
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 January 2012
Sec. Inflammation
Volume 3 - 2012 | https://doi.org/10.3389/fimmu.2012.00007
This article is part of the Research Topic Mast Cells in Immunity View all 14 articles
 Michael A. Shi
Michael A. Shi Guo-Ping Shi*
Guo-Ping Shi*Mast cells (MCs) play an important role in allergic hyperresponsiveness and in defending microorganism infections. Recent studies of experimental animals and humans have suggested that MCs participate in obesity and diabetes. MC distribution and activities in adipose tissues may vary, depending on the locations of different adipose tissues. In addition to releasing inflammatory mediators to affect adipose tissue extracellular matrix remodeling and to promote inflammatory cell recruitment and proliferation, MCs directly and indirectly interact and activate adipose tissue cells, including adipocytes and recruited inflammatory cells. Plasma MC protease levels are significantly higher in obese patients than in lean subjects. Experimental obese animals lose body weight after MC inactivation. MC functions in diabetes are even more complicated, and depend on the type of diabetes and on different diabetic complications. Both plasma MC proteases and MC activation essential immunoglobulin E levels are significant risk factors for human pre-diabetes and diabetes mellitus. MC stabilization prevents diet-induced diabetes and improves pre-established diabetes in experimental animals. MC depletion or inactivation can improve diet-induced type 2 diabetes and some forms of type 1 diabetes, but also can worsen other forms of type 1 diabetes, at least in experimental animals. Observations from animal and human studies have suggested beneficial effects of treating diabetic patients with MC stabilizers. Some diabetic patients may benefit from enhancing MC survival and proliferation – hypotheses that merit detailed basic researches and clinical studies.
Mast cells (MCs) are inflammatory cells similar to macrophages, neutrophils, and lymphocytes, but they localize near surfaces exposed to the environment – such as the skin, airway, and gastrointestinal tract, where pathogens, allergens, and other environment agents frequently reside (Metcalfe et al., 1997; Galli et al., 2005a). These cells therefore are first-line immune cells defending against bacterial and viral infections, and are essential in airway hypersensitivity, skin allergies, and food anaphylaxis. Recent studies, however, suggest that MCs appear in most tested tissues, and are more than just a first-line immune response. They are abundant in tumor tissues (Maltby et al., 2009), in atherosclerotic lesions and other vascular lesions (Kovanen, 2007; Sun et al., 2007a,b), and in inflamed white adipose tissue (WAT; Liu et al., 2009). MCs are derived from hematopoietic progenitor cells that migrate into virtually any tissue, where they complete their maturation (Metcalfe et al., 1997). In the microenvironment, MCs require IL3 for early proliferation (Mekori et al., 1993) and stem cell factor (SCF) from stromal cells to maintain viability and maturation (Nocka et al., 1990; Irani et al., 1992).
Obesity and diabetes are considered chronic inflammatory diseases, largely due to several recent seminal discoveries of inflammatory cells in WAT – including macrophages (Ochi et al., 1988; Weisberg et al., 2003; Xu et al., 2003), B cells (Winer et al., 2011), CD4+ T cells (Winer et al., 2009), CD8+ T cells (Nishimura et al., 2009), regulatory T cells (Treg; Feuerer et al., 2009), NK T cells (Ohmura et al., 2010), eosinophils (Wu et al., 2011), and MCs (Liu et al., 2009). These cells may interact directly through cell–cell contact, or indirectly by releasing cell mediators within adipose tissue. Relative to macrophages or lymphocytes, far fewer studies are available regarding MC functions in obesity and diabetes. This brief review summarizes what we have learned from experimental animals and observations from human studies.
Adipose tissue, and WAT in particular, provides structural support and serves as a reservoir for energy surplus, but it also regulates metabolism, blood pressure, immune responses, coagulation, and functions of other endocrine organs (Trayhurn, 2005). A remarkable heterogeneity exists in the severity of WAT inflammation in different types of fat depots in obese subjects (Murano et al., 2008; Altintas et al., 2011a). MCs are myeloid hematopoietic cells derived from hematopoietic stem cells in the bone marrow. Progenitor MCs leave the bone marrow and differentiate into different MC subtypes, corresponding to the tissue compartments into which they home (Nakano et al., 1985). Recent discoveries demonstrated that WAT also harbors mast lineage cells that can home to organs such as intestine and skin, where they fulfill their roles (Poglio et al., 2010). For example, mouse inguinal fat pads contain c-Kit+Thy-1loLin−Sca+ cells, which can differentiate into other cell types in vitro, such as functional mucosal MCs (Poglio et al., 2010). Therefore, WAT harbors not only MC maturation, but also the whole process of MC differentiation.
While WAT from obese humans and animals contains higher numbers of MCs than that from lean subjects in general, MC number, and functions can vary among fat pads from different locations. For example, whereas the difference in MC number in subcutaneous WAT between obese and lean subjects was not significant, MC numbers in visceral WAT increase significantly in obese mice compared with those in lean mice, and MC crown-like structures became prevalent in obese visceral WAT (Altintas et al., 2011a). The infrapatellar fat pad (IFP), also called Hoffa’s fat pad, is an adipose tissue depot located intracapsularly and extrasynovially in the knee joint, to secrete inflammatory cytokines (Tilg and Moschen, 2006; Clockaerts et al., 2010). In a recent study among patients in the Netherlands, IFPs from patients with osteoarthritis contained higher amounts of IL6 and adipsin, and higher numbers of MCs, than those from subcutaneous WAT from the same patient population. IFP TNF-α levels, which may come from MCs, correlate with body-mass index (BMI) among this osteoarthritis patient population (Klein-Wieringa et al., 2011). Unlike subcutaneous WAT or visceral WAT, epididymal fat masses from mice fed a high-fat diet (HFD) for 20 weeks correlate inversely with body weight and liver mass. These fat tissues contain abundant dead adipocytes, as well as MCs, macrophages, and apoptotic cells. Therefore, mouse mast cell protease-6 (mMCP-6, human tryptase homolog), macrophage marker F4/80, cell apoptosis marker cleaved caspase-3, and the levels of the cytokines TNF-α and IL10 mRNA and of protein, all increased in obese mouse epididymal fat (Altintas et al., 2011b). Reduced epididymal fat mass in mice with long-standing obesity therefore is accompanied by divergent distribution of crown-like structures, apoptotic cells, MCs, and macrophages (Altintas et al., 2011b).
White adipose tissue contains CD4+, CD8+, Treg, and NK T cells (Feuerer et al., 2009; Nishimura et al., 2009; Winer et al., 2009; Ohmura et al., 2010). We recently showed that CD8+ T cells and MCs localize together in mouse visceral WAT (Xu and Shi, 2012). Physical proximity between MCs and T cells proposes a bidirectional functional relationship between the two cell types (Mekori, 2004; Kalesnikoff and Galli, 2008). MCs degranulate in response to direct contact with T cells or T cell membrane preparations, and release MC mediators, such as TNF-α, IL8, and oncostatin M (Baram et al., 2001; Salamon et al., 2008). Separation of the two cell populations by a semipermeable porous membrane prevents MC activation (Inamura et al., 1998), suggesting that T cells activate MCs not necessarily by small molecule protein peptides, but instead by using membrane transfer as a mode in intercellular communication. Many cells, including monocytes (Shi and Shi, 2010) and T cells (Al-Nedawi et al., 2009; Théry et al., 2009), release microvesicles. A recent study using Centricon filtration, high-speed centrifugation, identification by electron microscopy, FACS (Annexin staining), and expression of integrin lymphocyte function-associated antigen 1 (LFA-1) demonstrated that human peripheral blood T cells use such microvesicles to activate human cord blood MC mitogen-activated protein kinase (MAPK) signaling pathway, followed by MC degranulation and release of IL8 and oncostatin M (Shefler et al., 2010). MC activation then boosts T cell proliferation by MC surface molecule LFA-1 (Sayed and Brown, 2007; Shefler et al., 2010; Figure 1).
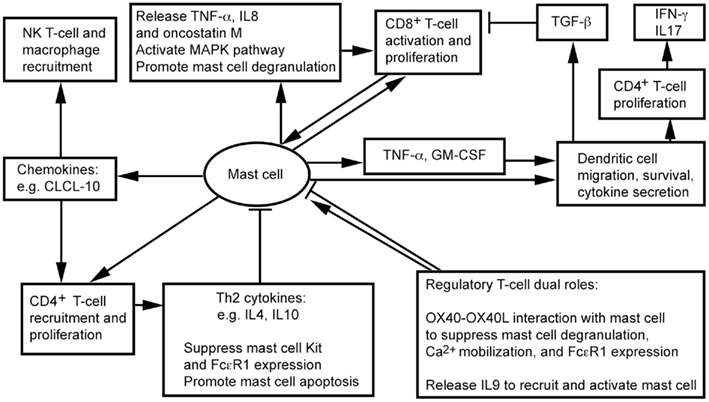
Figure 1. Possible interactions between mast cells and inflammatory cells in adipose tissues.
Regulatory T cells influences the inflammatory state of WAT, thereby increasing insulin sensitivity in mice (Feuerer et al., 2009). Although there is no direct evidence that Treg interacts with MCs in WAT, interaction between OX40 on Treg and OX40L on MCs suppresses MC degranulation and allergic responses (Gri et al., 2008; Piconese et al., 2009). In vitro interactions between human and mouse MCs and CD4+CD25+Foxp3+ Treg reduced MC degranulation and Ca2+ mobilization, without affecting overall cytokine secretion (Frossi et al., 2011). Anti-OX40L antibody blocks Treg-mediated MC stabilization (Gri et al., 2008). Treg also suppress MC expression of high-affinity IgE receptor FcεR1 (Kashyap et al., 2008). Treg may interact with MCs differently, however, under a different environment. In an allograft tolerance model, where MCs are essential in CD4+CD25+Foxp3+ Treg-dependent peripheral tolerance, Treg plays an immunosuppressive role by recruiting and activating MCs. When MC-deficient KitW-sh/W-sh mice do not induce tolerance, Treg produces IL9 to recruit and activate MCs in tolerant tissues to mediate regional immunosuppression. Anti-IL9 antibody neutralization leads to allograft rejection (Lu et al., 2006). In WAT, therefore, whether Treg activates MCs for immunosuppression or suppresses MC degranulation remains unknown.
NK T cells participate in WAT inflammation. Absence of NK T cells protects mice from diet-induced obesity (DIO), whereas NK T cell activation with α-galactosylceramide exacerbates glucose intolerance, macrophage infiltration, and WAT inflammatory cytokine expression (Ohmura et al., 2010). Although not tested in WAT, in mice infected with dengue virus, MCs express chemokine CXCL10 to trigger NK1.1+ cell infiltration to dengue virus-infected footpads – a mechanism of MC control viral infection within tissues – and limit viral spread to draining lymph nodes. Recruitment of NK1.1+ cells facilitates viral clearance (St. John et al., 2011). MCs may control NK T cell infiltration to WAT. We have shown that MC inactivation reduces macrophage infiltration to WAT (Liu et al., 2009). MCs also stimulate CD4+ T cell migration (Kashyap et al., 2008) and proliferation (Kotani et al., 2007), and enhance antigen-specific CD8+ T cell activation and proliferation – a process requiring direct interaction between MCs and T cells (Stelekati et al., 2009). While CD8+ T cells increased in obese WAT and exhibited adverse effects in obesity and diabetes (Nishimura et al., 2009), CD4+ T cells decreased in obese WAT (Winer et al., 2009). Th2 cytokines, such as IL4 and IL10, reduce the expression of Kit (SCF receptor) and FcεR1 (Ryan et al., 1998; Mirmonsef et al., 1999) in MCs, and promote MC apoptosis (Yeatman et al., 2000; Figure 1). High numbers of Th2 cells and low numbers of MCs therefore occur in WAT from lean subjects (Liu et al., 2009; Winer et al., 2009).
Dendritic cells (DCs) are the most effective professional antigen-presenting cells that prime T cells. DCs play an important role in atherogenesis (Tedgui et al., 2011), but their functions in obesity and diabetes remain unknown. Several lines of experiments – though most not related to obesity – suggest a controversial role of DCs in inflammation. DCs from obese mice show impaired antigen-presenting activity to activate CD8+ T cells, due to secretion of immunosuppressive transforming growth factor-β (TGF-β; Macia et al., 2006; Smith et al., 2009). Murine peritoneal MCs, however, directly contact immature DCs and induce their maturation with enhanced expression of DC co-stimulatory molecules. Co-culture of DCs and MCs releases the T cell modulating cytokines IFN-γ, IL2, IL6, and TGF-β. MCs also synergistically increase endotoxin-induced DC secretion of these cytokines, thereby inducing CD4+ T cell proliferation and the release of high levels of IFN-γ and IL17. MCs thus may indirectly promote Th1 and Th17 responses (Dudeck et al., 2011; Figure 1). In addition to affecting DC maturation, MCs release TNF-α to mediate DC migration (Suto et al., 2006). In a hapten FITC-induced cutaneous allergic contact hypersensitivity mouse model, MC deficiency (KitW-sh/W-sh mice) suppressed contact hypersensitivity, which can be repaired by local transfer of MCs from wild-type (WT) mice, but not those from Tnf−/− MCs. In the same model, FITC DC migration to draining lymph nodes or airway DC migration to local lymph nodes are impaired in KitW-sh/W-sh mice or Tnf−/− mice. In a mouse skin allograft tolerance model, MCs increased TNF-α-dependent DC accumulation in draining lymph nodes. MC production of GM-CSF also controls graft-derived DC survival. DCs that migrate from tolerant allografts to draining lymph nodes are tolerogenic and can suppress T cell responses (de Vries et al., 2011). MCs in WAT therefore may also regulate DC recruitment, maturation, and survival directly and indirectly (Figure 1). Consistent with this hypothesis, peripheral blood DC contents are significantly higher in obese patients with type 2 diabetes than in those without diabetes or lean patients, and DCs from obese and diabetic patients adhere much more efficiently to coronary smooth muscle cells (SMCs; Musilli et al., 2011).
Adipocytes, endothelial cells (ECs), and SMCs also are important components of WAT. We have shown that MCs release inflammatory cytokines to induce vascular EC and SMC expression of cysteinyl cathepsins, and therefore may promote angiogenesis and WAT growth in obese mice (Sun et al., 2007a,b; Liu et al., 2009; Figure 2). MCs and SMCs express thymic stromal lymphopoietin, which activates MCs to release inflammatory cytokines (e.g., IFN-γ, TNF-α, and IL1β) and a series of CCL chemokines (Kaur et al., 2011; Figure 2). Earlier studies showed that nerve growth factor and cyclooxygenase mediate prostaglandin D2 (PGD) production from MCs and T cells, respectively, and that PGD influences the differentiation of human fibroblasts into adipocytes (Marshall et al., 1999; Feldon et al., 2006). PGD can be metabolized into a final metabolite 15-deoxy-delta-12, 14-prostaglandin J2 (15-deoxy-delta PGJ2), which is a major endogenous ligand of peroxisome proliferator-activated receptor-γ (PPARγ; Kliewer et al., 1995; Herlong and Scott, 2006). Recent studies demonstrated that MC supernatant contains 15-deoxy-delta PGJ2 that induces pre-adipocyte 3T3-L1 cell adipogenesis (Figure 2). The specific PPARγ antagonist GW9662 blocks this MC activity (Tanaka et al., 2011). Using MC-deficient KitW/Wv mice, the investigators showed that reduced body weight in KitW/Wv mice can be reversed by WT bone marrow-derived mast cell (BMMC) transplantation, but not by transplantation of BMMC from hematopoietic PGD synthase (H-PGDS)-deficient mice. H-PGDS-deficient BMMC also failed to induce 3T3-L1 differentiation (Tanaka et al., 2011).
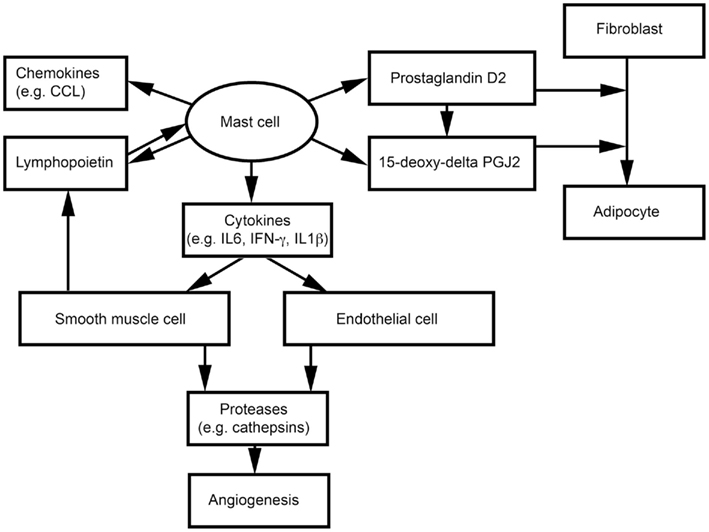
Figure 2. Possible interactions between mast cells and non-inflammatory cells in adipose tissues.
Increased inflammation and MC infiltration in obese WAT suggest an essential role of MCs in obesity and its associated complications. As discussed earlier, MCs may interact with inflammatory and non-inflammatory cells by direct cell–cell contact or by releasing inflammatory mediators. TNF-α, for example, is an important mediator of MCs (Zhang et al., 2011a,b) and is increased in obese rodents. Neutralization of TNF-α with a soluble TNF-α receptor-immunoglobulin G chimeric protein ameliorated insulin resistance (Hotamisligil et al., 1993), and obese mice lacking TNF-α demonstrated improved insulin sensitivity (Uysal et al., 1997). Hypoadiponectinemia associates with obesity and insulin resistance (Weyer et al., 2001). Adiponectin mRNA levels were increased in WAT from MC-null mice fed an HFD, although serum levels did not change (Tanaka et al., 2011).
Using KitW-sh/W-sh and KitW/Wv mice, we (Liu et al., 2009) and others (Tanaka et al., 2011) proved that MCs participate directly in obesity. While WT mice became obese after consuming an HFD, KitW-sh/W-sh and KitW/Wv mice were protected from DIO. Adoptive transfer of BMMC from WT or Tnf−/− mice, but not those from Il6−/−, Ifng−/−, or H-PGDS-deficient mice, partially reversed body weight gain in KitW-sh/W-sh and KitW/Wv mice. Pharmacological MC inactivation using cromolyn and ketotifen, two common MC stabilizers, yielded similar results. Both drugs not only prevented WT mice from DIO, but also reduced pre-established obesity (Liu et al., 2009), revealing a possible therapy for obese humans.
The role of MCs in human obesity has not been examined, but epidemiological studies consistently have shown a positive association between obesity and the risk of asthma (Beuther, 2009; Shore, 2010), and low-grade inflammation associated with obesity could be a pathogenetic mechanism linking obesity to asthma (Hersoug and Linneberg, 2007). In a Danish study, serum MC tryptase levels, skin prick test reactivity (atopy), methacholine bronchial hyperresponsiveness (BHR), BMI, and serum lipid levels were determined in 1216 patients. Serum tryptase levels were much higher in obese patients (BMI > 30) than in lean patients [BMI < 25; 4.4 (3.0–6.1; 25th–75th percentile) μg/L vs. 3.3 (2.3–4.9; 25th–75th percentile) μg/L, P < 0.0001]. Among this population, serum tryptase levels associated positively with male sex and smoking, and inversely with alcohol consumption. Tryptase levels, however, did not associate with atopy or BHR but positively associated with the symptoms of allergic respiratory disease and obesity (OR = 1.98, 95% CI = 1.25–3.14). Increasing BMI therefore associates with serum tryptase levels and the prevalence of allergic respiratory disease symptoms, and MC activity did not affect the association between BMI and asthma and rhinitis symptoms (Fenger et al., 2011). Similar conclusions were made in a Spanish study of 420 patients. Among those patients, serum tryptase associated positively with age and serum IgE levels, but inversely with female sex and heavy alcohol use. Tryptase levels appear significantly higher in non-atopic patients, overweight patients, or patients with metabolic syndromes, compared with those were atopic, of normal weight, or without metabolic syndrome.
In a multivariate analysis, the significance of metabolic syndrome association with tryptase levels dropped from P = 0.08 to P = 0.008 after adjusting for age, sex, atopy status, IgE, alcohol use, and smoking (Gonzalez-Quintela et al., 2010). These human studies strongly suggest a role of MC activation in obesity, but this hypothesis has not been tested directly.
We first reported that MCs participate directly in type 2 diabetes mellitus (T2DM). DIO mice also developed glucose intolerance and insulin resistance, which did not appear in KitW-sh/W-sh and KitW/Wv MC-deficient mice. MC stabilization with either cromolyn or ketotifen had results similar to those from MC-deficient mice. Genetic deficiency and pharmacological inactivation of MCs therefore prevented diet-induced T2DM in mice (Liu et al., 2009). Mechanistically, we found that MC-derived IL6 and IFN-γ were critical to both obesity and diabetes. Reduced T2DM symptoms (serum glucose, leptin, insulin levels, and glucose tolerance and insulin sensitivity) in MC-deficient mice can be reversed partially by adoptive transfer of BMMC from WT or Tnf−/− mice, but not those from Il6−/− and Ifng−/− mice. But different mechanisms may exist – for example, long-term culture in high-glucose medium augments IgE-induced MC activation (Anderson et al., 2010). Although not tested in our study, MC activation may be elevated in DIO mice. The most important finding of our study is that MC stabilization with cromolyn or ketotifen improved glucose tolerance and insulin sensitivity in DIO mice with pre-established T2DM. This finding has been confirmed in a patient with T2DM; less than 6 months of treatment with cromolyn reduced both plasma glucose and hemoglobin A1c levels to normal ranges (Shi, personal communication).
Although MC function in human type 1 diabetes mellitus (T1DM) has not been studied, several animal models have demonstrated rather complicated roles of MCs in T1DM. While accumulation of MCs is detrimental in animals with some T1DM models, MC depletion is an important mechanism of T1DM development in different experimental models.
Intravenous administration of alloxan induces T1DM in rats. These animals show decreased microvascular responses to inflammatory histamine (Fortes et al., 1983), decreased leukocyte–EC interaction (Fortes et al., 1991), and reduced MC degranulation (de Oliveira Barreto et al., 2003; Cavalher-Machado et al., 2004), but increased leukocyte apoptosis (Otton et al., 2004). These diabetic rats therefore are refractory to systemic anaphylactic shock, and show a selective reduction in the number of pleural MCs – a phenomenon also observed after treatment with the alternative diabetogenic agent streptozotocin (STZ; Diaz et al., 1996). Diabetic sensitized animals are clearly resistant to local and systemic allergic inflammatory responses (Carvalho et al., 2005). It was hypothesized that autoimmune T1DM is Th1-dependent and that allergic responses are Th2-dependent. The susceptibility to one disease might lead to refractoriness to the other (Huang, 1999). Children with T1DM and their siblings, therefore, are partially protected against asthma (Douek et al., 1999), but several studies suggest that this refractoriness associates with MC depletion or the inability to activate MCs, and overexpression of endogenous corticosteroids (Diaz et al., 1997; Carvalho et al., 2003). MC depletion in the intestinal tissue is important in the refractoriness of diabetic rats to anaphylactic shock. In alloxan-treated rats, ovalbumin (OVA) sensitization reduces total cell yield in lavage fluid after antigen challenge (Vianna and Garcia-Leme, 1995). In the same experimental model, bronchial segments from diabetic rats showed reduced contraction to OVA, and accompanied with a 50% reduction in degranulated MCs and histamine release, in addition to changes in MC ultrastructure (Ozsoy and Gül, 2005). Diabetes also influences immune cell production of IgE. Total and antigen-specific IgE augmentation was suppressed in diabetic sensitized rats (Carvalho et al., 2003). After transfer of spleen and lymph node immune cells from naïve mice to diabetic mice, immune cells lose their ability to form IgE. In contrast, immune cells from diabetic mice regain their ability to generate IgE after transfer to non-diabetic recipients (Ptak et al., 1983), suggesting that some factor(s) in the blood of diabetic mice affect immune cell IgE production. Diabetes also directly affects MC activities. Rats with alloxan-induced diabetes mount weaker allergen-evoked pleural MC degranulation and plasma leakage paralleled with reduction of MCs in the pleural cavity. Transfer of MCs from sensitized non-diabetic rats can restore allergen-evoked pleural protein extravasation in diabetic rats. In contrast, transfer of MCs from sensitized diabetic rats to naïve rats leads to a lower allergen-induced protein exudation. In vitro, purified MCs from diabetic rats are hyporesponsive to antigen and C48/80 stimulation (de Oliveira Barreto et al., 2003; Figure 3A).
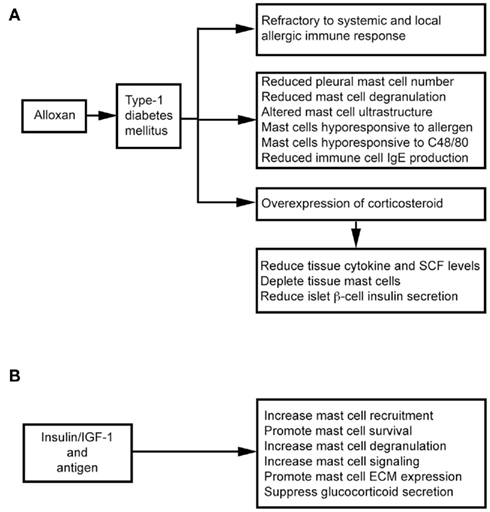
Figure 3. Mast cells in alloxan-induced type 1 diabetes mellitus. (A) Alloxan induces type 1 diabetes and suppresses mast cell activities. (B) Insulin therapy improves mast cell activities in alloxan-induced type 1 diabetes mellitus.
Alloxan administration induces glucocorticoid overexpression, which induces MC depletion in the skin, lungs, and intestines (Pipkorn et al., 1989; Goldsmith et al., 1990; Finotto et al., 1997). MC depletion and increased endogenous glucocorticoids are closely related (Diaz et al., 1997). Glucocorticoids exhibit anti-inflammatory activity by inhibiting tissue cytokine and SCF expression (Schleimer, 1993; Finotto et al., 1997), therefore participating in MC depletion in the intestines and other organs in diabetic rats (Pipkorn et al., 1989; Goldsmith et al., 1990; Finotto et al., 1997). Treatment with steroid receptor agonist RU486 (Carvalho Vde et al., 2009) or surgical bilateral adrenalectomy (Diaz et al., 2001) increases intestinal MC numbers and IL3 staining and IgE formation in diabetic mice – suggesting a negative correlation between MC–IgE system and serum glucocorticoid levels. Glucocorticoids also inhibit insulin secretion effectively through pancreatic β cells (Ludvik et al., 1993), and insulin treatment in alloxanated rats impairs the increase of systemic corticosterone. A balance between glucocorticoid and insulin is essential to T1DM (Diaz et al., 2001; Figure 3A).
Insulin therapy not only suppresses glucocorticoid secretion, but also stimulates several MC signaling pathways, including Lyn, Syk, Fyn, adapter protein Gab2, Akt, JNK, p38 kinase, SHIP1, and protein kinase C-θ. Antigen and insulin or insulin-like growth factor-1 (IGF-1) synergistically increased antigen-induced MC degranulation and cytoskeletal rearrangement and promoted MC survival in the absence of IL3 (Lessmann et al., 2006; Kettner et al., 2010). In diabetic rats, subcutaneous administration of insulin revealed an important role of insulin in MC extracellular matrix protein (ECM; e.g., laminin, fibronectin, and collagen) expression and MC recruitment (de F Carvalho et al., 2008; Figure 3B). MCs express integrin-type ECM receptors to mediate their activation and increase responses to antigen stimulation (Kitaura et al., 2005). Interaction between MCs and ECM is important to MC migration and distribution in tissues (Hamawy et al., 1994). Insulin treatment reverses alloxan-induced reductions in MC numbers, histamine production, and IgE release (Vianna and Garcia-Leme, 1995; Cavalher-Machado et al., 2004).
The role of MCs in T1DM can also vary by T1DM models. In 3-week-old rats with STZ-induced diabetes, the disease associates with increased mesenteric vessel fibrosis and MC numbers. Both mesenteric vessel fibrosis and chymase-positive MC numbers were reduced after rats received tranilast (Jones et al., 2004), suggesting a causative role of MCs in STZ-induced diabetes. Biobreeding (BB) rats and non-obese diabetic (NOD) mice spontaneously develop autoimmune T1DM (Kikutani and Makino, 1992; Wallis et al., 2009). DRlyp/lyp rats also develop spontaneous T1DM. Unless insulin is administered, death follows after T1DM onset as a result of hyperglycemia and ketoacidosis (Mordes et al., 1996). In BB rats, islet β cells express eotaxin (Geoffrey et al., 2006), which leads to infiltration of CCR3-expressing neutrophils and MCs to the pancreatic islets (Kukreja and Maclaren, 1999; Hessner et al., 2004). Treatment of DRlyp/lyp rats with cromolyn significantly delayed onset of T1DM relative to saline-treated control rats (Kukreja and Maclaren, 1999; Geoffrey et al., 2006). Mechanistically, MCs present antigens to activate T cells in major histocompatibility complex class I and class II pathways in rodents and humans (Frandji et al., 1993). T cells are primary mediators of human and rodent T1DM (Green et al., 1998; Mathis et al., 2001; Groen et al., 2003). In NOD mice and BB rats, early thymectomy prevents T1DM (Green et al., 1998; Mathis et al., 2001; Groen et al., 2003). MCs also promote T cell migration to inflammatory sites by producing chemokines or indirectly by increasing EC adhesion molecule expression (Galli et al., 2005b; Zhang et al., 2011b). As discussed earlier, MCs also affect DC maturation, migration, and activity, providing another pathway for T cell activation (Galli et al., 2005b; Figure 4). In NOD mice, however, anti-FcεR1 antibody delayed T1DM onset. Anti-FcεR1 was proposed to activate basophil and MC release of IL4 and histamine. Anti-FcεR1 antibody lost its ability to delay T1DM in Il4−/− NOD mice, but this ability was not affected by histamine receptor blocker. Basophils, therefore, were proposed to play a role in T1DM in NOD mice (Hübner et al., 2011).
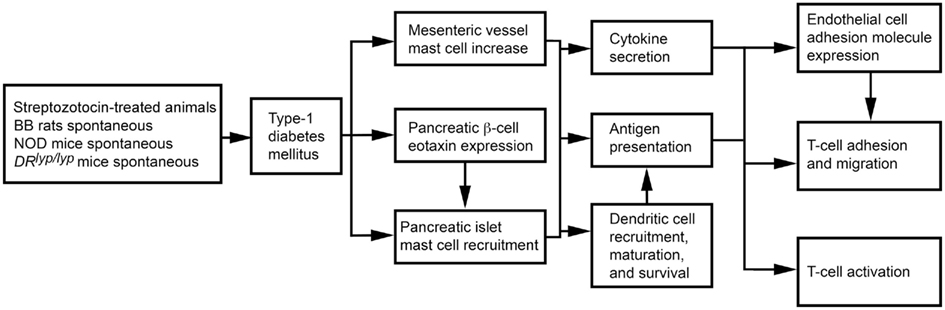
Figure 4. Mast cell activities in streptozotocin-induced and spontaneous type 1 diabetes mellitus.
Long-term diabetes associates with numerous complications, including nephropathy (Fowler, 2008) – a multistage clinical syndrome characterized by thickening of the glomerular basement membrane and mesangial expansion, with progression into glomerulosclerosis, tubular necrosis, and interstitial fibrosis, ultimately resulting in renal failure (Wolf and Ziyadeh, 1999, 2007). MCs are most abundant in the kidneys of diabetic patients with nephropathy (Okon and Stachura, 2007). An increase in renal MCs associates with fibrosis and ECM accumulation in the kidneys of diabetic nephropathy patients (Rüger et al., 1996). In kidneys from glomerulonephritis patients, MCs are clustered with T cells and macrophages in the interstitial (not in the glomerulus) and associate with α-SMA-positive interstitium and percentage of the interstitial fibrotic areas, suggesting a role of MCs in renal fibroproliferation and interstitial fibrosis (Tóth et al., 1999).
Mast cells produce TGF-β (Metcalfe, 2008), chymase, tryptase, cathepsin G, renin (Silver et al., 2004), and TNF-α (Bissonnette et al., 1995), as summarized in Figure 5. TGF-β is a fibrogenic cytokine linked to diabetic nephropathy in both animals and humans (Bollineni and Reddi, 1993; Sato et al., 1998). Elevated TGF-β has been implicated in the pathogenesis of diabetic nephropathy (Goldfarb and Ziyadeh, 2001). Treatment with an anti-TGF-β antibody (1D11; Benigni et al., 2006) or a soluble TGF-β type II receptor (aTβRII.Fc; Russo et al., 2007) reduced proteinuria, inhibited renal fibrosis, and produced renoprotective effects in rats with diabetic nephropathy. MC proteases contribute to renal fibrosis directly or indirectly. In diabetic nephropathy patients, MC chymase increases and associates with glomerulosclerosis, tubulointerstitial fibrosis, and vascular fibrosis (Ritz, 2003; Huang et al., 2007). MC chymase and cathepsin G convert angiotensin (Ang)-I to Ang-II, thereby activating TGF-β (Metcalfe et al., 1997; Doggrell and Wanstall, 2005; Helske et al., 2006). MC chymase and cathepsin G also activate MMP-9 (Ishida et al., 2008) to promote TGF-β signaling (Wilson et al., 2009). MC tryptase participates in the development of renal interstitial fibrosis by increasing ECM production (Kondo et al., 2001). MCs release histamine and tryptase to differentiate fibroblasts into α-smooth muscle actin-positive myofibroblasts (Gailit et al., 2001). In vitro, treatment of rat cardiac fibroblasts with MC tryptase induced fibroblast proliferation and collagen synthesis (Levick et al., 2009). Renin, also known as angiotensinogenase, converts angiotensinogen into Ang-I and regulates TGF-β expression in rat kidney mesangial cells, independent of Ang-II (Huang et al., 2007). Patients with diabetic nephropathy have elevated renin levels (Deinum et al., 1999). The renin inhibitor aliskiren attenuates glomerulosclerosis in diabetic transgenic (mRen-2) 27 rats with advanced diabetic nephropathy (Kelly et al., 2007). MC histamine stimulates H2 receptor-mediated release of renin from rat kidneys (Schwertschlag and Hackenthal, 1982), indirectly affects renal Ang-II production, and induces diabetic nephropathy. TNF-α expression increases in the glomeruli of rats and humans with diabetic nephropathy (Nakamura et al., 1993; Mahmoud et al., 2004). TNF-α overexpression induces renal hypertrophy (Navarro et al., 2003) and microalbuminuria (Kalantarinia et al., 2003) in rats with diabetic nephropathy. The TNF-α agonist TNFR:Fc prevents renal hypertrophy (Navarro et al., 2003). The soluble TNF-α receptor infliximab reduced urine albumin excretion in rats with diabetic nephropathy (Moriwaki et al., 2007).
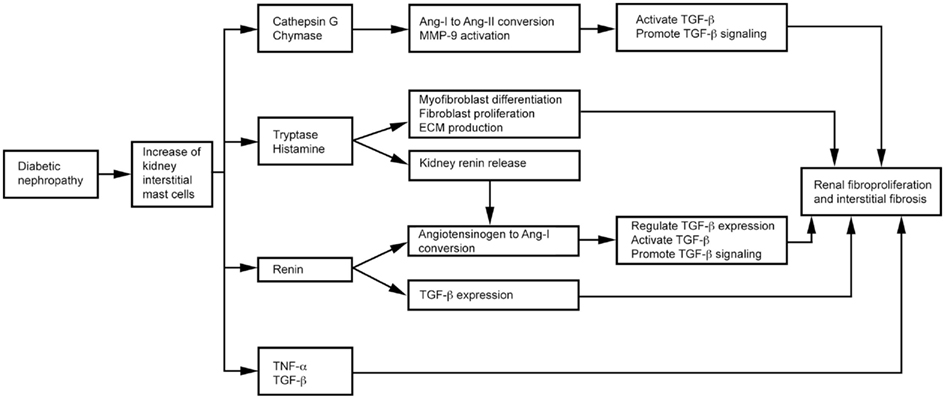
Figure 5. Mast cell functions in diabetic nephropathy.
Although MC functions in T1DM can be complicated, we anticipate a detrimental role of MCs in T2DM, based on our prior animal studies (Liu et al., 2009). Patients with T2DM are expected to have increased levels of plasma biomarkers of MC proteases and associated activation markers. We tested this hypothesis in our recent community screening study (Wang et al., 2012). In this study, we screened 3163 volunteers 55–75 years of age, from three neighborhood communities in Zhejiang Province, China. Of those, 1197 accepted the invitation for fasting plasma glucose (FPG) and 2-h oral glucose tolerance test (2 h-OGTT). Among the subjects, 807 had normal glucose tolerance (FPG < 5.6 mmol/L, 2 h-OGTT < 7.8 mmol/L), 267 had pre-diabetes (FPG ≥ 5.6 and <7.0 mmol/L or 2 h-OGTT ≥ 7.8 and <11.1 mmol/L), and 123 had T2DM (FPG ≥ 7.0 mmol/L or 2 h-OGTT ≥ 11.1 mmol/L). The Kruskal–Wallis test revealed significant differences in plasma IgE levels among patients with different glucose tolerance status (P = 0.008). A linear regression test showed significant correlations between plasma chymase (P = 0.030) and IgE (P = 0.022) and diabetes mellitus. Ordinal logistic regression analysis demonstrated that IgE was a significant risk factor of pre-diabetes and T2DM before (odds ratio: 1.674, P = 0.034) or after adjustment (odds ratio: 1.866, P = 0.015) of all tested T2DM risk factors. Although not statistically significant, plasma tryptase levels were also higher in T2DM patients than in those with pre-diabetes or with normal glucose levels (Wang et al., 2012). More sophisticated studies are required to test a role of MCs in diabetes or obesity, but this human study inspired us to explore MC participation in these metabolic diseases.
Observations from experimental animals and diabetic patients strongly suggest a direct participation of MCs in both obesity and diabetes, although the mechanisms by which MCs contribute to the pathogenesis of these metabolic diseases are rather complicated. Our animal study, using both cromolyn and ketotifen to treat pre-established obesity and T2DM in mice, encouraged us to develop novel medications for patients with those metabolic diseases by inactivating MCs. Indeed, in an early study to evaluate cromolyn applications in asthma patients, stabilization with a cromolyn nasal spray reduced body weight by more than 3 pounds in less than 3 months (Clarke and May, 1980). But other potential actions of cromolyn are unknown; reduced body weight (Clarke and May, 1980) or improved T2DM (Shi, personal communication) in humans do not necessarily relate only to MCs. Tranilast, for example, was developed as a MC stabilizer (Koda et al., 1985), and it reduces tubulointerstitial fibrosis, tubular atrophy, and albuminuria in rats with diabetic nephropathy (Mifsud et al., 2003) and retards the progression of advanced diabetic nephropathy in humans (Soma et al., 2002). But these tranilast activities may not result from MC stabilization only. In STZ-treated diabetic rats, tranilast reduced mesenteric weight, superior mesenteric artery wall-to-lumen ratio, matrix deposition, and expression of TGF-β, epidermal growth factor (EGF), and collagens – therefore attenuating vascular hypertrophy (Bonnet et al., 2003). In these mice, both ECs and MCs express endothelin, which constricts blood vessels. The endothelinA/B receptor antagonist bosentan had the same effects as tranilast, but bosentan did not prevent TGF-β overexpression in diabetic rats (Gilbert et al., 2000). Tranilast, therefore, may target cells other than MCs – such as ECs. Indeed, tranilast inhibits EC protein kinase C and nuclear factor-κB activities (Koyama et al., 1999; Spiecker et al., 2002). Tranilast also inhibits TGF-β release from fibroblasts and monocytes/macrophages (Suzawa et al., 1992; Yamada et al., 1994), and suppresses insulin secretion and induces glucose uptake from glucose-induced rat islets and a tolbutamide-treated rat islet cell line (INS-1E) without affecting their glucose transporter (Glut2) expression (Taguchi et al., 2008). We showed that MCs release inflammatory cytokines to induce EC adhesion molecule expression (Zhang et al., 2011b). MCs also interact with macrophages and induce expression of prostaglandin E2 and 6-keto-prostaglandin F1 during immediate hypersensitivity reactions (Jakschik et al., 1987). Tranilast activities, therefore, may associate indirectly with MC activity inhibition. More basic and clinical studies in this field will benefit greatly to patients suffering from these social diseases.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I thank Ms. Sara Karwacki for editorial assistance. Studies cited in this article are supported by National Institutes of Health grants HL60942, HL81090, and HL88547; and by an Established Investigator Award (0840118N) from the American Heart Association to Guo-Ping Shi.
Al-Nedawi, K., Meehan, B., and Rak, J. (2009). Microvesicles: messengers and mediators of tumor progression. Cell Cycle 8, 2014–2018.
Altintas, M. M., Azad, A., Nayer, B., Contreras, G., Zaias, J., Faul, C., Reiser, J., and Nayer, A. (2011a). Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. J. Lipid Res. 52, 480–488.
Altintas, M. M., Rossetti, M. M., Nayer, B., Puig, A., Zagallo, P., Ortega, L. M., Johnson, K. B., McNamara, G., Reiser, J., Mendez, A. J., and Nayer, A. (2011b). Apoptosis, mastocytosis, and diminished adipocytokine gene expression accompany reduced epididymal fat mass in long-standing diet-induced obese mice. Lipids Health Dis. 10, 198.
Anderson, E. K., Gutierrez, D. A., and Hasty, A. H. (2010). Adipose tissue recruitment of leukocytes. Curr. Opin. Lipidol. 21, 172–177.
Baram, D., Vaday, G. G., Salamon, P., Drucker, I., Hershkoviz, R., and Mekori, Y. A. (2001). Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNF-alpha. J. Immunol. 167, 4008–4016.
Benigni, A., Zoja, C., Campana, M., Corna, D., Sangalli, F., Rottoli, D., Gagliardini, E., Conti, S., Ledbetter, S., and Remuzzi, G. (2006). Beneficial effect of TGF-beta antagonism in treating diabetic nephropathy depends on when treatment is started. Nephron Exp. Nephrol. 104, e158–e168.
Bissonnette, E. Y., Enciso, J. A., and Befus, A. D. (1995). Inhibition of tumour necrosis factor-alpha (TNF-alpha) release from mast cells by the anti-inflammatory drugs, sodium cromoglycate and nedocromil sodium. Clin. Exp. Immunol. 102, 78–84.
Bollineni, J. S., and Reddi, A. S. (1993). Transforming growth factor-beta 1 enhances glomerular collagen synthesis in diabetic rats. Diabetes 42, 1673–1677.
Bonnet, F., Cao, Z., Cooper, M. E., Cox, A. J., Kelly, D. J., and Gilbert, R. E. (2003). Tranilast attenuates vascular hypertrophy, matrix accumulation and growth factor overexpression in experimental diabetes. Diabetes Metab. 29(Pt 1), 386–392.
Carvalho, V. F., Barreto, E. O., Cordeiro, R. S., Lagente, V., Martins, M. A., and e Silva, P. M. (2005). Mast cell changes in experimental diabetes: focus on attenuation of allergic events. Mem. Inst. Oswaldo Cruz 100(Suppl. 1), 121–125.
Carvalho, V. F., Barreto, E. O., Diaz, B. L., Serra, M. F., Azevedo, V., Cordeiro, R. S., Martins, M. A., and e Silva, P. M. (2003). Systemic anaphylaxis is prevented in alloxan-diabetic rats by a mechanism dependent on glucocorticoids. Eur. J. Pharmacol. 472, 221–227.
Carvalho Vde, F., Barreto Ede, O., Farias-Filho, F. A., Gomes, L. H., Mendonça Lde, L., Cordeiro, R. S., Martins, M. A., and Rodrigues e Silva, P. M. (2009). Reduced expression of IL-3 mediates intestinal mast cell depletion in diabetic rats: role of insulin and glucocorticoid hormones. Int. J. Exp. Pathol. 90, 148–155.
Cavalher-Machado, S. C., de Lima, W. T., Damazo, A. S., de Frias Carvalho, V., Martins, M. A., e Silva, P. M., and Sannomiya, P. (2004). Down-regulation of mast cell activation and airway reactivity in diabetic rats: role of insulin. Eur. Respir. J. 24, 552–558.
Clarke, C. W., and May, C. S. (1980). A comparison of the efficacy of ketotifen (HC 20-511) with sodium cromoglycate (SCG) in skin test positive asthma. Br. J. Clin. Pharmacol. 10, 473–476.
Clockaerts, S., Bastiaansen-Jenniskens, Y. M., Runhaar, J., Van Osch, G. J., Van Offel, J. F., Verhaar, J. A., De Clerck, L. S., and Somville, J. (2010). The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review. Osteoarthr. Cartil. 18, 876–882.
de F Carvalho, V., Campos, L. V., Farias-Filho, F. A., Florim, L. T., Barreto, E. O., Pirmez, C., Savino, W., Martins, M. A., and e Silva, P. M. (2008). Suppression of allergic inflammatory response in the skin of alloxan-diabetic rats: relationship with reduced local mast cell numbers. Int. Arch. Allergy Immunol. 147, 246–254.
de Oliveira Barreto, E., de Frias Carvalho, V., Diaz, B. L., Balduino, A., Cordeiro, R. S., Martins, M. A., and Rodrigues e Sílva, P. M. (2003). Adoptive transfer of mast cells abolishes the inflammatory refractoriness to allergen in diabetic rats. Int. Arch. Allergy Immunol. 131, 212–220.
de Vries, V. C., Pino-Lagos, K., Nowak, E. C., Bennett, K. A., Oliva, C., and Noelle, R. J. (2011). Mast cells condition dendritic cells to mediate allograft tolerance. Immunity 35, 550–561.
Deinum, J., Tarnow, L., van Gool, J. M., de Bruin, R. A., Derkx, F. H., Schalekamp, M. A., and Parving, H. H. (1999). Plasma renin and prorenin and renin gene variation in patients with insulin-dependent diabetes mellitus and nephropathy. Nephrol. Dial. Transplant. 14, 1904–1911.
Diaz, B., Barreto, E., Cordeiro, R., Perretti, M., Martins, M., and Silva, P. (2001). Enhanced serum glucocorticoid levels mediate the reduction of serosal mast cell numbers in diabetic rats. Life Sci. 68, 2925–2932.
Diaz, B. L., Serra, M. F., Alves, A. C., Pires, A. L., Corrêa, F. M., Cordeiro, R. S., Martins, M. A., and e Silva, P. M. (1996). Alloxan diabetes reduces pleural mast cell numbers and the subsequent eosinophil influx induced by allergen in sensitized rats. Int. Arch. Allergy Immunol. 111, 36–43.
Diaz, B. L., Serra, M. F., Barreto, E. O., Cordeiro, R. S., Martins, M. A., and e Silva, P. M. (1997). Antigen-induced pleural eosinophilia is suppressed in diabetic rats: role of corticosteroid hormones. Mem. Inst. Oswaldo Cruz 92(Suppl. 2), 219–222.
Doggrell, S. A., and Wanstall, J. C. (2005). Cardiac chymase: pathophysiological role and therapeutic potential of chymase inhibitors. Can. J. Physiol. Pharmacol. 83, 123–130.
Douek, I. F., Leech, N. J., Gillmor, H. A., Bingley, P. J., and Gale, E. A. (1999). Children with type-1 diabetes and their unaffected siblings have fewer symptoms of asthma. Lancet 353, 1850.
Dudeck, A., Suender, C. A., Kostka, S. L., von Stebut, E., and Maurer, M. (2011). Mast cells promote Th1 and Th17 responses by modulating dendritic cell maturation and function. Eur. J. Immunol. 41, 1883–1893.
Feldon, S. E., O’loughlin, C. W., Ray, D. M., Landskroner-Eiger, S., Seweryniak, K. E., and Phipps, R. P. (2006). Activated human T lymphocytes express cyclooxygenase-2 and produce proadipogenic prostaglandins that drive human orbital fibroblast differentiation to adipocytes. Am. J. Pathol. 169, 1183–1193.
Fenger, R. V., Linneberg, A., Vidal, C., Vizcaino, L., Husemoen, L. L., Aadahl, M., and Gonzalez-Quintela, A. (2011). Determinants of serum tryptase in a general population: the relationship of serum tryptase to obesity and asthma. Int. Arch. Allergy Immunol. 157, 151–158.
Feuerer, M., Herrero, L., Cipolletta, D., Naaz, A., Wong, J., Nayer, A., Lee, J., Goldfine, A. B., Benoist, C., Shoelson, S., and Mathis, D. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939.
Finotto, S., Mekori, Y. A., and Metcalfe, D. D. (1997). Glucocorticoids decrease tissue mast cell number by reducing the production of the c-kit ligand, stem cell factor, by resident cells: in vitro and in vivo evidence in murine systems. J. Clin. Invest. 99, 1721–1728.
Fortes, Z. B., Farsky, S. P., Oliveira, M. A., and Garcia-Leme, J. (1991). Direct vital microscopic study of defective leukocyte-endothelial interaction in diabetes mellitus. Diabetes 40, 1267–1273.
Fortes, Z. B., Garcia Leme, J., and Scivoletto, R. (1983). Influence of diabetes on the reactivity of mesenteric microvessels to histamine, bradykinin and acetylcholine. Br. J. Pharmacol. 78, 39–48.
Fowler, M. J. (2008). Microvascular and macrovascular complications of diabetes. Clin. Diabetes 26, 77–82.
Frandji, P., Oskéritzian, C., Cacaraci, F., Lapeyre, J., Peronet, R., David, B., Guillet, J. G., and Mécheri, S. (1993). Antigen-dependent stimulation by bone marrow-derived mast cells of MHC class II-restricted T cell hybridoma. J. Immunol. 151, 6318–6328.
Frossi, B., D’Incà, F., Crivellato, E., Sibilano, R., Gri, G., Mongillo, M., Danelli, L., Maggi, L., and Pucillo, C. E. (2011). Single-cell dynamics of mast cell-CD4+ CD25+ regulatory T cell interactions. Eur. J. Immunol. 41, 1872–1882.
Gailit, J., Marchese, M. J., Kew, R. R., and Gruber, B. L. (2001). The differentiation and function of myofibroblasts is regulated by mast cell mediators. J. Invest. Dermatol. 117, 1113–1119.
Galli, S. J., Kalesnikoff, J., Grimbaldeston, M. A., Piliponsky, A. M., Williams, C. M., and Tsai, M. (2005a). Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23, 749–786.
Galli, S. J., Nakae, S., and Tsai, M. (2005b). Mast cells in the development of adaptive immune responses. Nat. Immunol. 6, 135–142.
Geoffrey, R., Jia, S., Kwitek, A. E., Woodliff, J., Ghosh, S., Lernmark, A., Wang, X., and Hessner, M. J. (2006). Evidence of a functional role for mast cells in the development of type 1 diabetes mellitus in the BioBreeding rat. J. Immunol. 177, 7275–7286.
Gilbert, R. E., Rumble, J. R., Cao, Z., Cox, A. J., van Eeden, P., Allen, T. J., Kelly, D. J., and Cooper, M. E. (2000). Endothelin receptor antagonism ameliorates mast cell infiltration, vascular hypertrophy, and epidermal growth factor expression in experimental diabetes. Circ. Res. 86, 158–165.
Goldfarb, S., and Ziyadeh, F. N. (2001). TGF-beta: a crucial component of the pathogenesis of diabetic nephropathy. Trans. Am. Clin. Climatol. Assoc. 112, 27–32.
Goldsmith, P., McGarity, B., Walls, A. F., Church, M. K., Millward-Sadler, G. H., and Robertson, D. A. (1990). Corticosteroid treatment reduces mast cell numbers in inflammatory bowel disease. Dig. Dis. Sci. 35, 1409–1413.
Gonzalez-Quintela, A., Vizcaino, L., Gude, F., Rey, J., Meijide, L., Fernandez-Merino, C., Linneberg, A., and Vidal, C. (2010). Factors influencing serum total tryptase concentrations in a general adult population. Clin. Chem. Lab. Med. 48, 701–706.
Green, E. A., Eynon, E. E., and Flavell, R. A. (1998). Local expression of TNF-alpha in neonatal NOD mice promotes diabetes by enhancing presentation of islet antigens. Immunity 9, 733–743.
Gri, G., Piconese, S., Frossi, B., Manfroi, V., Merluzzi, S., Tripodo, C., Viola, A., Odom, S., Rivera, J., Colombo, M. P., and Pucillo, C. E. (2008). CD4+CD25+ regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity 29, 771–781.
Groen, H., Klatter, F., Pater, J., Nieuwenhuis, P., and Rozing, J. (2003). Temporary, but essential requirement of CD8+ T cells early in the pathogenesis of diabetes in BB rats as revealed by thymectomy and CD8 depletion. Clin. Dev. Immunol. 10, 141–151.
Hamawy, M. M., Mergenhagen, S. E., and Siraganian, R. P. (1994). Adhesion molecules as regulators of mast-cell and basophil function. Immunol. Today 15, 62–66.
Helske, S., Syväranta, S., Kupari, M., Lappalainen, J., Laine, M., Lommi, J., Turto, H., Mäyränpää, M., Werkkala, K., Kovanen, P. T., and Lindstedt, K. A. (2006). Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur. Heart J. 27, 1495–1504.
Herlong, J. L., and Scott, T. R. (2006). Positioning prostanoids of the D and J series in the immunopathogenic scheme. Immunol. Lett. 102, 121–131.
Hersoug, L. G., and Linneberg, A. (2007). The link between the epidemics of obesity and allergic diseases: does obesity induce decreased immune tolerance? Allergy 62, 1205–1213.
Hessner, M. J., Wang, X., Meyer, L., Geoffrey, R., Jia, S., Fuller, J., Lernmark, A., and Ghosh, S. (2004). Involvement of eotaxin, eosinophils, and pancreatic predisposition in development of type 1 diabetes mellitus in the BioBreeding rat. J. Immunol. 173, 6993–7002.
Hotamisligil, G. S., Shargill, N. S., and Spiegelman, B. M. (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91.
Huang, Y., Noble, N. A., Zhang, J., Xu, C., and Border, W. A. (2007). Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int. 72, 45–52.
Hübner, M. P., Larson, D., Torrero, M. N., Mueller, E., Shi, Y., Killoran, K. E., and Mitre, E. (2011). Anti-FcεR1 antibody injections activate basophils and mast cells and delay Type 1 diabetes onset in NOD mice. Clin. Immunol. 141, 205–217.
Inamura, N., Mekori, Y. A., Bhattacharyya, S. P., Bianchine, P. J., and Metcalfe, D. D. (1998). Induction and enhancement of Fc(epsilon)RI-dependent mast cell degranulation following coculture with activated T cells: dependency on ICAM-1- and leukocyte function-associated antigen (LFA)-1-mediated heterotypic aggregation. J. Immunol. 160, 4026–4033.
Irani, A. M., Nilsson, G., Miettinen, U., Craig, S. S., Ashman, L. K., Ishizaka, T., Zsebo, K. M., and Schwartz, L. B. (1992). Recombinant human stem cell factor stimulates differentiation of mast cells from dispersed human fetal liver cells. Blood 80, 3009–3021.
Ishida, K., Takai, S., Murano, M., Nishikawa, T., Inoue, T., Murano, N., Inoue, N., Jin, D., Umegaki, E., Higuchi, K., and Miyazaki, M. (2008). Role of chymase-dependent matrix metalloproteinase-9 activation in mice with dextran sodium sulfate-induced colitis. J. Pharmacol. Exp. Ther. 324, 422–426.
Jakschik, B. A., Rengers, T. A., and Pinski, J. R. (1987). Nature of the mast cell-macrophage interaction in immediate hypersensitivity. Adv. Prostaglandin Thromboxane Leukot. Res. 17A, 180–185.
Jones, S. E., Gilbert, R. E., and Kelly, D. J. (2004). Tranilast reduces mesenteric vascular collagen deposition and chymase-positive mast cells in experimental diabetes. J. Diabetes Complicat. 18, 309–315.
Kalantarinia, K., Awad, A. S., and Siragy, H. M. (2003). Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic rats. Kidney Int. 64, 1208–1213.
Kalesnikoff, J., and Galli, S. J. (2008). New developments in mast cell biology. Nat. Immunol. 9, 1215–1223.
Kashyap, M., Thornton, A. M., Norton, S. K., Barnstein, B., Macey, M., Brenzovich, J., Shevach, E., Leonard, W. J., and Ryan, J. J. (2008). Cutting edge: CD4 T cell-mast cell interactions alter IgE receptor expression and signaling. J. Immunol. 180, 2039–2043.
Kaur, D., Doe, C., Woodman, L., Wan, H., Sutcliffe, A., Hollins, F., and Brightling, C. (2011). Mast cell-airway smooth muscle crosstalk: the role of thymic stromal lymphopoietin. Chest. doi: 10.1378/chest.11-1782. [Epub ahead of print].
Kelly, D. J., Zhang, Y., Moe, G., Naik, G., and Gilbert, R. E. (2007). Aliskiren, a novel renin inhibitor, is renoprotective in a model of advanced diabetic nephropathy in rats. Diabetologia 50, 2398–2404.
Kettner, A., Di Matteo, M., and Santoni, A. (2010). Insulin potentiates Fc epsilon RI-mediated signaling in mouse bone marrow-derived mast cells. Mol. Immunol. 47, 1039–1046.
Kikutani, H., and Makino, S. (1992). The murine autoimmune diabetes model: NOD and related strains. Adv. Immunol. 51, 285–322.
Kitaura, J., Eto, K., Kinoshita, T., Kawakami, Y., Leitges, M., Lowell, C. A., and Kawakami, T. (2005). Regulation of highly cytokinergic IgE-induced mast cell adhesion by Src, Syk, Tec, and protein kinase C family kinases. J. Immunol. 174, 4495–4504.
Klein-Wieringa, I. R., Kloppenburg, M., Bastiaansen-Jenniskens, Y. M., Yusuf, E., Kwekkeboom, J. C., El-Bannoudi, H., Nelissen, R. G., Zuurmond, A., Stojanovic-Susulic, V., Van Osch, G. J., Toes, R. E., and Ioan-Facsinay, A. (2011). The infrapatellar fat pad of patients with osteoarthritis has an inflammatory phenotype. Ann. Rheum. Dis. 70, 851–857.
Kliewer, S. A., Lenhard, J. M., Willson, T. M., Patel, I., Morris, D. C., and Lehmann, J. M. (1995). A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83, 813–819.
Koda, A., Kurashina, Y., and Nakazawa, M. (1985). The inhibition mechanism of histamine release by N-(3,4-dimethoxycinnamoyl) anthranilic acid. Int. Arch. Allergy Appl. Immunol. 77, 244–245.
Kondo, S., Kagami, S., Kido, H., Strutz, F., Müller, G. A., and Kuroda, Y. (2001). Role of mast cell tryptase in renal interstitial fibrosis. J. Am. Soc. Nephrol. 12, 1668–1676.
Kotani, A., Hori, T., Fujita, T., Kambe, N., Matsumura, Y., Ishikawa, T., Miyachi, Y., Nagai, K., Tanaka, Y., and Uchiyama, T. (2007). Involvement of OX40 ligand+ mast cells in chronic GVHD after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 39, 373–375.
Kovanen, P. T. (2007). Mast cells: multipotent local effector cells in atherothrombosis. Immunol. Rev. 217, 105–122.
Koyama, S., Takagi, H., Otani, A., Suzuma, K., Nishimura, K., and Honda, Y. (1999). Tranilast inhibits protein kinase C-dependent signalling pathway linked to angiogenic activities and gene expression of retinal microcapillary endothelial cells. Br. J. Pharmacol. 127, 537–545.
Kukreja, A., and Maclaren, N. K. (1999). Autoimmunity and diabetes. J. Clin. Endocrinol. Metab. 84, 4371–4378.
Lessmann, E., Grochowy, G., Weingarten, L., Giesemann, T., Aktories, K., Leitges, M., Krystal, G., and Huber, M. (2006). Insulin and insulin-like growth factor-1 promote mast cell survival via activation of the phosphatidylinositol-3-kinase pathway. Exp. Hematol. 34, 1532–1541.
Levick, S. P., McLarty, J. L., Murray, D. B., Freeman, R. M., Carver, W. E., and Brower, G. L. (2009). Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 53, 1041–1047.
Liu, J., Divoux, A., Sun, J., Zhang, J., Clément, K., Glickman, J. N., Sukhova, G. K., Wolters, P. J., Du, J., Gorgun, C. Z., Doria, A., Libby, P., Blumberg, R. S., Kahn, B. B., Hotamisligil, G. S., and Shi, G. P. (2009). Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 15, 940–945.
Lu, L. F., Lind, E. F., Gondek, D. C., Bennett, K. A., Gleeson, M. W., Pino-Lagos, K., Scott, Z. A., Coyle, A. J., Reed, J. L., Van Snick, J., Strom, T. B., Zheng, X. X., and Noelle, R. J. (2006). Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 442, 997–1002.
Ludvik, B., Clodi, M., Kautzky-Willer, A., Capek, M., Hartter, E., Pacini, G., and Prager, R. (1993). Effect of dexamethasone on insulin sensitivity, islet amyloid polypeptide and insulin secretion in humans. Diabetologia 36, 84–87.
Macia, L., Delacre, M., Abboud, G., Ouk, T. S., Delanoye, A., Verwaerde, C., Saule, P., and Wolowczuk, I. (2006). Impairment of dendritic cell functionality and steady-state number in obese mice. J. Immunol. 177, 5997–6006.
Mahmoud, R. A., el-Ezz, S. A., and Hegazy, A. S. (2004). Increased serum levels of interleukin-18 in patients with diabetic nephropathy. Ital. J. Biochem. 53, 73–81.
Maltby, S., Khazaie, K., and McNagny, K. M. (2009). Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim. Biophys. Acta. 1796, 19–26.
Marshall, J. S., Gomi, K., Blennerhassett, M. G., and Bienenstock, J. (1999). Nerve growth factor modifies the expression of inflammatory cytokines by mast cells via a prostanoid-dependent mechanism. J. Immunol. 162, 4271–4276.
Mathis, D., Vence, L., and Benoist, C. (2001). Beta-cell death during progression to diabetes. Nature 414, 792–798.
Mekori, Y. A. (2004). The mastocyte: the “other” inflammatory cell in immunopathogenesis. J. Allergy Clin. Immunol. 114, 52–57.
Mekori, Y. A., Oh, C. K., and Metcalfe, D. D. (1993). IL-3-dependent murine mast cells undergo apoptosis on removal of IL-3. Prevention of apoptosis by c-kit ligand. J. Immunol. 151, 3775–3784.
Mifsud, S., Kelly, D. J., Qi, W., Zhang, Y., Pollock, C. A., Wilkinson-Berka, J. L., and Gilbert, R. E. (2003). Intervention with tranilast attenuates renal pathology and albuminuria in advanced experimental diabetic nephropathy. Nephron Physiol. 95, p83–p91.
Mirmonsef, P., Shelburne, C. P., Fitzhugh Yeatman, C. II, Chong, H. J., and Ryan, J. J. (1999). Inhibition of Kit expression by IL-4 and IL-10 in murine mast cells: role of STAT6 and phosphatidylinositol 3′-kinase. J. Immunol. 163, 2530–2539.
Mordes, J. P., Bortell, R., Doukas, J., Rigby, M., Whalen, B., Zipris, D., Greiner, D. L., and Rossini, A. A. (1996). The BB/Wor rat and the balance hypothesis of autoimmunity. Diabetes Metab. Rev. 12, 103–109.
Moriwaki, Y., Inokuchi, T., Yamamoto, A., Ka, T., Tsutsumi, Z., Takahashi, S., and Yamamoto, T. (2007). Effect of TNF-alpha inhibition on urinary albumin excretion in experimental diabetic rats. Acta Diabetol. 44, 215–218.
Murano, I., Barbatelli, G., Parisani, V., Latini, C., Muzzonigro, G., Castellucci, M., and Cinti, S. (2008). Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 49, 1562–1568.
Musilli, C., Paccosi, S., Pala, L., Gerlini, G., Ledda, F., Mugelli, A., Rotella, C. M., and Parenti, A. (2011). Characterization of circulating and monocyte-derived dendritic cells in obese and diabetic patients. Mol. Immunol. 49, 234–238.
Nakamura, T., Fukui, M., Ebihara, I., Osada, S., Nagaoka, I., Tomino, Y., and Koide, H. (1993). mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes 42, 450–456.
Nakano, T., Sonoda, T., Hayashi, C., Yamatodani, A., Kanayama, Y., Yamamura, T., Asai, H., Yonezawa, T., Kitamura, Y., and Galli, S. J. (1985). Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice. Evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J. Exp. Med. 162, 1025–1043.
Navarro, J. F., Mora, C., Maca, M., and Garca, J. (2003). Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am. J. Kidney Dis. 42, 53–61.
Nishimura, S., Manabe, I., Nagasaki, M., Eto, K., Yamashita, H., Ohsugi, M., Otsu, M., Hara, K., Ueki, K., Sugiura, S., Yoshimura, K., Kadowaki, T., and Nagai, R. (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 15, 914–920.
Nocka, K., Buck, J., Levi, E., and Besmer, P. (1990). Candidate ligand for the c-kit transmembrane kinase receptor: KL, a fibroblast derived growth factor stimulates mast cells and erythroid progenitors. EMBO J. 9, 3287–3294.
Ochi, M., Sawada, T., Kusunoki, T., and Hattori, T. (1988). Morphology and cell dynamics of adipose tissue in hypothalamic obese mice. Am. J. Physiol. 254(Pt 2), R740–R745.
Ohmura, K., Ishimori, N., Ohmura, Y., Tokuhara, S., Nozawa, A., Horii, S., Andoh, Y., Fujii, S., Iwabuchi, K., Onoé, K., and Tsutsui, H. (2010). Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler. Thromb. Vasc. Biol. 30, 193–199.
Okon, K., and Stachura, J. (2007). Increased mast cell density in renal interstitium is correlated with relative interstitial volume, serum creatinine and urea especially in diabetic nephropathy but also in primary glomerulonephritis. Pol. J. Pathol. 58, 193–197.
Otton, R., Soriano, F. G., Verlengia, R., and Curi, R. (2004). Diabetes induces apoptosis in lymphocytes. J. Endocrinol. 182, 145–156.
Ozsoy, N., and Gül, N. (2005). Characterization of the ultrastructure of gingival mast cells in alloxan-induced diabetic rats. Cell Biochem. Funct. 23, 333–337.
Piconese, S., Gri, G., Tripodo, C., Musio, S., Gorzanelli, A., Frossi, B., Pedotti, R., Pucillo, C. E., and Colombo, M. P. (2009). Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood 114, 2639–2648.
Pipkorn, U., Hammarlund, A., and Enerbäck, L. (1989). Prolonged treatment with topical glucocorticoids results in an inhibition of the allergen-induced weal-and-flare response and a reduction in skin mast cell numbers and histamine content. Clin. Exp. Allergy 19, 19–25.
Poglio, S., De Toni-Costes, F., Arnaud, E., Laharrague, P., Espinosa, E., Casteilla, L., and Cousin, B. (2010). Adipose tissue as a dedicated reservoir of functional mast cell progenitors. Stem Cells 28, 2065–2072.
Ptak, W., Rewicka, M., Strzyzewska, J., and Kollat, M. (1983). Alleviation of IgE-mediated immune reactions in hypoinsulinaemic and hyperglycaemic mice. Clin. Exp. Immunol. 52, 54–60.
Ritz, E. (2003). Chymase: a potential culprit in diabetic nephropathy? J. Am. Soc. Nephrol. 14, 1952–1954.
Rüger, B. M., Hasan, Q., Greenhill, N. S., Davis, P. F., Dunbar, P. R., and Neale, T. J. (1996). Mast cells and type VIII collagen in human diabetic nephropathy. Diabetologia 39, 1215–1222.
Russo, L. M., del Re, E., Brown, D., and Lin, H. Y. (2007). Evidence for a role of transforming growth factor (TGF)-beta1 in the induction of postglomerular albuminuria in diabetic nephropathy: amelioration by soluble TGF-beta type II receptor. Diabetes 56, 380–388.
Ryan, J. J., DeSimone, S., Klisch, G., Shelburne, C., McReynolds, L. J., Han, K., Kovacs, R., Mirmonsef, P., and Huff, T. F. (1998). IL-4 inhibits mouse mast cell Fc epsilon RI expression through a STAT6-dependent mechanism. J. Immunol. 161, 6915–6923.
Salamon, P., Shoham, N. G., Puxeddu, I., Paitan, Y., Levi-Schaffer, F., and Mekori, Y. A. (2008). Human mast cells release oncostatin M on contact with activated T cells: possible biologic relevance. J. Allergy Clin. Immunol. 121, 448–455.
Sato, H., Iwano, M., Akai, Y., Kurioka, H., Kubo, A., Yamaguchi, T., Hirata, E., Kanauchi, M., and Dohi, K. (1998). Increased excretion of urinary transforming growth factor beta 1 in patients with diabetic nephropathy. Am. J. Nephrol. 18, 490–494.
Sayed, B. A., and Brown, M. A. (2007). Mast cells as modulators of T-cell responses. Immunol. Rev. 217, 53–64.
Schleimer, R. P. (1993). An overview of glucocorticoid anti-inflammatory actions. Eur. J. Clin. Pharmacol. 45(Suppl. 1), S3–S7.
Schwertschlag, U., and Hackenthal, E. (1982). Histamine stimulates renin release from the isolated perfused rat kidney. Naunyn Schmiedebergs Arch. Pharmacol. 319, 239–242.
Shefler, I., Salamon, P., Reshef, T., Mor, A., and Mekori, Y. A. (2010). T cell-induced mast cell activation: a role for microparticles released from activated T cells. J. Immunol. 185, 4206–4212.
Shi, M. A., and Shi, G. P. (2010). Intracellular delivery strategies for microRNAs and potential therapies for human cardiovascular diseases. Sci. Signal. 3, pe40.
Shore, S. A. (2010). Obesity, airway hyperresponsiveness, and inflammation. J. Appl. Physiol. 108, 735–743.
Silver, R. B., Reid, A. C., Mackins, C. J., Askwith, T., Schaefer, U., Herzlinger, D., and Levi, R. (2004). Mast cells: a unique source of renin. Proc. Natl. Acad. Sci. U.S.A. 101, 13607–13612.
Smith, A. G., Sheridan, P. A., Tseng, R. J., Sheridan, J. F., and Beck, M. A. (2009). Selective impairment in dendritic cell function and altered antigen-specific CD8+ T-cell responses in diet-induced obese mice infected with influenza virus. Immunology 126, 268–279.
Soma, J., Sugawara, T., Huang, Y. D., Nakajima, J., and Kawamura, M. (2002). Tranilast slows the progression of advanced diabetic nephropathy. Nephron 92, 693–698.
Spiecker, M., Lorenz, I., Marx, N., and Darius, H. (2002). Tranilast inhibits cytokine-induced nuclear factor kappaB activation in vascular endothelial cells. Mol. Pharmacol. 62, 856–863.
St. John, A. L., Rathore, A. P., Yap, H., Ng, M. L., Metcalfe, D. D., Vasudevan, S. G., and Abraham, S. N. (2011). Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc. Natl. Acad. Sci. U.S.A. 108, 9190–9195.
Stelekati, E., Bahri, R., D’Orlando, O., Orinska, Z., Mittrücker, H. W., Langenhaun, R., Glatzel, M., Bollinger, A., Paus, R., and Bulfone-Paus, S. (2009). Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity 31, 665–676.
Sun, J., Sukhova, G. K., Wolters, P. J., Yang, M., Kitamoto, S., Libby, P., MacFarlane, L. A., Mallen-St Clair, J., and Shi, G. P. (2007a). Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat. Med. 13, 719–724.
Sun, J., Sukhova, G. K., Yang, M., Wolters, P. J., MacFarlane, L. A., Libby, P., Sun, C., Zhang, Y., Liu, J., Ennis, T. L., Knispel, R., Xiong, W., Thompson, R. W., Baxter, B. T., and Shi, G. P. (2007b). Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J. Clin. Invest. 117, 3359–3368.
Suto, H., Nakae, S., Kakurai, M., Sedgwick, J. D., Tsai, M., and Galli, S. J. (2006). Mast cell-associated TNF promotes dendritic cell migration. J. Immunol. 176, 4102–4112.
Suzawa, H., Kikuchi, S., Ichikawa, K., and Koda, A. (1992). Inhibitory action of tranilast, an anti-allergic drug, on the release of cytokines and PGE2 from human monocytes-macrophages. Jpn. J. Pharmacol. 60, 85–90.
Taguchi, S., Ozaki, N., Umeda, H., Mizutani, N., Yamada, T., and Oiso, Y. (2008). Tranilast inhibits glucose-induced insulin secretion from pancreatic beta-cells. Horm. Metab. Res. 40, 518–523.
Tanaka, A., Nomura, Y., Matsuda, A., Ohmori, K., and Matsuda, H. (2011). Mast cells function as an alternative modulator of adipogenesis through 15-deoxy-delta-12, 14-prostaglandin J2. Am. J. Physiol. Cell Physiol. 301, C1360–C1367.
Tedgui, A., Owens, A. P. III, and Mackman, N. (2011). Nobel prize in physiology or medicine: toll-like receptors, dendritic cells, and their roles in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 2767–2768.
Théry, C., Ostrowski, M., and Segura, E. (2009). Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 9, 581–593.
Tilg, H., and Moschen, A. R. (2006). Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 6, 772–783.
Tóth, T., Tóth-Jakatics, R., Jimi, S., Ihara, M., Urata, H., and Takebayashi, S. (1999). Mast cells in rapidly progressive glomerulonephritis. J. Am. Soc. Nephrol. 10, 1498–1505.
Trayhurn, P. (2005). Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol. Scand. 184, 285–293.
Uysal, K. T., Wiesbrock, S. M., Marino, M. W., and Hotamisligil, G. S. (1997). Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389, 610–614.
Vianna, E. O., and Garcia-Leme, J. (1995). Allergen-induced airway inflammation in rats. Role of insulin. Am. J. Respir. Crit. Care Med. 151(Pt 1), 809–814.
Wallis, R. H., Wang, K., Marandi, L., Hsieh, E., Ning, T., Chao, G. Y., Sarmiento, J., Paterson, A. D., and Poussier, P. (2009). Type 1 diabetes in the BB rat: a polygenic disease. Diabetes 58, 1007–1017.
Wang, Z., Zhang, H., Shen, X. H., Jin, K. L., Ye, G. F., Qian, L., Li, B., Zhang, Y. H., and Shi, G. P. (2012). Immunoglobulin E and mast cell proteases are potential risk factors of human pre-diabetes and diabetes mellitus. PLoS ONE 6, e28962.
Weisberg, S. P., McCann, D., Desai, M., Rosenbaum, M., Leibel, R. L., and Ferrante, A. W. Jr. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808.
Weyer, C., Funahashi, T., Tanaka, S., Hotta, K., Matsuzawa, Y., Pratley, R. E., and Tataranni, P. A. (2001). Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J. Clin. Endocrinol. Metab. 86, 1930–1935.
Wilson, T. J., Nannuru, K. C., and Singh, R. K. (2009). Cathepsin G-mediated activation of pro-matrix metalloproteinase 9 at the tumor-bone interface promotes transforming growth factor-beta signaling and bone destruction. Mol. Cancer Res. 7, 1224–1233.
Winer, D. A., Winer, S., Shen, L., Wadia, P. P., Yantha, J., Paltser, G., Tsui, H., Wu, P., Davidson, M. G., Alonso, M. N., Leong, H. X., Glassford, A., Caimol, M., Kenkel, J. A., Tedder, T. F., McLaughlin, T., Miklos, D. B., Dosch, H. M., and Engleman, E. G. (2011). B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 17, 610–617.
Winer, S., Chan, Y., Paltser, G., Truong, D., Tsui, H., Bahrami, J., Dorfman, R., Wang, Y., Zielenski, J., Mastronardi, F., Maezawa, Y., Drucker, D. J., Engleman, E., Winer, D., and Dosch, H. M. (2009). Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 15, 921–929.
Wolf, G., and Ziyadeh, F. N. (1999). Molecular mechanisms of diabetic renal hypertrophy. Kidney Int. 56, 393–405.
Wolf, G., and Ziyadeh, F. N. (2007). Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol. 106, p26–p31.
Wu, D., Molofsky, A. B., Liang, H. E., Ricardo-Gonzalez, R. R., Jouihan, H. A., Bando, J. K., Chawla, A., and Locksley, R. M. (2011). Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332, 243–247.
Xu, H., Barnes, G. T., Yang, Q., Tan, G., Yang, D., Chou, C. J., Sole, J., Nichols, A., Ross, J. S., Tartaglia, L. A., and Chen, H. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830.
Xu, J., and Shi, G. P. (2012). Emerging role of mast cells and macrophages in cardiovascular and metabolic diseases. Endocr. Rev. (in press).
Yamada, H., Tajima, S., Nishikawa, T., Murad, S., and Pinnell, S. R. (1994). Tranilast, a selective inhibitor of collagen synthesis in human skin fibroblasts. J. Biochem. 116, 892–897.
Yeatman, C. F. II, Jacobs-Helber, S. M., Mirmonsef, P., Gillespie, S. R., Bouton, L. A., Collins, H. A., Sawyer, S. T., Shelburne, C. P., and Ryan, J. J. (2000). Combined stimulation with the T helper cell type 2 cytokines interleukin (IL)-4 and IL-10 induces mouse mast cell apoptosis. J. Exp. Med. 192, 1093–1103.
Zhang, B., Alysandratos, K. D., Angelidou, A., Asadi, S., Sismanopoulos, N., Delivanis, D. A., Weng, Z., Miniati, A., Vasiadi, M., Katsarou-Katsari, A., Miao, B., Leeman, S. E., Kalogeromitros, D., and Theoharides, T. C. (2011a). Human mast cell degranulation and preformed TNF secretion require mitochondrial translocation to exocytosis sites: relevance to atopic dermatitis. J. Allergy Clin. Immunol. 127, 1522–1531.
Keywords: mast cell, obesity, diabetes, diabetic nephropathy, T cell, dendritic cell
Citation: Shi MA and Shi G-P (2012) Different roles of mast cells in obesity and diabetes: lessons from experimental animals and humans. Front. Immun. 3:7. doi: 10.3389/fimmu.2012.00007
Received: 14 December 2011; Accepted: 09 January 2012;
Published online: 25 January 2012.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Masato Kubo, Tokyo University of Science, JapanCopyright: © 2012 Shi and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Guo-Ping Shi, Cardiovascular Medicine, Brigham and Women’s Hospital, 77 Avenue Louis Pasteur, NRB-7 Boston, MA 02115, USA. e-mail:Z3NoaUByaWNzLmJ3aC5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.