


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 December 2012
Sec. B Cell Biology
Volume 3 - 2012 | https://doi.org/10.3389/fimmu.2012.00398
This article is part of the Research Topic PI3K signalling View all 12 articles
Understanding the mechanisms by which signal transduction pathways mediate changes in RNA abundance requires the examination of the fate of RNA from its transcription to its degradation. Evidence suggests that RNA abundance is partly regulated by post-transcriptional mechanisms affecting RNA decay and this in turn is modulated by some of the same signaling pathways that control transcription. Furthermore, the translation of mRNA is a key regulatory step that is influenced by signal transduction. These processes are regulated, in part, by RNA-binding proteins (RBPs) which bind to sequence-specific RNA elements. The function of RBPs is controlled and co-ordinated by phosphorylation. Based on the current literature we hypothesize that RBPs may be a point of convergence for the activity of different kinases such as phosphoinositide-3-kinase and mitogen-activated protein kinase which regulate RBP localization and function.
The response of cells to environmental stimuli frequently involves changes in gene expression. This may be controlled at multiple levels including the production of new RNA by transcription. Post-transcriptional regulation at the RNA level includes nuclear RNA processing (frequently a co-transcriptional process), as well as RNA export, decay, localization, and translation. These processes are integrated with changes in protein stability and function. Signaling pathways are a major mechanism for co-ordination of these distinct mechanisms (Schoenberg and Maquat, 2012).
Substantial evidence obtained over two decades has highlighted the importance of mRNA stability in gene regulation (Cheadle et al., 2005; Keene, 2007; Anderson, 2008; Hao and Baltimore, 2009). The half-life of different mRNAs can vary from 15 min to more than 24 h depending on the activation status of a cell, for example, the half-life for interleukin-2 (IL-2) mRNA is 17 min in non-stimulated T cells but upon activation with anti-CD3/CD28 the half-life for IL-2 mRNA is increased to 232 min (Raghavan et al., 2002, 2004; Yang et al., 2003). The mRNA half-lives in bacteria (Bernstein et al., 2002) and yeast (Wang et al., 2002) are mostly shorter in comparison to mammalian cells. The increased mRNA half-life correlates with increasing organismal complexity and a tendency for 3′ untranslated regions (UTR) within mRNA to become longer in more complex species (Mazumder et al., 2003; Dinger et al., 2011). Thus, post-transcriptional regulation of mRNA may be a more prevalent amongst complex multicellular organisms.
The difference in mRNA half-lives can lead to significant changes in the abundance of mRNA (Ross, 1995). This was illustrated in genome-wide studies which have shown that up to 50% of altered mRNA abundance in lymphocytes is due to the regulation of mRNA stability (Lam et al., 2001; Cheadle et al., 2005). The stability of mRNA is regulated by distinct sequences present in the coding and UTR of mRNA (Caput et al., 1986; Shaw and Kamen, 1986; Schoenberg and Maquat, 2012). Conservation of these sequences within the UTR region among different species further emphasizes their regulatory role.
The fate of RNA can be regulated by the interplay between sequences within the RNA (cis-acting) and trans-acting factors present in the nucleus and cytoplasm (Keene, 2007; Anderson, 2010; Elkon et al., 2010). Trans-acting factors such as non-coding RNA (Rinn and Chang, 2012), microRNA (Fabian and Sonenberg, 2012), and RNA-binding proteins (RBPs) have been reported to regulate mRNA stability and translation. Translation can also be regulated through control of the length of the poly(A) tail in the cytoplasm (Weill et al., 2012). The class of trans-acting factors we will focus on in this review are the RBPs. The function of RBPs can be controlled by different signaling pathways and several excellent reviews covering the detailed regulation of RBPs in response to stress pathways have been published (Eberhardt et al., 2007; Doller et al., 2008; Sandler and Stoecklin, 2008; Kim et al., 2010). Here we will discuss the function of selected RBPs at the molecular level and how they are being controlled by phosphoinositide-3-kinase (PI3K) and mitogen-activated protein kinases (MAPK) signaling.
It has been estimated that approximately 1000 RBPs are encoded in the mammalian genome (Keene, 2007; Araujo et al., 2012; Baltz et al., 2012; Castello et al., 2012). These play important roles in splicing, nuclear export, mRNA stability, localization, and translation. RBPs exert their function by physically interacting with RNA and can do so in a sequence-specific manner. Amongst the well-characterized sequences that bind RBPs are the adenine- and uridine-rich elements (ARE). RBPs that bind to ARE include KSRP as well as TTP (TIS11) and its homologs TIS11b (also called BRF-1; butyrate response factor-1), and TIS11d (BRF-2). These have been shown to promote ARE-dependent mRNA decay but may also affect translation. HuR and its close relatives have also been suggested to promote mRNA stability and to regulate translation (Srikantan and Gorospe, 2012; Yiakouvaki et al., 2012). The domain structures of these RBPs are depicted in Figure 1.
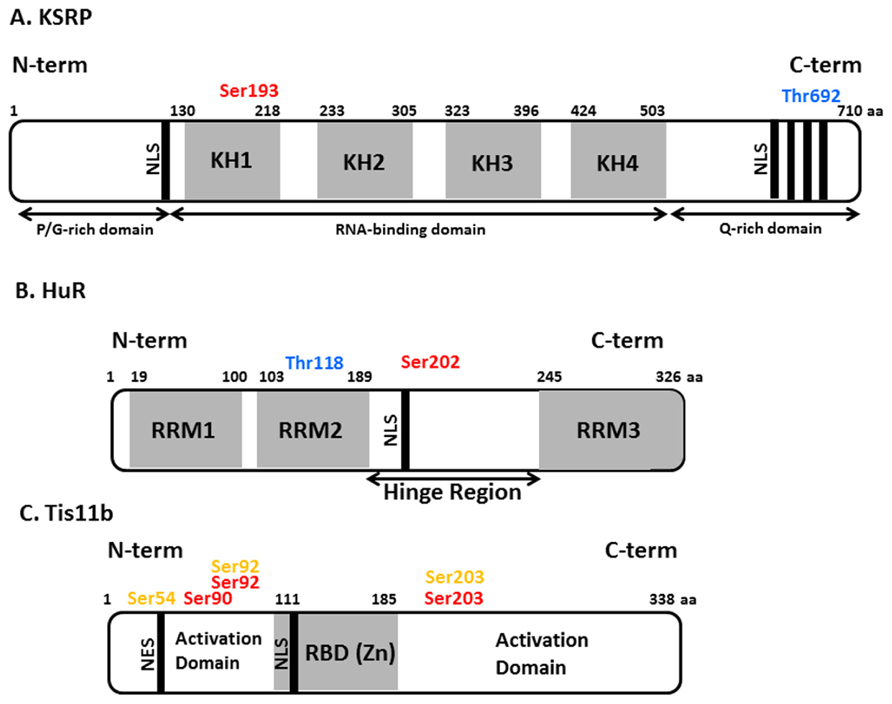
FIGURE 1. Schematic view of domains and phosphorylation sites in KSRP, HuR and Tis11b proteins. (A) KH domains and nuclear localization signals are represented in light gray and black colors respectively. The amino acid numbers are depicted on top of protein domain structure. Proline/glycine-rich and glutamine-rich domains on N-terminal and C-teriminal end respectively are marked. (B) Hinge region (containing nucleocytoplasmic sequence and nuclear localization signal, NLS); RNA recognition motifs (RRM) and their corresponding amino acid positions are depicted. (C) Activation domains on N- and C-terminal ends, NLS, nuclear export sequence (NES), and RBD (RNA-binding domain) are shown. The experimentally determined phosphorylation sites on RBPs are given colors in following manner, red: PKB phosphorylation sites, blue: p38 phosphorylation sites, yellow: MK2 phosphorylation sites.
Diverse stimuli (such as chemical and environmental mediators, reviewed in Eberhardt et al., 2007) induce signaling cascades which control RBP function by means of protein phosphorylation. However the details of how these signaling pathways control the abundance and RNA-binding properties of RBPs at molecular level are only beginning to be understood. Amongst the signaling kinases discussed below and summarized in Table 1 are PI3K, PKB (protein kinase B), mTOR (mammalian target of rapamycin), and p38 MAPK. Experimentally determined phosphorylation sites on the RBPs we will focus on are depicted in Figure 1. A more extensive list derived from phospho-proteomics studies is given in Table 2.
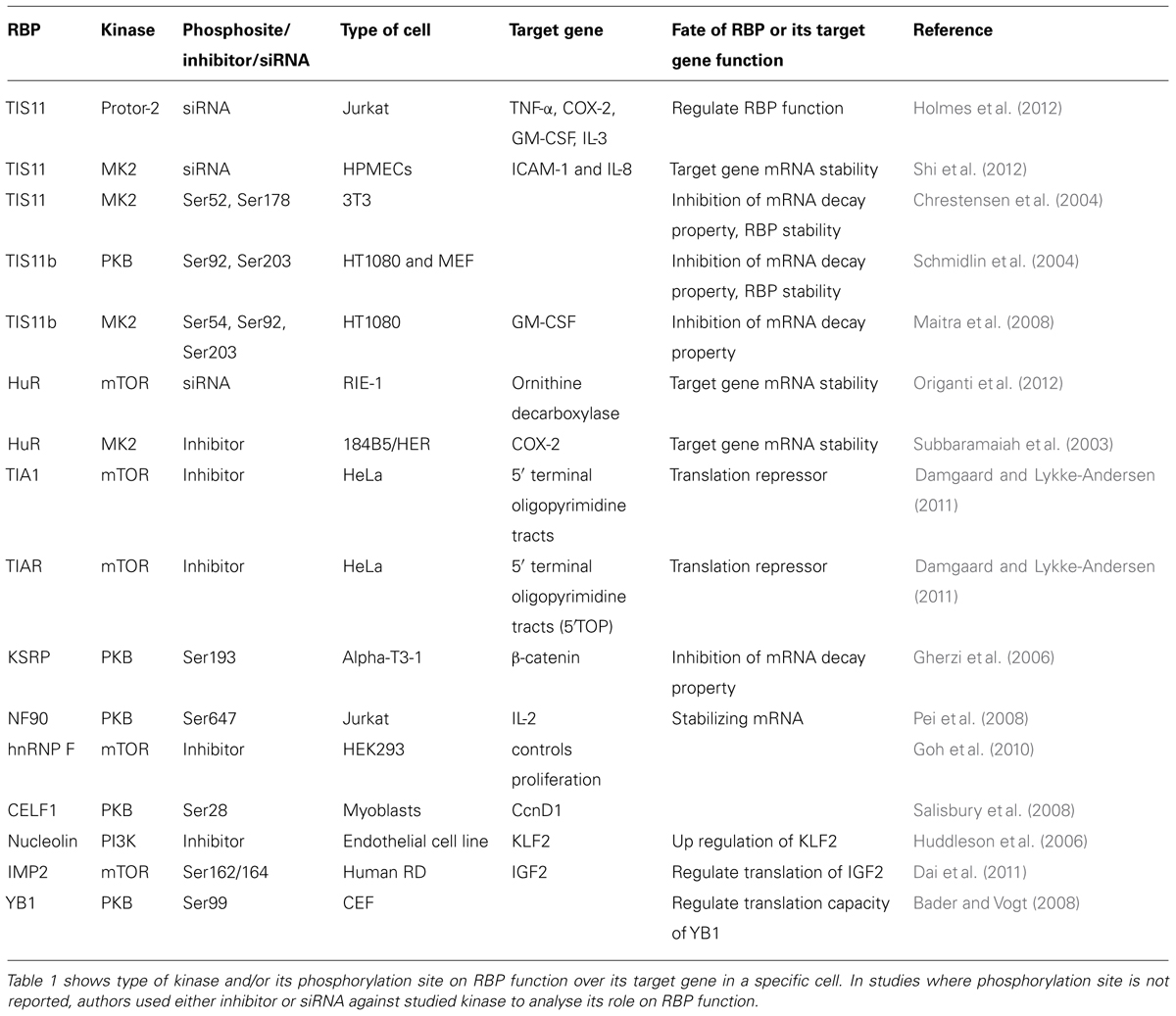
TABLE 1. Representative list of signaling kinases regulating RBP function.
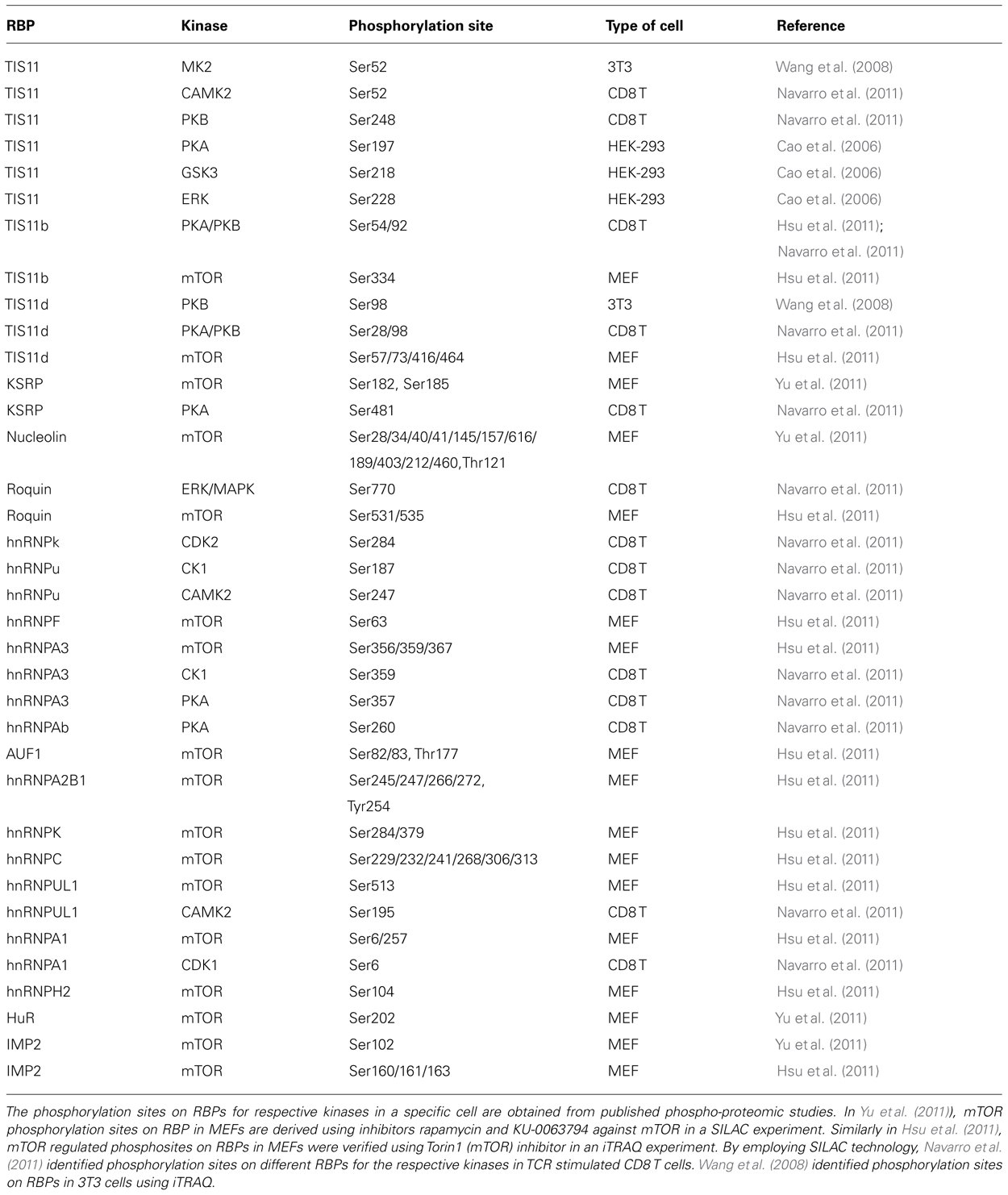
TABLE 2. Phosphorylation sites on RBP from phospho-proteomic studies.
The PI3K pathway plays an important role in controlling cell growth, differentiation, survival, chemotaxis, and metabolism. The activation of PI3K stimulates the generation of phosphatidylinositol 3,4,5 trisphosphate at the cell membrane, which mediates the recruitment of PKB (also known as AKT) and phosphoinositide-dependent kinase 1 (PDK1). PKB is then activated following phosphorylation by PDK1 at Thr308. Mammalian target of rapamycin complex 2 (mTORC2) phosphorylates PKB at hydrophobic motif (Ser473; Jacinto et al., 2006; Sarbassov et al., 2006; Facchinetti et al., 2008). PKB, which has three isoforms encoded by different genes, is involved in regulating cell proliferation and survival (Pearce et al., 2010). Genetic alterations in the PI3K pathway, such as mutations in catalytic subunits and loss of the negative regulator PTEN have been found in cancers (Kok et al., 2009). PI3K also regulates immunity and inflammation by controlling the recruitment and activation of immune cells. Thus, PI3K is an important signal integrator in maintaining immune homeostasis.
An early report implicated PI3K in the regulation of the stability of IL-3 mRNA by TIS11/TTP but did not identify phosphorylation sites within TTP (Ming et al., 2001). Subsequent studies using mass spectroscopy identified over 30 phosphorylation sites on TTP (Cao et al., 2006). Both Ser60 and Ser113 in human TTP are predicted to be PKB phosphorylation sites (Cao et al., 2007) but this has not been experimentally confirmed.
Following activation of the PI3K pathway the phosphorylation of TIS11b by PKB at Ser90, Ser92, and Ser203 facilitates its binding with 14-3-3. This sequesters TIS11b in the cytoplasm and inhibits its ability to promote mRNA decay (Schmidlin et al., 2004; Benjamin et al., 2006). Phosphorylation of these sites appears to increase the stability of the TIS11b protein which is consistent with a shorter half-life/increased degradation of TIS11b protein in PKBα knockout mouse embryo-derived fibroblasts (MEFs; Benjamin et al., 2006). Mutagenesis of Ser90, Ser92, and Ser203 in TIS11b uncoupled it from regulation by PKB and the mutated protein, which retained the ability to promote RNA decay, could no longer associate with 14-3-3 (Benjamin et al., 2006).
KSRP mediates mRNA destabilization by binding to AREs in target mRNAs. Phosphorylation of KSRP at Ser193 by PKB facilitates its binding to 14-3-3 which inhibits its interaction with the RNA decay machinery (in this case the exosome; Gherzi et al., 2006). In this way, KSRP is prevented from promoting the degradation of β-catenin mRNA (Gherzi et al., 2006). Phosphorylation at Ser193 creates a binding site for 14-3-3ζ in the N-terminal KH domain of KSRP (Diaz-Moreno et al., 2009) which, upon interaction with KSRP, promotes its nuclear localization. Thus, the availability of KSRP in cytoplasm and its ability to mediate mRNA decay is limited by phosphorylation. Interestingly, phosphorylation at Ser193 redirects the function of KSRP to become a regulator of the maturation of miRNA (Trabucchi et al., 2009). In C2C12 myoblasts PI3K-dependent phosphorylation of KSRP enhanced its ability to accelerate myogenic miRNA processing while attenuating its ability to promote myogenic mRNA decay. Thus, during myogenesis, KSRP appears to function as a dynamic switch controlling RNA regulated by PI3K (Briata et al., 2012).
The importance of the PI3K pathway in controlling mRNA decay was further illustrated by a recent study which demonstrated that approximately 20 out of 50 transcripts regulated by PI3K were affected at the level of mRNA stability (Graham et al., 2010). Using siRNA knockdown experiments TIS11b and KSRP were shown to be involved in stabilization of the mRNAs of down-regulated genes (Graham et al., 2010). Interestingly Graham et al., 2010) observed no effect on mRNA stability by RBPs such as AUF1 and HuR which are not known to be controlled by PI3K signaling, suggesting a key role of PI3K pathway in maintaining mRNA stability via TIS11b and KSRP.
The mTOR is a kinase component of signaling complexes which play very important roles in immune cell function (Powell et al., 2012). The mTORC1 complex can be activated in a PI3K-dependent or independent manner and is highly susceptible to inhibition by rapamycin. By contrast, mTORC2 which phosphorylates PKB Ser473 and contributes to its activation is much less sensitive to rapamycin (Jacinto et al., 2006; Sarbassov et al., 2006; Facchinetti et al., 2008). mTOR can also be activated in a PI3K-independent manner by spleen tyrosine kinase (SYK), as reported in follicular lymphoma cells (Leseux et al., 2006). In addition to SYK, the Erk pathway can also activate mTOR (Shaw and Cantley, 2006). Thus, mTOR activated in a PI3K-dependent or independent manner exerts effector functions via a number of targets including PKB (Powell et al., 2012).
In a phospho-proteomic study the Blenis Group reported Ser182 and Ser185 as phosphorylation sites on KSRP for mTOR. These findings were verified by treating MEFs with the mTOR inhibitors rapamycin and KU-0063794 (Table 2; Yu et al., 2011). The same phosphorylation sites in KSRP were predicted by PHOSIDA (www.phosida.com; the post-translational modification database which provides information on the sites of phosphorylation, N-glycosylation, and acetylation across nine different species; Gnad et al., 2007; Gnad et al., 2011). However Ser185 of KSRP has also been predicted to be a casein kinase 1 (CK1) phosphorylation site and the function of this phosphorylation is presently unknown.
The Blenis group also reported HuR Ser202 as a phosphorylation site for mTOR and this finding was substantiated using the mTOR inhibitors rapamycin and KU-0063794 in MEFs (Table 2; Yu et al., 2011). However, limited evidence exists for the functional regulation of HuR by mTOR as it has not yet been reported whether or not mTOR regulates HuR localization and function. Previously the Gorospe group had reported Ser202 as a target for cyclin-dependent kinase 1 (Cdk1; Kim et al., 2008). Phosphorylation of HuR by Cdk1 promoted its movement into the nucleus where HuR appeared to be associated with 14-3-3 proteins. A modified HuR protein with a non-phosphorylatable serine to alanine mutation resided predominantly in the cytoplasm. Unphosphorylated HuR bound poorly to 14-3-3, which increased the availability of HuR for stabilizing its target mRNAs (Kim et al., 2008). Thus, the function of HuR is modulated by Cdk1 during the cell cycle (Blethrow et al., 2008; Kim et al., 2008). In another very recent report CDK5 phosphorylation of HuR at Ser202 has been shown to regulate its function in cell cycle progression (Filippova et al., 2012). HuR regulates ornithine decarboxylase (ODC) mRNA stability (Nowotarski and Shantz, 2010) and the binding of HuR to the ODC transcript is decreased when mTORC1 signaling is inhibited using rapamycin, an mTORC1 inhibitor (Origanti et al., 2012). The mTOR mediated phosphorylation at Ser202 on HuR might be one mechanism through which mTOR regulates proliferation.
Protor2, a component of mTORC2 kinase has been shown to bind to TTP in Jurkat cells following treatment with carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone, an uncoupler of mitochondrial oxidative phosphorylation. The interaction between TTP and Protor2 was suggested to be necessary for enhancing TTP-mediated turnover of mRNAs such as IL-3, GM-CSF, COX-2, and TNF (Holmes et al., 2012). The siRNA knockdown of protor2 inhibited the localization of TTP to mRNA processing bodies (P-bodies), the sites where mRNA decay enzymes are concentrated (Parker and Sheth, 2007). Very recently mTOR has been reported to regulate iron homeostasis by modulating transferrin receptor 1 (TfR1) stability via TTP (Bayeva et al., 2012).
Mitogen-activated protein kinases are major regulatory hubs where inflammation and stress responses are regulated. Three major MAPK pathways are p38, JNK, and ERK. We discuss below the findings implicating p38 and its substrate MAPK activated protein kinase 2 (MK2) in regulating RBP activity.
The p38 pathway via MK2 regulates the mRNA decay property and the mRNA and protein expression of TTP (Dean et al., 2001; Tchen et al., 2004; Brook et al., 2006; Hitti et al., 2006). MK2 phosphorylates mouse TTP at Ser52 and Ser178 which stabilizes TTP protein (Brook et al., 2006). Upon dephosphorylation of these sites, TTP moves from the cytoplasm to the nucleus and undergoes degradation (Brook et al., 2006). MK2-mediated phosphorylation of TTP does not affect its binding to target mRNA, but inhibits the ability of TTP to recruit deadenylases to target mRNA for their degradation in cytoplasm (Carballo et al., 2001; Stoecklin et al., 2004; Clement et al., 2011). Further investigation of this demonstrated that carbon catabolite repressor protein 4-associated factor-1 (CAF1) was the major source of deadenylase activity responsible for TTP-directed deadenylation (Marchese et al., 2010). MK2 phosphorylation reduced the ability of TTP to promote deadenylation by inhibiting the recruitment of CAF1 deadenylase independently of 14-3-3. The Stoecklin group demonstrated that Not1, a component of carbon catabolite repressor protein 4 (Ccr4)-negative on TATA (NOT) complex, associates with TTP and is required for the decay of ARE-mRNAs (Sandler et al., 2011). Subsequently it has been reported that TTP regulates the translation of TNF mRNA at the endoplasmic reticulum (Tiedje et al., 2012). Phosphorylation of TTP by MK2 weakens its ability to bind to TNF mRNA and allowed HuR-binding to TNF mRNA which promoted its translation. Conflicting data exists on the ability of phospho-TTP to bind its target mRNA (Clement et al., 2011; Tiedje et al., 2012) The former group found that phosphorylation did not alter TTP binding to its target mRNA but the latter group found that phosphorylation reduced the affinity of TTP binding to its target mRNA.
TIS11b is also regulated by MK2. The phosphorylation of TIS11b at Ser54, Ser92, and Ser203 by MK2 inhibits the ability of TIS11b to promote ARE-mediated mRNA decay (Maitra et al., 2008). The phosphorylation-dependent inhibitory effects of MK2 on TIS11b do not seem to alter its ability to bind RNA or its association with mRNA decay enzymes. Furthermore, the MK2-mediated effects on TIS11b were independent of PKB (Maitra et al., 2008).
Several studies indicate that the function of HuR is regulated by the p38 pathway. The abundance of COX-2 mRNA is controlled by the p38-dependent regulation of the binding of HuR to the COX-2 3′ UTR (Subbaramaiah et al., 2003). In neuronal cell line p38 activation following treatment with anisomycin promotes the cytoplasmic accumulation of HuR where it interacts with and stabilizes the survival motor neuron (SMN) transcript (Farooq et al., 2009). In mouse splenic T cells, LFA-1 engagement activates p38 which promotes HuR translocation and stabilization of IFN-γ and TNF mRNA (Ramgolam et al., 2010). In none of these studies was it established whether HuR was directly phosphorylated by p38 or MK2. However, an independent study has reported that phosphorylation of HuR at Thr118 by p38 promotes its localization to the cytoplasm where it stabilizes p21 mRNA during the DNA damage response (Lafarga et al., 2009). HuR was found to be phosphorylated at Thr118 a site previously identified to be phosphorylated by Chk2 (Abdelmohsen et al., 2007). HuR regulated translation of TNF mRNA at the endoplasmic reticulum appeared to be mediated by the effects of the p38 pathway on TTP (Tiedje et al., 2012) and no evidence was found for p38-mediated phosphorylation of HuR. It was suggested that Thr118 was instead phosphorylated by Chk2 as a consequence of the over-expression system being used (Tiedje et al., 2012).
The p38 pathway also regulates KSRP. During C2C12 muscle cell differentiation the stability of p21, myogenin, and MyoD mRNA is regulated by p38-mediated phosphorylation of KSRP (Briata et al., 2005). p38 phosphorylates KSRP at Thr692 which renders KSRP unable to bind to ARE-containing transcripts thus promoting their stabilization. However, this phosphorylation event does not alter the ability of KSRP to interact with the mRNA degradation machinery (Briata et al., 2005). To our knowledge it has not yet been reported whether or not p38 regulates KSRP function in microRNA maturation.
In NIH 3T3 fibroblasts, the stability of TPA induced IL-3 mRNA is regulated by the p38 and PI3K pathways (Ming et al., 2001). The latter mediates its effects independently of p38 suggesting that p38 and PI3K pathways control IL-3 mRNA turnover by parallel mechanisms. Stabilization of IL-3 mRNA mediated by either of these two pathways is antagonized by TTP and this effect can be overcome by HuR when it is in collaboration with p38 but not with PI3K (Ming et al., 2001). This suggests that signaling pathways activated upon stimulation lead either to activation of stabilizing RBP (HuR) or inactivation of destabilizing RBP (TTP) thus preventing the degradation of transcripts (Ming et al., 2001). Furthermore, another study reported that in U87 glioblastoma cells, the regulation of cyclin D1 and c-Myc mRNA stability by TTP is controlled by p38 in a PKB-dependent manner (Marderosian et al., 2006), implying interdependent roles for p38 and PKB. Data from phospho-proteomic (Cao et al., 2006, 2007; Navarro et al., 2011) and in vitro (Chrestensen et al., 2004) studies suggest that MK2 and PKB phosphorylate TTP at Ser52/178 and Ser248 respectively (Table 2).
Both PKB and MK2 target the same phosphorylation sites on TIS11b (Ser92 and Ser203) and inhibit its mRNA decay activity (Benjamin et al., 2006; Maitra et al., 2008). The mRNA decay property of KSRP is also regulated by PKB and p38 (Briata et al., 2005; Gherzi et al., 2006), but in this instance the kinases do not use the same phosphorylation sites.
HuR provides a further example of a point of convergence. In this case the circumstances under which the mTOR would affect HuR have not been studied but the phosphorylation site (Ser202) has been shown to regulate the function of the protein. Similarly, p38-mediated phosphorylation of HuR at Thr118 targets a site previously shown to be targeted by Chk2 (Abdelmohsen et al., 2007). It is clear that multiple different kinases converge on HuR to regulate its function (Figure 2).
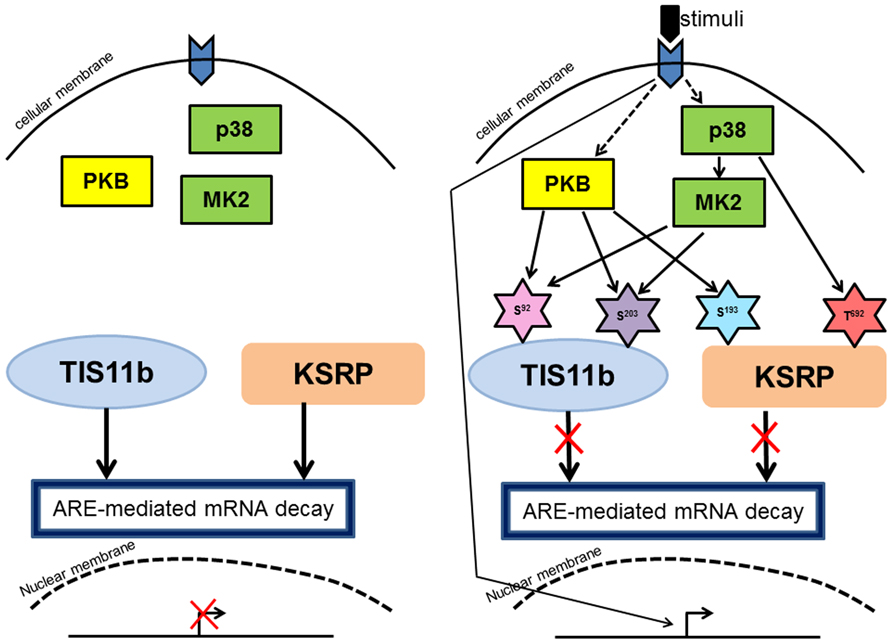
FIGURE 2. Convergence of signaling on RBP and its function. (A) In non-activated cells, RBPs (TIS11b and KSRP) are active in promoting mRNA decay of ARE-containing transcripts. (B) Upon activation, PKB and MK2 phosphorylate TIS11b at Ser92 and Ser203 and subsequently TIS11b is prevented in mediating ARE-mediated mRNA decay. Similarly, but not at same phosphorylation sites, KSRP is phosphorylated at Ser193 and Thr692 by PKB and p38 respectively. These phosphorylation events inhibit KSRP in mediating ARE-mediated mRNA decay. Transcription factors upon activation by signals from kinases drive transcription which is illustrated in the Figure 2. The phosphorylation sites are shown in star shape cartoons.
Current literature suggests that kinases control the function of RBPs, however RBPs have also been reported to regulate the expression of kinases suggesting a role for RBPs in feedback control over kinase expression. For example, the 68 kDa Src substrate associated during mitosis (Sam68) is an RBP reported to regulate alternative splicing of mTOR (Huot et al., 2012). In Sam68 knockout cells, intron five is retained in the mTOR transcript introducing a premature termination codon, which results in an unstable mRNA and subsequently reduced protein levels of mTOR. Consequently, the effector pathways of mTOR responsible for adipogenesis are deregulated in these mice, leading to a lean phenotype (Huot et al., 2012). Sam68 is also reported to associate with PI3K in insulin receptor signaling (Sanchez-Margalet and Najib, 2001) and this association may regulate the RNA-binding function of Sam68 (Taylor et al., 1995). Based on this information it is possible that PI3K is regulating RBP function via its downstream kinases and regulating the abundance of its downstream kinases (mTOR) via possible activation of Sam68. Conserved ARE elements in 3′ UTR of p38α, PKBα, PKBγ but not p38β, p38γ, and PKBβ, suggest that these proteins might be targets for regulation of ARE-mediated mRNA stability by RBPs (Gruber et al., 2011).
Gene expression is controlled at the post-transcriptional level by RBPs. However, regulation of the effector function of RBPs on RNA decay and translation is controlled by signals from protein kinases. These effects include inhibition of function by uncoupling from the RNA decay machinery and in some instances reassignment of function. Kinase-dependent relocation of RBP into different compartments of the cell seems to be a common theme amongst structurally diverse RBPs. RBPs represent a class of proteins upon which signaling by the PI3K and p38 pathways converge. Existing literature on this might indicate a potential redundancy of kinases phosphorylating the same serine or threonine amino acid in different cellular functions. A fuller understanding of the interplay between kinases, RBPs and target RNAs may provide important new insights into the dynamic regulation of gene expression.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Klaus Okkenhaug, Jessica McHugh, and Rebecca Newman for comments on the manuscript
Abdelmohsen, K., Pullmann, R. Jr., Lal, A., Kim, H. H., Galban, S., Yang, X., et al. (2007). Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 25, 543–557.
Anderson, P. (2010). Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol. 10, 24–35.
Araujo, P. R., Yoon, K., Ko, D., Smith, A. D., Qiao, M., Suresh, U., et al. (2012). Before it gets started: regulating translation at the 5′ UTR. Comp. Funct. Genomics 2012, 475731.
Bader, A. G., and Vogt, P. K. (2008). Phosphorylation by Akt disables the anti-oncogenic activity of YB-1. Oncogene 27, 1179–1182.
Baltz, A. G., Munschauer, M., Schwanhausser, B., Vasile, A., Murakawa, Y., Schueler, M., et al. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 46, 674–690.
Bayeva, M., Khechaduri, A., Puig, S., Chang, H. C., Patial, S., Blackshear, P. J., et al. (2012). mTOR regulates cellular iron homeostasis through Tristetraprolin. Cell Metab. 16, 645–657.
Benjamin, D., Schmidlin, M., Min, L., Gross, B., and Moroni, C. (2006). BRF1 protein turnover and mRNA decay activity are regulated by protein kinase B at the same phosphorylation sites. Mol. Cell. Biol. 26, 9497–9507.
Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S., and Cohen, S. N. (2002). Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 99, 9697–9702.
Blethrow, J. D., Glavy, J. S., Morgan, D. O., and Shokat, K. M. (2008). Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. U.S.A. 105, 1442–1447.
Briata, P., Forcales, S. V., Ponassi, M., Corte, G., Chen, C. Y., Karin, M., et al. (2005). p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol. Cell 20, 891–903.
Briata, P., Lin, W. J., Giovarelli, M., Pasero, M., Chou, C. F., Trabucchi, M., et al. (2012). PI3K/AKT signaling determines a dynamic switch between distinct KSRP functions favoring skeletal myogenesis. Cell Death Differ. 19, 478–487.
Brook, M., Tchen, C. R., Santalucia, T., Mcilrath, J., Arthur, J. S., Saklatvala, J., et al. (2006). Posttranslational regulation of tristetraprolin subcellular localization and protein stability by p38 mitogen-activated protein kinase and extracellular signal-regulated kinase pathways. Mol. Cell. Biol. 26, 2408–2418.
Cao, H., Deterding, L. J., and Blackshear, P. J. (2007). Phosphorylation site analysis of the anti-inflammatory and mRNA-destabilizing protein tristetraprolin. Expert Rev. Proteomics 4, 711–726.
Cao, H., Deterding, L. J., Venable, J. D., Kennington, E. A., Yates, J. R. III, Tomer, K. B., et al. (2006). Identification of the anti-inflammatory protein tristetraprolin as a hyperphosphorylated protein by mass spectrometry and site-directed mutagenesis. Biochem. J. 394, 285–297.
Caput, D., Beutler, B., Hartog, K., Thayer, R., Brown-Shimer, S., and Cerami, A. (1986). Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc. Natl. Acad. Sci. U.S.A. 83, 1670–1674.
Carballo, E., Cao, H., Lai, W. S., Kennington, E. A., Campbell, D., and Blackshear, P. J. (2001). Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J. Biol. Chem. 276, 42580–42587.
Castello, A., Fischer, B., Eichelbaum, K., Horos, R., Beckmann, B. M., Strein, C., et al. (2012). Insights into RNA biology from an Atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406.
Cheadle, C., Fan, J., Cho-Chung, Y. S., Werner, T., Ray, J., Do, L., et al. (2005). Control of gene expression during T cell activation: alternate regulation of mRNA transcription and mRNA stability. BMC Genomics 6:75. doi: 10.1186/1471-2164-6-75
Chrestensen, C. A., Schroeder, M. J., Shabanowitz, J., Hunt, D. F., Pelo, J. W., Worthington, M. T., et al. (2004). MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J. Biol. Chem. 279, 10176–10184.
Clement, S. L., Scheckel, C., Stoecklin, G., and Lykke-Andersen, J. (2011). Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol. Cell. Biol. 31, 256–266.
Dai, N., Rapley, J., Angel, M., Yanik, M. F., Blower, M. D., and Avruch, J. (2011). mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 25, 1159–1172.
Damgaard, C. K., and Lykke-Andersen, J. (2011). Translational coregulation of 5′TOP mRNAs by TIA-1 and TIAR. Genes Dev. 25, 2057–2068.
Dean, J. L., Wait, R., Mahtani, K. R., Sully, G., Clark, A. R., and Saklatvala, J. (2001). The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol. Cell. Biol. 21, 721–730.
Diaz-Moreno, I., Hollingworth, D., Frenkiel, T. A., Kelly, G., Martin, S., Howell, S., et al. (2009). Phosphorylation-mediated unfolding of a KH domain regulates KSRP localization via 14-3-3 binding. Nat. Struct. Mol. Biol. 16, 238–246.
Dinger, M. E., Gascoigne, D. K., and Mattick, J. S. (2011). The evolution of RNAs with multiple functions. Biochimie 93, 2013–2018.
Doller, A., Pfeilschifter, J., and Eberhardt, W. (2008). Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell. Signal. 20, 2165–2173.
Eberhardt, W., Doller, A., Akool El, S., and Pfeilschifter, J. (2007). Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 114, 56–73.
Elkon, R., Zlotorynski, E., Zeller, K. I., and Agami, R. (2010). Major role for mRNA stability in shaping the kinetics of gene induction. BMC Genomics 11:259. doi: 10.1186/1471-2164-11-259
Fabian, M. R., and Sonenberg, N. (2012). The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat. Struct. Mol. Biol. 19, 586–593.
Facchinetti, V., Ouyang, W., Wei, H., Soto, N., Lazorchak, A., Gould, C., et al. (2008). The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 27, 1932–1943.
Farooq, F., Balabanian, S., Liu, X., Holcik, M., and Mackenzie, A. (2009). p38 Mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum. Mol. Genet. 18, 4035–4045.
Filippova, N., Yang, X., King, P., and Nabors, L. B. (2012). Phosphoregulation of the RNA-binding protein Hu antigen R (HuR) by Cdk5 affects centrosome function. J. Biol. Chem. 287, 32277–32287.
Gherzi, R., Trabucchi, M., Ponassi, M., Ruggiero, T., Corte, G., Moroni, C., et al. (2006). The RNA-binding protein KSRP promotes decay of beta-catenin mRNA and is inactivated by PI3K-AKT signaling. PLoS Biol. 5:e5. doi: 10.1371/journal.pbio.0050005
Gnad, F., Gunawardena, J., and Mann, M. (2011). PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 39, D253–D260.
Gnad, F., Ren, S., Cox, J., Olsen, J. V., Macek, B., Oroshi, M., et al. (2007). PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, R250.
Goh, E. T., Pardo, O. E., Michael, N., Niewiarowski, A., Totty, N., Volkova, D., et al. (2010). Involvement of heterogeneous ribonucleoprotein F in the regulation of cell proliferation via the mammalian target of rapamycin/S6 kinase 2 pathway. J. Biol. Chem. 285, 17065–17076.
Graham, J. R., Hendershott, M. C., Terragni, J., and Cooper, G. M. (2010). mRNA degradation plays a significant role in the program of gene expression regulated by phosphatidylinositol 3-kinase signaling. Mol. Cell. Biol. 30, 5295–5305.
Gruber, A. R., Fallmann, J., Kratochvill, F., Kovarik, P., and Hofacker, I. L. (2011). AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 39, D66–D69.
Hao, S., and Baltimore, D. (2009). The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 10, 281–288.
Hitti, E., Iakovleva, T., Brook, M., Deppenmeier, S., Gruber, A. D., Radzioch, D., et al. (2006). Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol. Cell. Biol. 26, 2399–2407.
Holmes, B., Artinian, N., Anderson, L., Martin, J., Masri, J., Cloninger, C., et al. (2012). Protor-2 interacts with tristetraprolin to regulate mRNA stability during stress. Cell. Signal. 24, 309–315.
Hsu, P. P., Kang, S. A., Rameseder, J., Zhang, Y., Ottina, K. A., Lim, D., et al. (2011). The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322.
Huddleson, J. P., Ahmad, N., and Lingrel, J. B. (2006). Up-regulation of the KLF2 transcription factor by fluid shear stress requires nucleolin. J. Biol. Chem. 281, 15121–15128.
Huot, M. E., Vogel, G., Zabarauskas, A., Ngo, C. T., Coulombe-Huntington, J., Majewski, J., et al. (2012). The Sam68 STAR RNA-binding protein regulates mTOR alternative splicing during adipogenesis. Mol. Cell 46, 187–199.
Jacinto, E., Facchinetti, V., Liu, D., Soto, N., Wei, S., Jung, S. Y., et al. (2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137.
Keene, J. D. (2007). RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543.
Kim, H. H., Abdelmohsen, K., and Gorospe, M. (2010). Regulation of HuR by DNA damage response kinases. J. Nucleic Acids 2010, 981487.
Kim, H. H., Abdelmohsen, K., Lal, A., Pullmann, R., Jr., Yang, X., Galban, S., et al. (2008). Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev. 22, 1804–1815.
Kok, K., Geering, B., and Vanhaesebroeck, B. (2009). Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci. 34, 115–127.
Lafarga, V., Cuadrado, A., Lopez De Silanes, I., Bengoechea, R., Fernandez-Capetillo, O., and Nebreda, A. R. (2009). p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell. Biol. 29, 4341–4351.
Lam, L. T., Pickeral, O. K., Peng, A. C., Rosenwald, A., Hurt, E. M., Giltnane, J. M., et al. (2001). Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2, RESEARCH0041.
Leseux, L., Hamdi, S. M., Al Saati, T., Capilla, F., Recher, C., Laurent, G., et al. (2006). Syk-dependent mTOR activation in follicular lymphoma cells. Blood 108, 4156–4162.
Maitra, S., Chou, C. F., Luber, C. A., Lee, K. Y., Mann, M., and Chen, C. Y. (2008). The AU-rich element mRNA decay-promoting activity of BRF1 is regulated by mitogen-activated protein kinase-activated protein kinase 2. RNA 14, 950–959.
Marchese, F. P., Aubareda, A., Tudor, C., Saklatvala, J., Clark, A. R., and Dean, J. L. (2010). MAPKAP kinase 2 blocks tristetraprolin-directed mRNA decay by inhibiting CAF1 deadenylase recruitment. J. Biol. Chem. 285, 27590–27600.
Marderosian, M., Sharma, A., Funk, A. P., Vartanian, R., Masri, J., Jo, O. D., et al. (2006). Tristetraprolin regulates cyclin D1 and c-Myc mRNA stability in response to rapamycin in an Akt-dependent manner via p38 MAPK signaling. Oncogene 25, 6277–6290.
Mazumder, B., Seshadri, V., and Fox, P. L. (2003). Translational control by the 3′-UTR: the ends specify the means. Trends Biochem. Sci. 28, 91–98.
Ming, X. F., Stoecklin, G., Lu, M., Looser, R., and Moroni, C. (2001). Parallel and independent regulation of interleukin-3 mRNA turnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol. Cell. Biol. 21, 5778–5789.
Navarro, M. N., Goebel, J., Feijoo-Carnero, C., Morrice, N., and Cantrell, D. A. (2011). Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes. Nat. Immunol. 12, 352–361.
Nowotarski, S. L., and Shantz, L. M. (2010). Cytoplasmic accumulation of the RNA-binding protein HuR stabilizes the ornithine decarboxylase transcript in a murine nonmelanoma skin cancer model. J. Biol. Chem. 285, 31885–31894.
Origanti, S., Nowotarski, S. L., Carr, T. D., Sass-Kuhn, S., Xiao, L., Wang, J. Y., et al. (2012). Ornithine decarboxylase mRNA is stabilized in an mTORC1-dependent manner in Ras-transformed cells. Biochem. J. 442, 199–207.
Parker, R., and Sheth, U. (2007). P bodies and the control of mRNA translation and degradation. Mol. Cell 25, 635–646.
Pearce, L. R., Komander, D., and Alessi, D. R. (2010). The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22.
Pei, Y., Zhu, P., Dang, Y., Wu, J., Yang, X., Wan, B., et al. (2008). Nuclear export of NF90 to stabilize IL-2 mRNA is mediated by AKT-dependent phosphorylation at Ser647 in response to CD28 costimulation. J. Immunol. 180, 222–229.
Powell, J. D., Pollizzi, K. N., Heikamp, E. B., and Horton, M. R. (2012). Regulation of immune responses by mTOR. Annu. Rev. Immunol. 30, 39–68.
Raghavan, A., Dhalla, M., Bakheet, T., Ogilvie, R. L., Vlasova, I. A., Khabar, K. S., et al. (2004). Patterns of coordinate down-regulation of ARE-containing transcripts following immune cell activation. Genomics 84, 1002–1013.
Raghavan, A., Ogilvie, R. L., Reilly, C., Abelson, M. L., Raghavan, S., Vasdewani, J., et al. (2002). Genome-wide analysis of mRNA decay in resting and activated primary human T lymphocytes. Nucleic Acids Res. 30, 5529–5538.
Ramgolam, V. S., Degregorio, S. D., Rao, G. K., Collinge, M., Subaran, S. S., Markovic-Plese, S., et al. (2010). T cell LFA-1 engagement induces HuR-dependent cytokine mRNA stabilization through a Vav-1, Rac1/2, p38MAPK and MKK3 signaling cascade. PLoS ONE 5:e14450. doi: 10.1371/journal.pone.0014450
Rinn, J. L., and Chang, H. Y. (2012). Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166.
Salisbury, E., Sakai, K., Schoser, B., Huichalaf, C., Schneider-Gold, C., Nguyen, H., et al. (2008). Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp. Cell Res. 314, 2266–2278.
Sanchez-Margalet, V., and Najib, S. (2001). Sam68 is a docking protein linking GAP and PI3K in insulin receptor signaling. Mol. Cell. Endocrinol. 183, 113–121.
Sandler, H., Kreth, J., Timmers, H. T., and Stoecklin, G. (2011). Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res. 39, 4373–4386.
Sandler, H., and Stoecklin, G. (2008). Control of mRNA decay by phosphorylation of tristetraprolin. Biochem. Soc. Trans. 36, 491–496.
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., et al. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168.
Schmidlin, M., Lu, M., Leuenberger, S. A., Stoecklin, G., Mallaun, M., Gross, B., et al. (2004). The ARE-dependent mRNA-destabilizing activity of BRF1 is regulated by protein kinase B. EMBO J. 23, 4760–4769.
Schoenberg, D. R., and Maquat, L. E. (2012). Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 13, 246–259.
Shaw, G., and Kamen, R. (1986). A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46, 659–667.
Shaw, R. J., and Cantley, L. C. (2006). Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441, 424–430.
Shi, J. X., Su, X., Xu, J., Zhang, W. Y., and Shi, Y. (2012). MK2 posttranscriptionally regulates TNF-alpha-induced expression of ICAM-1 and IL-8 via tristetraprolin in human pulmonary microvascular endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L793–L799.
Stoecklin, G., Stubbs, T., Kedersha, N., Wax, S., Rigby, W. F., Blackwell, T. K., et al. (2004). MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 23, 1313–1324.
Subbaramaiah, K., Marmo, T. P., Dixon, D. A., and Dannenberg, A. J. (2003). Regulation of cyclooxgenase-2 mRNA stability by taxanes: evidence for involvement of p38, MAPKAPK-2, and HuR. J. Biol. Chem. 278, 37637–37647.
Taylor, S. J., Anafi, M., Pawson, T., and Shalloway, D. (1995). Functional interaction between c-Src and its mitotic target, Sam 68. J. Biol. Chem. 270, 10120–10124.
Tchen, C. R., Brook, M., Saklatvala, J., and Clark, A. R. (2004). The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J. Biol. Chem. 279, 32393–32400.
Tiedje, C., Ronkina, N., Tehrani, M., Dhamija, S., Laass, K., Holtmann, H., et al. (2012). The p38/MK2-driven exchange between Tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 8:e1002977. doi: 10.1371/journal.pgen.1002977
Trabucchi, M., Briata, P., Garcia-Mayoral, M., Haase, A. D., Filipowicz, W., Ramos, A., et al. (2009). The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459, 1010–1014.
Wang, Y., Liu, C. L., Storey, J. D., Tibshirani, R. J., Herschlag, D., and Brown, P. O. (2002). Precision and functional specificity in mRNA decay. Proc. Natl. Acad. Sci. U.S.A. 99, 5860–5865.
Wang, Z., Gucek, M., and Hart, G. W. (2008). Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. U.S.A. 105, 13793–13798.
Weill, L., Belloc, E., Bava, F. A., and Mendez, R. (2012). Translational control by changes in poly(A) tail length: recycling mRNAs. Nat. Struct. Mol. Biol. 19, 577–585.
Yang, E., Van Nimwegen, E., Zavolan, M., Rajewsky, N., Schroeder, M., Magnasco, M., et al. (2003). Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863–1872.
Yiakouvaki, A., Dimitriou, M., Karakasiliotis, I., Eftychi, C., Theocharis, S., and Kontoyiannis, D. L. (2012). Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J. Clin. Invest. 122, 48–61.
Keywords: RNA-binding proteins, PI3K, MAPK
Citation: Venigalla RKC and Turner M (2012) RNA-binding proteins as a point of convergence of the PI3K and p38 MAPK pathways. Front. Immun. 3:398. doi: 10.3389/fimmu.2012.00398
Received: 02 July 2012; Accepted: 10 December 2012;
Published online: 26 December 2012.
Edited by:
Klaus Okkenhaug, Babraham Institute, UKReviewed by:
Dinesh S. Rao, University of California, Los Angeles, USACopyright: © 2012 Venigalla and Turner. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Ram K. C. Venigalla and Martin Turner, Laboratory of Lymphocyte Signalling and Development, The Babraham Institute, Babraham Research Campus, Babraham CB22 3AT, UK. e-mail:cmFta3VtYXIudmVuaWdhbGxhQGJhYnJhaGFtLmFjLnVr;bWFydGluLnR1cm5lckBiYWJyYWhhbS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.