 Matthew Gumbleton1
Matthew Gumbleton1 William G. Kerr1,2,3*
William G. Kerr1,2,3*- 1Department of Microbiology and Immunology, State University of New York Upstate Medical University, Syracuse, NY, USA
- 2Department of Pediatrics, State University of New York Upstate Medical University, Syracuse, NY, USA
- 3Department of Chemistry, Syracuse University, Syracuse, NY, USA
Natural killer (NK) cells are important for host defense against malignancy and infection. At a cellular level NK cells are activated when signals from activating receptors exceed signaling from inhibitory receptors. At a molecular level NK cells undergo an education process to both prevent autoimmunity and acquire lytic capacity. Mouse models have shown important roles for inositol phospholipid signaling in lymphocytes. NK cells from mice with deletion in different members of the inositol phospholipid signaling pathway exhibit defects in development, NK cell repertoire expression and effector function. Here we review the current state of knowledge concerning the function of inositol phospholipid signaling components in NK cell biology.
Unlike T and B lymphocytes, natural killer (NK) cells do not rearrange antigen receptor genes in order to detect their cellular targets (Lanier, 1998). Rather, NK cells utilize an array of activating and inhibitory receptors with the latter largely detecting major histocompatibility complex (MHC) class I ligands, or in the case of 2B4, the signaling lymphocyte activation molecule (SLAM) family ligand CD48. Both activating and inhibitory NK receptors are stochastically expressed with frequencies in the NK compartment determined by their relative promoter strength, and in some cases, survival differences among NK subsets determined by the presence or absence of ligands their receptor array can detect and their relative affinity for that ligand (Manilay et al., 1999; Lowin-Kropf and Held, 2000; Wang et al., 2002; Fortenbery et al., 2010). Inhibitory receptors allow for the NK cell to recognize and ignore “healthy-self” cells while activating receptors enable the NK cell to recognize and lyse foreign or “damaged-self” cells or antibody bound cells. In some instances the NK cell may also produce inflammatory cytokines such as interferon (IFN)γ in response to target cell engagement (Vivier et al., 2011). Individual NK cells in the compartment can express different combinations of activating and inhibitory receptors, but also different levels of certain receptors (Bryceson et al., 2011). The final balance of activating and inhibitory receptors, and the presence or absence of ligands, determines a threshold for activation of an individual NK cell (Lanier, 1998; Bryceson et al., 2011; Vivier et al., 2011). In extreme cases the NK cell may even be anergized by unopposed activating signals (Raulet and Vance, 2006). This repertoire diversity in the NK cell compartment of an individual allows for a response to a diverse range of stimuli including an early response to virus-infected cells (Brandstadter and Yang, 2011) and surveillance for residual tumor cells (Vivier et al., 2008).
Phosphatidylinositol (PI) is a membrane lipid found in all cell types that can be phosphorylated to form phosphatidylinositol 3-monophosphate PI(3)P, PI(4)P, or PI(5)P. Each of these PIP species can be further phosphorylated by phosphoinositide 3-kinase (PI3K), PI4K, or PI5K to form PIP2 species. PI3K is able to phosphorylate PI(4,5)P2 to form PI(3,4,5)P3 (Berridge and Irvine, 1989; Rhee and Bae, 1997). PI(3,4)P2, PI(4,5)P2, and PI(3,4,5)P3 allow for recruitment to the plasma membrane of pleckstrin homology (PH) domain-containing proteins (several other domains are also able to recruit proteins to these lipids as well and will be discussed below) as shown in Figure 1 and Table 1. PI(4,5)P2 is also important in NK cell signaling by acting as the substrate for phospholipase C (PLC), which hydrolyzes PI(4,5)P2 into diacylglycerol (DAG), to activate PKC and inositol 1,4,5-trisphosphate [I(1,4,5)P3] which triggers release of intracellular Ca2+ stores. PIP2 and PIP3 can be modified by various phosphatases including inositol polyphosphate 4-phosphatase (INPP4) and SH2 domain-containing inositol-5-phosphatase (SHIP) or modified by phosphatase and tensin homologue deleted on chromosome 10 (PTEN) to create PI(3)P, PI(3,4)P2, or PI(4,5)P2, respectively. These activities can attenuate signaling pathways or, in the case of the SHIP product PI(3,4)P2, activate them by enabling recruitment of proteins with various PH domain-containing proteins to sites of signaling at the plasma membrane (Kerr, 2011). Here we will discuss the role of the above IP modifying enzymes in the context of NK cell biology.
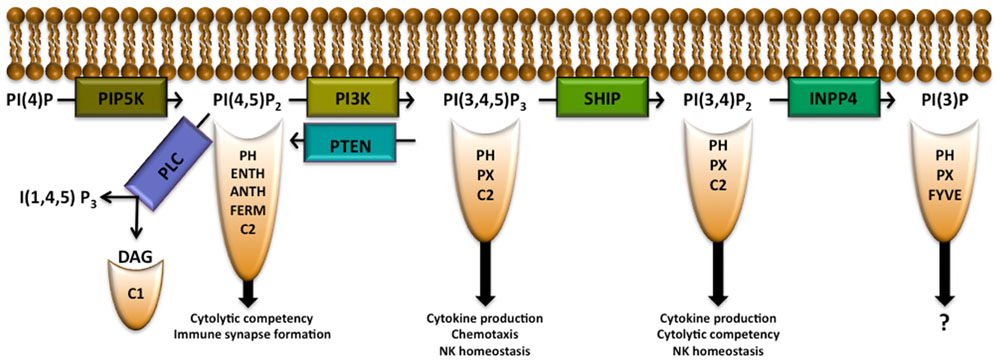
FIGURE 1. Inositol phospholipid family members allow for recruitment of proteins with indicated binding domains. Modification of the inositol phospholipids by the indicated enzymes allow for either the attenuation or promotion of signaling pathways that determine NK cell development, function, survival, and/or trafficking in the host. PIP5K PLC, phospholipase C; DAG, diacylglycerol; PIP5K, phosphatidylinositol 4-phosphate 5-kinase; PI3K, phosphoinositide 3-kinase; SHIP, SH2 domain-containing inositol-5-phosphatase; PTEN, phosphatase and tensin homologue deleted on chromosome 10; INPP4, inositol polyphosphate 4-phosphatase; PH, pleckstrin homology; ENTH, epsin N-terminal homology; ANTH, AP180 N-terminal homology.
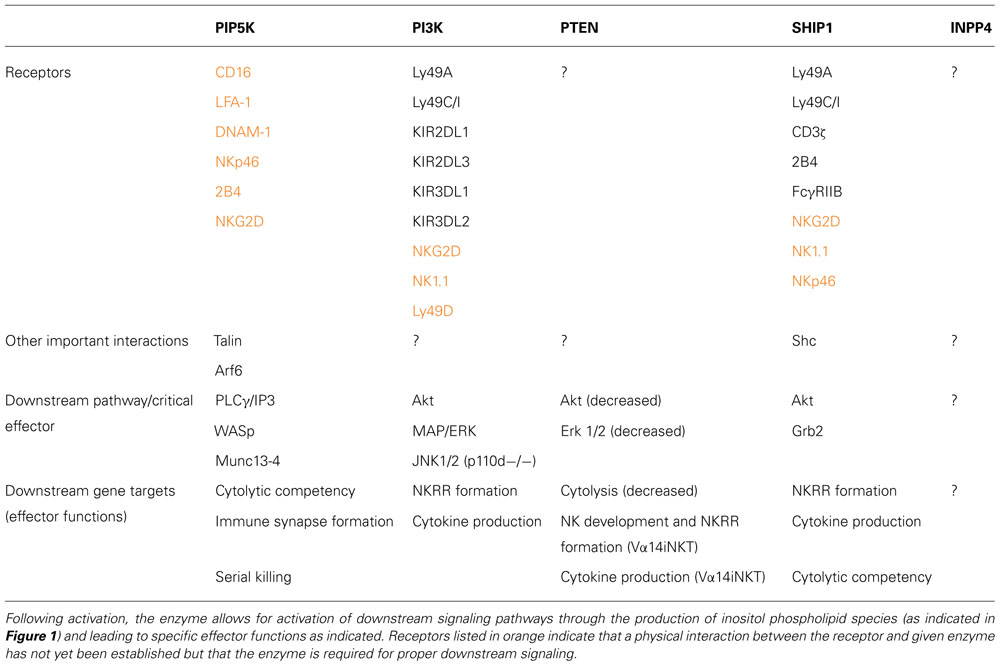
TABLE 1. PI modifying enzymes (PIP5K, PI3K, PTEN, SHIP1, and INPP4) are either recruited to or activated following signaling through the indicated NK cell receptor.
PIP5K
Phosphatidylinositol 4-phosphate can be phosphorylated by type I phosphatidylinositol phosphate kinases (PIPKIs) to form PI(4,5)P2. Three isoforms of phosphatidylinositol 4-phosphate 5-kinase have been described (PIP5Kα, PIP5Kβ, PIP5Kγ; Ishihara et al., 1996; Loijens and Anderson, 1996; Tolias et al., 1998) with PIP5Kα and PIP5Kγ playing important, non-redundant roles in cell signaling through the production of PI(4,5)P2 in NK cells (Micucci et al., 2008). PI(4,5)P2 is a major phosphoinositide at the plasma membrane (McLaughlin et al., 2002). It is believed that there are different pools of PI(4,5)P2 in cells, inside and outside of lipid rafts, that control different signaling pathways by allowing for localized changes in PI(4,5)P2 concentration (Pike and Casey, 1996; Hinchliffe et al., 1998; Martin, 2001; McLaughlin and Murray, 2005; Golebiewska et al., 2008; Johnson et al., 2008).
Following NK cell activation, ADP-ribosylation factor 6 (Arf6) and talin recruit PIP5Kα and PIP5Kγ to the immunological synapse, respectively (Di Paolo et al., 2002; Galandrini et al., 2005). Expression of both isozymes is required for cytolytic competency through the increase in PI(4,5)P2 they create at the plasma membrane (Vyas et al., 2001; Di Paolo et al., 2002; Ling et al., 2002; Galandrini et al., 2005; Mace et al., 2010). An important trait of NK cells is the ability to serially kill multiple target cells (Bhat and Watzl, 2007). PIP5Kγ may play a role in serial killing as it is required for the regulation of the soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) protein Munc13-4 that mediates lytic granule recycling. Hence, PIP5Kγ−/− NK cells are unable to serially lyse target cells as efficiently as wild type NK cells (Capuano et al., 2012). Interestingly, decreased levels of PIP5Kα or PIP5Kγ have no impact on IFNγ production and do not alter PI3K signaling in NK cells as measured by Akt activation and Vav-1 phosphorylation (Micucci et al., 2008). Thus, PIP5Ks are required for sustained cytolytic competence, but are dispensable for cytokine production by NK cells.
Having different pools of PI(4,5)P2 in the membrane may allow diverse cell functions to be compartmentalized via production of key second messengers [e.g., DAG, I(1,4,5)P3 and PI(3,4,5)P3] by selective recruitment of different signaling proteins with PI(4,5)P2 binding domains and the regulation of ion channels preferentially localized to these compartments (Gamper and Shapiro, 2007). There are several domains that enable PI(4,5)P2 binding by a protein: PH, epsin N-terminal homology (ENTH), AP180 N-terminal homology (ANTH), FERM, and C2 domains (Ferguson et al., 1995; Hamada et al., 2000; Ford et al., 2001). PH domains enable signaling proteins to selectively bind different PIP species while ENTH and ANTH domain-containing proteins have a higher affinity for PI(4,5)P2 than for other inositol phospholipids (Ford et al., 2001; Itoh et al., 2001). Wiskott–Aldrich syndrome protein (WASp), the clathrin adaptor AP1, the actin nucleating protein Arp2/3 AP180, and talin make use of ENTH and ANTH domains for recruitment to the plasma membrane. An NK cell is required to reorganize actin to form an immunological synapse before lysing a target cell (Rak et al., 2011). There is recent evidence that clathrin and AP1 aid in this process in T cells (Alvarez Arias et al., 2010; Calabia-Linares et al., 2011). After NK cell activation, increased PI(4,5)P2 levels recruit WASp to the membrane which in turn activates the actin nucleating protein complex Arp2/3 allowing for actin rearrangement and formation of the NK immunological synapse (Badour et al., 2003; Mace et al., 2010). Thus, PI(4,5)P2 also plays a critical role in actin reorganization and creation of the immune synapse.
PI3K
There are three different classes of PI3K enzymes; class I enzymes exist as a heterodimer between a catalytic subunit and a regulatory subunit. Class Ia PI3K enzymes are p110α (PI3KCA), p110β (PI3KCB), and p110δ (PI3KCD), which can pair with one of five regulatory subunits p85α, p55α, p50α (alternatively spliced from PIK3R1), P85β (PIK3R2), and p55γ (PIK3R3). There is one PI3K class Ib enzyme: p110γ (PI3KCG) which heterodimerizes with either p101 (PIK3R5) or p87 (PIK3R6). There are three class II enzymes (PI3K-C2α, PI3K-C2β, and PI3K-C2γ) that have a poorly defined role in cell signaling and do not heterodimerize with a regulatory subunit. One PI3K class III enzyme (VPS34) has recently been identified which heterodimerizes with its regulatory subunit VPS15 to catalyze the formation of PI(3)P from PI. Class I PI3K enzymes (in leukocytes primarily p110γ and p110δ) are the main enzymes responsible for the phosphorylation of the D-3 position of PI(4,5)P2 to create PI(3,4,5)P3 (Cantley, 2002; Saudemont and Colucci, 2009; Vanhaesebroeck et al., 2010), and thus this section will focus on class Ia enzymes.
Initial studies of PI3K enzymes in NK cells did not focus on individual PI3K subunits but instead were performed with broad-acting, non-selective PI3K inhibitors such as Ly294002 (Vlahos et al., 1994) and Wortmannin (Arcaro and Wymann, 1993). It was shown that PI3K is activated within 5 min of NK cell activation (Zhong et al., 2002) and that PI3K is required for antibody-dependent cellular cytotoxicity (ADCC; Kanakaraj et al., 1994) but is not required for the NK cell to make a “missing self” attack on MHC class I-deficient K562 cells (Bonnema et al., 1994). Other studies were able to show that PI3K expression in NK cells is required for lymphocyte function-associated antigen-1 (LFA-1) adherence to intercellular adhesion molecule-1 (ICAM-1)-expressing cells and thus, important for formation of the NK immune synapse (Barber and Long, 2003) and for facilitating signaling through various NK activating receptors (Barber et al., 2004). In addition, the 2B4 and killer cell immunoglobulin-like receptors (KIR; that sense self-ligands CD48 and MHC class I, respectively) can also recruit PI3K (Marti et al., 1998; Aoukaty and Tan, 2002; Eissmann et al., 2005), and this may enable these receptors to have self-licensing roles (Fortenbery et al., 2010).
The PI3K signaling cascade has emerged as an essential intracellular signaling pathway in NK cell biology. The spleen tyrosine kinase (Syk) is able to activate the PI3K–>Rac1–>PAK1–>MEK–>ERK signaling pathway leading to NK cell degranulation (Jiang et al., 2000, 2002, 2003). We believe that PI3K might also promote Bruton's tyrosine kinase (Btk) activation in NK cells given that increased PI(3,4,5)P3 levels in other hematopoietic cell types lead to Btk activation (Kawakami et al., 2000; Saito et al., 2001) and that Btk has recently been shown to be required for proper NK cell activation (Bao et al., 2012). Interestingly, Btk has been shown to regulate PIP5Ks [and thus PI(4,5)P2 production] in B cells (Saito et al., 2003). Thus, the interaction between Btk and the inositol phospholipid signaling pathway in NK cells merits further investigation.
Deletion of specific PI3K subunits has allowed for determining their individual contributions to inositol phospholipid signaling in NK cells. Awasthi et al. (2008) found that PIK3R1−/− NK cells (NK cells lacking p85α, p55α, and p50α) have a severely disrupted NK cell compartment. They showed that PIK3R1−/− NK cells are decreased in number in the bone marrow and liver but not the spleen. Moreover, NK cells that were present were cytolytically incompetent against both “missing-self” and NKG2D (an activating receptor expressed by both human and mouse NK cells) ligand-expressing target cells and had a skewed Ly49 receptor repertoire compared to wild type (WT) NK cells. This cytolytic defect could be due to improper formation of the NK immune synapse. Activation of NK cells via NKG2D requires interaction between the Rho guanosine triphosphatase Cdc42 (Carlin et al., 2011), the adaptor protein CrkL (Segovis et al., 2009) and DAP10 (Wu et al., 1999; Billadeau et al., 2003; Upshaw et al., 2006) with p85α required for proper formation of the immunological synapse. Thus, multiple inositol phospholipid signaling events are required for proper microtubule and actin cytoskeleton rearrangement.
The PI3K class Ia and class Ib subunits p110δ and p110γ seem to have non-redundant roles in NK cell signaling. In vitro cytolysis assays indicate that either p110δ (Kim et al., 2007; Saudemont et al., 2007; Tassi et al., 2007) or p110γ (Kim et al., 2007; Tassi et al., 2007) but not both (Kim et al., 2007; Tassi et al., 2007) enzymes are dispensable for target cell lysis. However, two papers have shown that there is decreased ability for NK cell rejection of tumor cell in vivo, at least in the case of p110δ deficiency (Saudemont et al., 2007; Guo et al., 2008). Further investigations are required to understand why p110δ is required for in vivo target cell lysis but not for NK cytolytic activity in vitro. Data regarding the requirement of p110δ and p110γ for cytokine production are more contradictory. Two studies found that p110δ is required for cytokine production (Kim et al., 2007; Guo et al., 2008) and one went on to show that p110γ is dispensable (Kim et al., 2007) for the production of cytokines including IFNγ, tumor necrosis factor (TNF)α, and granulocyte–macrophage colony-stimulating factor (GM-CSF). However, two different studies have shown that p110γ is in fact required for NK cell cytokine production (Tassi et al., 2007; Orr et al., 2009). At least part of the discrepancy may be due the use of different mouse genetic backgrounds. Kim et al. (2007) made use of B10D2 mice (MHC-H2d) background whereas most of the other mutants were on a C57BL/6 genetic background (MHC-H2b). Further, the mice used by Tassi et al. (2007) were incompletely backcrossed to the C57BL/6 background such that only ∼80% of all alleles were C57BL/6 homozygous, with other alleles remained from the original 129Sv background. This is potentially problematic as 129Sv mice have hyporesponsive NK cells and thus these 129Sv allelic loci may act as genetic modifiers of PI3K mutations (Belanger et al., 2008). For a more detailed and nuanced discussion of the differences observed in the different PI3K mutant studies please see Kerr and Colucci (2011). Interestingly, p110α, a PI3K isozyme found in many cell types but not in leukocytes, is required for the upregulation of the NKG2D ligand RAE-1 following murine cytomegalovirus (MCMV) infection (Tokuyama et al., 2011). Thus, PI3K may regulate NK cell behavior not only in a cell intrinsic manner, but also via regulation of activating ligands expressed by target cells.
Phosphoinositide 3-kinase is also required for NK cell chemotaxis to various chemokines. These include lymphotactin, CC-chemokine ligand (CCL)2, CCL5, IFN-inducible protein-10 (CXCL10) and stromal-derived factor-1 alpha (SDF-1α; al-Aoukaty et al., 1999). When the function of individual PI3K isoforms in NK chemotactic behavior was examined it was found that both p110δ and p110γ are required for chemotaxis to CXCL12 and CCL3 both in vitro and in vivo. However, only p110δ was found to be required for chemotaxis to CXCL10 and the G protein-coupled receptor (GPCR) sphingosine 1-phosphate receptor 5 (S1P5), a receptor known to influence NK cell tissue distribution. Additionally, p110δ was found to be sufficient to mediate NK cell extravasation to tumors and steady state NK cell distribution to the spleen, lymph nodes, and liver (Saudemont et al., 2009).
PTEN
Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is one of the most commonly mutated genes in human cancers and is the underlying genetic etiology of Cowden syndrome, a disease characterized by the development of multiple hamartomas (Lynch et al., 1997). PTEN reverses the PI3K reaction by hydrolyzing PI(3,4,5)P3 to PI(4,5)P2. It is unclear if this reaction contributes meaningfully to the PI(4,5)P2 pool or if the importance of PTEN rests solely on antagonizing PI(3,4,5)P3 production. PTEN has not been extensively investigated in the context of NK cells. One study found that PTEN-deficient Vα14iNKT cells, a subpopulation of NKT cells accounting for about half of NKT cells, are not able to produce IFNγ as efficiently as WT Vα14iNKT cells. Moreover, compared to mice without deletion of PTEN these mice were unable to mount an effective response to melanoma (Kishimoto et al., 2007). Recent preliminary work from the Caligiuri lab has indicated that PTEN may decrease NK cell activation through the attenuation of the Akt and ERK1/2 signaling pathways through decreased availability of PI(3,4,5)P3. NK-92 cells transduced with a lentivirus expressing PTEN have decreased cytotoxicity against target cells and primary NK cells over-expressing PTEN exhibit decreased CD107α surface expression upon stimulation (Briercheck et al., 2012). The data from the Caligiuri lab indicate that PTEN may play a conventional role in most NK cells by limiting Akt activation; however, perhaps at least in the Vα14iNKT subpopulation of NK cells PTEN plays a role in NK cell activation through the creation of PI(4,5)P2 pools. Thus, PTEN appears to have an important role in both NK types and thus should be investigated more thoroughly in the context of NK cell biology, perhaps through the creation of mice with NK-specific deletion of PTEN.
SHIP1
There are two paralogs of SHIP: SHIP1 (Damen et al., 1996; Kavanaugh et al., 1996; Kerr et al., 1996; Lioubin et al., 1996; Ono et al., 1996) which is expressed in hematopoietic cells, pluripotent stem cells (Tu et al., 2001) and osteoblast lineage cells (Hazen et al., 2009), and SHIP2 (Pesesse et al., 1997) which is expressed in a wide array of cell types and tissues. SHIP1 contains an N-terminal SH2 domain which allows it to bind to phosphotyrosine motifs, a inositol-5-phosphatase enzymatic domain allowing for removal of the 5′ phosphate from PI(3,4,5)P3 or I(1,3,4,5)P4 to produce PI(3,4)P2 and I(1,3,4)P3, respectively, and two C-terminal NPXY motifs which, when tyrosine phosphorylated, allow for PTB domain binding. SHIP1 also contains a C2 domain that binds its product PI(3,4)P2 triggering an allosteric change that can enhance SHIP1 enzyme activity (Ong et al., 2007), as well as a PH-like domain that recognizes its substrate PI(3,4,5)P3 (Ming-Lum et al., 2012). The conversion of PI(3,4,5)P3 to PI(3,4)P2 allows for the attenuation of signaling pathways where PH domain-containing PI3K effectors exhibit selective recruitment to PI(3,4,5)P3 while also enabling the activation of other PI3K effectors whose PH domains allow recruitment to PI(3,4)P2 (Kerr, 2011).
SH2 domain-containing inositol-5-phosphatase 1 has been shown to play an important role in NK cell biology in several different studies albeit with some discordant findings. SHIP1-deficient mice were initially shown to have increased NK cell numbers due to increased survival of certain subsets that expressed poly-specific Ly49 receptors resulting in a skewed NK receptor repertoire and thus an inability to reject an MHC-mismatched bone marrow transplant (Wang et al., 2002; Fortenbery et al., 2010). Subsequently it was shown that SHIP1−/− NK cells are hyporesponsive for target cell lysis on an H2b MHC-I background due to inappropriate recruitment of SHP-1 to 2B4 resulting in an imbalance of inhibitory signals (Wahle et al., 2006, 2007; Fortenbery et al., 2010). Mice that are deficient in both 2B4 and SHIP1 have restored ability to lyse target cells lending further evidence for the inhibitory dominance of 2B4 in SHIP1-deficient mice (Fortenbery et al., 2010). NK cells from SHIP1-deficient mice were also shown to produce IFNγ inefficiently following stimulation and to have inappropriate expression of Ly49B, a poly-specific MHC-I receptor (Scarpellino et al., 2007) normally expressed by myeloid cells (Fortenbery et al., 2010). Interestingly, unlike mice with an H2b MHC-I genetic background, SHIP1−/− mice on an H2d homozygous background are able to kill MHC-I-mismatched target cells with supernormal efficiency and MHC-matched target cells at levels comparable to WT NK cells (Fortenbery et al., 2010). This was proposed to occur due to increased NK licensing due to over-expression of Ly49A that was observed in the H2d SHIP−/− NK cell compartment. These studies led to the question of whether the NK defects in SHIP1−/− mice are NK cell intrinsic or due to the inflammatory milieu present in these mice. Banh et al. (2012) showed that based upon CD27 and CD11b expression NK cells from SHIP−/− mice are less mature than those from wild type mice. Contradictory to previous studies (Wang et al., 2002; Trotta et al., 2005), they showed that SHIP1-deficient mice have decreased numbers of NK cells and that NK cells from these mice do have decreased production of IFNγ when co-stimulated with interleukin (IL)-12 and IL-18. In bone marrow chimera experiments Banh et al. (2012) saw no difference in IFNγ production when NK activating receptors were cross-linked, but saw a difference only when NK cells were co-stimulated with IL-12 and IL-18 leading to the conclusion that SHIP1 does not play an intrinsic role in NK cell cytokine production. However, in a mouse model with NK cell-specific deletion of SHIP1 we saw a significant impairment in IFNγ production following activation receptor cross-linking (Gumbleton and Kerr, unpublished data). In another study, NK cells with over-expression of SHIP1 had decreased IFNγ production, and SHIP1-deficient mice produced greater IFNγ when stimulated with IL-12 and anti-CD16 antibody, indicating that perhaps SHIP1 plays an inhibitory role in the context of IL-12 co-stimulation (Parihar et al., 2005).
In contrast to mouse NK cells where an absence of SHIP1 leads to decreased IFNγ production and cellular cytotoxicity indicating SHIP1 plays a role in NK cell activation, in human NK cells SHIP1 was initially found to limit signaling from the CD16 receptor and thus decrease the ADCC response (Galandrini et al., 2002). Human NK cells are able to be dissected into two populations: CD56brightCD16negative/dim NK cells that produce cytokines at a high level and CD56dimCD16bright NK cells that produce cytokines inefficiently but instead have greater cytolytic activity. SHIP1 is expressed at a lower level in the CD56brightCD16negative/dim NK cell subset potentially providing a molecular basis for their comparatively high cytokine production (Trotta et al., 2005). The same group correlated this effect with the presence of MiR-155 and proposed MiR-155 as a regulator of SHIP1 expression and thus, a regulator of NK cell activity. They were also able to show that NK cells from mice deficient for the MiR-155 precursor, Bic, were not able to produce IFNγ as efficiently as NK cells from wild type mice when co-stimulated with IL-12 and IL-18 or with IL-12 and anti-CD16 antibody. While these results could be due to MiR-155 regulation of SHIP1, the impact of MiR155 on other key IP signaling proteins such as PTEN (Yamanaka et al., 2009) and the PI3K subunit p85α needs to be rigorously excluded before this MiR155-SHIP1 circuit in NK cells is confirmed. As discussed above PI3K enzymes are important in NK cell chemotaxis. SHIP1 has been shown to be important in the chemotaxis of other types of leukocytes (Kim et al., 1999; Nishio et al., 2007). Thus we believe it would be interesting to analyze the importance of SHIP1 on NK cell chemotaxis.
INPP4
Inositol polyphosphate 4-phosphatase (INPP4) catalyzes the removal of the D-4 phosphate from PI(3,4)P2 to form PI(3)P. There are two different isozymes of INPP4: INPP4A and INPP4B with α and β splice variants of both. INPP4B has recently been shown to function as a tumor suppressor indicating that both PI(3,4,5)P3 and PI(3,4)P2 can give positive growth signals and that SHIP1 in some instances could act as a proto-oncogene (Brooks et al., 2010; Fuhler et al., 2012). Similar to the way that SHIP1 is expressed largely in hematopoietic lineages, in B, NK, and mast cells only INPP4Bα mRNA is highly expressed with potentially very low levels of INPP4Aα also being expressed. Interestingly, INPP4B has been shown to have a prominent role in osteoclast function where SHIP1 is also known to inhibit OC resorptive behavior ex vivo (Ferron et al., 2011). Thus further studies of INPP4B in lymphocytes, including NK cells, seems merited.
Conclusion
While there is some controversy in specifics, there is overwhelming evidence to show that the inositol phospholipid signaling pathway plays a prominent role in the regulation of NK cell development and function. The PI3K pathway has a clear role in the regulation of actin skeleton rearrangement, the formation of the NK immune synapse, chemotaxis, cytokine production, and cytolytic competency. In summary of the data discussed above, PI(4,5)P2 is important for cytolytic competency while PI(3,4,5)P3 is important for cytokine production and PI(3,4)P2 may be important for both NK effector functions in several contexts. Given that PIP5K is required for immune synapse formation and PI3K isoforms are required for chemotaxis these properties warrant investigation in the context of SHIP deletion. Lastly, as mentioned above, both INPP4 and PTEN are important regulators in other hematopoietic cell types, and thus their role in NK cell biology should be examined.
Conflict of Interest Statement
William G. Kerr has patents pending and issued concerning the analysis and targeting of SHIP1 in disease. Matthew Gumbleton has no conflicts to disclose.
Acknowledgments
This work was supported in part by grants from the NIH (RO1 HL72523, R01 HL085580, R01 HL107127) and the Paige Arnold Butterfly Run. William G. Kerr is the Murphy Family Professor of Children's Oncology Research, an Empire Scholar of the State University of NY and a Senior Scholar of the Crohn's and Colitis Foundation.
References
al-Aoukaty, A., Rolstad, B., and Maghazachi, A. A. (1999). Recruitment of pleckstrin and phosphoinositide 3-kinase gamma into the cell membranes, and their association with G beta gamma after activation of NK cells with chemokines. J. Immunol. 162, 3249–3255.
Alvarez Arias, D. A., Mccarty, N., Lu, L., Maldonado, R. A., Shinohara, M. L., and Cantor, H. (2010). Unexpected role of clathrin adaptor AP-1 in MHC-dependent positive selection of T cells. Proc. Natl. Acad. Sci. U.S.A. 107, 2556–2561.
Aoukaty, A., and Tan, R. (2002). Association of the X-linked lymphoproliferative disease gene product SAP/SH2D1A with 2B4, a natural killer cell-activating molecule, is dependent on phosphoinositide 3-kinase. J. Biol. Chem. 277, 13331–13337.
Arcaro, A., and Wymann, M. P. (1993). Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 296(Pt 2), 297–301.
Awasthi, A., Samarakoon, A., Dai, X., Wen, R., Wang, D., and Malarkannan, S. (2008). Deletion of PI3K-p85alpha gene impairs lineage commitment, terminal maturation, cytokine generation and cytotoxicity of NK cells. Genes Immun. 9, 522–535.
Badour, K., Zhang, J., Shi, F., Mcgavin, M. K., Rampersad, V., Hardy, L. A., et al. (2003). The Wiskott–Aldrich syndrome protein acts downstream of CD2 and the CD2AP and PSTPIP1 adaptors to promote formation of the immunological synapse. Immunity 18, 141–154.
Banh, C., Miah, S. M., Kerr, W. G., and Brossay, L. (2012). Mouse natural killer cell development and maturation are differentially regulated by SHIP-1. Blood 120, 4583–4590.
Bao, Y., Zheng, J., Han, C., Jin, J., Han, H., Liu, Y., et al. (2012). Tyrosine kinase Btk is required for NK cell activation. J. Biol. Chem. 287, 23769–23778.
Barber, D. F., Faure, M., and Long, E. O. (2004). LFA-1 contributes an early signal for NK cell cytotoxicity. J. Immunol. 173, 3653–3659.
Barber, D. F., and Long, E. O. (2003). Coexpression of CD58 or CD48 with intercellular adhesion molecule 1 on target cells enhances adhesion of resting NK cells. J. Immunol. 170, 294–299.
Belanger, S., Tai, L. H., Anderson, S. K., and Makrigiannis, A. P. (2008). Ly49 cluster sequence analysis in a mouse model of diabetes: an expanded repertoire of activating receptors in the NOD genome. Genes Immun. 9, 509–521.
Berridge, M. J., and Irvine, R. F. (1989). Inositol phosphates and cell signalling. Nature 341, 197–205.
Bhat, R., and Watzl, C. (2007). Serial killing of tumor cells by human natural killer cells – enhancement by therapeutic antibodies. PLoS ONE 2:e326. doi: 10.1371/journal.pone.0000326
Billadeau, D. D., Upshaw, J. L., Schoon, R. A., Dick, C. J., and Leibson, P. J. (2003). NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 4, 557–564.
Bonnema, J. D., Karnitz, L. M., Schoon, R. A., Abraham, R. T., and Leibson, P. J. (1994). Fc receptor stimulation of phosphatidylinositol 3-kinase in natural killer cells is associated with protein kinase C-independent granule release and cell-mediated cytotoxicity. J. Exp. Med. 180, 1427–1435.
Brandstadter, J. D., and Yang, Y. (2011). Natural killer cell responses to viral infection. J. Innate Immun. 3, 274–279.
Briercheck, E., Trotta, R., Cole, J., Cole, T., Hartlage, A., Martin, C., et al. (2012). “Human natural killer (NK) cells: differential expression of phosphatase and tensin homologue deleted on chromosome ten (PTEN) during NK cell development regulates its cytolytic activity against leukemic target cells,” in 54th ASH Annual Meeting and Exposition, Atlanta, GA.
Brooks, R., Fuhler, G. M., Iyer, S., Smith, M. J., Park, M. Y., Paraiso, K. H., et al. (2010). SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J. Immunol. 184, 3582–3589.
Bryceson, Y. T., Chiang, S. C., Darmanin, S., Fauriat, C., Schlums, H., Theorell, J., et al. (2011). Molecular mechanisms of natural killer cell activation. J. Innate Immun. 3, 216–226.
Calabia-Linares, C., Robles-Valero, J., De La Fuente, H., Perez-Martinez, M., Martin-Cofreces, N., Alfonso-Perez, M., et al. (2011). Endosomal clathrin drives actin accumulation at the immunological synapse. J. Cell Sci. 124, 820–830.
Capuano, C., Paolini, R., Molfetta, R., Frati, L., Santoni, A., and Galandrini, R. (2012). PIP2-dependent regulation of Munc13-4 endocytic recycling: impact on the cytolytic secretory pathway. Blood 119, 2252–2262.
Carlin, L. M., Evans, R., Milewicz, H., Fernandes, L., Matthews, D. R., Perani, M., et al. (2011). A targeted siRNA screen identifies regulators of Cdc42 activity at the natural killer cell immunological synapse. Sci. Signal. 4, ra81.
Damen, J. E., Liu, L., Rosten, P., Humphries, R. K., Jefferson, A. B., Majerus, P. W., et al. (1996). The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc. Natl. Acad. Sci. U.S.A. 93, 1689–1693.
Di Paolo, G., Pellegrini, L., Letinic, K., Cestra, G., Zoncu, R., Voronov, S., et al. (2002). Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature 420, 85–89.
Eissmann, P., Beauchamp, L., Wooters, J., Tilton, J. C., Long, E. O., and Watzl, C. (2005). Molecular basis for positive and negative signaling by the natural killer cell receptor 2B4 (CD244). Blood 105, 4722–4729.
Ferguson, K. M., Lemmon, M. A., Schlessinger, J., and Sigler, P. B. (1995). Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83, 1037–1046.
Ferron, M., Boudiffa, M., Arsenault, M., Rached, M., Pata, M., Giroux, S., et al. (2011). Inositol polyphosphate 4-phosphatase B as a regulator of bone mass in mice and humans. Cell Metab. 14, 466–477.
Ford, M. G., Pearse, B. M., Higgins, M. K., Vallis, Y., Owen, D. J., Gibson, A., et al. (2001). Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 291, 1051–1055.
Fortenbery, N. R., Paraiso, K. H., Taniguchi, M., Brooks, C., Ibrahim, L., and Kerr, W. G. (2010). SHIP influences signals from CD48 and MHC class I ligands that regulate NK cell homeostasis, effector function, and repertoire formation. J. Immunol. 184, 5065–5074.
Fuhler, G. M., Brooks, R., Toms, B., Iyer, S., Gengo, E. A., Park, M. Y., et al. (2012). Therapeutic potential of SH2 domain-containing inositol-5′-phosphatase 1 (SHIP1) and SHIP2 inhibition in cancer. Mol. Med. 18, 65–75.
Galandrini, R., Micucci, F., Tassi, I., Cifone, M. G., Cinque, B., Piccoli, M., et al. (2005). Arf6: a new player in FcgammaRIIIA lymphocyte-mediated cytotoxicity. Blood 106, 577–583.
Galandrini, R., Tassi, I., Mattia, G., Lenti, L., Piccoli, M., Frati, L., et al. (2002). SH2-containing inositol phosphatase (SHIP-1) transiently translocates to raft domains and modulates CD16-mediated cytotoxicity in human NK cells. Blood 100, 4581–4589.
Gamper, N., and Shapiro, M. S. (2007). Target-specific PIP2 signalling: how might it work? J. Physiol. 582, 967–975.
Golebiewska, U., Nyako, M., Woturski, W., Zaitseva, I., and Mclaughlin, S. (2008). Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol. Biol. Cell 19, 1663–1669.
Guo, H., Samarakoon, A., Vanhaesebroeck, B., and Malarkannan, S. (2008). The p110 delta of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J. Exp. Med. 205, 2419–2435.
Hamada, K., Shimizu, T., Matsui, T., Tsukita, S., and Hakoshima, T. (2000). Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 19, 4449–4462.
Hazen, A. L., Smith, M. J., Desponts, C., Winter, O., Moser, K., and Kerr, W. G. (2009). SHIP is required for a functional hematopoietic stem cell niche. Blood 113, 2924–2933.
Hinchliffe, K. A., Ciruela, A., and Irvine, R. F. (1998). PIPkins1, their substrates and their products: new functions for old enzymes. Biochim. Biophys. Acta 1436, 87–104.
Ishihara, H., Shibasaki, Y., Kizuki, N., Katagiri, H., Yazaki, Y., Asano, T., et al. (1996). Cloning of cDNAs encoding two isoforms of 68-kDa type I phosphatidylinositol-4-phosphate 5-kinase. J. Biol. Chem. 271, 23611–23614.
Itoh, T., Koshiba, S., Kigawa, T., Kikuchi, A., Yokoyama, S., and Takenawa, T. (2001). Role of the ENTH domain in phosphatidylinositol-4,5-bisphosphate binding and endocytosis. Science 291, 1047–1051.
Jiang, K., Zhong, B., Gilvary, D. L., Corliss, B. C., Hong-Geller, E., Wei, S., et al. (2000). Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1, 419–425.
Jiang, K., Zhong, B., Gilvary, D. L., Corliss, B. C., Vivier, E., Hong-Geller, E., et al. (2002). Syk regulation of phosphoinositide 3-kinase-dependent NK cell function. J. Immunol. 168, 3155–3164.
Jiang, K., Zhong, B., Ritchey, C., Gilvary, D. L., Hong-Geller, E., Wei, S., et al. (2003). Regulation of Akt-dependent cell survival by Syk and Rac. Blood 101, 236–244.
Johnson, C. M., Chichili, G. R., and Rodgers, W. (2008). Compartmentalization of phosphatidylinositol 4,5-bisphosphate signaling evidenced using targeted phosphatases. J. Biol. Chem. 283, 29920–29928.
Kanakaraj, P., Duckworth, B., Azzoni, L., Kamoun, M., Cantley, L. C., and Perussia, B. (1994). Phosphatidylinositol-3 kinase activation induced upon Fc gamma RIIIA-ligand interaction. J. Exp. Med. 179, 551–558.
Kavanaugh, W. M., Pot, D. A., Chin, S. M., Deuter-Reinhard, M., Jefferson, A. B., Norris, F. A., et al. (1996). Multiple forms of an inositol polyphosphate 5-phosphatase form signaling complexes with Shc and Grb2. Curr. Biol. 6, 438–445.
Kawakami, Y., Kitaura, J., Satterthwaite, A. B., Kato, R. M., Asai, K., Hartman, S. E., et al. (2000). Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cell activation. J. Immunol. 165, 1210–1219.
Kerr, W. G. (2011). Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann. N. Y. Acad. Sci. 1217, 1–17.
Kerr, W. G., and Colucci, F. (2011). Inositol phospholipid signaling and the biology of natural killer cells. J. Innate Immun. 3, 249–257.
Kerr, W. G., Heller, M., and Herzenberg, L. A. (1996). Analysis of lipopolysaccharide-response genes in B-lineage cells demonstrates that they can have differentiation stage-restricted expression and contain SH2 domains. Proc. Natl. Acad. Sci. U.S.A. 93, 3947–3952.
Kim, C. H., Hangoc, G., Cooper, S., Helgason, C. D., Yew, S., Humphries, R. K., et al. (1999). Altered responsiveness to chemokines due to targeted disruption of SHIP. J. Clin. Invest. 104, 1751–1759.
Kim, N., Saudemont, A., Webb, L., Camps, M., Ruckle, T., Hirsch, E., et al. (2007). The p110delta catalytic isoform of PI3K is a key player in NK-cell development and cytokine secretion. Blood 110, 3202–3208.
Kishimoto, H., Ohteki, T., Yajima, N., Kawahara, K., Natsui, M., Kawarasaki, S., et al. (2007). The Pten/PI3K pathway governs the homeostasis of Valpha14iNKT cells. Blood 109, 3316–3324.
Ling, K., Doughman, R. L., Firestone, A. J., Bunce, M. W., and Anderson, R. A. (2002). Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature 420, 89–93.
Lioubin, M. N., Algate, P. A., Tsai, S., Carlberg, K., Aebersold, A., and Rohrschneider, L. R. (1996). p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev. 10, 1084–1095.
Loijens, J. C., and Anderson, R. A. (1996). Type I phosphatidylinositol-4-phosphate 5-kinases are distinct members of this novel lipid kinase family. J. Biol. Chem. 271, 32937–32943.
Lowin-Kropf, B., and Held, W. (2000). Positive impact of inhibitory Ly49 receptor-MHC class I interaction on NK cell development. J. Immunol. 165, 91–95.
Lynch, E. D., Ostermeyer, E. A., Lee, M. K., Arena, J. F., Ji, H., Dann, J., et al. (1997). Inherited mutations in PTEN that are associated with breast cancer, cowden disease, and juvenile polyposis. Am. J. Hum. Genet. 61, 1254–1260.
Mace, E. M., Zhang, J., Siminovitch, K. A., and Takei, F. (2010). Elucidation of the integrin LFA-1-mediated signaling pathway of actin polarization in natural killer cells. Blood 116, 1272–1279.
Manilay, J. O., Waneck, G. L., and Sykes, M. (1999). Levels of Ly-49 receptor expression are determined by the frequency of interactions with MHC ligands: evidence against receptor calibration to a “useful” level. J. Immunol. 163, 2628–2633.
Marti, F., Xu, C. W., Selvakumar, A., Brent, R., Dupont, B., and King, P. D. (1998). LCK-phosphorylated human killer cell-inhibitory receptors recruit and activate phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 95, 11810–11815.
Martin, T. F. (2001). PI(4,5)P(2) regulation of surface membrane traffic. Curr. Opin. Cell Biol. 13, 493–499.
McLaughlin, S., and Murray, D. (2005). Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438, 605–611.
McLaughlin, S., Wang, J., Gambhir, A., and Murray, D. (2002). PIP(2) and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175.
Micucci, F., Capuano, C., Marchetti, E., Piccoli, M., Frati, L., Santoni, A., et al. (2008). PI5KI-dependent signals are critical regulators of the cytolytic secretory pathway. Blood 111, 4165–4172.
Ming-Lum, A., Shojania, S., So, E., Mccarrell, E., Shaw, E., Vu, D., et al. (2012). A pleckstrin homology-related domain in SHIP1 mediates membrane localization during Fcgamma receptor-induced phagocytosis. FASEB J. 26, 3163–3177.
Nishio, M., Watanabe, K., Sasaki, J., Taya, C., Takasuga, S., Iizuka, R., et al. (2007). Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat. Cell Biol. 9, 36–44.
Ong, C. J., Ming-Lum, A., Nodwell, M., Ghanipour, A., Yang, L., Williams, D. E., et al. (2007). Small-molecule agonists of SHIP1 inhibit the phosphoinositide 3-kinase pathway in hematopoietic cells. Blood 110, 1942–1949.
Ono, M., Bolland, S., Tempst, P., and Ravetch, J. V. (1996). Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature 383, 263–266.
Orr, S. J., Quigley, L., and Mcvicar, D. W. (2009). In vivo expression of signaling proteins in reconstituted NK cells. J. Immunol. Methods 340, 158–163.
Parihar, R., Trotta, R., Roda, J. M., Ferketich, A. K., Tridandapani, S., Caligiuri, M. A., et al. (2005). Src homology 2-containing inositol 5′-phosphatase 1 negatively regulates IFN-gamma production by natural killer cells stimulated with antibody-coated tumor cells and interleukin-12. Cancer Res. 65, 9099–9107.
Pesesse, X., Deleu, S., De Smedt, F., Drayer, L., and Erneux, C. (1997). Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem. Biophys. Res. Commun. 239, 697–700.
Pike, L. J., and Casey, L. (1996). Localization and turnover of phosphatidylinositol 4,5-bisphosphate in caveolin-enriched membrane domains. J. Biol. Chem. 271, 26453–26456.
Rak, G. D., Mace, E. M., Banerjee, P. P., Svitkina, T., and Orange, J. S. (2011). Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol. 9:e1001151. doi: 10.1371/journal.pbio.1001151
Raulet, D. H., and Vance, R. E. (2006). Self-tolerance of natural killer cells. Nat. Rev. Immunol. 6, 520–531.
Rhee, S. G., and Bae, Y. S. (1997). Regulation of phosphoinositide-specific phospholipase C isozymes. J. Biol. Chem. 272, 15045–15048.
Saito, K., Scharenberg, A. M., and Kinet, J. P. (2001). Interaction between the Btk PH domain and phosphatidylinositol-3,4,5-trisphosphate directly regulates Btk. J. Biol. Chem. 276, 16201–16206.
Saito, K., Tolias, K. F., Saci, A., Koon, H. B., Humphries, L. A., Scharenberg, A., et al. (2003). BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19, 669–678.
Saudemont, A., and Colucci, F. (2009). PI3K signaling in lymphocyte migration. Cell Cycle 8, 3307–3310.
Saudemont, A., Garcon, F., Yadi, H., Roche-Molina, M., Kim, N., Segonds-Pichon, A., et al. (2009). p110gamma and p110delta isoforms of phosphoinositide 3-kinase differentially regulate natural killer cell migration in health and disease. Proc. Natl. Acad. Sci. U.S.A. 106, 5795–5800.
Saudemont, A., Okkenhaug, K., and Colucci, F. (2007). p110delta is required for innate immunity to transplantable lymphomas. Biochem. Soc. Trans. 35, 183–185.
Scarpellino, L., Oeschger, F., Guillaume, P., Coudert, J. D., Levy, F., Leclercq, G., et al. (2007). Interactions of Ly49 family receptors with MHC class I ligands in trans and cis. J. Immunol. 178, 1277–1284.
Segovis, C. M., Schoon, R. A., Dick, C. J., Nacusi, L. P., Leibson, P. J., and Billadeau, D. D. (2009). PI3K links NKG2D signaling to a CrkL pathway involved in natural killer cell adhesion, polarity, and granule secretion. J. Immunol. 182, 6933–6942.
Tassi, I., Cella, M., Gilfillan, S., Turnbull, I., Diacovo, T. G., Penninger, J. M., et al. (2007). p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity 27, 214–227.
Tokuyama, M., Lorin, C., Delebecque, F., Jung, H., Raulet, D. H., and Coscoy, L. (2011). Expression of the RAE-1 family of stimulatory NK-cell ligands requires activation of the PI3K pathway during viral infection and transformation. PLoS Pathog. 7:e1002265. doi: 10.1371/journal.ppat.1002265
Tolias, K. F., Rameh, L. E., Ishihara, H., Shibasaki, Y., Chen, J., Prestwich, G. D., et al. (1998). Type I phosphatidylinositol-4-phosphate 5-kinases synthesize the novel lipids phosphatidylinositol 3,5-bisphosphate and phosphatidylinositol 5-phosphate. J. Biol. Chem. 273, 18040–18046.
Trotta, R., Parihar, R., Yu, J., Becknell, B., Allard, J. II, Wen, J., et al. (2005). Differential expression of SHIP1 in CD56bright and CD56dim NK cells provides a molecular basis for distinct functional responses to monokine costimulation. Blood 105, 3011–3018.
Tu, Z., Ninos, J. M., Ma, Z., Wang, J. W., Lemos, M. P., Desponts, C., et al. (2001). Embryonic and hematopoietic stem cells express a novel SH2-containing inositol 5′-phosphatase isoform that partners with the Grb2 adapter protein. Blood 98, 2028–2038.
Upshaw, J. L., Arneson, L. N., Schoon, R. A., Dick, C. J., Billadeau, D. D., and Leibson, P. J. (2006). NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 7, 524–532.
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M., and Bilanges, B. (2010). The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341.
Vivier, E., Raulet, D. H., Moretta, A., Caligiuri, M. A., Zitvogel, L., Lanier, L. L., et al. (2011). Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–49.
Vivier, E., Tomasello, E., Baratin, M., Walzer, T., and Ugolini, S. (2008). Functions of natural killer cells. Nat. Immunol. 9, 503–510.
Vlahos, C. J., Matter, W. F., Hui, K. Y., and Brown, R. F. (1994). A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269, 5241–5248.
Vyas, Y. M., Mehta, K. M., Morgan, M., Maniar, H., Butros, L., Jung, S., et al. (2001). Spatial organization of signal transduction molecules in the NK cell immune synapses during MHC class I-regulated noncytolytic and cytolytic interactions. J. Immunol. 167, 4358–4367.
Wahle, J. A., Paraiso, K. H., Costello, A. L., Goll, E. L., Sentman, C. L., and Kerr, W. G. (2006). Cutting edge: dominance by an MHC-independent inhibitory receptor compromises NK killing of complex targets. J. Immunol. 176, 7165–7169.
Wahle, J. A., Paraiso, K. H., Kendig, R. D., Lawrence, H. R., Chen, L., Wu, J., et al. (2007). Inappropriate recruitment and activity by the Src homology region 2 domain-containing phosphatase 1 (SHP1) is responsible for receptor dominance in the SHIP-deficient NK cell. J. Immunol. 179, 8009–8015.
Wang, J. W., Howson, J. M., Ghansah, T., Desponts, C., Ninos, J. M., May, S. L., et al. (2002). Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science 295, 2094–2097.
Wu, J., Song, Y., Bakker, A. B., Bauer, S., Spies, T., Lanier, L. L., et al. (1999). An activating immunoreceptor complex formed by NKG2D and DAP10. Science 285, 730–732.
Yamanaka, Y., Tagawa, H., Takahashi, N., Watanabe, A., Guo, Y. M., Iwamoto, K., et al. (2009). Aberrant overexpression of microRNAs activate AKT signaling via down-regulation of tumor suppressors in natural killer-cell lymphoma/leukemia. Blood 114, 3265–3275.
Keywords: inositol phospholipid, PIP5K, SHIP, PTEN, PI3K, IFNγ, natural killer cells, INPP4
Citation: Gumbleton M and Kerr WG (2013) Role of inositol phospholipid signaling in natural killer cell biology. Front. Immunol. 4:47. doi: 10.3389/fimmu.2013.00047
Received: 30 November 2012; Accepted: 08 February 2013;
Published online: 06 March 2013.
Edited by:
Eric Vivier, Centre d'Immunologie de Marseille-Luminy, FranceReviewed by:
Akira Shibuya, University of Tsukuba, JapanJacques Zimmer, Centre de Recherche Public de la Santé, Luxembourg
Copyright: © 2013 Gumbleton and Kerr. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: William G. Kerr, Department of Microbiology and Immunology, State University of New York Upstate Medical University, 2204 Weiskotten Hall, 750 East Adams Street, Syracuse, NY 13210, USA. e-mail:a2VycndAdXBzdGF0ZS5lZHU=