 Sara L. Lawrence1
Sara L. Lawrence1 Susanne C. Feil1 Jessica K. Holien1 Michael J. Kuiper2 Larissa Doughty1
Susanne C. Feil1 Jessica K. Holien1 Michael J. Kuiper2 Larissa Doughty1 Olan Dolezal3
Olan Dolezal3 Terrence D. Mulhern4 Rodney K. Tweten5
Terrence D. Mulhern4 Rodney K. Tweten5 Michael W. Parker1,4*
Michael W. Parker1,4*- 1Biota Structural Biology Laboratory and Australian Cancer Research Foundation Rational Drug Discovery Centre, St. Vincent’s Institute of Medical Research, Fitzroy, VIC, Australia
- 2Victorian Life Sciences Computation Initiative, The University of Melbourne, Parkville, VIC, Australia
- 3CSIRO Materials Science and Engineering, Parkville, VIC, Australia
- 4Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, VIC, Australia
- 5Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA
The cholesterol-dependent cytolysins (CDCs) attack cells by punching large holes in their membranes. Lectinolysin from Streptococcus mitis is unique among CDCs due to the presence of an N-terminal lectin domain that enhances the pore-forming activity of the toxin. We recently determined the crystal structures of the lectin domain in complex with various glycans. These structures revealed the molecular basis for the Lewis antigen specificity of the toxin. Based on this information we have used in silico molecular modeling to design a mutant toxin, which we predicted would increase its specificity for Lewis y, an antigen found on the surface of cancer cells. Surprisingly, we found by surface plasmon resonance binding experiments that the resultant mutant lectin domain exhibited higher specificity for Lewis b antigens instead. We then undertook comparative crystallographic and molecular dynamics simulation studies of the wild-type and mutant lectin domains to understand the molecular basis for the disparity between the theoretical and experimental results. The crystallographic results revealed that the net number of interactions between Lewis y and wild-type versus mutant was unchanged whereas there was a loss of a hydrogen bond between mutant and Lewis b compared to wild-type. In contrast, the molecular dynamics studies revealed that the Lewis b antigen spent more time in the binding pocket of the mutant compared to wild-type and the reverse was true for Lewis y. The results of these simulation studies are consistent with the conclusions drawn from the surface plasmon resonance studies. This work is part of a program to engineer lectinolysin so that it will target and kill specific cells in human diseases.
Introduction
Lectinolysin (LLY) is a pore-forming toxin derived from some strains of Streptococcus mitis and S. pseudopneumoniae and is a member of the cholesterol-dependent cytolysin (CDC) family (Farrand et al., 2008). These pore-forming bacterial toxins are secreted as soluble monomers that assemble on the surface of cholesterol-rich cell membranes. The resultant pores are composed in excess of 30 monomers, and are greater than 150 Å in diameter (Hotze and Tweten, 2012). Perforation of the membrane by these pores results in cell lysis. The sequence similarity is high (40–80%) between CDC family members.
Streptococcus mitis can cause diseases such as infective endocarditis and septicemia (Hall and Baddour, 2002; Huang et al., 2002; Gowda et al., 2003; Kennedy et al., 2004). Serum isolates from Kawasaki disease patients were found to contain S. mitis human platelet aggregation factor (PAF or Sm-hPAF), so named because it was found to affect light scattering properties of human platelets, thought to be as a consequence of platelet aggregation (Ohkuni et al., 1997). Farrand et al. (2008) found that the Sm-hPAF gene sequence encodes a predicted CDC structure (Rossjohn et al., 1997; Polekhina et al., 2005), most closely related to intermedilysin (Farrand et al., 2008). Having established that PAF was in fact a member of the CDC family, it was renamed lectinolysin, because of its ability to bind carbohydrate (see below).
While LLY shares a number of characteristics typical of CDCs, it has a unique 162 amino acid N-terminal domain (LLYlec; Farrand et al., 2008). Sequence comparisons show that the LLYlec domain shares significant identity with fucose-binding proteins. Glycan array experiments revealed that LLY was highly selective for the difucosylated glycans Lewis y (Ley) and Lewis b (Leb) antigens (Farrand et al., 2008). Small-angle X-ray scattering (SAXS) modeling predicts that LLYlec is joined to CDC domain 1 so that the Le antigen-binding site is exposed on the outer surface of the pore (Feil et al., 2012).
Lewis antigens are blood group determinants with very rigid structures (Yuriev et al., 2005). Leb is a type 1 antigen and adsorbed onto the surface of blood cells. Ley is a type 2 antigen which, in healthy individuals, is expressed at low levels in tissues including epithelial cells and serum (Yuriev et al., 2005; Westwood et al., 2009). However, expression is elevated on the surface of a wide range of epithelial tumor cells including colon, lung, and ovary (Sakamoto et al., 1986; Miyake et al., 1992; Yin et al., 1996). Thus, Ley is a distinctive tumor marker and therefore a promising target for directing drugs to cancer cells.
Various approaches have been taken to produce Ley-specific cancer therapeutics, including the development of Ley-specific humanized monoclonal antibodies (Kitamura et al., 1994; Clarke et al., 2000a,b; Scott et al., 2000), and anti Ley T cells (Westwood et al., 2005). A chimeric approach has also been taken whereby a Ley-specific monoclonal antibody was conjugated to the CDC, listeriolysin O, to form an immunotoxin (Bergelt et al., 2009). LLY is a naturally occurring, potential oncotoxin, with in-built tumor targeting specificity for the Ley antigen. However, cross reactivity with Leb antigen decreases its usefulness as a specific, cancer-targeting molecule.
Previously we have determined the crystal structure of LLYlec, and its complexes with fucose and Leb and Ley antigens (Feil et al., 2012). The overall fold of the LLYlec domain exhibited similarities with fucolectin domains of the CBM family 98 glycoside hydrolase of Streptococcus pneumonia and Anguilla anguilla agglutinin. The binding of Ley and Leb to LLYlec was very similar with the exception of an additional hydrogen bond between Leb and amino acid residue Tyr62 (Feil et al., 2012).
Using these structures, we have explored the possibility of mutating LLYlec to develop a Ley-specific binding domain. In the work presented here a Y62H mutation was designed and constructed with the aim of removing the hydrogen bond between the N-acetylglucosamine (GlcNAc) of Leb and LLYlec and instead, gain a hydrogen bond with Ley. However, binding studies described here show that LLYlecY62H has a greater affinity for Leb over Ley, whereas LLYlecwt has a preference for Ley over Leb. In order to explore the molecular basis for this unexpected finding we determined the crystal structure of LLYlecY62H in complex with Leb and Ley. We then compared these structures with those of the wild-type lectin domain complexed with the same Lewis antigens. The crystallographic studies were complemented by molecular dynamics simulations in order to explore the importance of protein motion and interaction with the ligands over time.
Materials and Methods
In Silico Design of Mutants
The crystal structures of the lectin domain of LLY in complex with Leb antigen (PDB code: 3LEK) and Ley antigen (PDB code: 3LEG) were visually inspected using Pymol1. In silico mutations were created and assessed also using the Pymol program. Figures were created in Pymol and using Chemaxon software2.
Surface Plasmon Resonance Biosensor Binding Analysis
All surface plasmon resonance (SPR) experiments were performed at 25°C using a Biacore T200 instrument (GE Healthcare). Direct assays were performed with Ley and Leb antigens (Sigma) injected over mutant and wild-type LLYlec domain proteins immobilized on a CM5 chip (GE Healthcare). Immobilizations were performed in 1 × HBS-P running buffer [10 mM HEPES, 150 mM NaCl, 0.005% (w/v) Tween 20]. Binding experiments were performed in 1 × HBS-P + running buffer [10 mM HEPES, 150 mM NaCl, 0.05% (w/v) Tween 20] containing 1 mM MgCl2, 1 mM CaCl2, and 0.2 mg/ml bovine serum albumin.
LLYlecwt and LLYlecY62H proteins were immobilized in two separate channels on a CM5 chip using a standard amine coupling protocol. Briefly, the chip surface was activated with a single 5 min injection of freshly prepared 1:1 50 mM N-hydroxysuccinimide:200 mM 3-(N,N-dimethylamino)propyl-N-ethylcarbodiimide. Protein coupling was achieved by three 5 min injections of LLYlecwt or LLYlecY62H solution (50 μg/ml in 10 mM sodium acetate, pH 5.0). To deactivate residual reactive sites, lectin domain coupling was followed by a 5 min injection of 1 M ethanolamine (pH 8.5). Approximately 3000 response units (RU; 1 RU = 1 pg of protein/mm2) of LLYlecwt (channel 2) and 4000 RU of LLYlecY62H (channel 3) were coupled. Channel 1 was activated and blocked, as above, for use as a reference surface.
Leb and Ley were injected, in duplicate, in a twofold dilution series from 1 mM to 7.5 μM over the immobilized wild-type and mutant lectin domains. Thirty-second injections of Le antigen at 40 μl/min were followed by a 60-s dissociation period. Baseline returned to 0 almost immediately after the association phase ended, so regeneration of the surface was not required.
Binding data were processed and analyzed using Scrubber 2 software3 (version 2.0c). Rapid association and dissociation rates made data fitting to a kinetic model and subsequent calculation of kinetic rate constants ka and kd impractical. Consequently, equilibrium dissociation constants (KD) were derived by fitting binding responses at equilibrium to a 1:1 steady-state affinity model available within Scrubber.
Mutagenesis, Expression, and Purification of the LLYlec Domains
A mutant of LLYlec, LLYlecQ190C, with an N-terminal 6 × His tag and a TEV protease cleavage site for His-tag removal, was used in this study. The LLYlecQ190C coding region corresponds to LLY residues 38–190 (GenBank accession number AB051299.1). The C-terminal residue was mutated to cysteine (Q190C) for protein labeling studies. The point mutant displays wild-type activity and is located far from the Lewis antigen-binding site (Farrand et al., 2008). Henceforth, LLYlecQ190C will be described as LLYlecwt in this work. Expression was carried out in E. coli BL21, and the protein purified using nickel resin and size exclusion chromatography as previously described (Feil et al., 2012). LLYlecwt was mutated with QuikChangeTM Site-Directed Mutagenesis Kit (Stratagene) to create the Lewis antigen-binding site mutant LLYlecY62H. LLYlecwt and LLYlecY62H were purified to >95% purity, as determined by SDS gel electrophoresis. Proteins were concentrated to 10 mg/ml in 10 mM Tris–HCl pH 7.2, 10 mM NaCl, and 5 mM dithiothreitol, and were stored at -80°C.
Crystallization of the LLYlecY62H Mutant
All crystallizations were performed using the hanging drop vapor diffusion method at 21°C. 2 μl of protein was mixed with equal volume of precipitant and hung over 0.5 ml of well solution. LLYlecY62H crystallization conditions were determined by fine screening around the conditions that proved successful for the wild-type protein (Feil et al., 2012): 2 M MgSO4 and 100 mM Tris–HCl buffer pH 8.2–9.0. Optimization included the addition of the Hampton additive screen (Hampton Research, CA, USA) 10% (v/v) in the hanging drops. The optimal crystallization conditions were 2.4 M MgSO4 and 100 mM KCl with 100 mM Tris–HCl pH 8.75–9.0. Ley and Leb antigens were soaked into LLYlecY62H crystals by adding 1 μl 1 M MgSO4, 100 mM Tris–HCl pH 9.0, 100 mM KCl, 5 mM Ley or Leb to the crystal drop for 1 h at 21°C. Soaks were carried out immediately prior to cryoprotection and flash freezing in liquid nitrogen. Crystals were cryoprotected for X-ray data collection by adding glycerol in increments of 5% (v/v) to a final concentration of 20% (v/v) to the crystal drops.
Crystallographic Studies of the LLYlecY62H Mutant
Diffraction data were collected at the MX2 beamline at the Australian Synchrotron in Clayton, Victoria. The data collection was controlled using Blue-Ice software (McPhillips et al., 2002). The diffraction data were processed using the HKL2000 suite (Otwinowski and Minor, 1997). Model building was performed with COOT (Emsley and Cowtan, 2004) using the published wild-type crystal structure of LLYlec (PDB code: 1LE0) as a starting model. Data were refined with REFMAC 5 (Murshudov et al., 1997) from the CCP4 program suite (CCP4, 1994). Restrained positional and isotropic temperature factor refinement was employed until convergence and the refinement was monitored using the Rfree residual. The Lewis antigens were fully occupied in the binding sites of both structures and their temperature factors were very similar to those of the surrounding side-chains.
Molecular Modeling of the LLYlec Interaction with the Leb and Ley Antigens
To investigate the mechanism of association and disassociation between LLYlec and Lewis antigens four molecular models were used: two being the published crystal structures of LLYlecwt bound to either Leb or Ley antigen (Feil et al., 2012) and the other two being the crystal structures of LLYlecY62H, complexed to the same ligands, that are described here. The molecular dynamics program NAMD (Phillips et al., 2005) was used to model ligand disassociation of each complex with a 30-ns simulation from the initial starting bound conformation, repeated 100 times for a cumulative simulation of 3 μs for each system. Models were initially solvated with TIP3 water under periodic boundary conditions of dimensions 48 Å × 48 Å × 64 Å. Charges were neutralized with NaCl for a total ionic concentration of 150 mM. The bound calcium ion seen in the crystal structures was included in the simulations. Dynamic molecular modeling was conducted at a theoretical pH of 7.4. Each simulation run started with an equilibration phase of 0.5 ns, where the protein backbone and Lewis antigen was harmonically constrained to their starting positions at 310 K and an NPT ensemble (an isothermal and isobaric ensemble where the number of moles (N), pressure (P), and temperature (T) are conserved). Subsequent 30-ns production simulations were run with no constraints at 310 K under NVT ensembles [a canonical ensemble where the number of moles (N), volume (V), and temperature (T) are conserved]. Trajectory snapshots were captured every 100 ps. At the completion of 100 repeat simulations, all production runs per model were consolidated into a single trajectory and the frames root-mean-square (rms) deviation were centered, based on the LLY alpha carbon protein backbone. The Lewis antigen was then subject to clustering analysis, whereby the top 20 ligand conformational clusters with a cut-off of 1.5 Å were generated and averaged. Ligand clusters with a rms deviation value less than 3.5 Å from the initial position were considered to be in a bound conformation.
Results
In Silico Design of Mutants
The difference between Ley and Leb antigens is the core disaccharide linkage (Galβ1-4 versus Galβ1-3) with an opposing projection of the N-acetyl glucosamine (GlcNAc) N-acetyl and –CH2OH groups of the N-acetylglucosamine monosaccharide as shown in Figure 1. The overall Lewis determinants maintain the same conformation in both free and bound states.
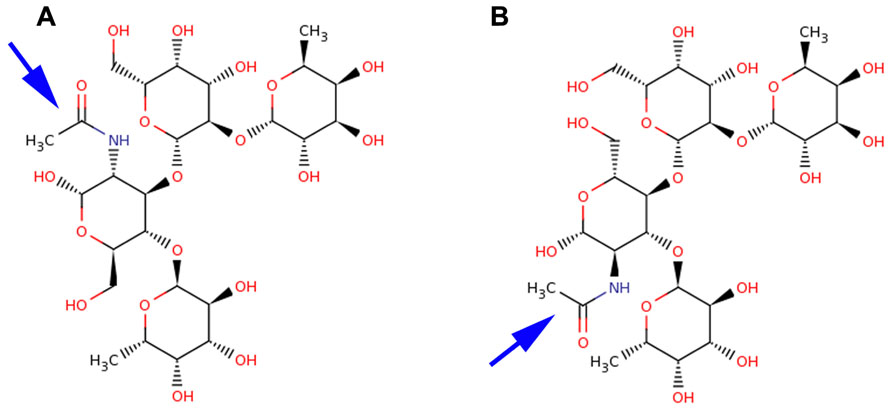
FIGURE 1. Structural comparison of Leb and Ley antigens. Lewis antigens Leb (A) and Ley (B) are structurally very similar. They differ in the core disaccharide linkages and the opposing projections of their N-acetyl (blue arrow) and CH2OH groups of the GlcNAc moiety.
In the crystal structure of LLYlecwt the GlcNAc of Leb is within hydrogen bonding distance (3.3 Å) of Tyr62 (Figure 2A; Feil et al., 2012). This interaction is not able to occur with Ley, thus suggesting that mutating Tyr62 may alter the affinity for Leb. Various mutations of this residue were created in silico. Mutation to a histidine residue would maintain structural similarity of the binding site whilst also creating the potential for a new interaction between the histidine side-chain and Ley as illustrated in Figure 2B. Replacement of Tyr62 with His was expected to remove the interaction between Leb and the hydroxyl group of Tyr62 (Figure 2A).
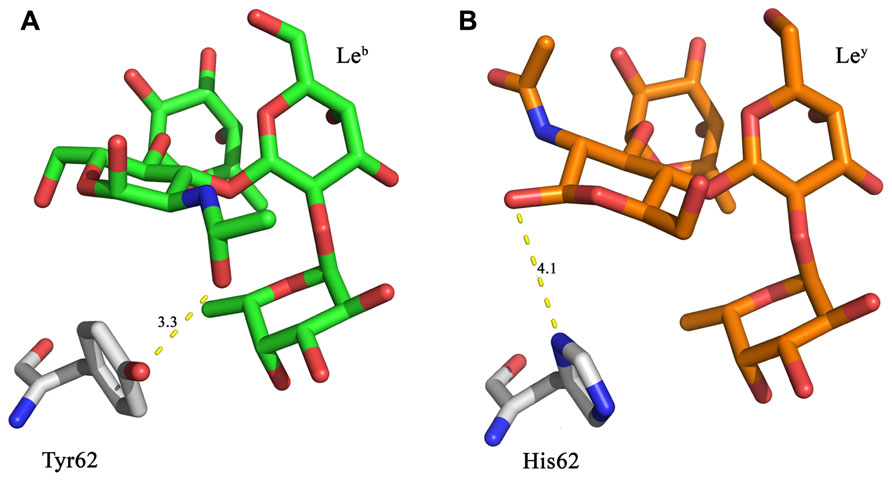
FIGURE 2. In silico LLYlecY62H mutant design. (A) Leb is within hydrogen bonding distance (3.3 Å) of Tyr62 from the LLYlecwt domain. (B) A Y62H mutation was designed to retain similar structural properties to tyrosine, with the possibility that it may also result in a hydrogen bond to the Ley antigen. Yellow dashed lines denote hydrogen bonds.
SPR Biosensor Binding Analysis
Purified LLYlecY62H yield was 20 mg/l E. coli BL21 culture in Luria–Bertani (LB) broth. This yield was equivalent to that found for LLYlecwt. The sensorgrams from SPR experiments between the Lewis antigens (Leb and Ley) and either wt or LLYlecY62H are shown in Figure 3. The estimated KD values are listed in Table 1. The data indicate low affinity binding interactions of the Lewis antigens with the lectin domains. The wild-type lectin domain (LLYlecwt) has more than threefold greater affinity for Ley over Leb. The LLYlecY62H mutant has a 1.6-fold greater preference for Leb over Ley. The mutation causes an overall 2.5-fold decrease in the lectin domain’s affinity for Ley and doubles its affinity for Leb over the wild-type domain. Overall, the data show that wild-type LLYlec preferentially binds Ley antigen while the mutant LLYlecY62H exhibits higher affinity for Leb than for Ley.
FIGURE 3. SPR measurements of immobilized LLYlecwt and LLYlecY62H. Ley antigen and Leb antigen were injected at concentrations of 7.6–1000 μM. (A) LLYlecwt vs Ley, (B) LLYlecY62H vs Ley, (C) LLYlecwt vs Leb, (D) LLYlecY62H vs Leb. Responses at equilibrium fitted to simple 1:1 binding isotherms for Ley (black lines) and Leb (blue lines) interacting with (E) LLYlecwt and (F) LLYlecY62H mutant. Duplicate binding data sets are shown.
TABLE 1. Estimated equilibrium dissociation constants (KD) for LLYlecY62H and LLYlecwt domains interacting with Ley and Leb antigens.
Crystal Structures of LLYlecY62H in Complex with Ley and Leb Antigens
LLYlecY62H crystallization was optimized in 100 mM Tris pH 8.75–9.0, 2.4 M MgSO4, and 100 mM KCl. Small bipyramidal crystals of dimensions 0.15 mm × 0.12 mm × 0.12 mm appeared in 6–8 weeks. After 8 months some crystals had grown to 0.25 mm × 0.2 mm × 0.2 mm. In contrast, LLYlec wt crystals appeared after 5 days in 100 mM Tris pH 8.2–8.4, 2.0 M MgSO4 with the same morphology as LLYlecY62H, but reach the same size within 10 days (Feil et al., 2012). Lewis antigens were soaked into crystals immediately prior to cryoprotection and freezing.
The crystal structures of the LLYlecY62H in complex with the Lewis antigens was performed as described in Materials and Methods. The crystal structures have been deposited in the PDB with codes: 4GWJ and 4GWI for the Lewis b and Lewis y complexes, respectively. Data statistics for the structures are listed in Table 2.
TABLE 2. Crystallographic data processing and refinement statistics for LLYlecwt and LLYlecY62H crystal structures in complex with Ley and Leb antigens.
LLYlec adopts an eight-stranded β-sandwich fold, composed of a five-stranded anti-parallel β-sheet on one side and a three-stranded anti-parallel β-sheet on the other side. Three short α-helices separate β-strands 1 and 2 and 3 and 4. There is one calcium ion in the structure that is in the same position as observed in structurally related fucolectin domains. There is also a metal ion that is covalently bound to His80, as well as five water molecules, all arranged in octahedral geometry. In our previous work we identified the ion as either a Ni2+ or Mg2+ ion (Feil et al., 2012) and here we have chosen the latter possibility. The Ley and Leb antigen-binding site is in a cleft at one end of the molecule. A more detailed description of the structure has been reported elsewhere (Feil et al., 2012). In the new mutant structures some additional N-terminal residues are observed (three extra in the Ley structure and one extra in the Leb structure), compared to the published wild-type structures (see Table 2).
The structures of the LLYlecwt-Ley (Figure 4A) and LLYlecY62H-Ley (Figure 4B) superimpose very closely, with a rms deviation of the alpha carbon atoms of 0.1 Å. A third metal ion binding site is observed in the mutant structure and we have tentatively identified it as a Mg2+ ion binding site due to the very high concentrations of this ion in the crystallization buffer. The Mg2+ ion is bound between the carboxylate of Asp97 and the side-chain carbonyl of Gln54 and four water molecules, all arranged in octahedral geometry around the metal ion. The water structure is the same as in LLYlecwt-Ley apart from a water molecule that is positioned between the hydroxyl group of Tyr62 and fucose 1 (Fuc 1; Figure 4A). This water molecule is shifted by 1.4 Å toward the NE2 of His62 in the mutant structure (Figure 4B), compared to wild-type (Figure 4A), to optimize its interaction with the His side-chain. LLYlecY62H forms the same 19 van der Waals interactions with Ley compared to the wild-type protein: 15 with Fuc 1, 1 with Gal, 2 with GlcNAc, and 1 with Fuc 2. The B-factor of Ley is 53.5 Å2 in the mutant (overall B-factor of protein is 29 Å2), compared to 38.2 Å2 in the wild-type protein (overall B-factor of protein complex is 21 Å2). Overall, there is no net change in the number of potential hydrogen binding and van der Waals interactions between wild-type and mutant Ley complexes.
FIGURE 4. LLYlecwt and LLYlecY62H domains in complex with Lewis antigens. Comparison between (A) the LLYlecwt-Ley complex (orange carbons, green Tyr62) and (B) the LLYlecY62H-Ley complex (purple carbons, green His62) shows that the water molecule shifts toward the histidine residue in LLYlecY62H-Ley. The water-mediated hydrogen bond to the hydroxyl group of Tyr62 seen in the LLYlecwt-Leb complex (cyan carbons, green Tyr62) (C) was lost in the LLYlecwt-Leb structure (magenta carbons, green Tyr62) (D) as this residue was replaced by His62. Hydrogen bonds are shown in gray and water molecules as black spheres. The fit of the ligands to the relevant 2Fo–Fc electron density map (dark green hash), contoured at 1σ, are shown.
Superposition of the structures of the LLYlecwt-Leb and LLYlecY62H-Leb gives a rms deviation of the alpha carbon atoms of 0.1 Å. In addition to the Ca2+ and Mg2+ ion binding sites seen in the wild-type structure there is the additional Mg2+ ion binding site as observed in the LLYlecY62H-Ley crystal structure. In the structure of LLYlecwt-Leb there is one hydrogen bond from the hydroxyl group of Tyr62 to the N-acetyl moiety of Leb and a second one via a water molecule to the hydroxyl group of Fuc 1 (Figure 4C). In the LLYlecY62H-Leb structure (Figure 4D) the bond from Tyr62 to the N-acetyl is replaced by a bifurcated hydrogen bond from His62 to the hydroxyl group of the GlcNAc ring and the hydroxyl of the N-acetyl moiety off the ring. Overall, there was a net loss of a potential hydrogen bond as the water-mediated hydrogen bond between Tyr62 and Fuc 1 seen in the LLYlecwt-Leb structure has been lost in LLYlecH62-Leb. The water molecules between the protein and Leb are conserved in wild-type and mutant structures. LLYlecY62H forms the same 23 van der Waals interactions with Leb as does the wild-type protein: 15 with Fuc 1, 1 with Gal, 2 with GlcNAc, and 4 with Fuc 2. The average B-factor of Leb is 43.8 Å2 in LLYlecY62H-Leb (overall B-factor of protein complex is 23 Å2) which is very similar to 36.7 Å2 observed in the wild-type complex (overall B-factor of protein complex is 18 Å2). Thus, overall, there is a loss of one potential hydrogen bond between Leb and mutant compared to wild-type.
Superposition of the crystal structures of LLYlecY62H-Leb and LLYlecY62H-Ley results in a rms deviation on the alpha carbons atoms of 0.1 Å. Fuc 1 and the Gal moieties of the Ley and Leb antigens superimpose quite well whereas GlcNAc and Fuc 2 are shifted. GlcNAc in Leb moves 0.6 Å toward the protein due to a water-mediated hydrogen bond between the oxygen of the GlcNAc-CH2OH group to the backbone oxygen of His62. Likewise, Fuc 2 is shifted by about 0.6 Å because the same interaction is not present in the Ley complex.
Molecular Simulations of Lewis Antigen Interactions
A total of 30,000 trajectory frames (100 × 30 ns) were collected for each Lewis antigen in complex with either LLYlecwt or the LLYlecY62H. In the wild-type simulations, the Ley antigen resided within the binding pocket more than the Leb antigen (Table 3). However, when Y62 was mutated to a His this trend was reversed, with Leb residing in the binding pocket more than Ley. These results are reflective of the SPR binding data, in that LLYlecwt has a higher affinity for Ley over Leb, whereas LLYlecY62H has a higher affinity for Leb over for Ley.

TABLE 3. Cumulative totals of bound Lewis antigen over the course of 100 independent, 30 ns simulations starting from the Lewis bound conformation.
Lewis antigen dissociation rates were plotted as a function of occupancy (defined as <3.5 Å deviation from the original binding site) for all 100 simulations (Figure 5). This indicated a rapid dissociation rate which supports the SPR data where the on and off rates were too fast to be calculated. There is an overall trend for the antigens to occupy the binding site less as the simulations progressed. However, it is important to note that the curves are not purely exponential and contain distinct areas of peaks and troughs. This is indicative of the ligands moving rapidly in and out of the binding site of both LLYlecwt and LLYlecY62H.
FIGURE 5. Molecular dynamics modeling of the association of Lewis antigens with the LLYlec binding sites. Traces are averaged for all 100 simulations. Overall, the Lewis antigens occupied the binding site of either the wild-type or LLYlecY62H less as the simulation progressed. However there are numerous areas of re-association, resulting in a spike in occupancy.
The simulation data reiterates the SPR finding that mutation of LLYlecTyr62 to His62 has changed the binding specificity from a preference for Ley by wild-type to a specificity for Leb by the LLYlecY62H mutant.
Discussion
The high level of expression of Ley on the surface of epithelial tumor cells and low level expression elsewhere makes cell-bound Ley a highly attractive target for anti-tumor agent delivery. In addition, the rigid nature and distinctive structure of Ley lends itself to the ready development of highly specific protein recognition domains, whether in antibodies or other glycan-binding proteins.
Previously, we published the crystal structures of LLYlecwt in complex with the Leb and Ley antigens (Feil et al., 2012). These structures, together with computational modeling, were used to design and construct a glycan-binding domain with altered specificity for the Lewis antigens. The LLYlecY62H mutation was suggested due to its structural similarity of His to Tyr and the potential to create a new interaction with Ley.
Surface plasmon resonance binding studies showed that LLYlecwt has a higher affinity for the Ley antigen over Leb. This relationship was reversed for the designed LLYlecY62H mutant, i.e., the Leb antigen had higher affinity than the Ley antigen. These results were unexpected, as the molecular modeling predicted that mutation of Tyr62 to His should decrease the number of hydrogen bonds with Leb and increase the number of potential hydrogen bonds with Ley. The expectation was that the mutation would lead to an increase in affinity for Ley and decrease in affinity for Leb, but in practice we found the reverse to be true.
To experimentally determine the interactions formed in the binding site as a result of the LLYlecY62H mutation, we solved the crystal structures of LLYlecY62H in complex with the Lewis antigens. In the LLYlecY62H-Ley complex, there was no additional bond formation between the Lewis antigen and His62, only a shift of a water molecule toward the His, as compared to its position relative to Tyr62 in the wild-type (Figures 4A,B). In the Leb complex there was a net loss of a potential hydrogen bond to His62 (Figures 4C,D). In summary, the crystal structures did not show the bonding as predicted from the in silico mutant model (Figure 2) but revealed a net decrease in hydrogen bonding interactions to Leb and no net change in bonding interactions with Ley. However, the SPR data revealed that the mutant had an increased affinity for Leb and a decreased affinity for Ley.
Crystal structures are a static average snapshot of what happens in solution, so we used molecular dynamics studies to simulate the lectin domain-Lewis antigen binding mechanism in solution. In these simulations it was observed that the amino acid residues around the Lewis antigen binding site are mobile, particularly Tyr62. Thus, the inferred hydrogen bonding interactions between protein and ligands observed in the crystal structures may not necessarily be persistent in solution. This may help explain the unexpected consequences of our mutation.
The molecular dynamics studies provided us with complementary results to the SPR data: both approaches showed that the association and dissociation rates with Lewis antigens are extremely rapid, and that the relative affinities between antigens and wild-type or LLYlecY62H domains are generally weak. The SPR data showed the strongest interaction being that of LLYlecwt to Ley with a KD of 78 μM and the weakest between LLYlecwt and Leb with a KD of 234.4 μM. Relative affinities of the wild-type or LLYlecY62H binding sites for antigens was measured in the simulations by calculating how much time the Lewis antigens spent in the binding pocket. These dynamics studies agreed with the general trend of the biological results, suggesting that there was a reversal of Lewis antigen affinity when Tyr62 was mutated to a His. Interestingly, and again in concurrence with SPR data, the dynamics studies also suggest that the Lewis antigens bind weakly and transiently to the LLYlec domain.
A recent example of protein engineering to alter Lewis antigen specificity is that of Norovirus virion protein 1 (NoV VP1), which specifically binds Leb (Kubota et al., 2012). However, NoV VP1 has a deeper binding site with more extensive contacts to the Lewis antigen structure than seen in LLYlec. A single residue (Gln) in the Leb binding site was mutated (to Asn) with the aim of increasing the affinity of the protein for Leb. The mutation increased the width of the binding site, allowing bonding between both fucose rings and amino acid side-chains. As expected, the mutant protein displayed a higher affinity for Leb.
In contrast, designing a high affinity Ley-specific LLYlec mutant is more challenging: the crystal structures show that the Lewis antigens sit in a shallow hydrophobic pocket and that the only hydrogen bonds are between the basic residues His85, Arg112, and Arg120, and the α1-2 linked fucose. Few contacts are made between LLYlec residues and other carbohydrate components of the Ley and Leb antigens (Feil et al., 2012). In addition, the structural differences between the Ley and Leb antigens are minor. These characteristics outline the challenges involved in redesigning LLYlec in order to increase its specificity for Ley. Nevertheless, the results described herein show that, despite the strong structural similarities between Lewis antigens Ley and Leb and the shallow binding pocket, it is possible to alter the substrate specificity of the LLY lectin domain.
It has previously been demonstrated that LLYlecwt domain binds fucose alone (Farrand et al., 2008; Feil et al., 2012) and that the presence of LLYlec enhances the pore-forming activity of LLY on platelets. Fucose is a commonly expressed carbohydrate, terminally decorating cell surface glycoproteins, including CD59 (Rudd et al., 1997; Wheeler et al., 2002), which is also the receptor for LLY (Wickham et al., 2011). This suggests the function of the lectin domain is perhaps as a “capture mechanism,” slowing down the passing monomers and increasing the local concentration of LLY in the microenvironment of the cell surface. A localized increase in LLY concentration would facilitate the oligomerization of LLY into prepores, prior to pore formation and consequent cell lysis. In this scenario, weak binding affinities for the fucose moieties, as demonstrated by SPR, would be beneficial as the monomers would need to be readily released for incorporation into assembling prepore oligomers. Further studies are required to confirm this mechanism.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was partly undertaken on the MX2 beamline at the Australian Synchrotron, Victoria, Australia. We thank Dr. Tom Caradoc-Davies and the other beamline staff for their assistance. We also thank Dr. Jerome Wielens for assistance with X-ray crystallographic data collection. This work was partly carried out in the Australian Cancer Research Foundation Rational Drug Discovery Centre and funding was received from the Victorian Government Operational Infrastructure Support Scheme to St. Vincent’s Institute. This research was supported by a Victorian Life Sciences Computation Initiative (VLSCI) grant number VR0021 on its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government, Australia. Rodney K. Tweten received support from Grant AI037657 from the National Institutes of Health. Susanne C. Feil was supported by a NHMRC Industry Fellowship. Michael W. Parker is an NHMRC Research Fellow.
Footnotes
References
Bergelt, S., Frost, S., and Lilie, H. (2009). Listeriolysin O as cytotoxic component of an immunotoxin. Protein Sci. 18, 1210–1220.
CCP4. (1994). Collaborative Computational Project. The CCP4 Suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763.
Clarke, K., Lee, F. T., Brechbiel, M. W., Smyth, F. E., Old, L. J., and Scott, A. M. (2000a). In vivo biodistribution of a humanized anti-Lewis Y monoclonal antibody (hu3S193) in MCF-7 xenografted BALB/c nude mice. Cancer Res. 60, 4804–4811.
Clarke, K., Lee, F. T., Brechbiel, M. W., Smyth, F. E., Old, L. J., and Scott, A. M. (2000b). Therapeutic efficacy of anti-Lewis(y) humanized 3S193 radioimmunotherapy in a breast cancer model: enhanced activity when combined with taxol chemotherapy. Clin. Cancer Res. 6, 3621–3628.
Emsley, P., and Cowtan, K. (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132.
Farrand, S., Hotze, E., Friese, P., Hollingshead, S. K., Smith, D. F., Cummings, R. D., et al. (2008). Characterization of a streptococcal cholesterol-dependent cytolysin with a lewis y and b specific lectin domain. Biochemistry 47, 7097–7107.
Feil, S. C., Lawrence, S., Mulhern, T. D., Holien, J. K., Hotze, E. M., Farrand, S., et al. (2012). Structure of the lectin regulatory domain of the cholesterol-dependent cytolysin lectinolysin reveals the basis for its lewis antigen specificity. Structure 20, 248–258.
Gowda, R. M., Ansari, A. W., and Khan, I. A. (2003). Complete endocardial cushion defect (complete atrioventricular canal) manifested in adult life by Streptococcus mitis endocarditis of the common atrioventricular valve. Int. J. Cardiol. 89, 109–110.
Hall, G. E., and Baddour, L. M. (2002). Apparent failure of endocarditis prophylaxis caused by penicillin-resistant Streptococcus mitis. Am. J. Med. Sci. 324, 51–53.
Hotze, E. M., and Tweten, R. K. (2012). Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta 1818, 1028–1038.
Huang, I. F., Chiou, C. C., Liu, Y. C., and Hsieh, K. S. (2002). Endocarditis caused by penicillin-resistant Streptococcus mitis in a 12-year-old boy. J. Microbiol. Immunol. Infect. 35, 129–132.
Kennedy, M. J., Jackson, M. A., and Kearns, G. L. (2004). Delayed diagnosis of penicillin-resistant Streptococcus mitis endocarditis following single-dose amoxicillin prophylaxis in a child. Clin. Pediatr. (Phila.) 43, 773–776.
Kitamura, K., Stockert, E., Garin-Chesa, P., Welt, S., Lloyd, K. O., Armour, K. L., et al. (1994). Specificity analysis of blood group Lewis-y (Le(y)) antibodies generatedagainst synthetic and natural Le(y) determinants. Proc. Natl. Acad. Sci. U.S.A. 91, 12957–12961.
Kubota, T., Kumagai, A., Ito, H., Furukawa, S., Someya, Y., Takeda, N., et al. (2012). Structural basis for the recognition of Lewis antigens by genogroup I norovirus. J Virol. 86, 11138–11150.
McPhillips, T. M., McPhillips, S. E., Chiu, H. J., Cohen, A. E., Deacon, A. M., Ellis, P. J., et al. (2002). Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron. Radiat. 9, 401–406.
Miyake, M., Taki, T., Hitomi, S., and Hakomori, S. (1992). Correlation of expression of H/Le(y)/Le(b) antigens with survival in patients with carcinoma of the lung. N. Engl. J. Med. 327, 14–18.
Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255.
Ohkuni, H., Todome, Y., Okibayashi, F., Watanabe, Y., Ohtani, N., Ishikawa, T., et al. (1997). Purification and partial characterization of a novel human platelet aggregation factor in the extracellular products of Streptococcus mitis, strain Nm-65. FEMS Immunol. Med. Microbiol. 17, 121–129.
Otwinowski, Z., and Minor, W. (1997). Processing of X-ray diffraction data collected in the oscillation mode. Methods Enzymol. 276, 307–326.
Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., et al. (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802.
Polekhina, G., Giddings, K. S., Tweten, R. K., and Parker, M. W. (2005). Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc. Natl. Acad. Sci. U.S.A. 102, 600–605.
Rossjohn, J., Feil, S. C., McKinstry, W. J., Tweten, R. K., and Parker, M. W. (1997). Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell 89, 685–692.
Rudd, P. M., Morgan, B. P., Wormald, M. R., Harvey, D. J., Van Den Berg, C. W., Davis, S. J., et al. (1997). The glycosylation of the complement regulatory protein, human erythrocyte CD59. J. Biol. Chem. 272, 7229–7244.
Sakamoto, J., Furukawa, K., Cordon-Cardo, C., Yin, B. W., Rettig, W. J., Oettgen, H. F., et al. (1986). Expression of Lewisa, Lewisb, X, and Y blood group antigens in human colonic tumors and normal tissue and in human tumor-derived cell lines. Cancer Res. 46, 1553–1561.
Scott, A. M., Geleick, D., Rubira, M., Clarke, K., Nice, E. C., Smyth, F. E., et al. (2000). Construction, production, and characterization of humanized anti-Lewis Y monoclonal antibody 3S193 for targeted immunotherapy of solid tumors. Cancer Res. 60, 3254–3261.
Westwood, J. A., Murray, W. K., Trivett, M., Haynes, N. M., Solomon, B., Mileshkin, L., et al. (2009). The Lewis-Y carbohydrate antigen is expressed by many human tumors and can serve as a target for genetically redirected T cells despite the presence of soluble antigen in serum. J. Immunother. 32, 292–301.
Westwood, J. A., Smyth, M. J., Teng, M. W., Moeller, M., Trapani, J. A., Scott, A. M., et al. (2005). Adoptive transfer of T cells modified with a humanized chimeric receptor gene inhibits growth of Lewis-Y-expressing tumors in mice. Proc. Natl. Acad. Sci. U.S.A. 102, 19051–19056.
Wheeler, S. F., Rudd, P. M., Davis, S. J., Dwek, R. A., and Harvey, D. J. (2002). Comparison of the N-linked glycans from soluble and GPI-anchored CD59 expressed in CHO cells. Glycobiology 12, 261–271.
Wickham, S. E., Hotze, E. M., Farrand, A. J., Polekhina, G., Nero, T. L., Tomlinson, S., et al. (2011). Mapping the intermedilysin-human CD59 receptor interface reveals a deep correspondence with the binding site on CD59 for complement binding proteins C8α and C9. J. Biol. Chem. 286, 20952–20962.
Yin, B. W., Finstad, C. L., Kitamura, K., Federici, M. G., Welshinger, M., Kudryashov, V., et al. (1996). Serological and immunochemical analysis of Lewis y (Ley) blood group antigen expression in epithelial ovarian cancer. Int. J. Cancer 65, 406–412.
Keywords: cholesterol-dependent cytolysins, Lewis antigens, molecular dynamics simulations, protein engineering, surface plasmon resonance, X-ray crystallography
Citation: Lawrence SL, Feil SC, Holien JK, Kuiper MJ, Doughty L, Dolezal O, Mulhern TD, Tweten RK and Parker MW (2012) Manipulating the Lewis antigen specificity of the cholesterol-dependent cytolysin lectinolysin. Front. Immun. 3:330. doi: 10.3389/fimmu.2012.00330
Received: 05 September 2012; Paper pending published: 15 September 2012;
Accepted: 17 October 2012; Published online: 05 November 2012.
Edited by:
Paul A. Ramsland, Burnet Institute, AustraliaReviewed by:
Brian V. Geisbrecht, University of Missouri-Kansas City, USAElizabeth Yuriev, Monash University, Australia
Copyright: © 2012 Lawrence, Feil, Holien, Kuiper, Doughty, Dolezal, Mulhern, Tweten and Parker. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Michael W. Parker, Biota Structural Biology Laboratory and ACRF Rational Drug Discovery Centre, St. Vincent’s Institute of Medical Research, 9 Princes Street, Fitzroy, VIC 3065, Australia. e-mail:bXBhcmtlckBzdmkuZWR1LmF1