequences depending on the local microenvironmental factors and presence of other circumstances. Here, we list out different cellular responses of lymphoid cells to HMGB1 in different conditions or settings (Table 2) which could act as a reference for readers to make comparisons or conduct experiments. Also, we summarize the common consequences in response to HMGB1 (Figure 2).
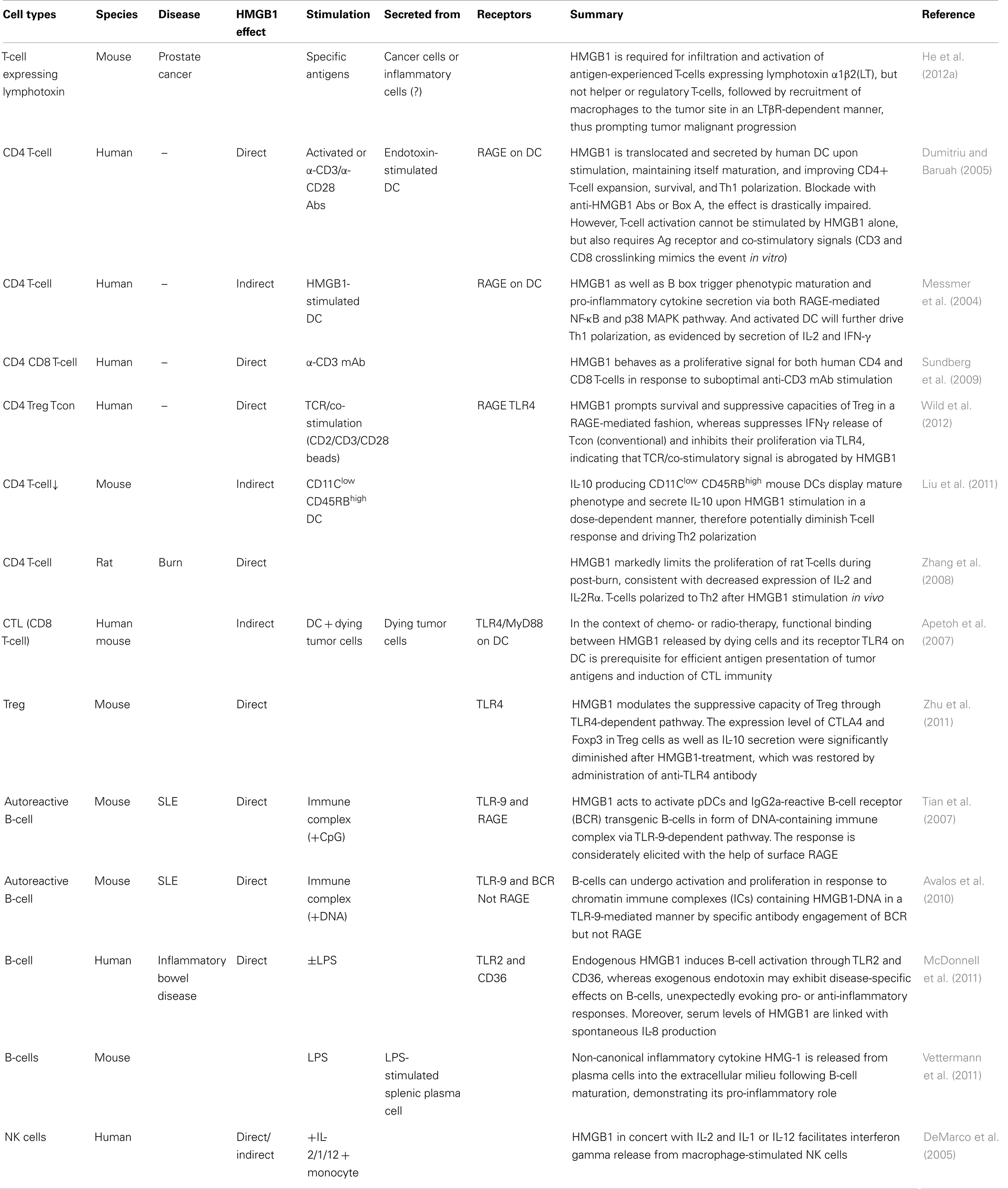
Table 2. Cellular responses of lymphoid cells to HMGB1 located differently.
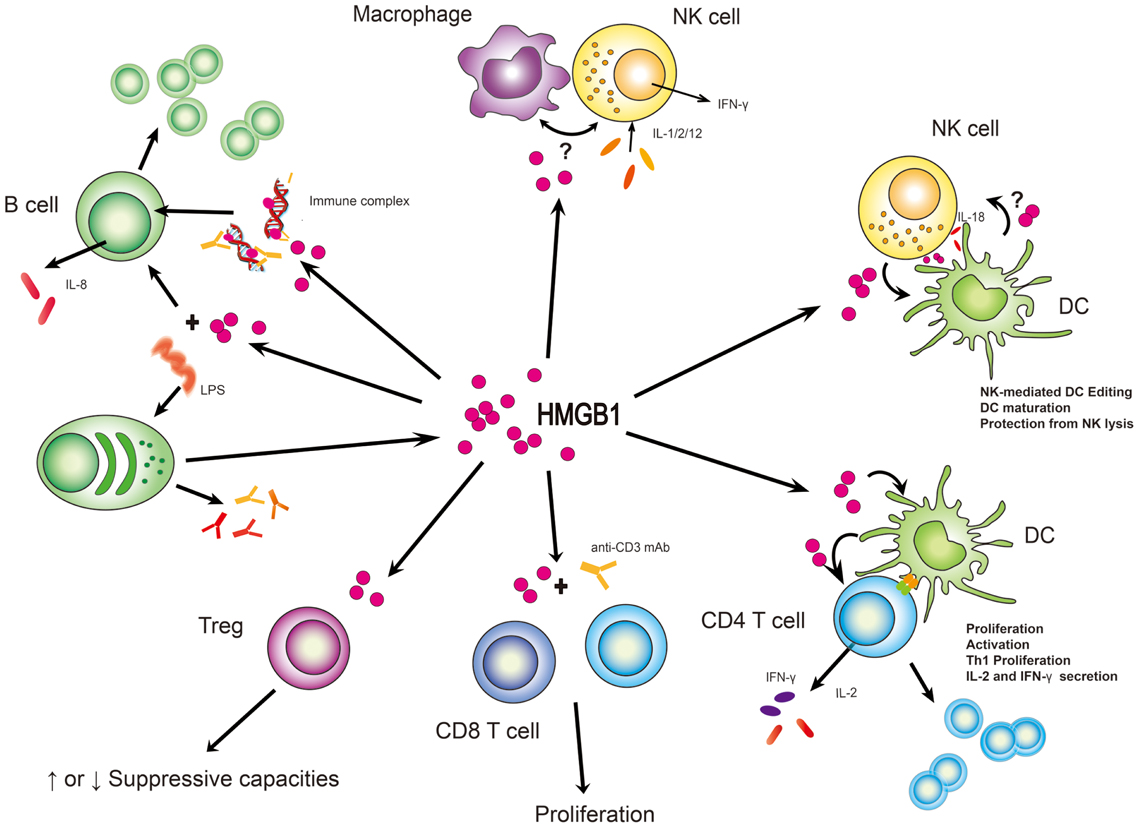
Figure 2. HMGB1 stimulates effector function from immune cells. In many instances, HMGB1’s function has been to subserve cell:cell interactions between lymphoid and myeloid cells. Its clearest definition has been with NK/macrophage or NK/DC interactions although suspected roles in B and T-cell activities are supported. What is unclear is whether these roles are cell type specific and whether HMGB1 might act in trans between a myeloid and lymphoid cell. “?” represents the state of uncertainty if HMGB1 plays a role in the setting of macrophage and NK cell interaction or HMGB1 is secreted from DCs to affect NK cell function.
HMGB1 and NK Cells
There is little information about the direct effects of HMGB1 on NK cells, with the exception of elevated secretion of IFN-γ by macrophage-stimulated NK cells in concert with other pro-inflammatory cytokines like IL-2 and IL-12 (DeMarco et al., 2005), work done by our group almost a decade ago. Nevertheless, whether it occurs depending on the activation of monocytes or in a direct NK-cell specific manner is still unclear. Further clarification as to which cell type is responsive to HMGB1 under these specific circumstance is needed and of significant interest, since little is known about the interaction between monocytes (macrophages) and NK cells, both of which are key sentinels and instigators of immune responses.
Natural killer cells do, however, secrete HMGB1. HMGB1 undergoes abundant, regulated secretion from activated NK cells into the immunological synapse during NK/iDC (immature DC) crosstalk, thus inducing maturation of DCs and limiting NK cell-mediated cytotoxicity of the DCs (Semino et al., 2005). The secretion of HMGB1 is markedly elevated following engagement of NKp30 (one type of natural cytotoxicity receptor or NCR) expressed on human NK cells, thereby triggering maturation of autologous DC (Semino et al., 2007). Whether or not DC can in turn secrete HMGB1 for further activation of NK cells and promote the quality of the crosstalk remains undefined. Specific cell types without HMGB1 expression would be needed to uncover the critical role of HMGB1 in intracellular communication.
High-mobility group box 1-mediated NK/DC crosstalk is important in the setting of HIV infection (Saïdi et al., 2008; Melki et al., 2010; Gougeon and Bras, 2011). DCHIV (infected by HIV virus) are resistant to the NK cell-induced editing process. Interestingly, HMGB1, essential for DC maturation presumably within secondary lymphoid tissues, also contributes to viral replication and DC persistence via up-regulation of apoptosis inhibitors against TRAIL (TNF-related apoptosis-inducing ligand)-mediated apoptosis.
Given that the cooperative dialog between NK cells and DCs is pivotal for sustaining innate immunity and initiating the subsequent adaptive immune response, it is worth investigating the detailed mechanism by which NK/DC crosstalk and its altered processes link to clinical manifestations of diseases, including cancer, autoimmune, and infectious diseases.
HMGB1 and T-Cells
Our knowledge of HMGB1 effector functions to T-cells are principally based on observations and inferences from the evaluation of T-cell subsets with the treatment of HMGB1 co-cultured with DCs. In the presence of other cytokines, HMGB1 can modify the fate of the overall immune response, promoting immunity or tolerance as demonstrated by targeting effector T-cells and regulatory T-cells (Tregs) reciprocally in response to individual stimuli. Differences that may have been found in terms of immunity or tolerance when comparing various observations might be, at least in part, due to differences in the experimental systems utilized (dosage, duration, and the presence of other factors) and the pharmacological inhibitors utilized to block these complex biological systems in vitro and in vivo. We know little about the effects of HMGB1 on naïve and memory T-cells as regards alteration in phenotype or cytokine proficiency among the defined T-cell subsets. In addition, the ability of HMGB1 to either recruit T-cells to sites of tissue damage or injury, thus allowing effector T-cell function, or to induce Treg infiltration and expansion is largely unknown.
Direct Effects on T-Cells
Acting as a pro-inflammatory cytokine, HMGB1, is not only released by stressed or necrotic tissues but also translocated and secreted by human DC following PAMP [endotoxin/lipopolysaccharide (LPS)] stimulation. It plays a critical role in promoting expansion, survival, and helper T (Th) 1 polarization of CD4+ T-cells (Dumitriu and Baruah, 2005; Jube et al., 2012). Similarly, HMGB1, is also a proliferative signal for both human CD4+ and CD8+ T-cells in response to suboptimal anti-CD3 mAb stimulation (Sundberg et al., 2009). The expression level of CTLA4 and Foxp3 in Treg cells as well as IL-10 secretion are significantly diminished following HMGB1 treatment. This is restored by administration of an anti-TLR4 antibody (Zhu et al., 2011). Altogether, HMGB1 is seemingly necessary for enhancing immunity through activation of effector T-cells and suppression of Treg’s. In contrast, HMGB1 can also promote migration and survival of Treg, whereas it suppresses IFNγ release of conventional T-cells and inhibits their proliferation via TLR4, indicating that the TCR/co-stimulatory signal is abrogated by HMGB1. Furthermore, HMGB1 elicits increased suppressive capacity of Treg when co-cultured with effector T-cells in a RAGE-dependent fashion. Additionally, several reports provide evidence suggesting that HMGB1 may contribute to Th17 cells proliferation and activation in the context of autoimmune disease, including rheumatoid arthritis, myocarditis, as well as acute allograft rejection (Duan et al., 2011; Su et al., 2011; He et al., 2012b; Shi et al., 2012).
When we examine immune responses in vivo, the findings are totally different. HMGB1 is essential for infiltration and activation of T-cells expressing lymphotoxin α1β2(LT) in mice with prostate cancer, therefore recruiting macrophages to promote tumor malignant progression (He et al., 2012a). This work further confirms the notion that HMGB1 can prompt progression of many types of cancers (Tang et al., 2010b). Surprisingly, neither T effectors nor Tregs are detected differentially between normal and cancerous tissues. The source of extracellular HMGB1 needs to be further characterized, whether arising from stressed tumor cells or recruited inflammatory cells, including NK cells or DCs, or all. HMGB1 markedly limits the proliferation of murine (rat) T-cells and induces Th2 polarization following burn injury, consistent with decreased expression of IL-2 and IL-2Rα (Zhang et al., 2008).
DC-Mediated Indirect Effects on T-Cells
High-mobility group box 1 is an inducer of DC maturation (Messmer et al., 2004; Rovere-Querini et al., 2004; Semino et al., 2005, 2007). Mature and activated DC will further drive Th1 polarization, as evidenced by secretion of IL-2 and IFN-γ (Messmer et al., 2004). It is worth noting that one of the promising mechanisms underlying the chemo- or radio-therapy-based antitumor responses is due to the functional binding between HMGB1 released by dying cells and one of its receptors, TLR4 expressed on DC, which allows for antigen presentation and subsequent cytotoxic CD8+ T-cell (CTL) effector function (Apetoh et al., 2007).
On the other hand, IL-10 producing CD11Clow CD45RBhigh mouse DCs also display a mature phenotype and secrete IL-10 following HMGB1 stimulation in a dose-dependent manner, thereby potentially diminishing T-cell responses with down-regulation of IL-2 and IL-2Rα and driving Th2 polarization, just the opposite in the case of CD11Chigh CD45RBlow DCs. This finding is in concordance with the potential of HMGB1 to polarize Th2 cells in rats following thermal injury (Zhang et al., 2008).
HMGB1 and B-Cells
Compared with T-cells, the role of HMGB1 in B-cells has not been fully delineated. Some studies have supported a role for HMGB1 in B-cell activation. In the form of immune complexes (ICs), HMGB1 promotes proliferation of autoreactive B-cells in response to endogenous TLR-9 ligands (e.g., DNA) (Tian et al., 2007; Avalos et al., 2010). This suggests are markedly immune-regulatory function in the pathogenesis of autoimmune diseases. TLR-9 is responsive to immune complex in intracellular endosomes, while the internalization of DNA may be mediated by RAGE which bound with HMGB1 (Tian et al., 2007) or by specific IgG and B-cell receptor interaction, followed by BCR engagement (Avalos et al., 2010). However, given that B-cell proliferation and Ig gene recombination share the same pathway but contrary states of molecules involved (e.g., FOXO degradation or dephosphorylation) and autoreactive antigen, the capacity of antibody production could be further investigated in terms of individual receptors of IC interaction, thus providing a comprehensive role for HMGB1 in B-cell activation. Furthermore, in the context of inflammatory bowel disease (IBD), enhanced serum levels of HMGB1 is accompanied by spontaneous IL-8 production by B-cells via interaction with TLR2 and CD36 (McDonnell et al., 2011). On the other hand, plasma cells release HMGB1 into the extracellular milieu following LPS-stimulated maturation (Vettermann et al., 2011), demonstrating its pro-inflammatory effects in promoting autoimmune disease and chronic inflammation.
Concluding Remarks
High-mobility group box 1, like other cytokines, is able to function as an agonist, an antagonist, to synergize with other factors and to have multiple pleiotropic functions on multiple cell types, including lymphoid cells. Unlike typical cytokines however, it interacts with a panoply of receptors, many of which are notably promiscuous with functions quite disparate from each other, depending upon the local microenvironment, location, and coordination with individual stimuli. In addition, unlike cytokines which interact with picogram or nanogram quantities to promote full receptor activation, HMGB1 requires, in many instances, microgram quantities in order to elicit a meaningful response in vitro. Increasing advances in understanding the role of HMGB1 in immunity have extended the knowledge and led to widespread acceptance of the notion that HMGB1 acts as a centrally important, potent, ubiquitous cytokine which exerts effect on both myeloid and lymphoid cells. It thus plays a multifaceted modulatory role in both innate and adaptive immune responses. Although there is much information about the diverse, sometimes even opposite effects of HMGB1 on various kinds of immune cells in culture, it is of great importance to understand the precise mechanism by which HMGB1 functions in vivo, in particular during altered pathology or physiology. In a complicated balance of guiding and choreographing disparate biologies, HMGB1, interspersed with DAMPs and PAMPs, develops the plot line and provides impetus to the emergent immune response. Improved understanding of when, where, which cell types produce/respond to HMGB1 and what levels at intimate cell:cell contact or released into tissues or systemically would provide a basis for suitable therapeutic implementation or interventions in the clinic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aderem, A., and Ulevitch, R. (2000). Toll-like receptors in the induction of the innate immune response. Nature 406. Available at: http://gene.bjmu.edu.cn/undereducation_education/download/Toll-like receptor in the induction of the innate immune response.pdf [accessed December 5, 2012].
Agrawal, A., and Schatz, D. G. (1997). RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell 89, 43–53.
Apetoh, L., Ghiringhelli, F., Tesniere, A., Obeid, M., Ortiz, C., Criollo, A., et al. (2007). Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059.
Avalos, A. M., Kiefer, K., Tian, J., Christensen, S., Shlomchik, M., Coyle, A. J., et al. (2010). RAGE-independent autoreactive B cell activation in response to chromatin and HMGB1/DNA immune complexes. Autoimmunity 43, 103–110.
Bonaldi, T., Talamo, F., Scaffidi, P., Ferrera, D., Porto, A., Bachi, A., et al. (2003). Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 22, 5551–5560.
Calogero, S., Grassi, F., Aguzzi, A., Voigtländer, T., Ferrier, P., Ferrari, S., et al. (1999). The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 22, 276–280.
Chen, G.-Y., Tang, J., Zheng, P., and Liu, Y. (2009). CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 323, 1722–1725.
Dai, Y., Wong, B., Yen, Y., Oettinger, M. A., Kwon, J., and Johnson, R. C. (2005). Determinants of HMGB proteins required to promote RAG1/2-recombination signal sequence complex assembly and catalysis during V(D)J recombination. Mol. Cell. Biol. 25, 4413–4425.
DeMarco, R. A., Fink, M. P., and Lotze, M. T. (2005). Monocytes promote natural killer cell interferon gamma production in response to the endogenous danger signal HMGB1. Mol. Immunol. 42, 433–444.
Duan, L., Wang, C.-Y., Chen, J., Gong, Q., Zhu, P., Zheng, F., et al. (2011). High-mobility group box 1 promotes early acute allograft rejection by enhancing IL-6-dependent Th17 alloreactive response. Lab. Invest. 91, 43–53.
Dumitriu, I., and Baruah, P. (2005). Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 174, 7506–7515.
Dumitriu, I. E., Baruah, P., Bianchi, M. E., Manfredi, A. A., and Rovere-Querini, P. (2005). Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur. J. Immunol. 35, 2184–2190.
Erlandsson Harris, H., and Andersson, U. (2004). Mini-review: the nuclear protein HMGB1 as a proinflammatory mediator. Eur. J. Immunol. 34, 1503–1512.
Gardella, S., Andrei, C., Ferrera, D., and Lotti, L. (2002). The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 3, 995–1001.
George, J., Motshwene, P. G., Wang, H., Kubarenko, A. V., Rautanen, A., Mills, T. C., et al. (2011). Two human MYD88 variants, S34Y and R98C, interfere with MyD88-IRAK4-myddosome assembly. J. Biol. Chem. 286, 1341–1353.
Goodwin, G., Rabbani, A., Nicolas, P., and Johns, E. (1977). The isolation of the high mobility group non-histone chromosomal protein HMG 14. FEBS Lett. 80, 413–416.
Gougeon, M.-L., and Bras, M. (2011). Natural killer cells, dendritic cells, and the alarmin high-mobility group box 1 protein: a dangerous trio in HIV-1 infection? Curr. Opin. HIV AIDS 6, 364–372.
He, Y., Zha, J., Wang, Y., Liu, W., Yang, X., and Yu, P. (2012a). Tissue damage-associated “danger signals” influence T cell responses that promote the progression of pre-neoplasia to cancer. Cancer Res. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23108142 [accessed November 17, 2012].
He, Z., Shotorbani, S. S., Jiao, Z., Su, Z., Tong, J., Liu, Y., et al. (2012b). HMGB1 promotes the differentiation of Th17 via up-regulating TLR2 and IL-23 of CD14+ monocytes from patients with rheumatoid arthritis. Scand. J. Immunol. 76, 483–490.
Ivanov, S., Dragoi, A.-M., Wang, X., Dallacosta, C., Louten, J., Musco, G., et al. (2007). A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 110, 1970–1981.
Jube, S., Rivera, Z. S., Bianchi, M. E., Powers, A., Wang, E., Pagano, I., et al. (2012). Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 72, 3290–3301.
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., et al. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036.
Lin, S.-C., Lo, Y.-C., and Wu, H. (2010). Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890.
Liu, Q., Yao, Y., Yan, Y., Dong, N., and Sheng, Z. (2011). High mobility group box 1 protein suppresses T cell-mediated immunity via CD11c(low)CD45RB(high) dendritic cell differentiation. Cytokine 54, 205–211.
Lotze, M. T., and Tracey, K. J. (2005). High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342.
McDonnell, M., Liang, Y., Noronha, A., Coukos, J., Kasper, D. L., Farraye, F. A., et al. (2011). Systemic Toll-like receptor ligands modify B-cell responses in human inflammatory bowel disease. Inflamm. Bowel Dis. 17, 298–307.
Medzhitov, R., and Janeway, C. A. (1997). Innate immunity: the virtues of a nonclonal system of recognition. Cell 91, 295–298.
Melki, M.-T., Saïdi, H., Dufour, A., Olivo-Marin, J.-C., and Gougeon, M.-L. (2010). Escape of HIV-1-infected dendritic cells from TRAIL-mediated NK cell cytotoxicity during NK-DC cross-talk – a pivotal role of HMGB1. PLoS Pathog. 6:e1000862. doi:10.1371/journal.ppat.1000862
Messmer, D., Yang, H., Telusma, G., Knoll, F., Li, J., Messmer, B., et al. (2004). High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 173, 307–313.
Motshwene, P. G., Moncrieffe, M. C., Grossmann, J. G., Kao, C., Ayaluru, M., Sandercock, A. M., et al. (2009). An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 284, 25404–25411.
Müller, S., Scaffidi, P., Degryse, B., Bonaldi, T., Ronfani, L., Agresti, A., et al. (2001). New EMBO members’ review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J. 20, 4337–4340.
Park, J. S., Gamboni-Robertson, F., He, Q., Svetkauskaite, D., Kim, J.-Y., Strassheim, D., et al. (2006). High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 290, C917–C924.
Park, J. S., Svetkauskaite, D., He, Q., Kim, J.-Y., Strassheim, D., Ishizaka, A., et al. (2004). Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279, 7370–7377.
Rovere-Querini, P., Capobianco, A., Scaffidi, P., Valentinis, B., Catalanotti, F., Giazzon, M., et al. (2004). HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 5, 825–830.
Rubartelli, A., and Lotze, M. T. (2007). Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 28, 429–436.
Saïdi, H., Melki, M.-T., and Gougeon, M.-L. (2008). HMGB1-dependent triggering of HIV-1 replication and persistence in dendritic cells as a consequence of NK-DC cross-talk. PLoS ONE 3:e3601. doi:10.1371/journal.pone.0003601
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195.
Semino, C., Angelini, G., Poggi, A., and Rubartelli, A. (2005). NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood 106, 609–616.
Semino, C., Ceccarelli, J., Lotti, L. V., Torrisi, M. R., Angelini, G., and Rubartelli, A. (2007). The maturation potential of NK cell clones toward autologous dendritic cells correlates with HMGB1 secretion. J. Leukoc. Biol. 81, 92–99.
Shi, Y., Sandoghchian Shotorbani, S., Su, Z., Liu, Y., Tong, J., Zheng, D., et al. (2012). Enhanced HMGB1 expression may contribute to Th17 cells activation in rheumatoid arthritis. Clin. Dev. Immunol. 2012, 295081.
Sims, G. P., Rowe, D. C., Rietdijk, S. T., Herbst, R., and Coyle, A. J. (2010). HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 28, 367–388.
Sparvero, L. J., Asafu-Adjei, D., Kang, R., Tang, D., Amin, N., Im, J., et al. (2009). RAGE (Receptor for Advanced Glycation End products), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 7, 17.
Su, Z., Sun, C., Zhou, C., Liu, Y., Zhu, H., Sandoghchian, S., et al. (2011). HMGB1 blockade attenuates experimental autoimmune myocarditis and suppresses Th17-cell expansion. Eur. J. Immunol. 41, 3586–3595.
Sundberg, E., Fasth, A. E. R., Palmblad, K., Harris, H. E., and Andersson, U. (2009). High mobility group box chromosomal protein 1 acts as a proliferation signal for activated T lymphocytes. Immunobiology 214, 303–309.
Tang, D., Kang, R., Coyne, C. B., Zeh, H. J., and Lotze, M. T. (2012). PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev. 249, 158–175.
Tang, D., Kang, R., Livesey, K. M., Cheh, C.-W., Farkas, A., Loughran, P., et al. (2010a). Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190, 881–892.
Tang, D., Kang, R., Zeh, H. J., and Lotze, M. T. (2010b). High-mobility group box 1 and cancer. Biochim. Biophys. Acta 1799, 131–140.
Tang, D., and Lotze, M. T. (2012). Tumor immunity times out: TIM-3 and HMGB1. Nat. Immunol. 13, 808–810.
Tang, D., Shi, Y., Kang, R., Li, T., Xiao, W., Wang, H., et al. (2007). Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol. 81, 741–747.
Thomas, J. O. (2001). HMG1 and 2: architectural DNA-binding proteins. Biochem. Soc. Trans. 29, 395–401.
Tian, J., Avalos, A. M., Mao, S.-Y., Chen, B., Senthil, K., Wu, H., et al. (2007). Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496.
Venereau, E., Casalgrandi, M., Schiraldi, M., Antoine, D. J., Cattaneo, A., De Marchis, F., et al. (2012). Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209, 1519–1528.
Vettermann, C., Castor, D., Mekker, A., Gerrits, B., Karas, M., and Jäck, H.-M. (2011). Proteome profiling suggests a pro-inflammatory role for plasma cells through release of high-mobility group box 1 protein. Proteomics 11, 1228–1237.
Wang, H., Bloom, O., Zhang, M., Vishnubhakat, J. M., Ombrellino, M., Che, J., et al. (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251.
Wild, C. A., Bergmann, C., Fritz, G., Schuler, P., Hoffmann, T. K., Lotfi, R., et al. (2012). HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int. Immunol. 24, 485–494.
Yanai, H., Ban, T., and Taniguchi, T. (2012). High-mobility group box family of proteins: ligand and sensor for innate immunity. Trends Immunol. 33, 633–640.
Yang, D., Chen, Q., Yang, H., Tracey, K. J., Bustin, M., and Oppenheim, J. J. (2007). High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J. Leukoc. Biol. 81, 59–66.
Zhang, L., Yao, Y., Dong, Y.-Q., Dong, N., Yu, Y., and Sheng, Z. (2008). Relationship between high-mobility group box 1 protein release and T-cell suppression in rats after thermal injury. Shock 30, 449–455.
Keywords: lymphocytes, HMGB1, TLR2, TLR4, RAGE, NK cells, T cells, B cells
Citation: Li G, Liang X and Lotze MT (2013) HMGB1: the central cytokine for all lymphoid cells. Front. Immunol. 4:68. doi: 10.3389/fimmu.2013.00068
Received: 04 January 2013; Accepted: 01 March 2013;
Published online: 20 March 2013.
Edited by:
Anna Rubartelli, IRCCS AOU San Martino IST, ItalyReviewed by:
Massimo Gadina, National Institutes of Health, USAPatrizia Rovere Querini, Ospedale San Raffaele and Vita-Salute University, Italy
Copyright: © 2013 Li, Liang and Lotze. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Michael T. Lotze, Division of Surgical Oncology, University of Pittsburgh Cancer Institute, 5517 Centre Avenue, Pittsburgh, PA 15213, USA. e-mail:bG90emVtdEB1cG1jLmVkdQ==