



95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 April 2012
Sec. Vaccines and Molecular Therapeutics
Volume 3 - 2012 | https://doi.org/10.3389/fimmu.2012.00079
Toll-like receptors (TLRs) are recognition molecules for multiple pathogens, including bacteria, viruses, fungi, and parasites. TLR2 forms heterodimers with TLR1 and TLR6, which is the initial step in a cascade of events leading to significant innate immune responses, development of adaptive immunity to pathogens and protection from immune sequelae related to infection with these pathogens. This review will discuss the current status of TLR2 mediated immune responses by recognition of pathogen-associated molecular patterns (PAMPS) on these organisms. We will emphasize both canonical and non-canonical responses to TLR2 ligands with emphasis on whether the inflammation induced by these responses contributes to the disease state or to protection from diseases.
The innate immune system contains germline-encoded pattern recognition receptors (PRRs) that detect pathogens threat and trigger prompt responses against them. PRRs recognize microbial components termed pathogen-associated molecular patterns (PAMPs), which play crucial roles in microbial pathogenesis, survival and replication. Examples of PAMPs include integral cell membrane and cell wall components, bacterial toxins, DNA RNA, etc. A cascade of events occurs following PAMP recognition by PRRs, which activate host defense mechanisms to prevent or fight off infections and initiate and enhance a subsequent adaptive immune response (West et al., 2006). A well-known group of PRRs discovered in the 1990s are the toll-like receptors (TLR; Gay and Gangloff, 2007), a family of very similar proteins containing leucine-rich repeats, that are widely expressed by a variety of cells in many animal species (Akira et al., 2006). TLRs and TOLLs (in the fruit fly) are evolutionarily ancient mediators of innate host defense (Leulier and Lemaitre, 2008). The most recent Nobel Prize in Medicine and Physiology was partially awarded for this discovery. To date, 13 TLRs have been identified, 10 human TLRs (TLR1-10), and 12 mouse TLRs (TLR1-9, TLR11, TLR12, mice do not express TLR10; Beutler, 2009).
TLR2 identification, molecular characterization, and cloning were first published in 1998, together with TLR1, TLR3, TLR4, and TLR5 (Rock et al., 1998). More than a decade of extensive research has demonstrated the importance of TLR2 in the vertebrate immunity. This receptor is the only TLR described so far to form functional heterodimers with more than two other types of TLRs. TLR2 also interacts with a large number of non-TLR molecules, allowing for recognition of a great number and variety of PAMPs (Zähringer et al., 2008). This diversity comprises different types of molecules from all microbial phyla including viruses, fungus, bacteria, and parasites. TLR2 expression has been detected in immune cells, endothelial, and epithelial cells (Flo et al., 2001; Brzezińska-Blaszczyk and Wierzbicki, 2010). This ubiquity is consistent with the wide range of roles and function of TLR2.
Toll-like receptor receptors are type I integral transmembrane glycoproteins composed of a conserved intracellular toll–interleukin-1 receptor (TIR) homology domain, a single transmembrane helix domain and a solenoid ectodomain (Kumar et al., 2009). The ectodomain is responsible for pathogen recognition and is composed of 16–28 diverse leucine-rich-repeat (LRR) modules (Akira et al., 2006). TLRs can be divided into six major families, according to the repeat number of LRRs and the motifs of two cysteine clusters flanking the LRRs. TLR2, together with TLR1, TLR6, and TLR10, are members of the TLR1 subfamily (Matsushima et al., 2007).
Ligand specific recognition and signaling through TLR2 occurs via heterodimerization with TLR1 or TLR6. TLR2/TLR1 and TLR2/TLR6 heterodimers are thought to be pre-formed on the cell surface. In the absence of TLR1 and TLR6, TLR2 homodimerization was proposed but it has not been observed with current techniques (Jin et al., 2007). The crystal structure of both heterodimers where determined, showing that LRR modules confers a horse-shoe shaped structure to the ectodomain and each heterodimer forms an “m” shaped complex with the ligand, stabilizing the two receptors. Without the ligand, the pre-formed heterodimers most likely have no interaction between their intracellular moieties and therefore there is no downstream signaling (Jin et al., 2007; Kang et al., 2009). New studies also describe the existence of TLR2/TLR10 pre-formed dimers, although their function is unclear (Guan et al., 2010). TLR1 and TLR10 were described to form dimers with TLRs from the same subfamily (Hasan et al., 2005), while TLR6 can form a heterodimer with TLR4 in response to endogenous ligands, promoting sterile inflammation (Stewart et al., 2010).
The wide array of TLR2 ligands includes molecules with diacyl and triacylglycerol moieties, proteins and polysaccharides (Table 1). The biochemical diversity of such ligand structures consequently raises questions about receptor specificity. For a long time the identity of most putative TLR ligands was doubted, due to potential lipoprotein contamination. A recent study claims that only lipoproteins/lipopeptides (LPs) are “true” TLR2 ligands, sensed at physiological concentrations by this receptor (Zähringer et al., 2008). To assess such potential contamination, treatment with lipoprotein lipase (Hashimoto et al., 2006, 2007), high temperatures (Liang et al., 2007a), and solvent washes (Ikeda et al., 2008) have been used, according to the molecular structure and physicochemical properties of the hypothetical TLR2 ligand. However, some of these methods were applied in the absence of proper ligand molecular structure analysis, leading to mistaken conclusions. An example is the use of lipoprotein lipase to assess lipoprotein contamination of diacylglycerol preparations, such as Lipoteichoic Acid (LTA), which are also cleaved by the lipase (Seo and Nahm, 2009).
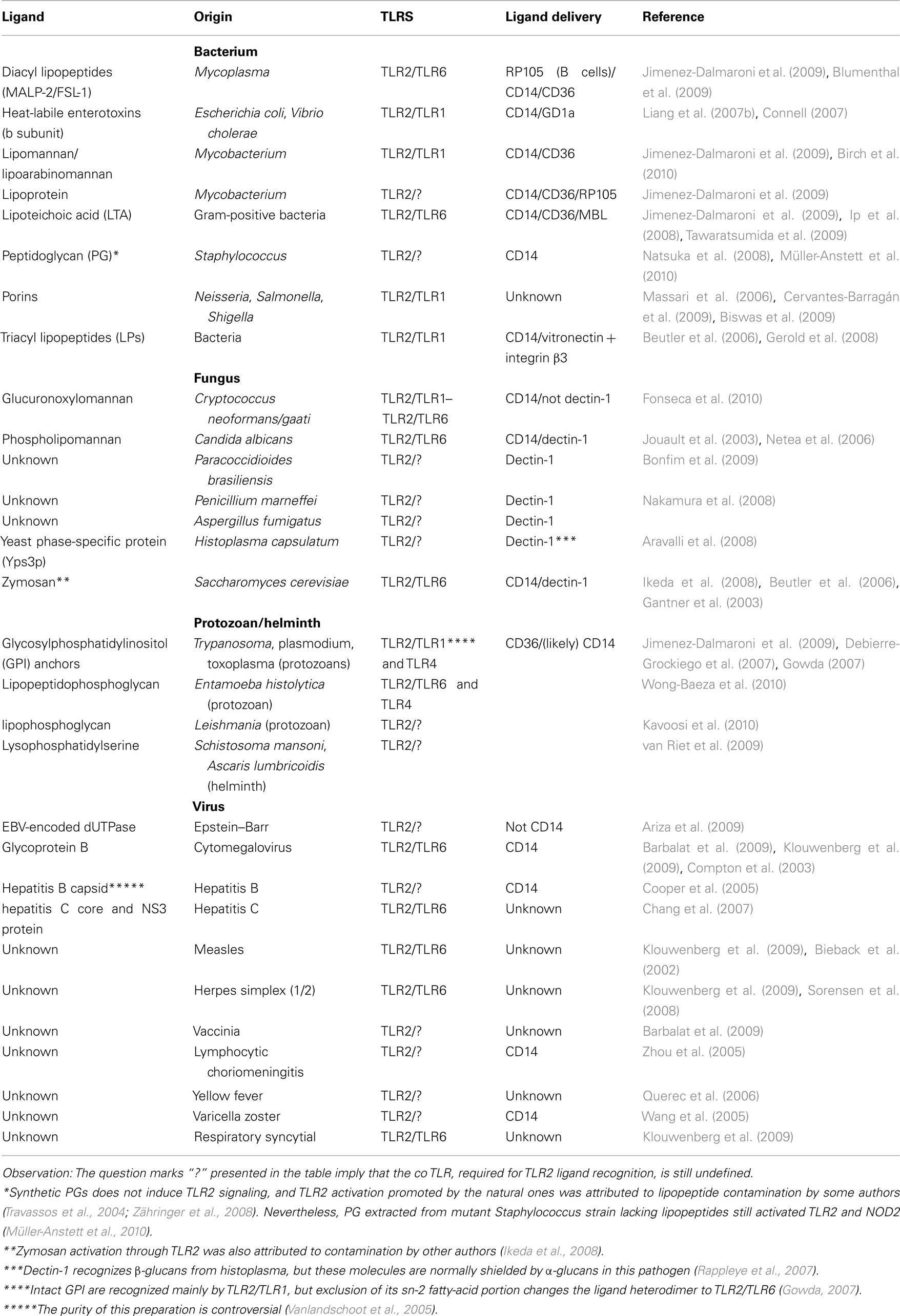
Table 1. TLR2 microbial ligands.
Endogenous ligands have also been described for TLR2, known as “alarmins” and indicating tissue damage, necrosis, or potential tumor cells. For example, human β-defensin-3, hyaluronan fragments, heat shock proteins, and high mobility group box 1 protein were identified as TLR2 endogenous ligands (Scheibner et al., 2006; Funderburg et al., 2007; Curtin et al., 2009; Erridge, 2010). Although it seems plausible that these molecules contribute to host responses to infections leading to this type of damage, there is no evidence, thus far, that endogenous ligands that engage TLR2 can modulate the course of microbial infections. Another complicating factor regarding studies with these TLR2 “endogenous ligands” is that contamination of the preparations with other non-endogenous TLR2 ligands was not always ruled out (Tsan and Gao, 2007).
The major TLR2 ligand characterized thus far are lipoproteins, ubiquitous to all bacteria and highly expressed in the outer membrane of Gram-positive bacteria. They present a unique NH3-terminal lipo-amino acid, N-acyl-S-diacylglycerol cysteine and usually three lipid chains (triacyl), except for those found in mycobacteria that can have two lipid chains (diacyl). These molecules have a variety of functions and are described in an extensive database (Babu et al., 2006). Triacyl LPs are recognized by TLR2/TLR1 while diacyl LPs are recognized by TLR2/TLR6 (Beutler et al., 2006). This has been described in detail by crystallographic techniques, demonstrating that the TLR1 ectodomain has a channel that binds the amide-bound lipid chain of the triacylated LP (Jin et al., 2007), while the same channel in TLR6 is obstructed by amino acid residues (Kang et al., 2009). TLR2 has a hydrophobic pocket that interacts with the remaining lipid chains in a less specific manner, allowing slight variations in length and chemical structure of lipid or hydrophobic moieties of its ligands (Kang et al., 2009). No matter which of the two dimers is activated, the classical signaling cascade triggered was found to be the same (Farhat et al., 2008), although the kinetics could be different depending on the ligands, and lead to different physiological outcomes (Depaolo et al., 2008; Long et al., 2009). TLR10 appears to recognize the same ligands as TLR1 and requires the same intracellular adaptor myeloid differentiation primary-response gene 88 (MyD88). However, the activated TLR2/TLR10 dimer could not trigger the common signaling cascade, suggesting a different role for this dimer yet to be defined (Guan et al., 2010). Recognition of lipopeptides through TLR2 in the absence of TLR1 or TLR6 participation was observed using either TLR-knockout mice or modified ligands (Buwitt-Beckmann, 2005). However, there is no demonstration that ligand recognition by TLR2 alone actually occurs at physiological concentrations in the course of an infection (Buwitt-Beckmann, 2005).
TLR2 forms heterodimers with its co-receptors, which increases the diversity of molecules recognized by the receptor. Recently, a number of accessory molecules and co-receptors have been described to concentrate microbial products on the cell surface or inside phagosomes to facilitate TLR2 responses. A new ligand complex was just proposed for activation by diacylglycerol ligands, including lipopeptides: CD36 may bind ligands and transfer them to the accessory molecule CD14, which, in turn, loads the ligand onto TLR2/TLR6 heterodimers (FSL-1, MALP-2, and LTA) or on TLR2/TLR1 (lipomannan) heterodimers (Jimenez-Dalmaroni et al., 2009). The ligand delivery occurs within lipid rafts, where CD14 and CD36 molecules are anchored, resulting in complex internalization to the Golgi apparatus, and this trafficking may be dependent on the TLR2 ligand. Despite this finding, activation does appear to occur at the cell surface and is independent from internalization (Triantafilou et al., 2006; Jimenez-Dalmaroni et al., 2009). The ectodomain of CD14 and CD36 is the active receptor moiety for ligand delivery (Jimenez-Dalmaroni et al., 2009). It is notable that, even though CD14 and CD36 are not an absolute requirement (Hoebe et al., 2005; Nakata et al., 2006) for TLR2 signaling, the role of these molecules is to enhance responses, lower the threshold of the concentrations needed for receptor recognition and signaling. A similar mechanism likely occurs with GPI anchors from some protozoan parasites, since they also contain the diacylglycerol moiety and are known to be TLR2 ligands. Accordingly, CD36−/− macrophages have an impaired cytokine response compared to wild-type macrophages when stimulated with Plasmodium falciparum GPI (Patel et al., 2007). Trypanosoma cruzi GPI needs CD14 to fully activate TLR2 (Almeida and Gazzinelli, 2001). However, this function of CD14 cannot be extended to all GPIs, since CD14 does not participate in Toxoplasma gondii GPI stimulation of TLR2 and TLR4. Instead, galectin-3 seems to deliver T. gondii GPIs for these TLRs (Debierre-Grockiego et al., 2010).
Interestingly, vitronectin, an extracellular matrix glycoprotein also present in the blood, has been reported as essential for triacyl LP engagement of TLR2. This protein, in its extended conformation, binds to triacyl LPs and is recognized by the integrin β3 receptor, which is part of the pre-formed TLR2/TLR1 signaling complex in resting monocytes (Gerold et al., 2008). CD14 (but not CD36) also concentrates and delivers triacyl LPs to TLR2/TLR1, without directly binding to the dimer (Hoebe et al., 2005; Nakata et al., 2006), and can contribute to the inflammatory response in phagocytes (Drage et al., 2009). Other researchers identified radioprotective 105 kDa (RP105) as a receptor able to bind mycobacterial lipoproteins, mostly TLR2/TLR1 agonists, acting as an accessory molecule for the TLR2 receptor complex in macrophages and improving the response against this pathogen. RP105 has an ectodomain related to the TLRs, but no intracellular moiety (Blumenthal et al., 2009). Further research is needed to define if the accessory mechanisms involved with triacyl LPs are complementary, non-concomitant, or overlapping.
So far, only the ganglioside GD1a has been shown to potentially have an accessory function in recognition of non-acetylated TLR2 ligands. It binds the β subunit of type IIb heat-labile enterotoxin of Escherichia coli, enabling this ligand to induce TLR2/TLR1 signaling within lipid rafts. GD1a does not appear to interact with triacyl molecules (Liang et al., 2007b). Since bacterial porins, which have been shown to be TLR2 ligands (Massari et al., 2002; Liu et al., 2008), are also oligomeric pore forming proteins that bind to the same dimer, there is a possibility that GD1a may also affect or enhance their signaling, but there is no experimental evidence supporting this hypothesis (Massari et al., 2006).
In regards to innate immune recognition of whole bacteria by TLRs, phagocytosis is an important step, forming phagosomes that could recruit TLRs and form different receptor complexes. The soluble molecule mannose binding lectin (MBL) was found to bind Staphylococcus aureus through membrane LTA and to synergize with TLR2/6, drastically increasing inflammatory responses upon complex internalization (Ip et al., 2008). The same complex is likely to occur with peptidoglycan, lipoarabinomannan, and lipophosphoglycan, since they were described to bind to MBL (Ip et al., 2009). CD36 may possibly maintain its ligand delivery role inside the phagosomes, because it is required for phagocytosis of S. aureus (Stuart et al., 2005). Lack of integrin α3β1 impairs release of IL-6 after Borrelia burgdorferi phagocytosis, due to weak activation of endosomal TLR2 (Marre et al., 2010). In addition, the β3 integrin was reported to facilitate host cell invasion by several bacterial pathogens and could also be linked to TLR2 triggering inside phagosomes (Gerold et al., 2008).
The only non-TLR molecule found to physically interact with TLR2 and induce cross-talk signaling was Dectin-1, the main receptor for β-glucans found on most fungi. Dectin-1 dependent signaling synergizes with both TLR2 and TLR4 for induction of tumor necrosis factor-α (TNF-α) in human primary peripheral blood mononuclear cells (PBMCs), when all three receptors are engaged and stimulated via their respective ligands (Ferwerda et al., 2008).
Finally, and of potential clinical importance, the responses induced by transmembrane TLR2 signaling has recently been found to be modulated by the presence of soluble TLR2 (Raby et al., 2009), in human plasma, milk, and amniotic fluid (Dulay et al., 2009). It is unclear what role this phenomenon may play in response to pathogens and defense against infections, but it could be postulated that varying levels of soluble TLR2 may positively or negatively modulate such responses.
Following ligand stimulation, TLR2 heterodimers generally initiate a MyD88-dependent intracellular signaling pathway, common to all TLRs except TLR3. This pathway induces nuclear translocation of nuclear factor-B (NF-B) to modulate gene transcription and consequent inflammatory cytokine production (Figure 1). The cascade also triggers serine/threonine-specific protein kinases (MAPKs) that can influence both transcription of inflammatory genes and mRNA stability of those transcripts, by activation protein 1 (AP-1) induction (Watters et al., 2007).
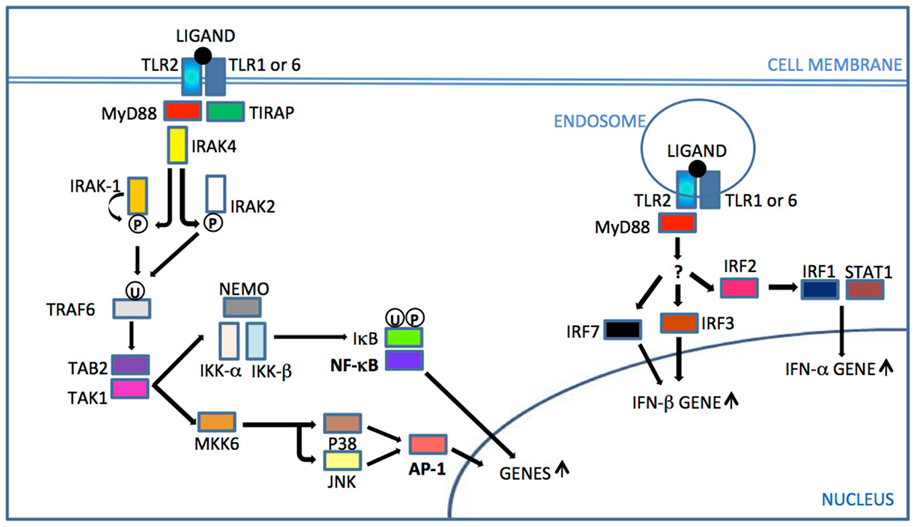
Figure 1. TLR2 signaling. After ligand recognition and consequent TLR2 dimer rearrangement, the TIR domain of TIRAP binds the TIR domain of TLR2 and recruits the adaptor protein MyD88. IRAKs are then recruited and IRAK 4 phosphorylates (P) IRAK1, which then initiates auto-phosphorylation. Phosphorylated IRAK1 dissociates from the complex and activates TRAF6. Since IRAK1 is rapidly degraded, IRAK2 also activates TRAF 6 in latter responses. Ubiquitinated (U) TRAF6 triggers the activation sequence TAB2 – TAK1 – IKK complex. IκB phosphorylation and ubiquitination by the IKK complex leads to its degradation and release of NF-κB translocation to the nucleus for gene up-regulation. TAK1 also activates MKK6 for subsequent JNK and p38 activation, leading to AP-1 activation that triggers gene transcription of cytokines and accessory molecules. Internalized receptor complex triggered by a viral ligand can activate by an unknown pathway IRF7/3 to IFN-β gene up-regulation or IRF2/IRF1/STAT1 for IFN-α gene up-regulation (Underhill et al., 1999; Watters et al., 2007; Liljeroos et al., 2008; Barbalat et al., 2009; Dietrich et al., 2010; Dunne et al., 2010). MyD88, myeloid differentiation primary-response gene 88; TIRAP, TIR adaptor protein; IRAK, interleukin-1 receptor associated kinase; TRAF, TNF receptor associated factor; TAK, transforming growth factor beta-activated kinase 1; TAB, TAK1-binding protein; MKK/JNK/P38, MAP kinases, NEMO/IKKs, kinase complex; NF-κB, nuclear factor-κB; IκB, kinase complex; AP, activator protein; IRF, interferon regulatory factor, STAT, signal transducer and activator of transcription 1.
Although the general signaling pathway is not unique, the phosphorylation and ubiquitination processes crucial for the cascade are not completely elucidated, especially the feedback loops. For example, IL-1 receptor associated kinase 1/4 (IRAK 1/4) were found to phosphorylate, ubiquitinate, and increase degradation of TIR domain containing adaptor protein (TIRAP also termed MyD88 associated ligand, MAL), indicating a possible down-regulation mechanism for TLR2 and TLR4 activation, in addition to their role in activating these pathways (Dunne et al., 2010). Sub-activating doses of TLR2 ligands may tolerize dendritic cells for TNF-α production, by a mechanism that involves disruption of signal transduction at the IRAK1 level (Albrecht et al., 2008). Bruton’s tyrosine kinase (Btk) and PI3 Kinase are likewise able to modulate TLR2 production of cytokines stimulated by LTA, but their place in the cascade is not clear (Liljeroos et al., 2007).
Since TLRs, nucleotide binding oligomerization domains (NODs), and Dectin-1 can induce NF-B translocation and MAPK p38 activation (O’Neill, 2008), intracellular cross talk for cytokine production synergy is likely to occur. In this context, Dectin-1 can synergize with TLR2 and TLR4 through an NF-B non-canonical pathway and augment cytokine expression induced by the cited TLRs (Gringhuis et al., 2009). Human PBMCs stimulated with heat killed Listeria monocytogenes (recognized through TLR2), plus a TLR7/8 ligand, demonstrated synergistic up-regulation of IFN-γ production and, to a minor extent, IFN-α production (Ghosh et al., 2007). Although with a different TLR2 ligand (Pam3CSK4), the opposite outcome of IFN-α was reported with human dendritic cells. In this case, TLR2 activation restrained the type I IFN (IFN-α/β) amplification loop used by TLR4 and TLR7/8 ligands (Wenink et al., 2009). The controversy can be partially explained by the fact that TLR2 depletes early IFN-α/β release induced by TLR9/TLR7, due to transient degradation of IRAK 1, but not later induction of type I IFN (Liu et al., 2012). The urokinase-type plasminogen activator receptor was recently described as important for optimal MAPK p38 activation and consequent inflammation induced by lipopeptides recognized by TLR2 (Liu et al., 2011). Mutations of the NOD2 gene results in defective release of Interleukin-10 (IL-10) from PBMC stimulated with TLR2 ligands, suggesting another avenue of cross-signaling (Netea et al., 2004a).
Type I interferon (IFN-β/α) production was recently found to be induced through TLR2 engagement. It was then necessary to postulate an additional signaling pathway to account for this phenomenon, as type I IFN synthesis requires activation of IFN regulatory factors (IRF), but not NF-B or AP-1, traditionally seen with TLR2 signaling. The stimulation occurs in the endosomal environment, which requires microbial or ligand endocytosis. In vitro stimulation of phagocytes with vaccinia virus (VV) and Lactobacillus acidophilus induced IFN-β production, while LTA from S. aureus stimulated IFN-α production in murine macrophages (Liljeroos et al., 2008; Dietrich et al., 2010; Weiss et al., 2010). It is possible that L. acidophilus also stimulates IFN-α due to LTA presence. Surprisingly, bacteria but not viruses induced TLR2-dependent production of IFN-β in dendritic cells, suggesting cell specificity related to the type of microbe. Although not yet elucidated, the newly discovered pathway was described as MyD88-dependent and requires IRF1/7 for IFN-β and IRF1/2 for IFN-α release (Figure 1). Activators of transcription 1 (STAT1), STAT 3, PI3, and Btk were also reported as part of the signaling cascade required for IFN-α release induced by LTA. TRIF adapter is not required for TLR2 activation of interferon production, as it is for TLR4 and TLR3 (Barbalat et al., 2009).
The intracellular cross-talk scenarios are essential for understanding host–microbe interactions, since most invaders have multiple molecules recognized by multiple innate immune receptors. Investigators have still not been able to relate most of what has been learned about intracellular regulation of pathogens with infection outcomes in the host, and this must be the focus for future studies.
In order to assess the role for TLR2 in immune protection or pathogenesis of infections, a number of techniques have been developed. The vast majority of researchers have used inbred mice as the in vivo model, limiting individual gene variability and allowing for examination of knockout or transgenic mice. However, limiting genetic variability also limits immune response variability. In addition, another important consideration in interpreting results is the phenotypic divergence between mice and humans, including the level of TLR2 expression and which cells express TLR2. Comparison of murine and human TLR2 sequences has revealed higher correlation between intracellular domains (84%) than between extracellular domains (65%; Grabiec et al., 2004), the latter being involved with ligand recognition. Regarding cellular expression, one example of the divergence between mice and humans is the fact that murine T lymphocytes express TLR2 in a constitutive manner while human T cells do not (Rehli, 2002). These discrepancies may contribute to the differential pathology and immune response observed in mouse models of infectious diseases as compared to humans. In addition, there is evidence of associations between human TLR2 polymorphisms with disease course and susceptibility, but other factors, such as ethnicity, location, environmental factors, and exposure to infectious agents contribute to the conflicting results regarding these associations (Corr and O’Neill, 2009). Specific descriptions of TLR2 polymorphisms are included in the following sections. To our knowledge, it is unclear whether TLR1 or TLR6 can activate host immune response in the absence of TLR2 involvement. Nevertheless, we cannot assume that TLR1 or TLR6 polymorphisms that lead to alteration in receptor function are impacting TLR2 recognition and activation. There is evidence that TLR1 and TLR6 polymorphism do exist in humans that affect responses to infectious diseases (especially mycobacteria; Kesh et al., 2005; Bhide et al., 2009; Shey et al., 2010). However, for sake of clarity, this review will not address TLR1 or TLR6 polymorphisms and will only focus on TLR2 polymorphisms.
In vitro models are mostly based on the use of murine cells, human cells, or cell lines derived from both species, including dendritic cells, macrophages, and human PBMCs. The use of human PBMC is not free of criticism as well, since these cells might not replicate the responses of tissue resident cells in a specific infected organ (von Herrath and Nepom, 2005).
All models have discrepancies from true biological responses, so the conclusions based on these models need to be examined closely to insure that there is no over-interpretation. We shall examine the role of TLR2 in infections by pathogen type: viruses, fungi, protozoan/helminthes, and bacteria.
Viruses are biological organisms that only replicate inside host cells. Cell adhesion and entrance usually occurs without specific PAMP recognition. In fact, almost all viral PAMPs are nucleic acid-based, which are not exposed until after host cell entry, and TLRs that recognize these PAMPs are almost exclusively intracellular. For this reason, host immune recognition of viruses relies mainly on a complex combination of endosomal TLRs and cytosolic non-TLR receptors, activated by viral nucleic acids uncommon to the host (Thompson and Iwasaki, 2008). Type I interferon (IFN) seems to be a key innate immune factor against viruses since IFN α/β receptor knockout mice are extremely vulnerable to multiple viral infections (Müller et al., 1994). For some types of viruses, type II IFN (IFN-γ) is also essential to assure viral clearance (Zhang et al., 2008). Type I IFN, mainly produced by dendritic cells, can prime natural killer (NK) cells, essential for early infection control (Szomolanyi-Tsuda et al., 2006), and induce maturation of T helper 1 (Th1) cells (Joffre et al., 2009). The production of neutralizing antibodies and activation of cytotoxic T lymphocytes (CTL) are likewise important for a specific and effective antiviral immune response (Koyama et al., 2008). However, despite these observations, there is mounting evidence of TLR2 involvement in responses to viral infections.
IFN-β production by murine monocytes can be triggered by TLR2 following stimulation with virus particles like VV or murine Cytomegalovirus (CMV; Barbalat et al., 2009). TLR2 is also involved in NK cell response, as demonstrated by impaired NK cell activity and increased murine CMV load in TLR2 knockout mice (Szomolanyi-Tsuda et al., 2006). In humans, malfunction of TLR2 caused by homozygosis for R753Q polymorphism contributes to CMV infection after liver transplantation (Kang et al., 2012). VV was recently shown to directly stimulate NK cells via the TLR2–MyD88 signaling pathway (Martinez et al., 2010), in addition to directly promoting CD8 T cell survival, allowing cloning expansion and memory cell establishment in mice (Quigley et al., 2009). TLR2/TLR6 were identified as important murine receptors to control murine respiratory syncytial virus (RSV; Murawski et al., 2009). Based on these studies, TLR2 seems to have an overall protective role in VV, CMV, and RSV infections.
Lack of functional viral Hemagglutinin impairs TLR2 response to measles virus, resulting in diminished IL-6 secretion by murine monocytes and measles receptor CD150 (Bieback et al., 2002). Varicella zoster virus (VZV) virulent strain induces IL-6 production by human monocytes through TLR2 and CD14, but blocks TLR2 mediated production of IL-12 in human dendritic cells to evade the Th1 driven immune response. The attenuated Varicella strain used for the current vaccine can not block IL-12 production, which suggests that Th1 driven immune responses are important for protection against VZV. However, the mechanisms of TLR2 blockade or IL-12 inhibition have not been clarified (Wang et al., 2005; Gutzeit et al., 2010). dUTPase from Epstein–Barr Virus also induces IL-6 production through TLR2 in TLR2 over-expressing HEK293 cells, but CD14 does not act as the co-receptor (Ariza et al., 2009). Yellow fever-attenuated virus, used as the current Yellow Fever vaccine, is capable of activating multiple TLRs and induces a balanced Th1/Th2 response in murine dendritic cells. However, in TLR2 knockout mice, the Th1 responses are considerably intensified while IL-6 and IL-12 production is decreased (Querec et al., 2006). Although hepatitis B virus capsid particles was also found to prime human THP-1 macrophages for production of TNF-α, IL-6, and IL-12p40 via TLR2 (Cooper et al., 2005), the lack of purity of this preparation makes these results controversial (Vanlandschoot et al., 2005).
Hepatitis C virus (HCV) can be recognized by the host by the TLR2/TLR6 heterodimer by recognition of the HCV core and NS3 proteins, along with recognition of viral genome by TLR3, 7, and 8 (Chang et al., 2007; Moriyama et al., 2007). HCV TLR2 recognition activates human monocytes and macrophages to secrete mainly IL-10 and TNF-α, but inhibits macrophage-dependent dendritic cell differentiation and function (Chang et al., 2007). Accordingly, patients with chronic hepatitis were also found to have increased expression of TLR2 in PBMC, correlated with augmented circulating TNF-α and hepatic necroinflammatory activity (Riordan et al., 2006; Wang et al., 2010). Activation of PBMCs from chronic HCV patients induced by TLR2/TLR4 ligands resulted in decreased IL-6 production when compared to healthy subjects, probably caused by TLR2-induced tolerance (Chung et al., 2011). Divergent results demonstrated that the TLR2 polymorphism R753Q impairs TLR2 recognition of HCV core and NS3 proteins and increases the risk of allograft failure after liver transplantation for chronic hepatitis C patients with this mutation (Brown et al., 2010). This is an example of the contrasting roles TLR2 may play in infectious processes in humans.
The ability of viruses to stimulate cells can differ from species or viral type, but also according to the murine strain, organ, or cell type infected. TLR2/TLR9 double-knockout mice infected intravaginallly with herpes simplex (HSV) 2 had significant higher viral loads in the brain than single knockouts or wild-type mice. Viral loads in the vaginal mucosa or liver, on the other hand, did not differ between groups of mice. The observations lead to a synergistic protective role for both TLRs in the brain, benefic for encephalitis prevention, but not effective for general decrease of HSV2 viral loads (Sorensen et al., 2008). TLR2/TLR9 were shown to have a more prominent protective role for HSV1, as TLR2/TLR9 double-knockout mice all died after intranasal infection with HSV1 EK strain, as compared to 90% survival of infected wild-type mice or TLR2 knockout mice and 40% survival of infected TLR9 knockout mice. Another experiment pointed out a deleterious role for TLR2 in mice infected with HSV1 KOS strain, in which TLR2 knockout mice were more resistance to infection as compared to WT mice. Nevertheless, these apparently contradictory results were observed using different HSV1 strains and viral loads, inoculated through different routes (Krug et al., 2004; Lima et al., 2010). More work is obviously needed to truly dissect the dual property of TLR2 to be involved in both HSV disease protection and disease induction. TLR2 may also be involved in inflammation related to morbidity and mortality caused by lymphocytic choriomeningitis virus (LCMV); TLR2 is essential for induced monocyte chemotactic protein 1 (MCP-1), RANTES, and TNF-α, in mouse central nervous system glial cells (Zhou et al., 2008; Lee et al., 2012).
The common immune response measured by most studies when examining TLR2 function has been increase in IL-6 and TNF-α production during acute processes, while IL-10 was consistently found in chronic infections. IL-6 secreted by dendritic cells drives Th2/Th17 T cell differentiation (Wenink et al., 2009). Considering that type I IFN is released upon virus stimulation and constrains Th17 differentiation in mice (Guo et al., 2008), IL-6 in the context of viral infection most likely drives Th2 responses. When the Th1 phenotype is dominant, TNF-α act as an enhancer of IFN-γ killing through activation of cellular immune responses. In contrast, when Th2 or Th0 is dominant, TNF-α causes tissue damage (Hernandez-Pando and Rook, 1994; Rizzardi et al., 1998). In this context, TLR2 activation may improve viral clearance when the Th1 response is fully functional and dominant, during the initial stage of infection. TLR2 can also promote Th2 responses, which are important for adequate antibody production and protect the host by inhibition of exacerbated Th1 response.
To our knowledge, none of the viral particles were tested for TLR2 induction of regulatory T (Treg) cells or Th17 cell function or activation, which could help to elucidate outcomes of TLR2 receptor activation and reveal new information regarding the Th1/Th2 bias studied so far. It is clear that TLR2 function and its role in viral immunity are co-dependent on other PRRs and the subsequent adaptive immune response. Therefore, isolated analyses can be insufficient to determine the true role of TLR2 in each infection type.
In relation to recognition, the examples of viruses mentioned above are variable in relation to species, genetic material and disease outcomes, but all viruses assumed to be recognized by TLR2 are enveloped virus. Some viruses do not have their TLR2 ligands defined. TLR2/6 was the only TLR2 heterodimer described for viral ligands, but the heterodimer needed for TLR2 recognition of many other viral ligands has not been discerned. The involvement of the TLR2/6 heterodimer is consistent with the cytokine profile described above, since the same heterodimer has been found to drive IL-10 production in dendritic cells (Depaolo et al., 2008). Studies on TLR2 accessory molecules involved with viral recognition has also been minimal and only revealed a role for CD14.
As opposed to viruses, pathogenic fungi are facultative intracellular or extracellular eukaryotic organisms and therefore do not depend on the host cell to replicate. Some fungal species are dimorphic and change between different forms, for example, yeast or conidia to hyphae, and consequently modify the way they are recognized by the host. In addition, cell wall composition varies among species and serotypes, which can also change the way these cells are processed by the immune system (McKenzie et al., 2010). Fungal infections remain generally localized when controlled by a regular immune response, but systemic dissemination more often occurs when the host is immunocompromised, has its physical barriers disrupted or has its microbiota altered (Chai et al., 2009a). Dendritic cells usually trigger adaptive immune response against fungal invasion, while direct clearance is performed by neutrophils, monocytes, and macrophages. All of these cells express dectin-1, TLR2, and TLR4, receptors associated with fungal recognition (Hohl et al., 2006). Most yeast can be phagocytized by macrophages through Dectin-1 receptor recognition, whereas TLR2 helps initiate the concomitant inflammatory state (Zak and Aderem, 2009). The two receptors, when stimulated, mutually synergize their cytokine and immune responses to promote a consistent pathogen clearance (Zak and Aderem, 2009). In contrast, phagocytosis of hyphae is believed to occur without independent PRR recognition and leads to Th2/Treg cells bias (Romani, 2004).
The role of TLR2 in response to one of the most common fungi involved in human disease, Candida albicans, is unclear and results have been somewhat contradictory. The TLR2 ligand phospholipomannan is well defined and present in hyphae and yeast forms (Li et al., 2009), while the outcomes triggered by it are not quite clearly understood. Human keratinocytes stimulated with phospholipomannan produce IL-6 and IL-8, necessary for activation and recruitment of neutrophils and macrophages in situ (Li et al., 2009). Studies with TLR2 knockout mice showed both resistance (Netea et al., 2004b) or susceptibility to primary or secondary systemic C. albicans infection (Gil et al., 2005; Hise et al., 2009), while TLR2 knockout macrophages showed impaired fungal clearance (Blasi et al., 2005), with both impairment or enhancement of pro-inflammatory cytokine production (especially TNF-α; Blasi et al., 2005; Gil and Gozalbo, 2006). The absence of TLR2 also decreased chemotaxis rate of murine neutrophils, together with decreased fungal killing mechanisms and survival of these cells (Tessarolli et al., 2009). TLR2 is probably not directly involved with humoral response after secondary C. albicans systemic infection, since this parameter was not altered in TLR2 knockout mice (Villamón et al., 2004). On the other hand, Dectin-1 and TLR2 were directly related with protection against oral candidiasis through an effect on IL-1 β release, but only when both receptors are engaged (Hise et al., 2009). Other factors can influence TLR2 mediated effects and contribute to fluctuations in cytokine levels in candidiasis, for example high concentrations of vitamin D3 in mice can decrease TLR2 surface expression (Khoo et al., 2011).
Similar to C. albicans, Aspergillus fumigatus recognition varies with morphology. Both conidium and hyphae are recognized by TLR2 (Chai et al., 2009b), but hyphae are not recognized by TLR4 and result in IL-10 production through TLR2 activation (Netea et al., 2003). Murine neutrophils, on the other hand, have increased fungicidal activity and release pro-inflammatory cytokines when activated by TLR2. It has been shown that TLR2 is essential for protection in immunocompromised mice but not immunocompetent mice (Chignard et al., 2007).
Paracoccidioides brasiliensis is recognized by TLR2, and pulmonary infection of TLR2 knockout mice leads to Th17 skewed immunity and increased (Loures et al., 2009) or similar (Calich et al., 2008) lung inflammation as compared to wild-type mice. Even so, fungal burden is either diminished or the same as wild-type mice (Calich et al., 2008; Loures et al., 2009), with equal mortality rates demonstrating that overall outcomes are not necessarily affected by TLR2 signaling.
Cryptococcal recognition by TLR2 occurs through the polysaccharide glucuronoxylomannan, and the level of NF-κB activation differs based on polysaccharide effective diameters of the varying cryptococcal species (Fonseca et al., 2010). Although TLR2 was found to be important for control of Cryptococcus neoformans murine infection (Yauch et al., 2004), this is still quite controversial (Nakamura et al., 2006). TLR2 was also shown to play a major role in protection against Penicillium marneffei, due to stimulation of murine bone marrow-derived dendritic cells to produce IL-12p40 upon fungal recognition (Nakamura et al., 2008). Histoplasma capsulatum is recognized by mice TLR2 through the cell wall protein Yps3p, resulting in activation of NF-κB (Aravalli et al., 2008). Regardless, TLR2 knockout mice had no alteration in inflammatory cytokine production induced by this pathogen (Lin et al., 2010).
Generation of a dominant TH1-cell response is required for most protective immune responses to fungi (Romani, 2004). The infection models mentioned above did not evaluate fungal morphology during the course of infection, which can account for the varied immune responses and survival rates described. Other TLRs and Dectin-1 expression were also not measured in parallel, and these parameters are able to alter the profile of TLR2 responses. The shielding of β-glucans can impair Dectin-1 recognition and consequent loss of synergy with TLR2, which allows for a preferential induction of an anti-inflammatory response when stimulated by fungal particles (Netea et al., 2008). In addition, it has been shown that different murine strains have different dectin-1 isoforms, which may impact on TLR2 function (Heinsbroek et al., 2008). However, it is safe to say that the absence of TLR2 influences macrophage and neutrophil anti-fungal activity, mainly through an effect on TNF-α production. This cytokine is important for induction of fungicidal activity (Chignard et al., 2007). The relevance of this effect varies according to the infectious microenvironment and with Dectin-1 function. When TLR2 responses were found to be detrimental to the host, it was due to an overwhelming suppression of the early inflammatory response and consequent increased fungal burden. Since almost all fungi are recognized to some extent by TLR2 and are recognized by TLR4 as well, it would be meaningful to evaluate the synergism between these two TLRs in the context of fungal infections, as bacterial ligands have been shown to synergistically induce IL-10 when both TLRs are engaged (Hirata et al., 2008). To date, TLR2 polymorphisms have not been shown to be related to susceptibility or protection to fungal infections, but it is also true that these studies are scarce in the literature.
Protozoa are a group of intracellular or extracellular eukaryotic organisms that can cause human disease and depending on its life cycle, may mainly cause chronic infections (Sacks and Sher, 2002). TLR2 is activated by glycosylphosphatidylinositols (GPIs) present on some of these protozoa. The level of the inflammatory response induced is directly linked to GPI lipid and carbohydrate content (Almeida and Gazzinelli, 2001). P. falciparum, one of the plasmodia that cause malaria, can trigger TNF-α and IL-6 production in murine macrophages by means of TLR2/TLR1 or TLR2/TLR6 activation with GPI. This GPI is only recognized in merozoites, the erythrocyte infective form of the protozoan (Wong-Baeza et al., 2010; Zhu et al., 2011). GPI can also modulate internalization of malaria-parasitized erythrocytes. Still, when inside the erythrocyte, the parasite does not allow for display of its TLR ligands to murine and human macrophages, resulting in a non-inflammatory phagocytosis of these erythrocytes and delayed cytokine responses (Erdman et al., 2009). No TLR2 polymorphisms are related with P. falciparum infections (Greene et al., 2009). However, the presence of human heterozygotes for a TIRAP polymorphism (TIRAP S180L) has been related to protection of malaria. This adaptor protein mediates TLR2 and TLR4 signaling whereas its polymorphic variant severely decreases IL-6 production triggered by these TLRs, suggesting that a normal TLR2 response would be detrimental to the infected human host (Khor et al., 2007).
Toxoplasma gondii GPIs from the highly dividing tachyzoite form, also trigger TLR2 for TNF-α and MCP-1 production. TLR2 knockout mice have increased susceptibility to infection caused by high doses of T. gondii (Mun et al., 2003). However, the use of lower doses leads to an increase parasite burden only in double TLR2/4 knockout mice (Debierre-Grockiego et al., 2007), suggesting a protective role for TLR2 in synergy with TLR4. Strong IL-12 induction through TLR11 is protective and part of the murine immune response, but since this gene is not functional in humans, other TLRs can have a different role in this host (Yarovinsky et al., 2005). A human embryonic intestinal epithelial cell line recognizes the parasite through TLR2, resulting in production of IL-8 and other chemokines important for neutrophil and phagocyte recruitment (Ju et al., 2009). TLR2 mediated signaling by GPIs from T. cruzi infective trypomastigotes results in IL-12, TNF-α, and nitric oxide production by murine macrophages (Campos et al., 2004). The T. cruzi protein 52 (Tc52) can induce IL-6 and IL-8 in a TLR2-dependent manner (Wong-Baeza et al., 2010). TLR2 stimulation leads to IL-12 and TNF-α production in T. cruzi infections, in addition to cooperating with TLR9 to enhance this production. T. gondii infection of TLR2/TLR9 double-knockout mice had increased susceptibility to T. cruzi infection. The higher parasite burden of double knockouts was related to low IL-12 and IFN-γ levels (Bafica et al., 2006). Surprisingly T. cruzi IL-10 modulation of murine bone marrow-derived dendritic cells was found to be independent of TLR2 (Poncini et al., 2010). An interesting cooperation between TLR2 and the bradykinin receptor in T. cruzi murine subcutaneous infection has been reported, where TLR2 induces leak of plasma that leads to a kininogen accumulation. This molecule is degraded by T. cruzi proteases to kinins recognizable by the bradykinin receptor, with consequent induction of IL-12 production by murine dendritic cells (Monteiro et al., 2006).
Entamoeba and Leishmania are protozoa genera that have lipopeptidophosphoglycans (LPPG) and lipophosphoglycans (LPG), respectively, which have been identified as TLR2 ligands. Entamoeba LPPG, also recognized by TLR4, leads to TNF-α, IL-12, IL-10, and nitric oxide release in phagocytes (Wong-Baeza et al., 2010). It also stimulates murine NK Cells for production of IFN-γ through TLR2, which is directly related with protection from amebic liver abscess (Lotter et al., 2009). Membrane LPG from Leishmania up-regulates the same cytokine milieu as LPPG in J774 murine macrophage cell line (Kavoosi et al., 2010). However, Leishmania brasiliensis infection of TLR2 knockout mice presented higher levels of IFN-γ and resistance to infection in vivo when compared to wild-type mice, while DCs from knockout mice had up-regulated IL-12 secretion in vitro (Vargas-Inchaustegui et al., 2009). This is likely associated with a diminished Th2 environment that may be normally induced by TLR2 stimulation, as this has been shown to be associated with increased protection from leishmaniasis.
Human parasitic helminths are complex eukaryotic organisms that mostly live asymptomatically inside the host, with infection usually associated with tolerogenic responses (Maizels and Yazdanbakhsh, 2003). TLR2 is known to be activated by lysophosphatidylserine of two genera of this group: Schistosoma and Ascaris. There is no information regarding the relevance of TLR2 in Ascaris infection. S. mansoni eggs fraction containing lysophosphatidylserine activate DCs via TLR2 to induce a Th2 response and regulatory T-cell development (van der Kleij et al., 2002). Conversely, S. mansoni infection of MyD88−/− mice had a normal Th2 response while other research has shown that SEA induces a Th2 bias in a TLR2 independent manner. It is likely that the SEA used in the latter experiments lacked lysophosphatidylserine to explain such contradictory results (Kane et al., 2008).
As expected, the effect of TLR2 mediated stimulation is dependent on the cellular environment in which activation occurs. Parasite clearance is related to early IFN-γ synthesis by NK cells, which, in turn, is mostly induced by IL-12 produced by dendritic cells (Campos et al., 2004; Patel et al., 2007). TLR2 ligands derived from parasites induce IFN-γ directly in NK cells and IL-12 in dendritic cells, but production of the latter is more efficient with concomitant TLR4 or TLR9 stimulation. Nevertheless, lack of TLR2 does not increase parasite burden or decrease Th1 responses, suggesting a redundant role in early infection. TNF-α induced via TLR2, on the other hand, may be involved with pathology seen with these infections, when induced at later time points during the course of disease. This may explain why TLR2 knockout mice or human TLR2 polymorphism can lead to an attenuated or resolved infection. Another possibility is that TLR2-driven Th2 responses are involved with a more severe disease state, as commonly seen in leishmaniasis (Deak et al., 2010). It is likely that a defective early IFN-γ response allows for TLR2-induced Th2 skewing and consequent suppression of IL-12 production. Parasites can infect cells and replicate inside them without TLR recognition, whereas the infected cell is frequently phagocytized in a non-inflammatory process, which may allow for delayed recognition and TLR2 mediated responses to these pathogens. Curiously, TLR2 mediated regulation of IL-10 during parasitic infections has only been to demonstrated for schistosomiasis, showing that the Th2 skewing outcome was more relevant than the alteration of the Th1/Th2 profiles in TLR2 context in this disease.
Bacteria are prokaryotic unicellular organisms with varying strategies for energy production, survival, infectivity, and self-protection. Bacterial cells are limited by a common phospholipid bilayer membrane with inserted functional proteins, covered by a peptidoglycan cell wall. Some species have an outer membrane while others have a capsule or are able to sporulate, which alters the way they are recognized by the host (Silhavy et al., 2010). Lipoproteins, important TLR2 ligands, are produced by all bacteria. Due to the high number of bacterial pathogenic species, they are analyzed in the present review by their genera and Gram staining groups. The genera to be explored are the ones that include the main human pathogens.
The Gram-positive bacterium cell wall contains a thick peptidoglycan layer, combined with teichoic acids and extracellular proteins. Lipoteichoic acid (LTA), as well as lipoproteins and peptidoglycan, can differentially activate TLR2 dependent on the bacteria of origin (Ryu et al., 2009; Müller-Anstett et al., 2010). The pathogenic bacteria considered here are from the genera Corynebacterium, Nocardia, Bacillus, Listeria, Staphylococcus, Clostridium, Enterococus, Streptococus, Mycobacterium, and Mycoplasma. The last two genera do not stain as Gram-positive because they have different cell wall structure, but are regularly grouped with Gram-positive bacteria due to the genetic background (Silhavy et al., 2010). So far, all, Gram-positive bacteria activate TLR2 to some extent. Murine phagocytes with impaired TLR2 function, either by using blocking antibodies or gene knockouts, when exposed to the above bacteria or their cell wall components have diminished or absent production of TNF-α or/and IL-6, as opposed to normal cells (Takeuchi et al., 1999, 2000; Torres et al., 2004; Bafica et al., 2005; Leendertse et al., 2008; Love et al., 2010).
Various infection models have helped to characterize some functions of TLR2 in recognition of Gram-positive bacteria. Respiratory infection models of TLR2 knockout mice with Mycoplasma and Mycobacterium showed increased bacterial burden, tissue damage, and death. This potentially protective role for TLR2 in mycobacterial infection was confirmed in humans, where TLR2 polymorphisms that decrease TLR2 expression were found to predispose people to tuberculosis (Yim et al., 2006) and non-tuberculous mycobacterial lung disease (Yim et al., 2008). Conversely, protection from respiratory infection with Streptococcus pneumoniae only requires TLR2 when the TLR4 ligand pneumolysin is not expressed, indicating redundancy in recognition and protective responses to these bacteria (Dessing et al., 2008). Interestingly, middle ear infections (Han et al., 2009) and meningitis (Letiembre et al., 2005) caused by Streptococcus are exacerbated when TLR2 is absent, which may indicate that pneumolysin may be differentially expressed depending on the environment and strain variant. Intraperitoneal inoculation of Enterococcus (Leendertse et al., 2008), Listeria (Torres et al., 2004), and Staphylococcus (Mullaly and Kubes, 2006) caused similar disease and inflammatory states in TLR2 knockout as compared to wild-type mice, while, in contrast, intravenous inoculation of these TLR2 KO mice with Listeria (Torres et al., 2004) or Staphylococcus (Takeuchi et al., 2000) resulted in death and bacterial burden as compared to wild-type mice. This suggests that the presence of TLR2, for these organisms at least, triggers an immune response that prevents dissemination. However, once dissemination occurs, TLR2 stimulation is no longer occurs or is protective. Regardless of strain variations, the route of infection seems quite important in regards to protection induced by TLR2 toward Gram-positive bacterial infections, at least in mice.
Human polymorphism studies found no relationship between TLR2 gene polymorphism R753Q and serious pneumococcal (Moens et al., 2007; Yuan et al., 2008), streptococcal (Liadaki et al., 2011), staphylococcal infection (Moore et al., 2004), or altered cytokine patterns in Gram-positive septic patients (Woehrle et al., 2008)’ even though this polymorphism prevents TLR2 from responding to LTA. However, one functional allele is enough to compensate for this deficiency, since heterozygous subjects respond to infections the same way as the ones without the polymorphism (von Aulock et al., 2004). A TLR2-16933AA promoter human polymorphism has been associated with increased prevalence of Gram-positive bacterial sepsis but not an increase in true shock or mortality (Sutherland et al., 2005). Accordingly, blood monocytes from septic patients had higher TLR2 and CD14 expression than healthy individuals, but mortality was associated with down-regulation of the same receptors and consequent cytokine induction (Schaaf et al., 2009).
These studies demonstrates that TLR2 is important, if not essential, for control of Gram-positive infections in general, but its absence can be compensated by activation of other receptors by some genera.
The Gram-negative bacterium cell wall contains a thin peptidoglycan layer with pore forming proteins, covered by an extra layer of lipopolysaccharides (LPS). However, unlike Gram-positive bacteria, they do not produce the TLR2 ligand lipoteichoic acid. LPS is an important TLR ligand present in all Gram-negative bacteria, with its molecular composition varying among the genera. These variation leads to different receptor activation and subsequent inflammation (Munford and Varley, 2006).
Campylobacter, Bordetella, Shigella, Escherichia, Haemophilus, Salmonella, Neisseria, Klebsiella, and Leptospira produce the typical LPS, a hexaacylated form capable of activate inflammation through TLR4 (Munford and Varley, 2006). All of the above genera can stimulate cells via TLR2. Escherichia, Haemophilus, Salmonella, Neisseria, Klebsiella, and Leptospira have all been examined in animal models of infection to determine the role of TLR2 in protection or pathology. In this context, TLR2 was not essential for resolution of infection for these bacteria, while TLR4 played a much greater role (Lorenz et al., 2005; Spiller et al., 2007; Sjölinder et al., 2008; Fedele et al., 2010; Moranta et al., 2010; Pore et al., 2010; Seibert et al., 2010). Nevertheless, TLR2 knockout mice had higher bacterial burdens during the course of infections with Klebsiella and Salmonella, but with mortality or antibody responses were not altered as compared to wild-type mice (Spiller et al., 2007; Seibert et al., 2010). TLR2/TLR4 double-knockout mice were even more susceptible to infection with Leptospira and Klebsiella, as compared to TLR4 single knockout mice, demonstrating an important role for TLR2 recognition in control of these infection and a potential synergistic function with TLR4, demonstrating another potential redundancy of immune system activation (Spiller et al., 2007; Chassin et al., 2009). Regarding animal models of sepsis, Escherichia and Salmonella trigger excessive inflammation and subsequent mice death via TLR4 and TLR2. IFN-γ induced by TLR4 stimulation increases TLR2 up-regulation and affects the immunopathology, a probable reason why TLR4/TLR2 double-knockout mice all survived infection with these organisms after antibiotic therapy as opposed to single knockouts or wild-type mice, where antibiotics could not rescue these animals (Spiller et al., 2008). This demonstrates a pathologic effect of the inflammatory response induced by bacteria through these TLRs.
The under-acylated forms of LPS are weak inducers of inflammation, some of them capable of competing with typical LPS for TLR4 and therefore acting as anti-inflammatory agents (Munford and Varley, 2006). Gram-negative bacteria that produce these forms of LPS generally evade TLR4-driven immune response and the host relies on other receptors to recognize and react to the threat. Porphyromonas and Coxiella infection resolution are partially dependent on TLR4, but pro-inflammatory cytokine production is dependent on TLR2. The establishment of Porphyromonas chronic infection and inflammation appears to be affected by TLR2, where the absence or presence of TLR2 has been associated with both deleterious and protective effects (Shin et al., 2010). Chlamydia murine infections have TLR2 dependent immune pathology, although TLR2 human polymorphisms have not been associated with the disease (Karimi et al., 2009). It has also been shown that TLR2 mediates morbidity and mortality in murine models of infection with Burkholderia species, as TLR2 knockout mice have decreased pathology and increased survival as compared to wild-type mice (Wiersinga et al., 2007). TLR2 can mediate protection as well, i.e., in murine infection models of Legionella, Francisella, Citrobacter, and Borrelia. The resolution of these infections or consequent inflammation are TLR4 independent, while lack of TLR2 appears to be associated with higher bacterial burden (Archer and Roy, 2006; Abplanalp et al., 2009; Dickinson et al., 2010), tissue damage (Gibson et al., 2008), or decreased survival (Fuse et al., 2007; Gibson et al., 2008; Abplanalp et al., 2009; Dickinson et al., 2010). TLR2 mice orally infected with Yersinia develop a gut barrier defect and diminished intestinal clearance, demonstrating a role for TLR2 in full protection from this infection, but this may not just be specific for this bowel pathogen (Dessein et al., 2009).
Overall, the role of TLR2 in Gram-negative infections seems to be directly related with the dominant PRR activated by the bacterium. Often, at least in animal models, an excessive bacterial burden increases TLR2 sensitivity and its role in pathology. It is important to note that infection models often use attenuated or heat killed bacteria, which can change the predominant roles of participating PRRs. Accordingly, heat killed Treponema signals through TLR2, as opposed to live bacteria which suppresses activation by this receptor (Shin et al., 2010).
Bartonella quintana, Helicobacter pylori, Moraxella catarrhalis, Pasteurella multocida, Rickettsia akari, and Vibrio vulnificus are also recognized by TLR2 with subsequent induction of many inflammation related cytokines or chemokines: TNF-α, IL-6, IL-10 (phagocytes), or IL-8 (mucosal epithelial cells), depending on which cells were examined (Slevogt et al., 2007; Matera et al., 2008; Hildebrand et al., 2009; Peek et al., 2010; Lee et al., 2010; Quevedo-Diaz et al., 2010). Surprisingly, no relationship to disease has been studied or confirmed (Oliveira et al., 2008).
As discussed, there are a large variety of microbial pathogens in various phyla that express components that are recognized by TLR2. These molecules then trigger various innate immune responses that are involved with protection from these organisms and are also involved with inflammatory sequelae that are associated with disease caused by these microbes. The outcomes of ligand recognition were shown to be dependent on the recognition dimer (TLR2/TLR1 or TLR2/TLR6), and co-receptors. It is important to remember, however, that the innate immune system does not recognize microbial TLR2 ligands in a vacuum, and many of these pathogens express ligands recognized by other PRRs, i.e., LPS and TLR4, RNA and TLR3, CpG DNA motifs and TLR9, etc. Therefore, the responses to these organisms are complex and not easily predicted. It needs to be mentioned, though it is beyond the scope of this review, that many investigators have taken advantage of the ability of some of these microbial derived molecules to stimulate cells via TLR2, or other TLRs, to utilize this ability in at the development of vaccine adjuvants (Wetzler, 2010; Duthie et al., 2011).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all the members of the Wetzler Lab, the Section of Infectious Diseases and the Department of Microbiology at the Boston University School of Medicine for their discussions and thoughts on this topic.
Abplanalp, A. L., Morris, I. R., Parida, B. K., Teale, J. M., and Berton, M. T. (2009). TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS ONE 4, e7920. doi:10.1371/journal.pone.0007920
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Albrecht, V., Hofer, T. P., Foxwell, B., Frankenberger, M., and Ziegler-Heitbrock, L. (2008). Tolerance induced via TLR2 and TLR4 in human dendritic cells: role of IRAK-1. BMC Immunol. 9, 69. doi:10.1186/1471-2172-9-69
Almeida, I. C., and Gazzinelli, R. T. (2001). Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J. Leukoc. Biol. 70, 467–477.
Aravalli, R. N., Hu, S., Woods, J. P., and Lokensgard, J. R. (2008). Histoplasma capsulatum yeast phase-specific protein Yps3p induces toll-like receptor 2 signaling. J. Neuroinflammation 5, 30.
Archer, K. A., and Roy, C. R. (2006). MyD88-dependent responses involving toll-like receptor 2 are important for protection and clearance of Legionella pneumophila in a mouse model of legionnaires’ disease. Infect. Immun. 74, 3325–3333.
Ariza, M. E., Glaser, R., Kaumaya, P. T., Jones, C., and Williams, M. V. (2009). The EBV-encoded dUTPase activates NF-{kappa}B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 182, 851–859.
Babu, M. M., Priya, M. L., Selvan, A. T., Madera, M., Gough, J., Aravind, L., and Sankaran, K. (2006). A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. J. Bacteriol. 188, 2761–2773.
Bafica, A., Santiago, H. C., Goldszmid, R., Ropert, C., Gazzinelli, R. T., and Sher, A. (2006). Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J. Immunol. 177, 3515–3519.
Bafica, A., Scanga, C. A., Feng, C. G., Leifer, C., Cheever, A., and Sher, A. (2005). TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J. Exp. Med. 202, 1715–1724.
Barbalat, R., Lau, L., Locksley, R. M., and Barton, G. M. (2009). Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 10, 1200–1207.
Beutler, B., Jiang, Z., Georgel, P., Crozat, K., Croker, B., Rutschmann, S., Du, X., and Hoebe, K. (2006). Genetic analysis of host resistance: toll-like receptor signaling and immunity at large. Annu. Rev. Immunol. 24, 353–389.
Bhide, M. R., Mucha, R., Mikula, I. J. r., Kisova, L., Skrabana, R., Novak, M., and Mikula, I. S. r. (2009). Novel mutations in TLR genes cause hyporesponsiveness to Mycobacterium avium subsp. paratuberculosis infection. BMC Genet. 10, 21. doi:10.1186/1471-2156-10-21
Bieback, K., Lien, E., Klagge, I. M., Avota, E., Schneider-Schaulies, J., Duprex, W. P., Wagner, H., Kirschning, C. J., ter Meulen, V., and Schneider-Schaulies, S. (2002). Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 76, 8729–8736.
Birch, H. L., Alderwick, L. J., Appelmelk, B. J., Maaskant, J., Bhatt, A., Singh, A., Nigou, J., Eggeling, L., Geurtsen, J., and Besra, G. S. (2010). A truncated lipoglycan from mycobacteria with altered immunological properties. Proc. Natl. Acad. Sci. U.S.A. 107, 2634–2639.
Biswas, A., Banerjee, P., and Biswas, T. (2009). Porin of Shigella dysenteriae directly promotes toll-like receptor 2-mediated CD4+ T cell survival and effector function. Mol. Immunol. 46, 3076–3085.
Blasi, E., Mucci, A., Neglia, R., Pezzini, F., Colombari, B., Radzioch, D., Cossarizza, A., Lugli, E., Volpini, G., Del Giudice, G., and Peppoloni, S. (2005). Biological importance of the two toll-like receptors, TLR2 and TLR4, in macrophage response to infection with Candida albicans. FEMS Immunol. Med. Microbiol. 44, 69–79.
Blumenthal, A., Kobayashi, T., Pierini, L. M., Banaei, N., Ernst, J. D., Miyake, K., and Ehrt, S. (2009). RP105 facilitates macrophage activation by Mycobacterium tuberculosis lipoproteins. Cell Host Microbe 5, 35–46.
Bonfim, C. V., Mamoni, R. L., and Blotta, M. H. S. L. (2009). TLR-2, TLR-4 and Dectin-1 expression in human monocytes and neutrophils stimulated by Paracoccidioides brasiliensis. Med. Mycol. 47, 722.
Brown, R. A., Gralewski, J. H., Eid, A. J., Knoll, B. M., Finberg, R. W., and Razonable, R. R. (2010). R753Q single-nucleotide polymorphism impairs toll-like receptor 2 recognition of hepatitis C virus core and nonstructural 3 proteins. Transplantation 89, 811–815.
Brzezińska-Blaszczyk, E., and Wierzbicki, M. (2010). Mast cell toll-like receptors (TLRs). Postepy Hig. Med. Dosw. (Online) 64, 11–21.
Buwitt-Beckmann, U. (2005). TLR1- and TLR6-independent recognition of bacterial lipopeptides. J. Biol. Chem. 281, 9049–9057.
Calich, V. L., Pina, A., Felonato, M., Bernardino, S., Costa, T. A., and Loures, F. V. (2008). Toll-like receptors and fungal infections: the role of TLR2, TLR4 and MyD88 in paracoccidioidomycosis. FEMS Immunol. Med. Microbiol. 53, 1–7.
Campos, M. A., Closel, M., Valente, E. P., Cardoso, J. E., Akira, S., Alvarez-Leite, J. I., Ropert, C., and Gazzinelli, R. T. (2004). Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J. Immunol. 172, 1711–1718.
Cervantes-Barragán, L., Gil-Cruz, C., Pastelin-Palacios, R., Lang, K. S., Isibasi, A., Ludewig, B., and López-Macías, C. (2009). TLR2 and TLR4 signaling shapes specific antibody responses to Salmonella typhi antigens. Eur. J. Immunol. 39, 126–135.
Chai, L. Y., Netea, M. G., Vonk, A. G., and Kullberg, B. J. (2009a). Fungal strategies for overcoming host innate immune response. Med. Mycol. 47, 227–236.
Chai, L. Y., Kullberg, B. J., Vonk, A. G., Warris, A., Cambi, A., Latgé, J. P., Joosten, L. A., van der Meer, J. W., and Netea, M. G. (2009b). Modulation of toll-like receptor 2 (TLR2) and TLR4 responses by Aspergillus fumigatus. Infect. Immun. 77, 2184–2192.
Chang, S., Dolganiuc, A., and Szabo, G. (2007). Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 82, 479–487.
Chassin, C., Picardeau, M., Goujon, J. M., Bourhy, P., Quellard, N., Darche, S., Badell, E., d’Andon, M. F., Winter, N., Lacroix-Lamandé, S., Buzoni-Gatel, D., Vandewalle, A., and Werts, C. (2009). TLR4- and TLR2-mediated B cell responses control the clearance of the bacterial pathogen, leptospira interrogans. J. Immunol. 183, 2669–2677.
Chignard, M., Balloy, V., Sallenave, J. M., and Si-Tahar, M. (2007). Role of toll-like receptors in lung innate defense against invasive aspergillosis. Distinct impact in immunocompetent and immunocompromized hosts. Clin. Immunol. 124, 238–243.
Chung, H., Watanabe, T., Kudo, M., and Chiba, T. (2011). Correlation between hyporesponsiveness to toll-like receptor ligands and liver dysfunction in patients with chronic hepatitis C virus infection. J. Viral Hepat. 18, e561–e567.
Compton, T., Kurt-Jones, E. A., Boehme, K. W., Belko, J., Latz, E., Golenbock, D. T., and Finberg, R. W. (2003). Human cytomegalovirus activates inflammatory cytokine responses via CD14 and toll-like receptor 2. J. Virol. 77, 4588–4596.
Connell, T. D. (2007). Cholera toxin, LT-I, LT-IIa and LT-IIb: the critical role of ganglioside binding in immunomodulation by type I and type II heat-labile enterotoxins. Expert Rev. Vaccines 6, 821–834.
Cooper, A., Tal, G., Lider, O., and Shaul, Y. (2005). Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J. Immunol. 175, 3165–3176.
Corr, S. C., and O’Neill, L. A. J. (2009). Genetic variation in toll-like receptor signalling and the risk of inflammatory and immune diseases. J. Innate Immun. 1, 350–357.
Curtin, J. F., Liu, N., Candolfi, M., Xiong, W., Assi, H., Yagiz, K., Edwards, M. R., Michelsen, K. S., Kroeger, K. M., Liu, C., Muhammad, A. K., Clark, M. C., Arditi, M., Comin-Anduix, B., Ribas, A., Lowenstein, P. R., and Castro, M. G. (2009). HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 6, e1000010. doi:10.1371/journal.pmed.1000010
Deak, E., Jayakumar, A., Cho, K. W., Goldsmith-Pestana, K., Dondji, B., Lambris, J. D., and McMahon-Pratt, D. (2010). Murine visceral leishmaniasis: IgM and polyclonal B-cell activation lead to disease exacerbation. Eur. J. Immunol. 40, 1355–1368.
Debierre-Grockiego, F., Campos, M. A., Azzouz, N., Schmidt, J., Bieker, U., Resende, M. G., Mansur, D. S., Weingart, R., Schmidt, R. R., Golenbock, D. T., Gazzinelli, R. T., and Schwarz, R. T. (2007). Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J. Immunol. 179, 1129–1137.
Debierre-Grockiego, F., Niehus, S., Coddeville, B., Elass, E., Poirier, F., Weingart, R., Schmidt, R. R., Mazurier, J., Guérardel, Y., and Schwarz, R. T. (2010). Binding of Toxoplasma gondii glycosylphosphatidylinositols to galectin-3 is required for their recognition by macrophages. J. Biol. Chem. 285, 32744–32750.
Depaolo, R. W., Tang, F., Kim, I., Han, M., Levin, N., Ciletti, N., Lin, A., Anderson, D., Schneewind, O., and Jabri, B. (2008). Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe 4, 350–361.
Dessein, R., Gironella, M., Vignal, C., Peyrin-Biroulet, L., Sokol, H., Secher, T., Lacas-Gervais, S., Gratadoux, J. J., Lafont, F., Dagorn, J. C., Ryffel, B., Akira, S., Langella, P., Nùñez, G., Sirard, J. C., Iovanna, J., Simonet, M., and Chamaillard, M. (2009). Toll-like receptor 2 is critical for induction of Reg3β expression and intestinal clearance of Yersinia pseudotuberculosis. Gut 58, 771–776.
Dessing, M. C., Florquin, S., Paton, J. C., and van der Poll, T. (2008). Toll-like receptor 2 contributes to antibacterial defence against pneumolysin-deficient pneumococci. Cell. Microbiol. 10, 237–246.
Dickinson, G. S., Piccone, H., Sun, G., Lien, E., Gatto, L., and Alugupalli, K. R. (2010). Toll-like receptor 2 deficiency results in impaired antibody responses and septic shock during Borrelia hermsii infection. Infect. Immun. 78, 4579–4588.
Dietrich, N., Lienenklaus, S., Weiss, S., and Gekara, N. O. (2010). Murine toll-like receptor 2 activation induces type I interferon responses from endolysosomal compartments. PLoS ONE 5, e10250. doi:10.1371/journal.pone.0010250
Drage, M. G., Pecora, N. D., Hise, A. G., Febbraio, M., Silverstein, R. L., Golenbock, D. T., Boom, W. H., and Harding, C. V. (2009). TLR2 and its co-receptors determine responses of macrophages and dendritic cells to lipoproteins of Mycobacterium tuberculosis. Cell. Immunol. 258, 29–37.
Dulay, A. T., Buhimschi, C. S., Zhao, G., Oliver, E. A., Mbele, A., Jing, S., and Buhimschi, I. A. (2009). Soluble TLR2 is present in human amniotic fluid and modulates the intraamniotic inflammatory response to infection. J. Immunol. 182, 7244–7253.
Dunne, A., Carpenter, S., Brikos, C., Gray, P., Strelow, A., Wesche, H., Morrice, N., and O’Neill, L. A. (2010). IRAK1 and IRAK4 promote phosphorylation, ubiquitination and degradation of MyD88 adapter-like (MAL). J. Biol. Chem. 285, 18276–18282.
Duthie, M. S., Windish, H. P., Fox, C. B., and Reed, S. G. (2011). Use of defined TLR ligands as adjuvants within human vaccines. Immunol. Rev. 239, 178–196.
Erdman, L. K., Cosio, G., Helmers, A. J., Gowda, D. C., Grinstein, S., and Kain, K. C. (2009). CD36 and TLR interactions in inflammation and phagocytosis: implications for malaria. J. Immunol. 183, 6452–6459.
Erridge, C. (2010). Endogenous ligands of TLR2 and TLR4: agonists or assistants? J. Leukoc. Biol. 87, 989–999.
Farhat, K., Riekenberg, S., Heine, H., Debarry, J., Lang, R., Mages, J., Buwitt-Beckmann, U., Röschmann, K., Jung, G., Wiesmüller, K. H., and Ulmer, A. J. (2008). Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J. Leukoc. Biol. 83, 692–701.
Fedele, G., Spensieri, F., Palazzo, R., Nasso, M., Cheung, G. Y., Coote, J. G., and Ausiello, C. M. (2010). Bordetella pertussis commits human dendritic cells to promote a Th1/Th17 response through the activity of adenylate cyclase toxin and MAPK-pathways. PLoS ONE 5, e8734. doi:10.1371/journal.pone.0008734
Ferwerda, G., Meyer-Wentrup, F., Kullberg, B. J., Netea, M. G., and Adema, G. J. (2008). Dectin-1 synergizes with TLR2 and TLR4 for cytokine production in human primary monocytes and macrophages. Cell. Microbiol. 10, 2058–2066.
Flo, T. H., Halaas, O., Torp, S., Ryan, L., Lien, E., Dybdahl, B., Sundan, A., and Espevik, T. (2001). Differential expression of toll-like receptor 2 in human cells. J. Leukoc. Biol. 69, 474–481.
Fonseca, F. L., Nohara, L. L., Cordero, R. J., Frases, S., Casadevall, A., Almeida, I. C., Nimrichter, L., and Rodrigues, M. L. (2010). Immunomodulatory effects of serotype B glucuronoxylomannan from Cryptococcus gattii correlate with polysaccharide diameter. Infect. Immun. 78, 3861–3870.
Funderburg, N., Lederman, M. M., Feng, Z., Drage, M. G., Jadlowsky, J., Harding, C. V., Weinberg, A., and Sieg, S. F. (2007). Human β-defensin-3 activates professional antigen-presenting cells via toll-like receptors 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 104, 18631–18635.
Fuse, E. T., Tateda, K., Kikuchi, Y., Matsumoto, T., Gondaira, F., Azuma, A., Kudoh, S., Standiford, T. J., and Yamaguchi, K. (2007). Role of toll-like receptor 2 in recognition of Legionella pneumophila in a murine pneumonia model. J. Med. Microbiol. 56(Pt 3), 305–312.
Gantner, B. N., Simmons, R. M., Canavera, S. J., Akira, S., and Underhill, D. M. (2003). Collaborative induction of inflammatory responses by Dectin-1 and toll-like receptor 2. J. Exp. Med. 197, 1107–1117.
Gay, N. J., and Gangloff, M. (2007). Structure and function of toll receptors and their ligands. Annu. Rev. Biochem. 76, 141–165.
Gerold, G., Ajaj, K. A., Bienert, M., Laws, H. J., Zychlinsky, A., and de Diego, J. L. (2008). A toll-like receptor 2-integrin [beta]3 complex senses bacterial lipopeptides via vitronectin. Nat. Immunol. 9, 761–768.
Ghosh, T. K., Mickelson, D. J., Solberg, J. C., Lipson, K. E., Inglefield, J. R., and Alkan, S. S. (2007). TLR-TLR cross talk in human PBMC resulting in synergistic and antagonistic regulation of type-1 and 2 interferons, IL-12 and TNF-[alpha]. Int. Immunopharmacol. 7, 1111–1121.
Gibson, D. L., Ma, C., Rosenberger, C. M., Bergstrom, K. S., Valdez, Y., Huang, J. T., Khan, M. A., and Vallance, B. A. (2008). Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell. Microbiol. 10, 388–403.
Gil, M. L., Fradelizi, D., and Gozalbo, D. (2005). TLR2: for or against Candida albicans? Trends Microbiol. 13, 298–299.
Gil, M. L., and Gozalbo, D. (2006). TLR2, but not TLR4, triggers cytokine production by murine cells in response to Candida albicans yeasts and hyphae. Microbes Infect. 8, 2299–2304.
Grabiec, A., Meng, G., Fichte, S., Bessler, W., Wagner, H., and Kirschning, C. J. (2004). Human but not murine toll-like receptor 2 discriminates between tri-palmitoylated and tri-lauroylated peptides. J. Biol. Chem. 279, 48004–48012.
Greene, J. A., Moormann, A. M., Vulule, J., Bockarie, M. J., Zimmerman, P. A., and Kazura, J. W. (2009). Toll-like receptor polymorphisms in malaria-endemic populations. Malar. J. 8, 50.
Gringhuis, S. I., den Dunnen, J., Litjens, M., van der Vlist, M., Wevers, B., Bruijns, S. C., and Geijtenbeek, T. B. (2009). Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-[kappa]B activation through Raf-1 and Syk. Nat. Immunol. 10, 203–213.
Guan, Y., Ranoa, D. R., Jiang, S., Mutha, S. K., Li, X., Baudry, J., and Tapping, R. I. (2010). Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J. Immunol. 184, 5094–5103.
Guo, B., Chang, E. Y., and Cheng, G. (2008). The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J. Clin. Invest. 118, 1680–1690.
Gutzeit, C., Raftery, M. J., Peiser, M., Tischer, K. B., Ulrich, M., Eberhardt, M., Stockfleth, E., Giese, T., Sauerbrei, A., Morita, C. T., and Schönrich, G. (2010). Identification of an important immunological difference between virulent varicella-zoster virus and its avirulent vaccine: viral disruption of dendritic cell instruction. J. Immunol. 185, 488–497.
Han, F., Yu, H., Tian, C., Li, S., Jacobs, M. R., Benedict-Alderfer, C., and Zheng, Q. Y. (2009). Role for toll-like receptor 2 in the immune response to Streptococcus pneumoniae infection in mouse otitis media. Infect. Immun. 77, 3100–3108.
Hasan, U., Chaffois, C., Gaillard, C., Saulnier, V., Merck, E., Tancredi, S., Guiet, C., Brière, F., Vlach, J., Lebecque, S., Trinchieri, G., and Bates, E. E. (2005). Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J. Immunol. 174, 2942–2950.
Hashimoto, M., Furuyashiki, M., Kaseya, R., Fukada, Y., Akimaru, M., Aoyama, K., Okuno, T., Tamura, T., Kirikae, T., Kirikae, F., Eiraku, N., Morioka, H., Fujimoto, Y., Fukase, K., Takashige, K., Moriya, Y., Kusumoto, S., and Suda, Y. (2007). Evidence of immunostimulating lipoprotein existing in the natural lipoteichoic acid fraction. Infect. Immun. 75, 1926–1932.
Hashimoto, M., Tawaratsumida, K., Kariya, H., Aoyama, K., Tamura, T., and Suda, Y. (2006). Lipoprotein is a predominant toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int. Immunol. 18, 355–362.
Heinsbroek, S. E., Taylor, P. R., Martinez, F. O., Martinez-Pomares, L., Brown, G. D., and Gordon, S. (2008). Stage-specific sampling by pattern recognition receptors during Candida albicans phagocytosis. PLoS Pathog. 4, e1000218.
Hernandez-Pando, R., and Rook, G. A. (1994). The role of TNF-alpha in T-cell-mediated inflammation depends on the Th1/Th2 cytokine balance. Immunology 82, 591–595.
Hildebrand, D., Heeg, K., and Kubatzky, K. F. (2009). The Pasteurella Multocida toxin (PMT) induced differentiation of haematopoietic progenitor cells in macrophages and B cells. Cell Commun. Signal. 7(Suppl. 1), :A47.
Hirata, N., Yanagawa, Y., Ebihara, T., Seya, T., Uematsu, S., Akira, S., Hayashi, F., Iwabuchi, K., and Onoé, K. (2008). Selective synergy in anti-inflammatory cytokine production upon cooperated signaling via TLR4 and TLR2 in murine conventional dendritic cells. Mol. Immunol. 45, 2734–2742.
Hise, A. G., Tomalka, J., Ganesan, S., Patel, K., Hall, B. A., Brown, G. D., and Fitzgerald, K. A. (2009). An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5, 487–497.
Hoebe, K., Georgel, P., Rutschmann, S., Du, X., Mudd, S., Crozat, K., Sovath, S., Shamel, L., Hartung, T., Zähringer, U., and Beutler, B. (2005). CD36 is a sensor of diacylglycerides. Nature 433, 523–527.
Hohl, T. M., Rivera, A., and Pamer, E. G. (2006). Immunity to fungi. Curr. Opin. Immunol. 18, 465–472.
Ikeda, Y., Adachi, Y., Ishii, T., Miura, N., Tamura, H., and Ohno, N. (2008). Dissociation of toll-like receptor 2-mediated innate immune response to zymosan by organic solvent-treatment without loss of Dectin-1 reactivity. Biol. Pharm. Bull. 31, 13–18.
Ip, W. K., Takahashi, K., Ezekowitz, R. A., and Stuart, L. M. (2009). Mannose-binding lectin and innate immunity. Immunol. Rev. 230, 9–21.
Ip, W. K., Takahashi, K., Moore, K. J., Stuart, L. M., and Ezekowitz, R. A. (2008). Mannose-binding lectin enhances toll-like receptors 2 and 6 signaling from the phagosome. J. Exp. Med. 205, 169–181.
Jimenez-Dalmaroni, M. J., Xiao, N., Corper, A. L., Verdino, P., Ainge, G. D., Larsen, D. S., Painter, G. F., Rudd, P. M., Dwek, R. A., Hoebe, K., Beutler, B., and Wilson, I. A. (2009). Soluble CD36 ectodomain binds negatively charged diacylglycerol ligands and acts as a co-receptor for TLR2. PLoS ONE 4, e7411.
Jin, M. S., Kim, S. E., Heo, J. Y., Lee, M. E., Kim, H. M., Paik, S. G., Lee, H., and Lee, J. O. (2007). Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082.
Joffre, O., Nolte, M. A., Spörri, R., and Reis e Sousa, C. (2009). Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol. Rev. 227, 234–247.
Jouault, T., Ibata-Ombetta, S., Takeuchi, O., Trinel, P. A., Sacchetti, P., Lefebvre, P., Akira, S., and Poulain, D. (2003). Candida albicans phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 188, 165–172.
Ju, C. H., Chockalingam, A., and Leifer, C. A. (2009). Early response of mucosal epithelial cells during Toxoplasma gondii infection. J. Immunol. 183, 7420–7427.
Kane, C. M., Jung, E., and Pearce, E. J. (2008). Schistosoma mansoni egg antigen-mediated modulation of toll-like receptor (TLR)-induced activation occurs independently of TLR2, TLR4, and MyD88. Infect. Immun. 76, 5754–5759.
Kang, J. Y., Nan, X., Jin, M. S., Youn, S. J., Ryu, Y. H., Mah, S., Han, S. H., Lee, H., Paik, S. G., and Lee, J. O. (2009). Recognition of lipopeptide patterns by toll-like receptor 2-toll-like receptor 6 heterodimer. Immunity 31, 873–884.
Kang, S. H., Abdel-Massih, R. C., Brown, R. A., Dierkhising, R. A., Kremers, W. K., and Razonable, R. R. (2012). Homozygosity for the toll-like receptor 2 R753Q single-nucleotide polymorphism is a risk factor for cytomegalovirus disease after liver transplantation. J. Infect. Dis. 205, 639–646.
Karimi, O., Ouburg, S., de Vries, H. J., Peña, A. S., Pleijster, J., Land, J. A., and Morré, S. A. (2009). TLR2 haplotypes in the susceptibility to and severity of chlamydia trachomatis infections in Dutch women. Drugs Today 45(Suppl. B) 67–74.
Kavoosi, G., Ardestani, S. K., Kariminia, A., and Alimohammadian, M. H. (2010). Leishmania major lipophosphoglycan: discrepancy in toll-like receptor signaling. Exp. Parasitol. 124, 214–218.
Kesh, S., Mensah, N. Y., Peterlongo, P., Jaffe, D., Hsu, K., van den Brink, M., O’Reilly, R., Pamer, E., Satagopan, J., and Papanicolaou, G. A. (2005). TLR1 and TLR6 polymorphisms are associated with susceptibility to invasive aspergillosis after allogeneic stem cell transplantation. Ann. N. Y. Acad. Sci. 1062, 95–103.
Khoo, A. L., Chai, L. Y., Koenen, H. J., Kullberg, B. J., Joosten, I., van der Ven, A. J., and Netea, M. G. (2011). 1,25-Dihydroxyvitamin D3 modulates cytokine production induced by Candida albicans: impact of seasonal variation of immune responses. J. Infect. Dis. 203, 122–130.
Khor, C. C., Chapman, S. J., Vannberg, F. O., Dunne, A., Murphy, C., Ling, E. Y., Frodsham, A. J., Walley, A. J., Kyrieleis, O., Khan, A., Aucan, C., Segal, S., Moore, C. E., Knox, K., Campbell, S. J., Lienhardt, C., Scott, A., Aaby, P., Sow, O. Y., Grignani, R. T., Sillah, J., Sirugo, G., Peshu, N., Williams, T. N., Maitland, K., Davies, R. J., Kwiatkowski, D. P., Day, N. P., Yala, D., Crook, D. W., Marsh, K., Berkley, J. A., O’Neill, L. A., and Hill, A. V. (2007). A mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat. Genet. 39, 523–528.
Klouwenberg, K. P., Tan, L., Werkman, W., van Bleek, G. M., and Coenjaerts, F. (2009). The role of toll-like receptors in regulating the immune response against respiratory syncytial virus. Crit. Rev. Immunol. 29, 531–550.
Koyama, S., Ishii, K. J., Coban, C., and Akira, S. (2008). Innate immune response to viral infection. Cytokine 43, 336–341.
Krug, A., Luker, G. D., Barchet, W., Leib, D. A., Akira, S., and Colonna, M. (2004). Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103, 1433–1437.
Kumar, H., Kawai, T., and Akira, S. (2009). Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388, 621–625.
Lee, B. C., Kim, M. S., Choi, S. H., and Kim, T. S. (2010). Involvement of capsular polysaccharide via a TLR2/NF-kappaB pathway in Vibrio vulnificus-induced IL-8 secretion of human intestinal epithelial cells. Int. J. Mol. Med. 25, 581–591.
Lee, C. C., Avalos, A. M., and Ploegh, H. L. (2012). Accessory molecules for toll-like receptors and their function. Nat. Rev. Immunol. 12, 168–179.
Leendertse, M., Willems, R. J., Giebelen, I. A., van den Pangaart, P. S., Wiersinga, W. J., de Vos, A. F., Florquin, S., Bonten, M. J., and van der Poll, T. (2008). TLR2-dependent MyD88 signaling contributes to early host defense in murine Enterococcus faecium peritonitis. J. Immunol. 180, 4865–4874.
Letiembre, M., Echchannaoui, H., Ferracin, F., Rivest, S., and Landmann, R. (2005). Toll-like receptor-2 deficiency is associated with enhanced brain TNF gene expression during pneumococcal meningitis. J. Neuroimmunol. 168, 21–33.
Leulier, F., and Lemaitre, B. (2008). Toll-like receptors – taking an evolutionary approach. Nat. Rev. Genet. 9, 165–178.
Li, M., Chen, Q., Shen, Y., and Liu, W. (2009). Candida albicans phospholipomannan triggers inflammatory responses of human keratinocytes through toll-like receptor 2. Exp. Dermatol. 18, 603–610.
Liadaki, K., Petinaki, E., Skoulakis, C., Tsirevelou, P., Klapsa, D., Germenis, A. E., and Speletas, M. (2011). Toll-like receptor 4 gene (TLR4), but not TLR2, polymorphisms modify the risk of tonsillar disease due to Streptococcus pyogenes and Haemophilus influenzae. Clin. Vaccine Immunol. 18, 217–222.
Liang, S., Wang, M., Triantafilou, K., Triantafilou, M., Nawar, H. F., Russell, M. W., Connell, T. D., and Hajishengallis, G. (2007a). The A subunit of type IIb enterotoxin (LT-IIb) suppresses the proinflammatory potential of the B subunit and its ability to recruit and interact with TLR2. J. Immunol. 178, 4811–4819.
Liang, S., Wang, M., Tapping, R. I., Stepensky, V., Nawar, H. F., Triantafilou, M., Triantafilou, K., Connell, T. D., and Hajishengallis, G. (2007b). Ganglioside GD1a is an essential coreceptor for toll-like receptor 2 signaling in response to the B subunit of type IIb enterotoxin. J. Biol. Chem. 282, 7532–7542.
Liljeroos, M., Vuolteenaho, R., Morath, S., Hartung, T., Hallman, M., and Ojaniemi, M. (2007). Bruton’s tyrosine kinase together with PI 3-kinase are part of toll-like receptor 2 multiprotein complex and mediate LTA induced toll-like receptor 2 responses in macrophages. Cell. Signal. 19, 625–633.
Liljeroos, M., Vuolteenaho, R., Rounioja, S., Henriques-Normark, B., Hallman, M., and Ojaniemi, M. (2008). Bacterial ligand of TLR2 signals stat activation via induction of IRF1/2 and interferon-alpha production. Cell. Signal. 20, 1873–1881.
Lima, G. K., Zolini, G. P., Mansur, D. S., Freire Lima, B. H., Wischhoff, U., Astigarraga, R. G., Dias, M. F., das Graças Almeida Silva, M., Béla, S. R., do Valle Antonelli, L. R., Arantes, R. M., Gazzinelli, R. T., Báfica, A., Kroon, E. G., and Campos, M. A. (2010). Toll-like receptor (TLR) 2 and TLR9 expressed in trigeminal ganglia are critical to viral control during herpes simplex virus 1 infection. Am. J. Pathol. 177, 2433–2445.
Lin, J. S., Huang, J. H., Hung, L. Y., Wu, S. Y., and Wu-Hsieh, B. A. (2010). Distinct roles of complement receptor 3, Dectin-1, and sialic acids in murine macrophage interaction with histoplasma yeast. J. Leukoc. Biol. 88, 95–106.
Liu, G., Yang, Y., Yang, S., Banerjee, S., De Freitas, A., Friggeri, A., Davis, K. I., and Abraham, E. (2011). The receptor for urokinase regulates TLR2 mediated inflammatory responses in neutrophils. PLoS ONE 6, e25843. doi:10.1371/journal.pone.0025843
Liu, X., Wetzler, L. M., and Massari, P. (2008). The PorB Porin from commensal Neisseria lactamica induces Th1 and Th2 immune responses to ovalbumin in mice and is a potential immune adjuvant. Vaccine 26, 786–796.
Liu, Y. C., Simmons, D. P., Li, X., Abbott, D. W., Boom, W. H., and Harding, C. V. (2012). TLR2 signaling depletes IRAK1 and inhibits induction of type I IFN by TLR7/9. J. Immunol. 188, 1019–1026.
Long, E. M., Millen, B., Kubes, P., and Robbins, S. M. (2009). Lipoteichoic acid induces unique inflammatory responses when compared to other toll-like receptor 2 ligands. PLoS ONE 4, e5601. doi:10.1371/journal.pone.0005601
Lorenz, E., Chemotti, D. C., Jiang, A. L., and McDougal, L. D. (2005). Differential involvement of toll-like receptors 2 and 4 in the host response to acute respiratory infections with wild-type and mutant Haemophilus influenzae strains. Infect. Immun. 73, 2075–2082.
Lotter, H., González-Roldán, N., Lindner, B., Winau, F., Isibasi, A., Moreno-Lafont, M., Ulmer, A. J., Holst, O., Tannich, E., and Jacobs, T. (2009). Natural killer T cells activated by a lipopeptidophosphoglycan from Entamoeba histolytica are critically important to control amebic liver abscess. PLoS Pathog. 5, e1000434. doi:10.1371/journal.ppat.1000434
Loures, F. V., Pina, A., Felonato, M., and Calich, V. L. (2009). TLR2 is a negative regulator of Th17 cells and tissue pathology in a pulmonary model of fungal infection. J. Immunol. 183, 1279–1290.
Love, W., Dobbs, N., Tabor, L., and Simecka, J. W. (2010). Toll-like receptor 2 (TLR2) plays a major role in innate resistance in the lung against murine mycoplasma. PLoS ONE 5, e10739. doi:10.1371/journal.pone.0010739
Maizels, R. M., and Yazdanbakhsh, M. (2003). Immune regulation by helminth parasites: cellular and molecular mechanisms. Nat. Rev. Immunol. 3, 733–744.
Marre, M. L., Petnicki-Ocwieja, T., DeFrancesco, A. S., Darcy, C. T., and Hu, L. T. (2010). Human integrin α3β1 regulates TLR2 recognition of lipopeptides from endosomal compartments. PLoS ONE 5, e12871. doi:10.1371/journal.pone.0012871
Martinez, J., Huang, X., and Yang, Y. (2010). Direct TLR2 signaling is critical for NK cell activation and function in response to Vaccinia viral infection. PLoSPathogen 6, e1000811.
Massari, P., Henneke, P., Ho, Y., Latz, E., Golenbock, D. T., and Wetzler, L. M. (2002). Cutting edge: immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J. Immunol. 168, 1533–1537.
Massari, P., Visintin, A., Gunawardana, J., Halmen, K. A., King, C. A., Golenbock, D. T., and Wetzler, L. M. (2006). Meningococcal porin PorB binds to TLR2 and requires TLR1 for signaling. J. Immunol. 176, 2373–2380.
Matera, G., Liberto, M. C., Joosten, L. A., Vinci, M., Quirino, A., Pulicari, M. C., Kullberg, B. J., Van der Meer, J. W., Netea, M. G., and Focà, A. (2008). The Janus face of Bartonella quintana recognition by toll-like receptors (TLRs): a review. Eur. Cytokine Netw. 19, 113–118.
Matsushima, N., Tanaka, T., Enkhbayar, P., Mikami, T., Taga, M., Yamada, K., and Kuroki, Y. (2007). Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genomics 8, 124. doi:10.1186/1471-2164-8-124
McKenzie, C. G., Koser, U., Lewis, L. E., Bain, J. M., Mora-Montes, H. M., Barker, R. N., Gow, N. A., and Erwig, L. P. (2010). Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infect. Immun. 78, 1650–1658.
Moens, L., Verhaegen, J., Pierik, M., Vermeire, S., De Boeck, K., Peetermans, W. E., and Bossuyt, X. (2007). Toll-like receptor 2 and toll-like receptor 4 polymorphisms in invasive pneumococcal disease. Microbes Infect. 9, 15–20.
Monteiro, A. C., Schmitz, V., Svensjo, E., Gazzinelli, R. T., Almeida, I. C., Todorov, A., de Arruda, L. B., Torrecilhas, A. C., Pesquero, J. B., Morrot, A., Bouskela, E., Bonomo, A., Lima, A. P., Müller-Esterl, W., and Scharfstein, J. (2006). Cooperative activation of TLR2 and Bradykinin B2 receptor is required for induction of type 1 immunity in a mouse model of subcutaneous infection by Trypanosoma cruzi. J. Immunol. 177, 6325–6335.
Moore, C. E., Segal, S., Berendt, A. R., Hill, A. V., and Day, N. P. (2004). Lack of association between toll-like receptor 2 polymorphisms and susceptibility to severe disease caused by Staphylococcus aureus. Clin. Diagn. Lab. Immunol. 11, 1194–1197.
Moranta, D., Regueiro, V., March, C., Llobet, E., Margareto, J., Larrarte, E., Garmendia, J., and Bengoechea, J. A. (2010). Klebsiella pneumoniae capsule polysaccharide impedes the expression of beta-defensins by airway epithelial cells. Infect. Immun. 78, 1135–1146.
Moriyama, M., Kato, N., Otsuka, M., Shao, R. X., Taniguchi, H., Kawabe, T., and Omata, M. (2007). Interferon-beta is activated by hepatitis C virus NS5B and inhibited by NS4A, NS4B, and NS5A. Hepatol. Int. 1, 302–310.
Mullaly, S. C., and Kubes, P. (2006). The role of TLR2 in vivo following challenge with Staphylococcus aureus and prototypic ligands. J. Immunol. 177, 8154–8163.
Müller, U., Steinhoff, U., Reis, L. F., Hemmi, S., Pavlovic, J., Zinkernagel, R. M., and Aguet, M. (1994). Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921.
Müller-Anstett, M. A., Müller, P., Albrecht, T., Nega, M., Wagener, J., Gao, Q., Kaesler, S., Schaller, M., Biedermann, T., and Götz, F. (2010). Staphylococcal peptidoglycan co-localizes with Nod2 and TLR2 and activates innate immune response via both receptors in primary murine keratinocytes. PLoS ONE 5, e13153.
Mun, H. S., Aosai, F., Norose, K., Chen, M., Piao, L. X., Takeuchi, O., Akira, S., Ishikura, H., and Yano, A. (2003). TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int. Immunol. 15, 1081–1087.
Munford, R. S., and Varley, A. W. (2006). Shield as signal: lipopolysaccharides and the evolution of immunity to Gram-negative bacteria. PLoS Pathog. 2, e67. doi:10.1371/journal.ppat.0020067
Murawski, M. R., Bowen, G. N., Cerny, A. M., Anderson, L. J., Haynes, L. M., Tripp, R. A., Kurt-Jones, E. A., and Finberg, R. W. (2009). Respiratory syncytial virus activates innate immunity through toll-like receptor 2. J. Virol. 83, 1492–1500.
Nakamura, K., Miyagi, K., Koguchi, Y., Kinjo, Y., Uezu, K., Kinjo, T., Akamine, M., Fujita, J., Kawamura, I., Mitsuyama, M., Adachi, Y., Ohno, N., Takeda, K., Akira, S., Miyazato, A., Kaku, M., and Kawakami, K. (2006). Limited contribution of toll-like receptor 2 and 4 to the host response to a fungal infectious pathogen, Cryptococcus neoformans. FEMS Immunol. Med. Microbiol. 47, 148–154.
Nakamura, K., Miyazato, A., Koguchi, Y., Adachi, Y., Ohno, N., Saijo, S., Iwakura, Y., Takeda, K., Akira, S., Fujita, J., Ishii, K., Kaku, M., and Kawakami, K. (2008). Toll-like receptor 2 (TLR2) and Dectin-1 contribute to the production of IL-12p40 by bone marrow-derived dendritic cells infected with Penicillium marneffei. Microbes Infect. 10, 1223–1227.
Nakata, T., Yasuda, M., Fujita, M., Kataoka, H., Kiura, K., Sano, H., and Shibata, K. (2006). CD14 directly binds to triacylated lipopeptides and facilitates recognition of the lipopeptides by the receptor complex of toll-like receptors 2 and 1 without binding to the complex. Cell. Microbiol. 8, 1899–1909.
Natsuka, M., Uehara, A., Yang, S., Echigo, S., and Takada, H. (2008). A polymer-type water-soluble peptidoglycan exhibited both toll-like receptor 2- and NOD2-agonistic activities, resulting in synergistic activation of human monocytic cells. Innate Immun. 14, 298–308.
Netea, M. G., Brown, G. D., Kullberg, B. J., and Gow, N. A. (2008). An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 6, 67–78.
Netea, M. G., Kullberg, B. J., de Jong, D. J., Franke, B., Sprong, T., Naber, T. H., Drenth, J. P., and Van der Meer, J. W. (2004a). NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur. J. Immunol. 34, 2052–2059.
Netea, M. G., Sutmuller, R., Hermann, C., Van der Graaf, C. A., Van der Meer, J. W., van Krieken, J. H., Hartung, T., Adema, G., and Kullberg, B. J. (2004b). Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J. Immunol. 172, 3712–3718.
Netea, M. G., van der Meer, J. W., and Kullberg, B. J. (2006). Both TLR2 and TLR4 are involved in the recognition of Candida albicans. Reply to ‘TLR2, but not TLR4, triggers cytokine production by murine cells in response to Candida albicans yeasts and hyphae’ by Gil and Gozalbo, microbes and infection 8 (2006) 2823–2824. Microbes Infect. 8, 2821–2822.
Netea, M. G., Warris, A., Van der Meer, J. W., Fenton, M. J., Verver-Janssen, T. J., Jacobs, L. E., Andresen, T., Verweij, P. E., and Kullberg, B. J. (2003). Aspergillus fumigatus evades immune recognition during germination through loss of toll-like receptor-4-mediated signal transduction. J. Infect. Dis. 188, 320–326.
Oliveira, S. C., de Oliveira, F. S., Macedo, G. C., de Almeida, L. A., and Carvalho, N. B. (2008). The role of innate immune receptors in the control of Brucella abortus infection: toll-like receptors and beyond. Microbes Infect. 10, 1005–1009.
O’Neill, L. A. (2008). When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity 29, 12–20.
Patel, S. N., Lu, Z., Ayi, K., Serghides, L., Gowda, D. C., and Kain, K. C. (2007). Disruption of CD36 impairs cytokine response to Plasmodium falciparum glycosylphosphatidylinositol and confers susceptibility to severe and fatal malaria in vivo. J. Immunol. 178, 3954–3961.
Peek, R. M. Jr., Fiske, C., and Wilson, K. T. (2010). Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 90, 831–858.
Poncini, C. V., Giménez, G., Pontillo, C. A., Alba-Soto, C. D., de Isola, E. L., Piazzón, I., and Cappa, S. M. (2010). Central role of extracellular signal-regulated kinase and toll-like receptor 4 in IL-10 production in regulatory dendritic cells induced by Trypanosoma cruzi. Mol. Immunol. 47, 1981–1988.
Pore, D., Mahata, N., Pal, A., and Chakrabarti, M. K. (2010). 34 kDa MOMP of Shigella flexneri promotes TLR2 mediated macrophage activation with the engagement of NF-kappaB and P38 MAP kinase signaling. Mol. Immunol. 47, 1739–1746.
Querec, T., Bennouna, S., Alkan, S., Laouar, Y., Gorden, K., Flavell, R., Akira, S., Ahmed, R., and Pulendran, B. (2006). Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 203, 413–424.
Quevedo-Diaz, M. A., Song, C., Xiong, Y., Chen, H., Wahl, L. M., Radulovic, S., and Medvedev, A. E. (2010). Involvement of TLR2 and TLR4 in cell responses to Rickettsia akari. J. Leukoc. Biol. 88, 675–685.
Quigley, M., Martinez, J., Huang, X., and Yang, Y. (2009). A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following Vaccinia viral infection. Blood 113, 2256–2264.
Raby, A. C., Le Bouder, E., Colmont, C., Davies, J., Richards, P., Coles, B., George, C. H., Jones, S. A., Brennan, P., Topley, N., and Labéta, M. O. (2009). Soluble TLR2 reduces inflammation without compromising bacterial clearance by disrupting TLR2 triggering. J. Immunol. 183, 506–517.
Rappleye, C. A., Eissenberg, L. G., and Goldman, W. E. (2007). Histoplasma capsulatum α-(1,3)-glucan blocks innate immune recognition by the β-glucan receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 1366–1370.
Rehli, M. (2002). Of mice and men: species variations of toll-like receptor expression. Trends Immunol. 23, 375–378.
Riordan, S. M., Skinner, N. A., Kurtovic, J., Locarnini, S., McIver, C. J., Williams, R., and Visvanathan, K. (2006). Toll-like receptor expression in chronic hepatitis C: correlation with pro-inflammatory cytokine levels and liver injury. Inflamm. Res. 55, 279–285.
Rizzardi, G. P., Marriott, J. B., Cookson, S., Lazzarin, A., Dalgleish, A. G., and Barcellini, W. (1998). Tumour necrosis factor (TNF) and TNF-related molecules in HIV-1+ individuals: relationship with in vitro Thl/Th2-type response. Clin. Exp. Immunol. 114, 61–65.
Rock, F. L., Hardiman, G., Timans, J. C., Kastelein, R. A., and Bazan, J. F. (1998). A family of human receptors structurally related to Drosophila toll. Proc. Natl. Acad. Sci. U.S.A. 95, 588–593.
Ryu, Y. H., Baik, J. E., Yang, J. S., Kang, S. S., Im, J., Yun, C. H., Kim, D. W., Lee, K., Chung, D. K., Ju, H. R., and Han, S. H. (2009). Differential Immunostimulatory effects of Gram-positive bacteria due to their lipoteichoic acids. Int. Immunopharmacol. 9, 127–133.
Sacks, D., and Sher, A. (2002). Evasion of innate immunity by parasitic protozoa. Nat. Immunol. 3, 1041–1047.
Schaaf, B., Luitjens, K., Goldmann, T., van Bremen, T., Sayk, F., Dodt, C., Dalhoff, K., and Droemann, D. (2009). Mortality in human sepsis is associated with downregulation of toll-like receptor 2 and CD14 expression on blood monocytes. Diagn. Pathol. 4, 12.
Scheibner, K. A., Lutz, M. A., Boodoo, S., Fenton, M. J., Powell, J. D., and Horton, M. R. (2006). Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 177, 1272–1281.
Seibert, S. A., Mex, P., Köhler, A., Kaufmann, S. H., and Mittrücker, H. W. (2010). TLR2-, TLR4- and Myd88-independent acquired humoral and cellular immunity against Salmonella enterica Serovar typhimurium. Immunol. Lett. 127, 126–134.
Seo, H. S., and Nahm, M. H. (2009). Lipoprotein lipase and hydrofluoric acid deactivate both bacterial lipoproteins and lipoteichoic acids, but platelet-activating factor-acetylhydrolase degrades only lipoteichoic acids. Clin. Vaccine Immunol. 16, 1187–1195.
Shey, M. S., Randhawa, A. K., Bowmaker, M., Smith, E., Scriba, T. J., de Kock, M., Mahomed, H., Hussey, G., Hawn, T. R., and Hanekom, W. A. (2010). Single nucleotide polymorphisms in toll-like receptor 6 are associated with altered lipopeptide- and mycobacteria-induced interleukin-6 secretion. Genes Immun. 11, 561–572.
Shin, J. E., Kim, Y. S., Oh, J. E., Min, B. M., and Choi, Y. (2010). Treponema denticola suppresses expression of human {beta}-defensin-3 in gingival epithelial cells through inhibition of the toll-like receptor 2 axis. Infect. Immun. 78, 672–679.
Silhavy, T. J., Kahne, D., and Walker, S. (2010). The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2, a000414.
Sjölinder, H., Mogensen, T. H., Kilian, M., Jonsson, A. B., and Paludan, S. R. (2008). Important role for toll-like receptor 9 in host defense against meningococcal sepsis. Infect. Immun. 76, 5421–5428.
Slevogt, H., Seybold, J., Tiwari, K. N., Hocke, A. C., Jonatat, C., Dietel, S., Hippenstiel, S., Singer, B. B., Bachmann, S., Suttorp, N., and Opitz, B. (2007). Moraxella catarrhalis is internalized in respiratory epithelial cells by a trigger-like mechanism and initiates a TLR2- and partly NOD1-dependent inflammatory immune response. Cell. Microbiol. 9, 694–707.
Sorensen, L. N., Reinert, L. S., Malmgaard, L., Bartholdy, C., Thomsen, A. R., and Paludan, S. R. (2008). TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J. Immunol. 181, 8604–8612.
Spiller, S., Dreher, S., Meng, G., Grabiec, A., Thomas, W., Hartung, T., Pfeffer, K., Hochrein, H., Brade, H., Bessler, W., Wagner, H., and Kirschning, C. J. (2007). Cellular recognition of trimyristoylated peptide or enterobacterial lipopolysaccharide via both TLR2 and TLR4. J. Biol. Chem. 282, 13190–13198.
Spiller, S., Elson, G., Ferstl, R., Dreher, S., Mueller, T., Freudenberg, M., Daubeuf, B., Wagner, H., and Kirschning, C. J. (2008). TLR4-induced IFN- production increases TLR2 sensitivity and drives Gram-negative sepsis in mice. J. Exp. Med. 205, 2177–2177.
Stewart, C. R., Stuart, L. M., Wilkinson, K., van Gils, J. M., Deng, J., Halle, A., Rayner, K. J., Boyer, L., Zhong, R., Frazier, W. A., Lacy-Hulbert, A., El Khoury, J., Golenbock, D. T., and Moore, K. J. (2010). CD36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, U155–U75.
Stuart, L. M., Deng, J., Silver, J. M., Takahashi, K., Tseng, A. A., Hennessy, E. J., Ezekowitz, R. A., and Moore, K. J. (2005). Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J. Cell Biol. 170, 477–485.
Sutherland, A. M., Walley, K. R., and Russell, J. A. (2005). Polymorphisms in CD14, mannose-binding lectin, and toll-like receptor-2 are associated with increased prevalence of infection in critically Ill adults. Crit. Care Med. 33, 638–644.
Szomolanyi-Tsuda, E., Liang, X., Welsh, R. M., Kurt-Jones, E. A., and Finberg, R. W. (2006). Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J. Virol. 80, 4286–4291.
Takeuchi, O., Hoshino, K., and Akira, S. (2000). Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 165, 5392–5396.
Takeuchi, O., Hoshino, K., Kawai, T., Sanjo, H., Takada, H., Ogawa, T., Takeda, K., and Akira, S. (1999). Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity 11, 443–451.
Tawaratsumida, K., Furuyashiki, M., Katsumoto, M., Fujimoto, Y., Fukase, K., Suda, Y., and Hashimoto, M. (2009). Characterization of N-terminal structure of TLR2-activating lipoprotein in Staphylococcus aureus. J. Biol. Chem. 284, 9147–9152.
Tessarolli, V., Gasparoto, T. H., Lima, H. R., Figueira, E. A., Garlet, T. P., Torres, S. A., Garlet, G. P., Da Silva, J. S., and Campanelli, A. P. (2009). Absence of TLR2 influences survival of neutrophils after infection with Candida albicans. Med. Mycol. 48, 129–140.
Thompson, J. M., and Iwasaki, A. (2008). Toll-like receptors regulation of viral infection and disease. Adv. Drug Deliv. Rev. 60, 786–794.
Torres, D., Barrier, M., Bihl, F., Quesniaux, V. J., Maillet, I., Akira, S., Ryffel, B., and Erard, F. (2004). Toll-like receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect. Immun. 72, 2131–2139.
Travassos, L. H., Girardin, S. E., Philpott, D. J., Blanot, D., Nahori, M. A., Werts, C., and Boneca, I. G. (2004). Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 5, 1000–1006.
Triantafilou, M., Gamper, F. G., Haston, R. M., Mouratis, M. A., Morath, S., Hartung, T., and Triantafilou, K. (2006). Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J. Biol. Chem. 281, 31002–31011.
Tsan, M. F., and Gao, B. (2007). Pathogen-associated molecular pattern contamination as putative endogenous ligands of toll-like receptors. J. Endotoxin Res. 13, 6–14.
Underhill, D. M., Ozinsky, A., Hajjar, A. M., Stevens, A., Wilson, C. B., Bassetti, M., and Aderem, A. (1999). The toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401, 811–815.
van der Kleij, D., Latz, E., Brouwers, J. F., Kruize, Y. C., Schmitz, M., Kurt-Jones, E. A., Espevik, T., de Jong, E. C., Kapsenberg, M. L., Golenbock, D. T., Tielens, A. G., and Yazdanbakhsh, M. (2002). A novel host-parasite lipid cross-talk. J. Biol. Chem. 277, 48122–48129.
van Riet, E., Everts, B., Retra, K., Phylipsen, M., van Hellemond, J. J., Tielens, A. G., van der Kleij, D., Hartgers, F. C., and Yazdanbakhsh, M. (2009). Combined TLR2 and TLR4 ligation in the context of bacterial or helminth extracts in human monocyte derived dendritic cells: molecular correlates for Th1/Th2 polarization. BMC Immunol. 10, 9. doi:10.1186/1471-2172-10-19
Vanlandschoot, P., Leroux-Roels, G., Cooper, A., Tal, G., and Shaul, Y. (2005). The role of heparan sulfate and TLR2 in cytokine induction by hepatitis B virus capsids. J. Immunol. 175, 6253–6255.
Vargas-Inchaustegui, D. A., Tai, W., Xin, L., Hogg, A. E., Corry, D. B., and Soong, L. (2009). Distinct roles for MyD88 and TLR2 during Leishmania braziliensis infection in mice. Infect. Immun. 77, 2948–2956.
Villamón, E., Gozalbo, D., Roig, P., O’Connor, J. E., Ferrandiz, M. L., Fradelizi, D., and Gil, M. L. (2004). Toll-like receptor 2 is dispensable for acquired host immune resistance to Candida albicans in a murine model of disseminated candidiasis. Microbes Infect. 6, 542–548.
von Aulock, S., Schroder, N. W. J., Traub, S., Gueinzius, K., Lorenz, E., Hartung, T., Schumann, R. R., and Hermann, C. (2004). Heterozygous toll-like receptor 2 polymorphism does not affect lipoteichoic acid-induced chemokine and inflammatory responses. Infect. Immun. 72, 1828–1831.
von Herrath, M. G., and Nepom, G. T. (2005). Lost in translation: barriers to implementing clinical immunotherapeutics for autoimmunity. J. Exp. Med. 202, 1159–1162.
Wang, J. P., Kurt-Jones, E. A., Shin, O. S., Manchak, M. D., Levin, M. J., and Finberg, R. W. (2005). Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via toll-like receptor 2. J. Virol. 79, 12658–12666.
Wang, J. P., Zhang, Y., Wei, X., Li, J., Nan, X. P., Yu, H. T., Li, Y., Wang, P. Z., and Bai, X. F. (2010). Circulating toll-like receptor (TLR) 2, TLR4, and regulatory T cells in patients with chronic hepatitis C. APMIS 118, 261–270.
Watters, T. M., Kenny, E. F., and O’Neill, L. A. (2007). Structure, function and regulation of the toll//IL-1 receptor adaptor proteins. Immunol. Cell Biol. 85, 411–419.
Weiss, G., Rasmussen, S., Zeuthen, L. H., Nielsen, B. N., Jarmer, H., Jespersen, L., and Frøkiaer, H. (2010). Lactobacillus acidophilus induces virus immune defence genes in murine dendritic cells by a toll-like receptor-2-dependent mechanism. Immunology 131, 268–281.
Wenink, M. H., Santegoets, K. C., Broen, J. C., van Bon, L., Abdollahi-Roodsaz, S., Popa, C., Huijbens, R., Remijn, T., Lubberts, E., van Riel, P. L., van den Berg, W. B., and Radstake, T. R. (2009). TLR2 promotes Th2/Th17 responses via TLR4 and TLR7/8 by abrogating the type I IFN amplification loop. J. Immunol. 183, 6960–6970.
West, A. P., Koblansky, A. A., and Ghosh, S. (2006). Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 22, 409–437.
Wetzler, L. M. (2010). Innate immune function of the neisserial porins and the relationship to vaccine adjuvant activity. Future Microbiol. 5, 749–758.
Wiersinga, W. J., Wieland, C. W., Dessing, M. C., Chantratita, N., Cheng, A. C., Limmathurotsakul, D., Chierakul, W., Leendertse, M., Florquin, S., de Vos, A. F., White, N., Dondorp, A. M., Day, N. P., Peacock, S. J., and van der Poll, T. (2007). Toll-like receptor 2 impairs host defense in Gram-negative sepsis caused by Burkholderia pseudomallei (Melioidosis). PLoS Med. 4, e248. doi:10.1371/journal.pmed.0040248
Woehrle, T., Du, W., Goetz, A., Hsu, H. Y., Joos, T. O., Weiss, M., Bauer, U., Brueckner, U. B., and Marion Schneider, E. (2008). Pathogen specific cytokine release reveals an effect of TLR2 Arg753Gln during candida sepsis in humans. Cytokine 41, 322–329.
Wong-Baeza, I., Alcántara-Hernández, M., Mancilla-Herrera, I., Ramírez-Saldívar, I., Arriaga-Pizano, L., Ferat-Osorio, E., López-Macías, C., and Isibasi, A. (2010). The role of lipopeptidophosphoglycan in the immune response to Entamoeba histolytica. J. Biomed. Biotechnol. 2010, 254521.
Yarovinsky, F., Zhang, D., Andersen, J. F., Bannenberg, G. L., Serhan, C. N., Hayden, M. S., Hieny, S., Sutterwala, F. S., Flavell, R. A., Ghosh, S., and Sher, A. (2005). TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308, 1626–1629.
Yauch, L. E., Mansour, M. K., Shoham, S., Rottman, J. B., and Levitz, S. M. (2004). Involvement of CD14, toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect. Immun. 72, 5373–5382.
Yim, J. J., Kim, H. J., Kwon, O. J., and Koh, W. J. (2008). Association between microsatellite polymorphisms in intron II of the human toll-like receptor 2 gene and nontuberculous mycobacterial lung disease in a Korean population. Hum. Immunol. 69, 572–576.
Yim, J. J., Lee, H. W., Lee, H. S., Kim, Y. W., Han, S. K., Shim, Y. S., and Holland, S. M. (2006). The association between microsatellite polymorphisms in intron II of the human toll-like receptor 2 gene and tuberculosis among Koreans. Genes Immun. 7, 150–155.
Yuan, F. F., Marks, K., Wong, M., Watson, S., de Leon, E., McIntyre, P. B., and Sullivan, J. S. (2008). Clinical relevance of TLR2, TLR4, CD14 and Fc Gamma RIIA gene polymorphisms in Streptococcus pneumoniae infection. Immunol. Cell Biol. 86, 268–270.
Zähringer, U., Lindner, B., Inamura, S., Heine, H., and Alexander, C. (2008). TLR2 – promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213, 205–224.
Zhang, S. Y., Boisson-Dupuis, S., Chapgier, A., Yang, K., Bustamante, J., Puel, A., Picard, C., Abel, L., Jouanguy, E., and Casanova, J. L. (2008). Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol. Rev. 226, 29–40.
Zhou, S., Halle, A., Kurt-Jones, E. A., Cerny, A. M., Porpiglia, E., Rogers, M., Golenbock, D. T., and Finberg, R. W. (2008). Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J. Neuroimmunol. 194, 70–82.
Zhou, S., Kurt-Jones, E. A., Mandell, L., Cerny, A., Chan, M., Golenbock, D. T., and Finberg, R. W. (2005). MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 35, 822–830.
Keywords: TLR2, TLR1, TLR6, TLR2 ligands, polymorphisms, co-receptors
Citation: Oliveira-Nascimento L, Massari P and Wetzler LM (2012) The role of TLR2 in infection and immunity. Front. Immun. 3:79. doi: 10.3389/fimmu.2012.00079
Received: 06 December 2011; Accepted: 28 March 2012;
Published online: 18 April 2012.
Edited by:
Gregoire S. Lauvau, Albert Einstein College of Medicine, USAReviewed by:
Laurel L. Lenz, National Jewish Health, USACopyright: © 2012 Oliveira-Nascimento, Massari and Wetzler. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Lee M. Wetzler, Section of Infectious Diseases, Department of Medicine, Boston University School of Medicine, 650 Albany Street, Boston, MA 02118, USA. e-mail:bHdldHpsZXJAYnUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.