
- Department of Immunology and Infection, London School of Hygiene and Tropical Medicine, London, UK
Natural killer (NK) cells are large granular lymphocytes that express a diverse array of germline encoded inhibitory and activating receptors for MHC Class I and Class I-like molecules, classical co-stimulatory ligands, and cytokines. The ability of NK cells to be very rapidly activated by inflammatory cytokines, to secrete effector cytokines, and to kill infected or stressed host cells, suggests that they may be among the very early responders during infection. Recent studies have also identified a small number of pathogen-derived ligands that can bind to NK cell surface receptors and directly induce their activation. Here we review recent studies that have begun to elucidate the various pathways by which viral, bacterial, and parasite pathogens activate NK cells. We also consider two emerging themes of NK cell–pathogen interactions, namely their contribution to adaptive immune responses and their potential to take on regulatory and immunomodulatory functions.
Introduction
The innate immune system, comprising large populations of cells expressing germ line-encoded receptors for non-self molecules, is designed to provide rapid but generic responses to foreign organisms. Innate immune cells initiate, amplify, and direct the subsequent adaptive response via diverse contact-dependent and cytokine-mediated signals; vaccine adjuvants take direct advantage of this capability (Pashine et al., 2005). Among these innate cell populations, natural killer (NK) cells – first recognized and named for their ability to kill malignant or transformed cells (Kiessling et al., 1975) – are now known to play an important role in the control of intracellular pathogens including viruses, bacteria, and protozoa (Korbel et al., 2004; Lodoen and Lanier, 2006) and two distinct pathways of NK cell activation are now recognized (Figure 1). In the direct “missing-self” pathway, NK cell activation results from their interaction with somatic cells that lack MHC Class I ligands for NK inhibitory receptors (Karre et al., 1986; Karre, 2008). In the absence of inhibitory signals, signals from activating receptors (recognizing endogenous stress molecules or pathogen-encoded ligands) lead to induction of NK cell cytotoxic function and cytokine release. The indirect pathway of NK cell activation – mediated by myeloid accessory cells responding to pathogens via diverse pattern recognition receptors by cytokine release and upregulation of co-stimulatory receptors (reviewed in Newman and Riley, 2007) and augmented by antigen-specific T cells (Horowitz et al., 2010a; Evans et al., 2011) over-rides the inhibitory signals provided by MHC Class I competent cells. The various combinations of NK cell receptors that induce different modes of NK cell effector function have been reviewed (Bryceson et al., 2006; Lanier, 2008) and are summarized in Figure 2.
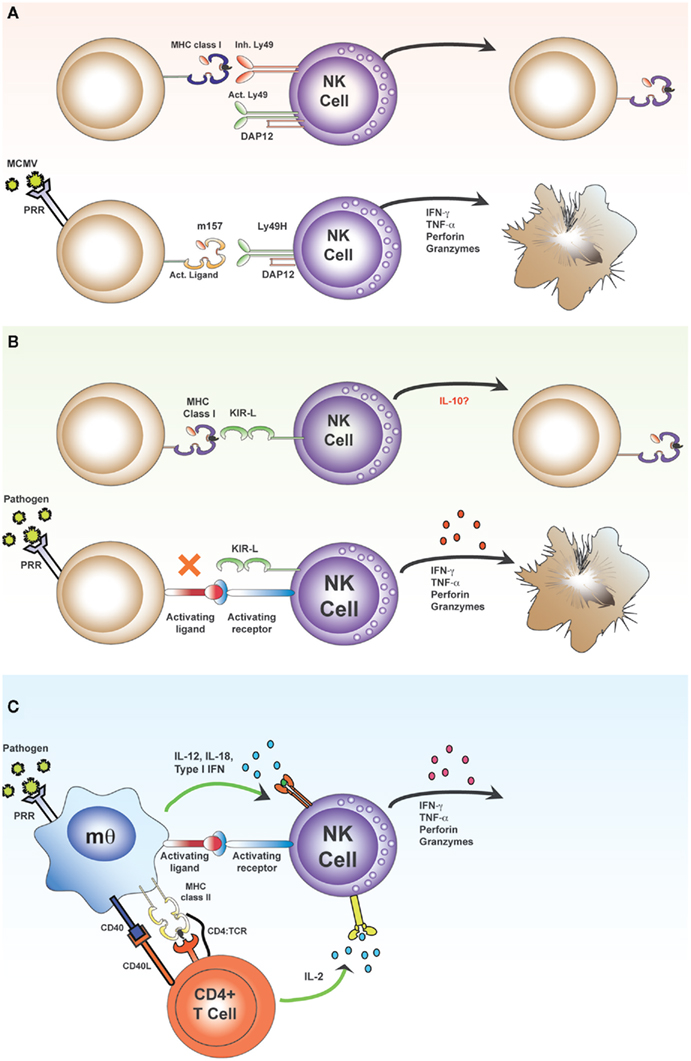
Figure 1. Direct and indirect activation of NK cells. The “classical” pathway of NK cell activation (A,B) results from skewing of the balance between signals transmitted via inhibitory and activating receptors. Inhibitory receptors, such as the Ly49 lectin-like receptors in mice (A) and the killer cell immunoglobulin-like receptors (KIR) in humans (B) bind MHC class I molecules. These MHC class I molecules may contain either self or foreign (antigenic) peptides within the peptide binding groove and there is increasing evidence that the nature of the bound peptide may influence the affinity of receptor–MHC interactions. In the absence of appropriate inhibitory interactions (due to down regulation of MHC or expression of foreign MHC molecules which provide no ligands for polymorphic inhibitory receptors), activating signals may predominate, leading to production of effector molecules, and direct killing of target cells. Activating ligands include stress-induced ligands on infected or transformed cells and virus-encoded ligands. For example, during infection with murine cytomegalovirus (MCMV), the activating receptor Ly49H recognizes the MCMV-derived MHC class I-like molecule, m157, which leads to direct killing of MCMV-infected target cells [(A); lower panel]. The “indirect” pathway of NK cell activation results from contact-dependent and soluble signals derived from accessory cells and T cells transmitted to activating receptors on NK cells (C). Myeloid accessory cells recognize pathogens via PRRs, secrete cytokines, and upregulate co-stimulatory molecules. Accessory cells also present antigen to CD4+ T cells and provide T cell co-stimulation; activated T cells secrete IL-2 which synergizes with accessory cell signals to potentiate the activation of NK cells.
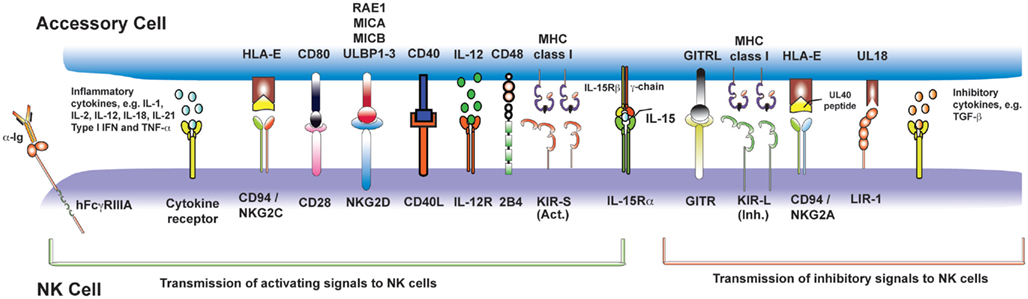
Figure 2. Nature killer cell activating signals from myeloid accessory cells and T cells. Following recognition of pathogen by PRRs, accessory cells (in blue, above) become activated and transmit signals (both contact-dependent and -independent) to activating receptors on NK cells (in purple, below). This figure summarizes those for which there is the strongest experimental evidence, but is not exhaustive.
The purpose of this article is to review recent developments in our understanding of how different pathogens induce NK cell activation; as such this article represents an updating of our previous article on this subject (Newman and Riley, 2007). We review recently published studies which extend our understanding of NK cell activation by viruses, bacteria, and protozoa as well as recent data revealing that NK cells may also influence the outcome of fungal and helminth infections.
NK Cells and Viruses
The role of NK cells in controlling viral infections has been long recognized (Biron and Brossay, 2001). Many viruses modify the surface membrane of their host cell, leading to down regulation of MHC class I molecules and/or upregulation of host-derived stress-related molecules which mark the infected cell as “abnormal” and lead to NK cell-mediated killing. The role of accessory cell-derived co-stimulatory signals in NK cell activation during viral infection has also been long recognized (Biron et al., 1999), indicating that both the direct and indirect pathways of NK cell activation are likely to contribute to their control of viral infections.
Direct Activation of NK Cells during Viral Infection
Evidence for direct NK cell activation during viral infection came from the discovery that influenza hemagglutinin binds to NKp46 on human NK cells and induces cytotoxic activity (Mandelboim et al., 2001). At more or less the same time, it was found that the m157 glycoprotein of murine cytomegalovirus (MCMV) binds to the NK cell activating receptor Ly49H and that strains of mice, which express Ly49H are highly resistant to MCMV infection. Ly49H binding of m157 is entirely sufficient for NK cell activation and MCMV elimination (Daniels et al., 2001; Arase et al., 2002; Smith et al., 2002). Mouse NK cells can also be induced to secrete IFN-γ by interaction of the activating receptor Ly49P with H2-Dk of MCMV-infected cells; recognition of MCMV-infected cells is dependent on the viral protein m04, but m04 alone is not sufficient for activation of Ly49P+ NK cells (Kielczewska et al., 2009).
Viral envelope proteins of two flaviviruses (West Nile virus and Dengue virus) bind to NKp44 (Hershkovitz et al., 2009) however, as NKp44 is only expressed on IL-2-activated NK cells (Vitale et al., 1998), it is likely that the NK cells also require signals from accessory cells in order to respond to these two viruses.
NK cells mediate antiviral activity in the very early phase of infection with poxviruses such as vaccinia virus (VV) and ectromelia virus (ECTV; Parker et al., 2007). During ECTV infection, the activating receptor NKG2D induces NK cytotoxicity and controls virus dissemination (Fang et al., 2008) but during VV infection cytotoxicity is regulated by NKp30, NKp44, and NKp46 (Chisholm and Reyburn, 2006). Poxvirus hemagglutinins are ligands for NKp30 and NKp46; hemagglutinin blocks NKp30-dependent activation but induces NKp46-dependent activation (Jarahian et al., 2011). NK cells also recognize ECTV infected cells by binding of the CD94–NKG2E heterodimer to the MHC class Ib molecule Qa-1b (Fang et al., 2011); CD94 expression is essential for resistance to ECTV as NK cells from CD94-deficient mice are unable to prevent virus dissemination (Fang et al., 2011).
There is accumulating evidence that direct interactions between MHC Class I/Class 1-like molecules on target cells and MHC receptors on NK cells influence the outcome of chronic virus infections. In humans, the polymorphic killer cell immunoglobulin-like receptors (KIRs) bind to HLA Class 1 molecules and transduce (mainly) inhibitory signals. The strength of the interaction between particular KIRs and their particular HLA/peptide ligands determines the threshold for NK cell activation and thus, potentially, viral clearance (Alter et al., 2011a; Fadda et al., 2011). Thus, expression of the inhibitory NK cell receptor KIR2DL3 in combination with its ligand HLA-C1 has been associated with clearance of acute hepatitis C virus (HCV) infection (Khakoo et al., 2004) and with positive outcomes of treatment of chronic HCV infection with pegylated IFNα and ribavirin. Individuals expressing an alternate receptor, KIR2DL2, tend to respond less well to treatment (Yoon et al., 2009), which may reflect the stronger binding affinity of KIR2DL2 to HLA-C1, leading to greater inhibition of NK cells.
Recent studies have demonstrated that the nature of the peptide bound within the HLA Class I groove can influence KIR–HLA interactions. HLA-C1 binding peptides can modify binding to KIR2DL2 and KIR2DL3 with significant knock-on effects on NK cell activation (Fadda et al., 2011). Moreover, non-synonymous polymorphisms in the HIV-1 sequence are reported to select for increased recognition and binding by KIR2DL2 in chronically HIV-infected donors (Alter et al., 2011a). Finally, the recent suggestion that the terminal D0 domain of KIR3DL2 binds CpG DNA, leading to KIR internalization and shuttling of the CpG to early endosomes where it binds to TLR9 and activates the NK cell offers an intriguing new route by which a variety of microparasites, including viruses, might directly activate NK cells (Sivori et al., 2011).
The roles of other NK cell surface receptors during viral infections remain somewhat unclear. Reports that NK cell effector functions can be inhibited by cross linking of CD81 (a receptor for HCV viral entry into cells) on NK cells by HCV envelope protein 2 (Crotta et al., 2002, 2010; Tseng and Klimpel, 2002) have been challenged by a recent report showing that HCV envelope protein 2, as part of intact and infectious HCV virions, does not alter NK cell function (Yoon et al., 2009). Several studies have investigated NK receptor expression in chronic HCV virus infection – with rather mixed results (reviewed in Cheent and Khakoo, 2011). Recently it was reported that expression of the activating receptor NKG2D is upregulated on peripheral blood NK cells of individuals with acute HCV infection (Amadei et al., 2010) although this did not correlate with the outcome of infection (i.e., chronic or self-limited infection), NK cells of patients with acute HCV produced more IFN-γ upon IL-12 stimulation than those of healthy controls suggesting that they are maintained in a partially activated state (Amadei et al., 2010). On the other hand, the percentages of NKG2D+ (as well as NKp30+ and NKp46+) peripheral blood NK cells are reportedly decreased in patients with acute, chronic, and resolved HCV infections whilst NKG2A+ and CD94+ NK cells were found to be raised in acute and chronic HCV infection but not in resolved infection (Alter et al., 2011b). Moreover, despite increased expression of the NKG2D ligands MICA and MICB on hepatocytes, chronic HCV infection is reportedly associated with down modulation of NKG2D on circulating NK cells leading to decreased NK cell-mediated cytotoxic killing and IFN-γ production (Sene et al., 2010). Further studies are required to determine whether the phenotype of these peripheral NK cells reflects the phenotype of NK cells trafficking into the infected liver.
Indirect Activation of NK Cells during Viral Infection
The indirect pathway of NK cell activation is well-described for many viruses, including herpes viruses such as MCMV and herpes simplex virus-1 (HSV-1), and involves secretion of type I interferons (IFNs) and IL-12 from plasmacytoid dendritic cells (pDCs; Dzionek et al., 2001; Dalod et al., 2003; Krug et al., 2004a; Zucchini et al., 2008; Steinberg et al., 2009; Amadei et al., 2010). NK cell IFN-γ production appears to be particularly dependent upon sustained IL-12 production whereas NK cell cytotoxicity can be maintained in the presence of either IFN-α or IFN-β (Steinberg et al., 2009). In the early phase of MCMV infection, IFN-α and IL-12 production is dependent on MyD88/TLR9 signaling (Delale et al., 2005; Zucchini et al., 2008). In the later phases of infection, IFN-α production (and hence NK cell cytotoxicity) is maintained in an increasingly pDC- and MyD88-independent manner whereas NK cell IFN-γ production is impaired in the latter phase of infection due to reduced availability of IL-12 (Delale et al., 2005). The availability of mice expressing the diphtheria toxin receptor under control of the pDC-specific promoter BDCA-2 – in which pDCs are selectively depleted by administration of diphtheria toxin – has revealed that pDC depletion during the early stages of MCMV infection leads to reduced IFN-α/β production and enhanced MCMV burden (Dzionek et al., 2001; Swiecki et al., 2010). Accordingly, pDCs seem to be involved in activation of NK cells in the early phase of infection, but expansion of Ly49H+ NK cells in the later phase of infection is independent of pDCs. For HSV-1, IFN-α, and IL-12 production from pDCs is dependent on MyD88/TLR9 signaling but mice lacking either MyD88 or TLR9 are able to control HSV-1 infection (Krug et al., 2004a), suggesting that alternate pathways are available for NK cell activation. Depletion of pDCs during infection with the rhabdovirus vesicular stomatitis virus (VSV) also allows for significantly enhanced viral replication in the very early phase of infection, suggesting an important role for pDCs in clearance of diverse viral infections (Swiecki et al., 2010). Nevertheless, CD11b+ myeloid (m) DCs can also mediate IFN-α/β-dependent NK cell activation during MCMV infection (Andoniou et al., 2005); this appears to be TLR-9 independent (Andoniou et al., 2005) but requires TLR-2 and/or TLR-3 signaling (Tabeta et al., 2004; Szomolanyi-Tsuda et al., 2006; Barbalat et al., 2009).
The importance of indirect NK cell activation by cytokines in controlling virus infections is evident from the evolution of immune evasion strategies to subvert this pathway. For example, poxviruses can produce IL-18 binding proteins which inactivate host IL-18; viral mutants lacking IL-18 binding proteins induce higher levels of NK cell IFN-γ and cytotoxicity (Born et al., 2000; Reading and Smith, 2003).
During chronic HCV infection, NK cells express higher than normal levels of activation markers such as CD122 (the IL-2 receptor sub-unit responsible for IL-2 and IL-15 signaling), CD69 and NKp44 (Oliviero et al., 2009; Ahlenstiel et al., 2010), and IFN-α induces expression of the TNF-related apoptosis-inducing ligand (TRAIL; CD253) on liver NK cells (Ochi et al., 2004). TRAIL+ NK cells can kill autologous HCV-infected hepatocytes, a function which is enhanced by poly(I:C) treatment, although this may contribute to liver pathology in chronically infected individuals (Ochi et al., 2004). TRAIL+ NK cells have also been observed in the livers of patients with chronic hepatitis B virus (HBV) infection; TRAIL expression on NK cells was temporally associated with liver inflammation and hepatocyte expression of a TRAIL death-inducing receptor (Dunn et al., 2007) but an understanding of the pathways leading to TRAIL upregulation on NK cells remains incomplete.
Taken together, these studies support the concept that pDCs and mDCs can mount a coordinated response to viral infection by production of type I IFNs, IL-12, and chemokines leading to recruitment and activation of NK cells (Takeda et al., 2003; Krug et al., 2004b). However, there are also reports suggesting that, during MCMV infection, cross-talk between pDCs and mDCs can inhibit IL-12 production, leading to suppression of IFN-γ-producing NK cells (Dalod et al., 2003; Amadei et al., 2010) and a recent study demonstrated that antagonizing IFN-β1, IL-12, and IFN-γ can prevent HCMV-induced down regulation of NKG2D on NK cells and thus reverse suppression of NKG2D-mediated cytotoxicity (Muntasell et al., 2010). Inhibition of NK cell function during chronic HCV infection appears to be mediated by binding of the HCV non-structural protein 5A to TLR4 on monocytes, inducing a switch from IL-12 to IL-10 secretion and induction of TGF-β; NKG2D expression and NK cell function could be restored with exogenous IL-15 (Sene et al., 2010). This is in line with reports of a positive feedback loop of type-1 IFN and IL-15 production leading to NK cell cytotoxicity and production of IFN-γ, which becomes impaired during chronic HCV infection (Jinushi et al., 2003). In chronic HBV infection, liver NK cell IFN-γ production and non-cytolytic antiviral functions are also inhibited by IL-10 and/or TGF-β (Peppa et al., 2010), possibly emanating from Kupffer cells (Tu et al., 2008), suggesting that selective modulation of NK cell function in the liver both prevents viral clearance (perhaps as a means to prevent immune-mediated liver damage). Suppression of NK cell IFN-γ secretion is correlated with high HBV viral load; interventions to reduce viral load appear to restore NK cell function and may facilitate clearance of the infection (Tjwa et al., 2011). Taken together, these studies reflect an emerging picture of the ability of the innate immune response to self-regulate during chronic infection with cells of myeloid origin guiding both activation and inhibition of NK cell function.
One of the most interesting recent developments in NK cell biology – the concept that NK cells may acquire some form of immunological memory – has emerged directly from studies of viral infections. Although first observed during studies of delayed hypersensitivity (O’Leary et al., 2006), enhanced NK cell function during secondary, compared to primary, antigen exposure has now been described for several viral infections; secondary NK cell responses are virus-specific, and are independent of adaptive T and B lymphocytes, but are restricted to a CXCR6+ and/or Thy1+ population of hepatic NK cells (Paust et al., 2010; Gillard et al., 2011). The molecular basis for these adaptive NK cell response is currently entirely unclear and there is, as yet, no evidence for similar responses in humans. However, induction of NK cell “memory” has been characterized in more detail during MCMV infection. NK cells expressing the Ly49H receptor, which binds the MCMV m157 protein, expand during primary MCMV infection and persist at high frequency in both lymphoid and non-lymphoid tissues; upon reinfection these “primed” or “memory” NK cells proliferate, degranulate, and secrete IFN-γ to a much greater extent than “naïve” NK cells and confer protective immunity to MCMV (Sun et al., 2009). The mechanism by which NK cells acquire this type of memory phenotype is an area of ongoing research but the fact that a similar phenotype is observed among NK cells primed by TLR-activated DCs and IL-15 (Lucas et al., 2007) or activated with a cocktail of cytokines (IL-12, IL-18, and IL-15; Cooper et al., 2009), that NK cell activation during acute HCV infection (Pelletier et al., 2010) and in response to influenza A virus (He et al., 2004) is highly correlated with T cell activation, and that the frequency of IFN-γ-producing NK cells among re-stimulated PBMCs is enhanced after influenza vaccination (Long et al., 2008), all tend to suggest that NK cells increase in frequency and/or undergo functional maturation after exposure to viral infection and/or the concomitant cytokine-mediated inflammation. Studies with protozoal pathogens (Bihl et al., 2010; Horowitz et al., 2010b; McCall et al., 2010; discussed in more detail below) have revealed that this “priming” of NK cells is not limited to viral infections and led to the hypothesis that IL-2 from antigen-specific T cells allows NK cells to contribute to the effector arm of adaptive immune responses (Figure 3). In support of this notion, we have shown that NK cell responses (CD69 expression, IFN-γ production, expression of the degranulation marker LAMP-1 (CD107a), and release of perforin granules) to inactivated rabies virus (RV) are augmented in an antigen-specific, T cell, and IL-2 dependent manner by vaccination (Horowitz et al., 2010b). Importantly, we observed that NK cells respond much more quickly than do CD8+ T cells after re-exposure to the vaccine antigen and that NK cells represent more than 70% of IFN-γ-secreting and cytotoxic cells during the first 24 h after re-exposure to RV. The logical conclusion of all of these observations is that antigen-specific IL-2 secretion from T cells may recruit NK cells as effectors of adaptive immunity and, thus, that NK cell responses can be potentiated by vaccination. Whether specific subsets of human NK cells can be preferentially reactivated by viruses expressing ligands for particular NK cell receptors (as suggested by the responsiveness of Ly49H+ murine NK cells to MCMV m157; Sun et al., 2009) remains to be seen but, if they can, then the potential for IL-2 from antigen-specific T cells to synergize with enhanced direct activation of “memory” NK cells by pathogen-derived ligands may lead to even more potent NK cell responses during secondary infections.
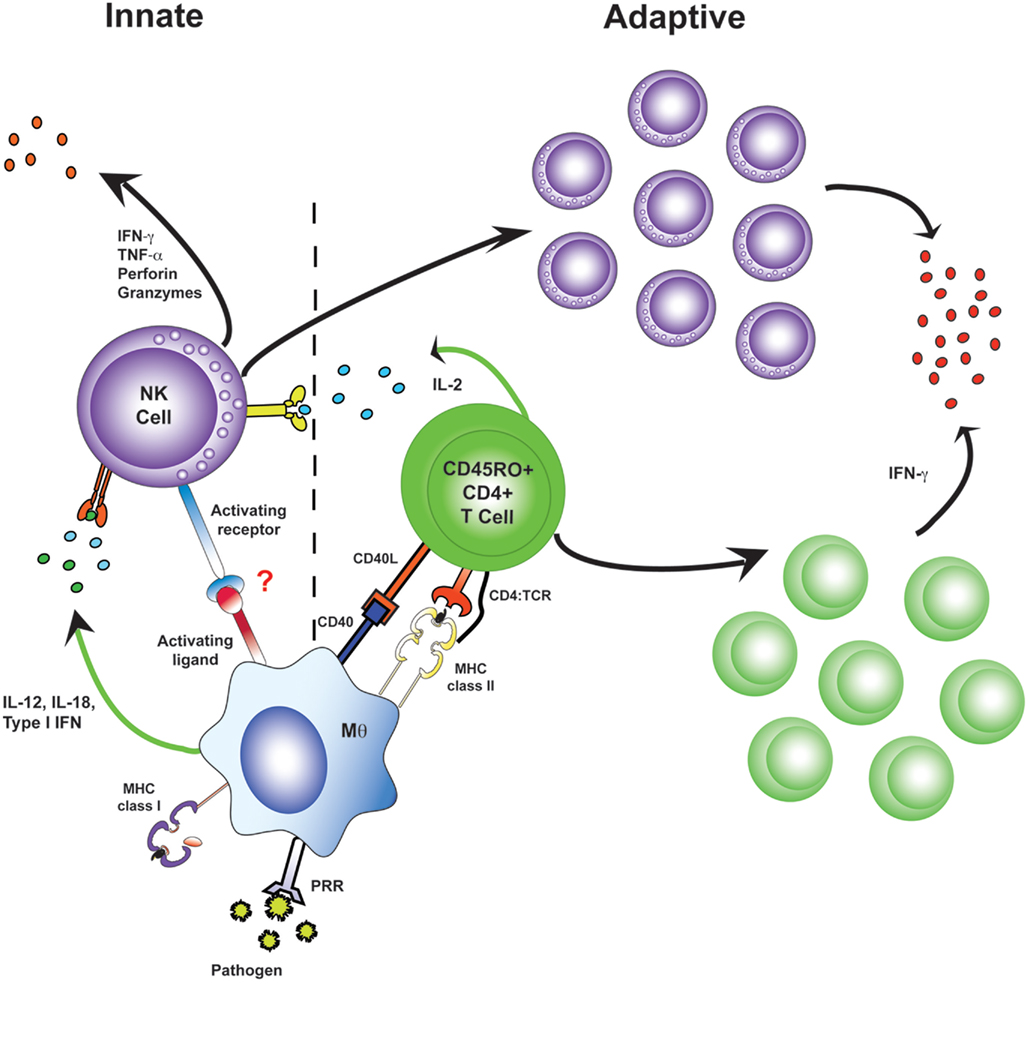
Figure 3. Antigen-specific effector memory CD4+ T cells recruit NK cells as effector cells in adaptive immune responses. During primary infection, NK cells receive innate activation signals from myeloid accessory cells and make very limited responses (left). Upon re-exposure to a pathogen, antigen-specific CD4+ T cells are a potent and immediate source of IL-2. Accessory cell signals synergize with this IL-2, resulting in rapid and robust “adaptive” NK cell responses, including NK cell proliferation, secretion of effector molecules, and killing of affected target cells (right).
NK Cells and Bacterial Infections
Bacteria provide numerous potent ligands for TLRs and other pattern recognition receptors and induce very characteristic inflammatory responses. Thus, there is considerable potential for indirect activation of NK cells during bacterial infection and for NK cell-derived IFN-γ to enhance phagocytosis of extracellular bacteria or infected host cells by macrophages. Whether bacteria also possess ligands for NK cell surface receptors, and can thus mediate direct NK cell activation, is however less well characterized.
Direct Activation of NK Cells by Bacteria?
Direct binding of Mycobacterium bovis BCG (BCG), Nocardia farcinica, and Pseudomonas aeruginosa cell wall-derived proteins to NKp44 – and consequent NK cell activation – has recently been reported (Esin et al., 2008) however blocking anti-NKp44 antibodies did not inhibit the activation of NK cells. Thus, as previously discussed for West Nile and Dengue viruses, it is quite likely that accessory cell- and cytokine-mediated upregulation of NKp44 is a consequence rather than a cause of NK cell activation, perhaps serving to potentiate rather than initiate the NK cell response. Marcenaro et al. (2008) provide evidence that highly purified human NK cells directly recognize M. bovis BCG, leading to their activation and expression of CD69 and CD25, release of IFN-γ and TNF, and killing of target cells; NK responses were inhibited by blocking antibodies specific to TLR-2, and the authors suggest that NK cells may bear a functional TLR-2 receptor. However, the bacterial ligands and NK receptors involved have not been identified and these findings are somewhat at odds with the findings of other studies (cited below) which demonstrate essential roles for accessory cell-derived stimuli in NK activation by BCG.
Indirect Activation of NK Cells by Bacteria
Indirect activation of NK cells by bacteria is well-described in numerous systems including Listeria monocytogenes, Staphylococcus aureus, Lactobacillus johnsonii, Mycobacterium tuberculosis, and M. bovis BCG infections and requires TLR-mediated activation of mDCs and monocytes to secrete IL-12, IL-18, and type-1 interferons (Newman and Riley, 2007). NK cell activation by L. monocytogenes (following binding of listeriolysin O to TLR-2; Nishibori et al., 1996; Ito et al., 2005; Datta et al., 2006) is mediated through production of IL-1β, IL-12, IL-18, and TNF-α from macrophages and mDCs (Wherry et al., 1991; Tripp et al., 1993; Hunter et al., 1995; Nomura et al., 2002; Humann and Lenz, 2010) but was thought to be independent of direct contact with APCs (Wherry et al., 1991). More recent data indicate that contact between NK cells and DCs is required (Humann and Lenz, 2010), although the contact-dependent signals have not been characterized. IL-18 is required for optimal type-1 inflammatory responses, constraint of bacterial growth, and prevention of neutrophil-mediated lung damage in M. tuberculosis infected mice (Schneider et al., 2010). Given reports of the effect of NK cell depletion on dissemination of murine M. tuberculosis infections (Feng et al., 2006) and of the ability of bovine NK cells to constrain replication of M. bovis inside macrophages (Denis et al., 2007), it is likely that NK cell-mediated control of Mycobacterium spp. infections may also be impaired in the absence of IL-18. Certainly, we recently observed that optimal activation of human NK cells by M. bovis BCG requires IL-12 and IL-18, as well as IL-2 from CD4+ T cells (Evans et al., 2011), supporting and extending previously published findings (Korbel et al., 2008; Zhou et al., 2009). On the other hand, NK cells which inhibit growth of M. tuberculosis via secretion of IL-22 appear to require exogenous IL-15 and IL-23 but not IL-12 or IL-18 (Dhiman et al., 2009). It is not yet clear how any of these findings can be reconciled with those of Marcenaro et al., 2008; above) but differences in NK responses to virulent and avirulent strains of mycobacteria may provide some clues.
Lastly, building on studies initially performed with protozoal infections (Maroof et al., 2008), studies of acute systemic infections with L. monocytogenes and Yersinia pestis have found that NK cells can be a significant source of the regulatory cytokine, IL-10. IL-10 secretion from NK cells suppresses IL-12 secretion by DCs and thus blocks further NK cell activation (Perona-Wright et al., 2009); localized infections with an attenuated strain of Y. pestis or with influenza virus do not induce this IL-10 response suggesting that NK cell-derived IL-10 may be required to prevent immune pathology during systemic infections (Figure 4). The precise signals that lead to IL-10 secretion by NK cells remain to be determined although, by analogy with studies with HCMV (Romo et al., 2011), it would seem likely that modulation of signals provided by accessory cells may play a role.
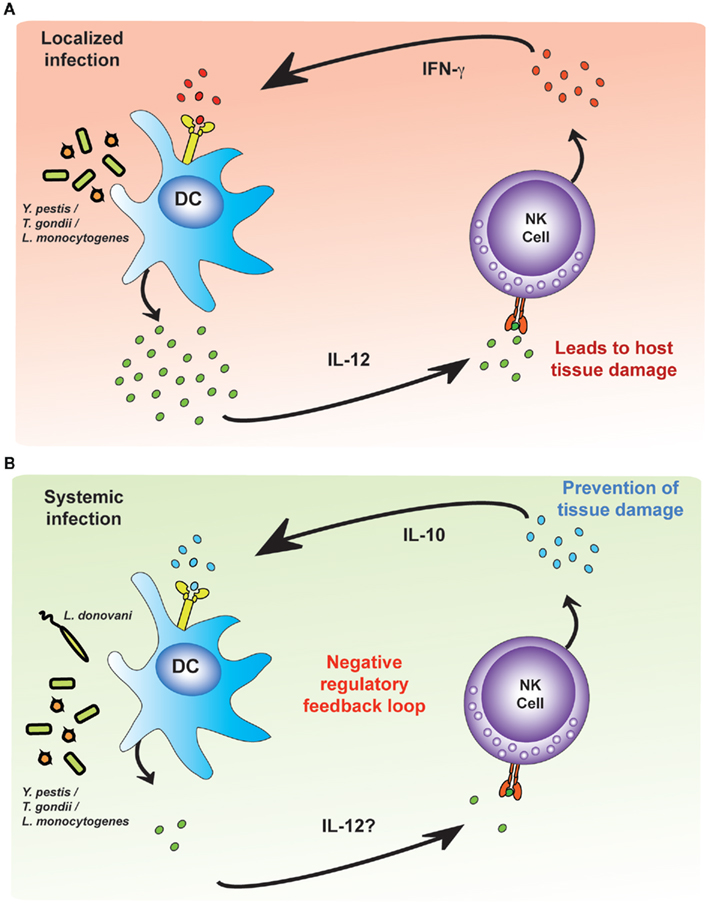
Figure 4. Environmental cues modulate pro-inflammatory and regulatory NK cell responses. Dendritic cells (DCs) infected with intracellular bacteria produce IL-12. (A) During localized infections, NK cells respond to IL-12 by producing IFN-γ, which amplifies the DC IL-12 response, creating an unregulated feedback loop. The pathogen is killed or contained but at the expense of significant tissue damage. (B) During systemic infection, as yet uncharacterized differences in the environmental cues provided by accessory cells (but possibly influenced by the concentration of IL-12) lead to NK cell production of IL-10. NK cell-derived IL-10 limits tissue damage but may also allow the infection to persist.
NK Cells and Protozoal Infections
Like bacteria, protozoan parasites induce strong inflammatory responses and activate NK cells via the indirect pathway; rapid NK cell-derived IFN-γ production is necessary for effective clearance of many protozoan infections (reviewed in Korbel et al., 2004). There is, as yet, no strong evidence that protozoa can directly activate NK cells.
In mice infected with Plasmodium chabaudi AS (malaria), IL-12 (Stevenson et al., 1995; Mohan et al., 1997; but not IL-15; Ing and Stevenson, 2009) enhances NK cell cytotoxicity, IFN-γ and TNF-α production, facilitates clearance of infection, and primes DCs for efficient presentation of antigen to CD4+ T cells. Rapid and potent human NK cell IFN-γ responses are induced in vitro by P. falciparum-infected red blood cells (iRBC) in an accessory cell-, IL-12-, and IL-18-dependent manner (Artavanis-Tsakonas and Riley, 2002; Artavanis-Tsakonas et al., 2003; Baratin et al., 2005). Having observed that NK cell activation by P. falciparum iRBC is dependent on IL-2 and that – although myeloid cells were essential for NK cell activation – the NK cell response within mixed peripheral blood mononuclear cells could not be recapitulated by addition of myeloid cells (plastic adherent monocytes, macrophages, and DCs) to purified NK cells (Newman et al., 2006), we found that NK cell activation is highly dependent on IL-2 from antigen-specific CD4+ T cells (Horowitz et al., 2010a). Thus, as described above for RV and BCG, IL-2 from memory CD4+ T cells synergizes with myeloid accessory cell-derived stimuli leading to optimal NK cell responses to P. falciparum iRBC. In line with these observations, NK cell responses to P. falciparum iRBC are potentiated (in a T cell dependent manner) in individuals recovering from primary malaria infections (McCall et al., 2010) and NK cell responses to a malaria sporozoite antigen are enhanced by vaccination (Horowitz et al., submitted for publication).
Natural killer cell dependency on T cell-derived signals has also been recently reported for Leishmania major infection in mice. It has long been known that DC-derived IL-12 and IL-18 are required for induction of NK cell IFN-γ production and cytotoxicity by L. major (Scharton-Kersten et al., 1995; Wei et al., 1999); this response is mediated by TLR-9 (Liese et al., 2007). It is now clear that NK cell responses to L. major also require both IL-2 and CD40–CD40L co-stimulation from primed, antigen-specific CD4+ T cells (Bihl et al., 2010). Indeed, the DC and T cell pathways of NK cell activation by L. major may both depend on TLR-9 since treatment of L. major-infected mice with the TLR-9 agonist CpG leads to very rapid secretion of IL-2 as well as robust production of IFN-γ by NK cells (Laabs et al., 2009). Similarly, both mDCs and pDCs secrete IL-12 in a TLR-9-dependent manner during L. infantum infection; although pDCs also produce IFN-α/β, only the mDCs are able to internalize the parasite. It is not yet known whether murine NK cells can bind and internalize CpG for binding to TLR-9 (as recently reported for human NK cells; Sivori et al., 2011). Depletion of pDCs has no effect on NK cell IFN-γ production or cytotoxicity but depletion of mDCs abolishes the NK cell response, providing direct in vivo evidence of NK cell dependency on signals from mDCs (Schleicher et al., 2007). A recent study of murine L. donovani infection has revealed for the first time that the anti-leishmanial role of splenic NK cells is dependent upon physical contact between NK cells and the infected macrophage (Manna et al., 2010). However, although NK cells made sustained contact with infected macrophages their anti-leishmanial role was non-cytolytic and confined to TNF-α production (Manna et al., 2010), suggesting that the contact-dependent signals are required to fully activate the NK cell rather than for the NK cell to deliver a death signal to the macrophage.
Natural killer cells can themselves be infected by T. gondii (Persson et al., 2009): NK cells lyse infected DCs in a perforin-dependent manner leading to egress of viable T. gondii that rapidly infect the surrounding NK cells offering a potential route for immune evasion. In T. gondii-infected mice, MyD88 signaling is required for IL-12 secretion by DCs and consequent NK cell activation (Hou et al., 2011). Toxoplasma profilin is a known ligand for TLR-11 (a receptor that is non-functional in humans) and a potent activator of murine DCs (Yarovinsky et al., 2005). Surprisingly, in the absence of TLR-11 NK cells of T. gondii-infected mice secrete very high levels of IFN-γ and there is extensive destruction of pancreatic tissue and fat necrosis (Yarovinsky et al., 2008) suggesting that TLR-11 signaling is required to regulate the NK cell response and prevent immunopathology. This is reminiscent of the role of DCs in regulating NK cell responses to viruses, as described above. The recent demonstration that NK cells are a major source of IL-17 during early T. gondii infection, that this response is largely dependent on APC-derived IL-6, IL-23, and TGF-β and that it is inhibited by exogenous IL-2, IL-15, and IL-27 (Passos et al., 2010) raises the possibility that TLR-11 signaling in DCs is required to prevent excessive NK IL-17-mediated immune pathology, but this hypothesis is, as yet, untested.
NK Cell Responses to Helminths
Effective control of helminth infections typically depends on a highly polarized type-2 immune response, characterized by secretion of IL-4, IL-5, and IL-13, secretion of IgE antibodies and activation of mast cells (Allen and Maizels, 2011). Relatively few studies have examined the potential for NK cells to either contribute to, or to modulate, immune responses to helminths but this possibility has recently begun to attract the attention of helminth immunologists. Excretory/secretory (ES) products of the nematode Necator americanus (but not comparable products of other nematodes) have been reported to bind selectively to CD56bright NK cells and, in the presence of sub-optimal concentrations of IL-2 and IL-12, to induce NK cell IFNγ production (Hsieh et al., 2004) suggesting that protein or glycoprotein components of the N. americanus ES may be able to bind to activating NK cell receptors and lower the NK cell threshold for activation. NK cells from N. americanus-infected individuals spontaneously produce IFN-γ ex vivo but fail to bind exogenous biotin-labeled ES, suggesting that their circulating NK cells may have already bound (and been activated by) endogenous ES ligands (Teixeira-Carvalho et al., 2008).
In murine gastro-intestinal nematode infections there is accumulating evidence that, when there is a disruption of Th2 immunity, NK cells (particularly those of male mice) may become an important source of the protective type-2 cytokine, IL-13 (McDermott et al., 2005; Hepworth and Grencis, 2009). Depletion of Th2 CD4+ T cells, the main source of IL-13, results in complete abrogation of worm expulsion, but depletion of NK cells results in delayed expulsion only in male mice. There is no information, as yet, regarding the signals that induce NK cells to secrete IL-13 although it may be relevant that the transcription factor E4bp4, which plays an essential role in NK cell ontogeny (Gascoyne et al., 2009) has recently been shown to regulate IL-13 production (Motomura et al., 2011). NK cells have also been reported to be activated during murine infection with the filarial nematode Litomosoides sigmodontis, where they influence the balance of type-1 and type-2 cytokines (Korten et al., 2002), and to be an early source of IFN-γ in Fasciola hepatica infection of rats (Tliba et al., 2002), but again there is no information on the pathways of NK cell activation in these infections.
Fungal Activation of NK Cells
Investigation of the role of NK cells in fungal infections is in its very early stages but killing of diverse fungal species by NK cells has been reported. NK cells are the main source of IFN-γ in the early phase of Aspergillus fumigatus infection and are required to induce killing of A. fumigatus by macrophages; depletion of either NK cells or IFN-γ leads to an increase in fungal load (Park et al., 2009). CCL2-dependent recruitment of NK cells is crucial in clearing A. fumigatus infection in neutropenic mice (Morrison et al., 2003). A. fumigatus hyphae are able to activate NK cells – although the mechanism of activation has not been described – and NK cells can kill A. fumigatus hyphae in a perforin-dependent manner (Schmidt et al., 2011). However, it is also reported that A. fumigatus hyphae modulate NK cell secretion of IFN-γ and GM-CSF (Schmidt et al., 2011). NK cells are thought to mediate perforin-dependent killing of Cryptococcus neoformans (Ma et al., 2004); perforin release is mediated by PI3K-ERK1/2 signaling (Wiseman et al., 2007). Finally, resistance to Candida albicans is correlated with NK cell activation and NK cells are able to activate splenic macrophages to phagocytose C. albicans (Algarra et al., 2002). To date however, the mechanisms of activation of NK cells by fungi have not been elucidated.
Concluding Remarks
There is now a strong body evidence to implicate NK cells in effective control of a diverse array of pathogens, including viruses, bacteria, protozoa, helminths, and fungi. Whilst many of these infections can be contained in the absence of NK cells, clearance of these organisms is almost always more efficient and more complete in the presence of a functional NK cell response. NK cell activation by pathogens occurs predominantly via the indirect pathway – involving cytokines and cell contact-dependent signals from accessory cells. Whilst there are many reports of direct NK cell activation by binding of pathogen-derived molecules to NK cell surface receptors, in only very few of these cases (MCMV and pox virus activation of murine NK cells and activation of human NK cells by influenza virus) have both the pathogen ligand and the NK cell receptor been defined at the gene and protein levels (Mandelboim et al., 2001; Arase et al., 2002; Smith et al., 2002; Chisholm and Reyburn, 2006). Validation of other putative pathogen-derived ligands for NK cells awaits similar biochemical and genetic information. Nevertheless, even where direct activation of NK cells by pathogens has been confirmed, the NK cell response is further augmented by external cytokine and co-stimulatory signals.
Two particularly interesting themes to have emerged from recent studies of NK cell–pathogen interactions are the role of T cell-derived IL-2 in amplifying NK cell responses – with obvious implications for augmentation of NK cell effector responses by vaccination – and the potential for NK cells to take on regulatory or immunomodulatory roles, via secretion of IL-10 or IL-13. These observations suggest that we have not yet realized the full potential of NK cells to influence the outcome of infectious diseases and suggest that further exploration of the molecular basis of indirect NK cell activation – particularly during helminth and fungal infections – could be extremely rewarding.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahlenstiel, G., Titerence, R. H., Koh, C., Edlich, B., Feld, J. J., Rotman, Y., Ghany, M. G., Hoofnagle, J. H., Liang, T. J., Heller, T., and Rehermann, B. (2010). Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology 138, 325–335 e321–322.
Algarra, I., Ortega, E., Serrano, M. J., Alvarez de Cienfuegos, G., and Gaforio, J. J. (2002). Suppression of splenic macrophage Candida albicans phagocytosis following in vivo depletion of natural killer cells in immunocompetent BALB/c mice and T-cell-deficient nude mice. FEMS Immunol. Med. Microbiol. 33, 159–163.
Allen, J. E., and Maizels, R. M. (2011). Diversity and dialogue in immunity to helminths. Nat. Rev. Immunol. 11, 375–388.
Alter, G., Heckerman, D., Schneidewind, A., Fadda, L., Kadie, C. M., Carlson, J. M., Oniangue-Ndza, C., Martin, M., Li, B., Khakoo, S. I., Carrington, M., Allen, T. M., and Altfeld, M. (2011a). HIV-1 adaptation to NK-cell-mediated immune pressure. Nature 476, 96–100.
Alter, G., Jost, S., Rihn, S., Reyor, L. L., Nolan, B. E., Ghebremichael, M., Bosch, R., Altfeld, M., and Lauer, G. M. (2011b). Reduced frequencies of NKp30+NKp46+, CD161+, and NKG2D+ NK cells in acute HCV infection may predict viral clearance. J. Hepatol. 55, 278–288.
Amadei, B., Urbani, S., Cazaly, A., Fisicaro, P., Zerbini, A., Ahmed, P., Missale, G., Ferrari, C., and Khakoo, S. I. (2010). Activation of natural killer cells during acute infection with hepatitis C virus. Gastroenterology 138, 1536–1545.
Andoniou, C. E., van Dommelen, S. L., Voigt, V., Andrews, D. M., Brizard, G., Asselin-Paturel, C., Delale, T., Stacey, K. J., Trinchieri, G., and Degli-Esposti, M. A. (2005). Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat. Immunol. 6, 1011–1019.
Arase, H., Mocarski, E. S., Campbell, A. E., Hill, A. B., and Lanier, L. L. (2002). Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326.
Artavanis-Tsakonas, K., Eleme, K., McQueen, K. L., Cheng, N. W., Parham, P., Davis, D. M., and Riley, E. M. (2003). Activation of a subset of human NK cells upon contact with Plasmodium falciparum-infected erythrocytes. J. Immunol. 171, 5396–5405.
Artavanis-Tsakonas, K., and Riley, E. M. (2002). Innate immune response to malaria: rapid induction of IFN-gamma from human NK cells by live Plasmodium falciparum-infected erythrocytes. J. Immunol. 169, 2956–2963.
Baratin, M., Roetynck, S., Lépolard, C., Falk, C., Sawadogo, S., Uematsu, S., Akira, S., Ryffel, B., Tiraby, J. G., Alexopoulou, L., Kirschning, C. J., Gysin, J., Vivier, E., and Ugolini, S. (2005). Natural killer cell and macrophage cooperation in MyD88-dependent innate responses to Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 102, 14747–14752.
Barbalat, R., Lau, L., Locksley, R. M., and Barton, G. M. (2009). Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 10, 1200–1207.
Bihl, F., Pecheur, J., Bréart, B., Poupon, G., Cazareth, J., Julia, V., Glaichenhaus, N., and Braud, V. M. (2010). Primed antigen-specific CD4+ T cells are required for NK cell activation in vivo upon Leishmania major infection. J. Immunol. 185, 2174–2181.
Biron, C. A., and Brossay, L. (2001). NK cells and NKT cells in innate defense against viral infections. Curr. Opin. Immunol. 13, 458–464.
Biron, C. A., Nguyen, K. B., Pien, G. C., Cousens, L. P., and Salazar-Mather, T. P. (1999). Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17, 189–220.
Born, T. L., Morrison, L. A., Esteban, D. J., VandenBos, T., Thebeau, L. G., Chen, N., Spriggs, M. K., Sims, J. E., and Buller, R. M. (2000). A poxvirus protein that binds to and inactivates IL-18, and inhibits NK cell response. J. Immunol. 164, 3246–3254.
Bryceson, Y. T., March, M. E., Ljunggren, H. G., and Long, E. O. (2006). Activation, coactivation, and costimulation of resting human natural killer cells. Immunol. Rev. 214, 73–91.
Cheent, K., and Khakoo, S. I. (2011). Natural killer cells and hepatitis C: action and reaction. Gut 60, 268–278.
Chisholm, S. E., and Reyburn, H. T. (2006). Recognition of vaccinia virus-infected cells by human natural killer cells depends on natural cytotoxicity receptors. J. Virol. 80, 2225–2233.
Cooper, M. A., Elliott, J. M., Keyel, P. A., Yang, L., Carrero, J. A., and Yokoyama, W. M. (2009). Cytokine-induced memory-like natural killer cells. Proc. Natl. Acad. Sci. U.S.A. 106, 1915–1919.
Crotta, S., Brazzoli, M., Piccioli, D., Valiante, N. M., and Wack, A. (2010). Hepatitis C virions subvert natural killer cell activation to generate a cytokine environment permissive for infection. J. Hepatol. 52, 183–190.
Crotta, S., Stilla, A., Wack, A., D’Andrea, A., Nuti, S., D’Oro, U., Mosca, M., Filliponi, F., Brunetto, R. M., Bonino, F., Abrignani, S., and Valiante, N. M. (2002). Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med. 195, 35–41.
Dalod, M., Hamilton, T., Salomon, R., Salazar-Mather, T. P., Henry, S. C., Hamilton, J. D., and Biron, C. A. (2003). Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J. Exp. Med. 197, 885–898.
Daniels, K. A., Devora, G., Lai, W. C., O’Donnell, C. L., Bennett, M., and Welsh, R. M. (2001). Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J. Exp. Med. 194, 29–44.
Datta, S. K., Okamoto, S., Hayashi, T., Shin, S. S., Mihajlov, I., Fermin, A., Guiney, D. G., Fierer, J., and Raz, E. (2006). Vaccination with irradiated Listeria induces protective T cell immunity. Immunity 25, 143–152.
Delale, T., Paquin, A., Asselin-Paturel, C., Dalod, M., Brizard, G., Bates, E. E., Kastner, P., Chan, S., Akira, S., Vicari, A., Biron, C. A., Trinchieri, G., and Brière, F. (2005). MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J. Immunol. 175, 6723–6732.
Denis, M., Keen, D. L., Parlane, N. A., Storset, A. K., and Buddle, B. M. (2007). Bovine natural killer cells restrict the replication of Mycobacterium bovis in bovine macrophages and enhance IL-12 release by infected macrophages. Tuberculosis (Edinb.) 87, 53–62.
Dhiman, R., Indramohan, M., Barnes, P. F., Nayak, R. C., Paidipally, P., Rao, L. V., and Vankayalapati, R. (2009). IL-22 produced by human NK cells inhibits growth of Mycobacterium tuberculosis by enhancing phagolysosomal fusion. J. Immunol. 183, 6639–6645.
Dunn, C., Brunetto, M., Reynolds, G., Christophides, T., Kennedy, P. T., Lampertico, P., Das, A., Lopes, A. R., Borrow, P., Williams, K., Humphreys, E., Afford, S., Adams, D. H., Bertoletti, A., and Maini, M. K. (2007). Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J. Exp. Med. 204, 667–680.
Dzionek, A., Sohma, Y., Nagafune, J., Cella, M., Colonna, M., Facchetti, F., Günther, G., Johnston, I., Lanzavecchia, A., Nagasaka, T., Okada, T., Vermi, W., Winkels, G., Yamamoto, T., Zysk, M., Yamaguchi, Y., and Schmitz, J. (2001). BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 194, 1823–1834.
Esin, S., Batoni, G., Counoupas, C., Stringaro, A., Brancatisano, F. L., Colone, M., Maisetta, G., Florio, W., Arancia, G., and Campa, M. (2008). Direct binding of human NK cell natural cytotoxicity receptor NKp44 to the surfaces of mycobacteria and other bacteria. Infect. Immun. 76, 1719–1727.
Evans, J. H., Horowitz, A., Mehrabi, M., Wise, E. L., Pease, J. E., Riley, E. M., and Davis, D. M. (2011). A distinct subset of human NK cells expressing HLA-DR expand in response to IL-2 and can aid immune responses to BCG. Eur. J. Immunol. 41, 1924–1933.
Fadda, L., Borhis, G., Ahmed, P., Cheent, K., Pageon, S. V., Cazaly, A., Stathopoulos, S., Middleton, D., Mulder, A., Claas, F. H., Elliott, T., Davis, D. M., Purbhoo, M. A., and Khakoo, S. I. (2011). Peptide antagonism as a mechanism for NK cell activation. Proc. Natl. Acad. Sci. U.S.A. 107, 10160–10165.
Fang, M., Lanier, L. L., and Sigal, L. J. (2008). A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog. 4, e30. doi: 10.1371/journal.ppat.0040030
Fang, M., Orr, M. T., Spee, P., Egebjerg, T., Lanier, L. L., and Sigal, L. J. (2011). CD94 is essential for NK cell-mediated resistance to a lethal viral disease. Immunity 34, 579–589.
Feng, C. G., Kaviratne, M., Rothfuchs, A. G., Cheever, A., Hieny, S., Young, H. A., Wynn, T. A., and Sher, A. (2006). NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J. Immunol. 177, 7086–7093.
Gascoyne, D. M., Long, E., Veiga-Fernandes, H., de Boer, J., Williams, O., Seddon, B., Coles, M., Kioussis, D., and Brady, H. J. (2009). The basic leucine zipper transcription factor E4BP4 is essential for natural killer cell development. Nat. Immunol. 10, 1118–1124.
Gillard, G. O., Bivas-Benita, M., Hovav, A. H., Grandpre, L. E., Panas, M. W., Seaman, M. S., Haynes, B. F., and Letvin, N. L. (2011). Thy1+ Nk cells from vaccinia virus-primed mice confer protection against vaccinia virus challenge in the absence of adaptive lymphocytes. PLoS Pathog. 7, e1002141. doi: 10.1371/journal.ppat.1002141
He, X. S., Draghi, M., Mahmood, K., Holmes, T. H., Kemble, G. W., Dekker, C. L., Arvin, A. M., Parham, P., and Greenberg, H. B. (2004). T cell-dependent production of IFN-gamma by NK cells in response to influenza A virus. J. Clin. Invest. 114, 1812–1819.
Hepworth, M. R., and Grencis, R. K. (2009). Disruption of Th2 immunity results in a gender-specific expansion of IL-13 producing accessory NK cells during helminth infection. J. Immunol. 183, 3906–3914.
Hershkovitz, O., Rosental, B., Rosenberg, L. A., Navarro-Sanchez, M. E., Jivov, S., Zilka, A., Gershoni-Yahalom, O., Brient-Litzler, E., Bedouelle, H., Ho, J. W., Campbell, K. S., Rager-Zisman, B., Despres, P., and Porgador, A. (2009). NKp44 receptor mediates interaction of the envelope glycoproteins from the West Nile and dengue viruses with NK cells. J. Immunol. 183, 2610–2621.
Horowitz, A., Newman, K. C., Evans, J. H., Korbel, D. S., Davis, D. M., and Riley, E. M. (2010a). Cross-talk between T cells and NK cells generates rapid effector responses to Plasmodium falciparum-infected erythrocytes. J. Immunol. 184, 6043–6052.
Horowitz, A., Behrens, R. H., Okell, L., Fooks, A. R., and Riley, E. M. (2010b). NK cells as effectors of acquired immune responses: effector CD4+ T cell-dependent activation of NK cells following vaccination. J. Immunol. 185, 2808–2818.
Hou, B., Benson, A., Kuzmich, L., DeFranco, A. L., and Yarovinsky, F. (2011). Critical coordination of innate immune defense against Toxoplasma gondii by dendritic cells responding via their Toll-like receptors. Proc. Natl. Acad. Sci. U.S.A. 108, 278–283.
Hsieh, G. C., Loukas, A., Wahl, A. M., Bhatia, M., Wang, Y., Williamson, A. L., Kehn, K. W., Maruyama, H., Hotez, P. J., Leitenberg, D., Bethony, J., and Constant, S. L. (2004). A secreted protein from the human hookworm Necator americanus binds selectively to NK cells and induces IFN-gamma production. J. Immunol. 173, 2699–2704.
Humann, J., and Lenz, L. L. (2010). Activation of naive NK cells in response to Listeria monocytogenes requires IL-18 and contact with infected dendritic cells. J. Immunol. 184, 5172–5178.
Hunter, C. A., Chizzonite, R., and Remington, J. S. (1995). IL-1 beta is required for IL-12 to induce production of IFN-gamma by NK cells. A role for IL-1 beta in the T cell-independent mechanism of resistance against intracellular pathogens. J. Immunol. 155, 4347–4354.
Ing, R., and Stevenson, M. M. (2009). Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect. Immun. 77, 770–782.
Ito, Y., Kawamura, I., Kohda, C., Tsuchiya, K., Nomura, T., and Mitsuyama, M. (2005). Seeligeriolysin O, a protein toxin of Listeria seeligeri, stimulates macrophage cytokine production via Toll-like receptors in a profile different from that induced by other bacterial ligands. Int. Immunol. 17, 1597–1606.
Jarahian, M., Fiedler, M., Cohnen, A., Djandji, D., Hämmerling, G. J., Gati, C., Cerwenka, A., Turner, P. C., Moyer, R. W., Watzl, C., Hengel, H., and Momburg, F. (2011). Modulation of NKp30- and NKp46-mediated natural killer cell responses by poxviral hemagglutinin. PLoS Pathog. 7, e1002195. doi: 10.1371/journal.ppat.1002195
Jinushi, M., Takehara, T., Tatsumi, T., Kanto, T., Groh, V., Spies, T., Suzuki, T., Miyagi, T., and Hayashi, N. (2003). Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J. Immunol. 171, 5423–5429.
Karre, K., Ljunggren, H. G., Piontek, G., and Kiessling, R. (1986). Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678.
Khakoo, S. I., Thio, C. L., Martin, M. P., Brooks, C. R., Gao, X., Astemborski, J., Cheng, J., Goedert, J. J., Vlahov, D., Hilgartner, M., Cox, S., Little, A. M., Alexander, G. J., Cramp, M. E., O’Brien, S. J., Rosenberg, W. M., Thomas, D. L., and Carrington, M. (2004). HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305, 872–874.
Kielczewska, A., Pyzik, M., Sun, T., Krmpotic, A., Lodoen, M. B., Munks, M. W., Babic, M., Hill, A. B., Koszinowski, U. H., Jonjic, S., Lanier, L. L., and Vidal, S. M. (2009). Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J. Exp. Med. 206, 515–523.
Kiessling, R., Klein, E., and Wigzell, H. (1975). “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 5, 112–117.
Korbel, D. S., Finney, O. C., and Riley, E. M. (2004). Natural killer cells and innate immunity to protozoan pathogens. Int. J. Parasitol. 34, 1517–1528.
Korbel, D. S., Schneider, B. E., and Schaible, U. E. (2008). Innate immunity in tuberculosis: myths and truth. Microbes Infect. 10, 995–1004.
Korten, S., Volkmann, L., Saeftel, M., Fischer, K., Taniguchi, M., Fleischer, B., and Hoerauf, A. (2002). Expansion of NK cells with reduction of their inhibitory Ly-49A, Ly-49C, and Ly-49G2 receptor-expressing subsets in a murine helminth infection: contribution to parasite control. J. Immunol. 168, 5199–5206.
Krug, A., Luker, G. D., Barchet, W., Leib, D. A., Akira, S., and Colonna, M. (2004a). Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103, 1433–1437.
Krug, A., French, A. R., Barchet, W., Fischer, J. A., Dzionek, A., Pingel, J. T., Orihuela, M. M., Akira, S., Yokoyama, W. M., and Colonna, M. (2004b). TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21, 107–119.
Laabs, E. M., Wu, W., and Mendez, S. (2009). Vaccination with live Leishmania major and CpG DNA promotes interleukin-2 production by dermal dendritic cells and NK cell activation. Clin. Vaccine Immunol. 16, 1601–1606.
Lanier, L. L. (2008). Up on the tightrope: natural killer cell activation and inhibition. Nat. Immunol. 9, 495–502.
Liese, J., Schleicher, U., and Bogdan, C. (2007). TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur. J. Immunol. 37, 3424–3434.
Lodoen, M. B., and Lanier, L. L. (2006). Natural killer cells as an initial defense against pathogens. Curr. Opin. Immunol. 18, 391–398.
Long, B. R., Michaelsson, J., Loo, C. P., Ballan, W. M., Vu, B. A., Hecht, F. M., Lanier, L. L., Chapman, J. M., and Nixon, D. F. (2008). Elevated frequency of gamma interferon-producing NK cells in healthy adults vaccinated against influenza virus. Clin. Vaccine Immunol. 15, 120–130.
Lucas, M., Schachterle, W., Oberle, K., Aichele, P., and Diefenbach, A. (2007). Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26, 503–517.
Ma, L. L., Wang, C. L., Neely, G. G., Epelman, S., Krensky, A. M., and Mody, C. H. (2004). NK cells use perforin rather than granulysin for anticryptococcal activity. J. Immunol. 173, 3357–3365.
Mandelboim, O., Lieberman, N., Lev, M., Paul, L., Arnon, T. I., Bushkin, Y., Davis, D. M., Strominger, J. L., Yewdell, J. W., and Porgador, A. (2001). Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409, 1055–1060.
Manna, P. P., Chakrabarti, G., and Bandyopadhyay, S. (2010). Innate immune defense in visceral leishmaniasis: cytokine mediated protective role by allogeneic effector cell. Vaccine 28, 803–810.
Marcenaro, E., Ferranti, B., Falco, M., Moretta, L., and Moretta, A. (2008). Human NK cells directly recognize Mycobacterium bovis via TLR2 and acquire the ability to kill monocyte-derived DC. Int. Immunol. 20, 1155–1167.
Maroof, A., Beattie, L., Zubairi, S., Svensson, M., Stager, S., and Kaye, P. M. (2008). Posttranscriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity 29, 295–305.
McCall, M. B., Roestenberg, M., Ploemen, I., Teirlinck, A., Hopman, J., de Mast, Q., Dolo, A., Doumbo, O. K., Luty, A., van der Ven, A. J., Hermsen, C. C., and Sauerwein, R. W. (2010). Memory-like IFN-gamma response by NK cells following malaria infection reveals the crucial role of T cells in NK cell activation by P. falciparum. Eur. J. Immunol. 40, 3472–3477.
McDermott, J. R., Humphreys, N. E., Forman, S. P., Donaldson, D. D., and Grencis, R. K. (2005). Intraepithelial NK cell-derived IL-13 induces intestinal pathology associated with nematode infection. J. Immunol. 175, 3207–3213.
Mohan, K., Moulin, P., and Stevenson, M. M. (1997). Natural killer cell cytokine production, not cytotoxicity, contributes to resistance against blood-stage Plasmodium chabaudi AS infection. J. Immunol. 159, 4990–4998.
Morrison, B. E., Park, S. J., Mooney, J. M., and Mehrad, B. (2003). Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J. Clin. Invest. 112, 1862–1870.
Motomura, Y., Kitamura, H., Hijikata, A., Matsunaga, Y., Matsumoto, K., Inoue, H., Atarashi, K., Hori, S., Watarai, H., Zhu, J., Taniguchi, M., and Kubo, M. (2011). The transcription factor E4BP4 regulates the production of IL-10 and IL-13 in CD4+ T cells. Nat. Immunol. 12, 450–459.
Muntasell, A., Magri, G., Pende, D., Angulo, A., and Lopez-Botet, M. (2010). Inhibition of NKG2D expression in NK cells by cytokines secreted in response to human cytomegalovirus infection. Blood 115, 5170–5179.
Newman, K. C., Korbel, D. S., Hafalla, J. C., and Riley, E. M. (2006). Cross-talk with myeloid accessory cells regulates human natural killer cell interferon-gamma responses to malaria. PLoS Pathog. 2, e118. doi: 10.1371/journal.ppat.0020118
Newman, K. C., and Riley, E. M. (2007). Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nat. Rev. Immunol. 7, 279–291.
Nishibori, T., Xiong, H., Kawamura, I., Arakawa, M., and Mitsuyama, M. (1996). Induction of cytokine gene expression by listeriolysin O and roles of macrophages and NK cells. Infect. Immun. 64, 3188–3195.
Nomura, T., Kawamura, I., Tsuchiya, K., Kohda, C., Baba, H., Ito, Y., Kimoto, T., Watanabe, I., and Mitsuyama, M. (2002). Essential role of interleukin-12 (IL-12) and IL-18 for gamma interferon production induced by listeriolysin O in mouse spleen cells. Infect. Immun. 70, 1049–1055.
Ochi, M., Ohdan, H., Mitsuta, H., Onoe, T., Tokita, D., Hara, H., Ishiyama, K., Zhou, W., Tanaka, Y., and Asahara, T. (2004). Liver NK cells expressing TRAIL are toxic against self hepatocytes in mice. Hepatology 39, 1321–1331.
O’Leary, J. G., Goodarzi, M., Drayton, D. L., and von Andrian, U. H. (2006). T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 7, 507–516.
Oliviero, B., Varchetta, S., Paudice, E., Michelone, G., Zaramella, M., Mavilio, D., De Filippi, F., Bruno, S., and Mondelli, M. U. (2009). Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 137, 1151–1160, 1160.e1151–e1157.
Park, S. J., Hughes, M. A., Burdick, M., Strieter, R. M., and Mehrad, B. (2009). Early NK cell-derived IFN-{gamma} is essential to host defense in neutropenic invasive aspergillosis. J. Immunol. 182, 4306–4312.
Parker, A. K., Parker, S., Yokoyama, W. M., Corbett, J. A., and Buller, R. M. (2007). Induction of natural killer cell responses by ectromelia virus controls infection. J. Virol. 81, 4070–4079.
Pashine, A., Valiante, N. M., and Ulmer, J. B. (2005). Targeting the innate immune response with improved vaccine adjuvants. Nat. Med. 11, S63-68.
Passos, S. T., Silver, J. S., O’Hara, A. C., Sehy, D., Stumhofer, J. S., and Hunter, C. A. (2010). IL-6 promotes NK cell production of IL-17 during toxoplasmosis. J. Immunol. 184, 1776–1783.
Paust, S., Gill, H. S., Wang, B. Z., Flynn, M. P., Moseman, E. A., Senman, B., Szczepanik, M., Telenti, A., Askenase, P. W., Compans, R. W., and von Andrian, U. H. (2010). Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 11, 1127–1135.
Pelletier, S., Drouin, C., Bédard, N., Khakoo, S. I., Bruneau, J., and Shoukry, N. H. (2010). Increased degranulation of natural killer cells during acute HCV correlates with the magnitude of virus-specific T cell responses. J. Hepatol. 53, 805–816.
Peppa, D., Micco, L., Javaid, A., Kennedy, P. T., Schurich, A., Dunn, C., Pallant, C., Ellis, G., Khanna, P., Dusheiko, G., Gilson, R. J., and Maini, M. K. (2010). Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLoS Pathog. 6, e1001227. doi: 10.1371/journal.ppat.1001227
Perona-Wright, G., Mohrs, K., Szaba, F. M., Kummer, L. W., Madan, R., Karp, C. L., Johnson, L. L., Smiley, S. T., and Mohrs, M. (2009). Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe 6, 503–512.
Persson, C. M., Lambert, H., Vutova, P. P., Dellacasa-Lindberg, I., Nederby, J., Yagita, H., Ljunggren, H. G., Grandien, A., Barragan, A., and Chambers, B. J. (2009). Transmission of Toxoplasma gondii from infected dendritic cells to natural killer cells. Infect. Immun. 77, 970–976.
Reading, P. C., and Smith, G. L. (2003). Vaccinia virus interleukin-18-binding protein promotes virulence by reducing gamma interferon production and natural killer and T-cell activity. J. Virol. 77, 9960–9968.
Romo, N., Magri, G., Muntasell, A., Heredia, G., Baía, D., Angulo, A., Guma, M., and López-Botet, M. (2011). Natural killer cell-mediated response to human cytomegalovirus-infected macrophages is modulated by their functional polarization. J. Leukoc. Biol. 90, 717–726.
Scharton-Kersten, T., Afonso, L. C., Wysocka, M., Trinchieri, G., and Scott, P. (1995). IL-12 is required for natural killer cell activation and subsequent T helper 1 cell development in experimental leishmaniasis. J. Immunol. 154, 5320–5330.
Schleicher, U., Liese, J., Knippertz, I., Kurzmann, C., Hesse, A., Heit, A., Fischer, J. A., Weiss, S., Kalinke, U., Kunz, S., and Bogdan, C. (2007). NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J. Exp. Med. 204, 893–906.
Schmidt, S., Tramsen, L., Hanisch, M., Latgé, J. P., Huenecke, S., Koehl, U., and Lehrnbecher, T. (2011). Human natural killer cells exhibit direct activity against Aspergillus fumigatus hyphae, but not against resting conidia. J. Infect. Dis. 203, 430–435.
Schneider, B. E., Korbel, D., Hagens, K., Koch, M., Raupach, B., Enders, J., Kaufmann, S. H., Mittrücker, H. W., and Schaible, U. E. (2010). A role for IL-18 in protective immunity against Mycobacterium tuberculosis. Eur. J. Immunol. 40, 396–405.
Sene, D., Levasseur, F., Abel, M., Lambert, M., Camous, X., Hernandez, C., Pène, V., Rosenberg, A. R., Jouvin-Marche, E., Marche, P. N., Cacoub, P., and Caillat-Zucman, S. (2010). Hepatitis C virus (HCV) evades NKG2D-dependent NK cell responses through NS5A-mediated imbalance of inflammatory cytokines. PLoS Pathog. 6, e1001184. doi: 10.1371/journal.ppat.1001184
Sivori, S., Falco, M., Carlomagno, S., Romeo, E., Soldani, C., Bensussan, A., Viola, A., Moretta, L., and Moretta, A. (2011). A novel KIR-associated function: evidence that CpG DNA uptake and shuttling to early endosomes is mediated by KIR3DL2. Blood 116, 1637–1647.
Smith, H. R., Heusel, J. W., Mehta, I. K., Kim, S., Dorner, B. G., Naidenko, O. V., Iizuka, K., Furukawa, H., Beckman, D. L., Pingel, J. T., Scalzo, A. A., Fremont, D. H., and Yokoyama, W. M. (2002). Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. U.S.A. 99, 8826–8831.
Steinberg, C., Eisenächer, K., Gross, O., Reindl, W., Schmitz, F., Ruland, J., and Krug, A. (2009). The IFN regulatory factor 7-dependent type I IFN response is not essential for early resistance against murine cytomegalovirus infection. Eur. J. Immunol. 39, 1007–1018.
Stevenson, M. M., Tam, M. F., Wolf, S. F., and Sher, A. (1995). IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. J. Immunol. 155, 2545–2556.
Sun, J. C., Beilke, J. N., and Lanier, L. L. (2009). Adaptive immune features of natural killer cells. Nature 457, 557–561.
Swiecki, M., Gilfillan, S., Vermi, W., Wang, Y., and Colonna, M. (2010). Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity 33, 955–966.
Szomolanyi-Tsuda, E., Liang, X., Welsh, R. M., Kurt-Jones, E. A., and Finberg, R. W. (2006). Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J. Virol. 80, 4286–4291.
Tabeta, K., Georgel, P., Janssen, E., Du, X., Hoebe, K., Crozat, K., Mudd, S., Shamel, L., Sovath, S., Goode, J., Alexopoulou, L., Flavell, R. A., and Beutler, B. (2004). Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. U.S.A. 101, 3516–3521.
Teixeira-Carvalho, A., Fujiwara, R. T., Stemmy, E. J., Olive, D., Damsker, J. M., Loukas, A., Corrêa-Oliveira, R., Constant, S. L., and Bethony, J. M. (2008). Binding of excreted and/or secreted products of adult hookworms to human NK cells in Necator americanus-infected individuals from Brazil. Infect. Immun. 76, 5810–5816.
Tjwa, E. T., van Oord, G. W., Hegmans, J. P., Janssen, H. L., and Woltman, A. M. (2011). Viral load reduction improves activation and function of natural killer cells in patients with chronic hepatitis B. J. Hepatol. 54, 209–218.
Tliba, O., Chauvin, A., Le Vern, Y., Boulard, C., and Sbille, P. (2002). Evaluation of the hepatic NK cell response during the early phase of Fasciola hepatica infection in rats. Vet. Res. 33, 327–332.
Tripp, C. S., Wolf, S. F., and Unanue, E. R. (1993). Interleukin 12 and tumor necrosis factor alpha are costimulators of interferon gamma production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. Proc. Natl. Acad. Sci. U.S.A. 90, 3725–3729.
Tseng, C. T., and Klimpel, G. R. (2002). Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 195, 43–49.
Tu, Z., Bozorgzadeh, A., Pierce, R. H., Kurtis, J., Crispe, I. N., and Orloff, M. S. (2008). TLR-dependent cross talk between human Kupffer cells and NK cells. J. Exp. Med. 205, 233–244.
Vitale, M., Bottino, C., Sivori, S., Sanseverino, L., Castriconi, R., Marcenaro, E., Augugliaro, R., Moretta, L., and Moretta, A. (1998). NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 187, 2065–2072.
Wei, X. Q., Leung, B. P., Niedbala, W., Piedrafita, D., Feng, G. J., Sweet, M., Dobbie, L., Smith, A. J., and Liew, F. Y. (1999). Altered immune responses and susceptibility to Leishmania major and Staphylococcus aureus infection in IL-18-deficient mice. J. Immunol. 163, 2821–2828.
Wherry, J. C., Schreiber, R. D., and Unanue, E. R. (1991). Regulation of gamma interferon production by natural killer cells in scid mice: roles of tumor necrosis factor and bacterial stimuli. Infect. Immun. 59, 1709–1715.
Wiseman, J. C., Ma, L. L., Marr, K. J., Jones, G. J., and Mody, C. H. (2007). Perforin-dependent cryptococcal microbicidal activity in NK cells requires PI3K-dependent ERK1/2 signaling. J. Immunol. 178, 6456–6464.
Yarovinsky, F., Hieny, S., and Sher, A. (2008). Recognition of Toxoplasma gondii by TLR11 prevents parasite-induced immunopathology. J. Immunol. 181, 8478–8484.
Yarovinsky, F., Zhang, D., Andersen, J. F., Bannenberg, G. L., Serhan, C. N., Hayden, M. S., Hieny, S., Sutterwala, F. S., Flavell, R. A., Ghosh, S., and Sher, A. (2005). TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308, 1626–1629.
Yoon, J. C., Shiina, M., Ahlenstiel, G., and Rehermann, B. (2009). Natural killer cell function is intact after direct exposure to infectious hepatitis C virions. Hepatology 49, 12–21.
Zhou, H., Fan, Y. Y., and Wu, C. Y. (2009). Bacillus Calmette-Guerin enhances the function of human nature killer cells by inducing IL-12 production and IL-12R expression. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 25, 976–979.
Keywords: NK cells, infection, activation, bacteria, viruses, parasites
Citation: Horowitz A, Stegmann KA and Riley EM (2012) Activation of natural killer cells during microbial infections. Front. Immun. 2:88. doi: 10.3389/fimmu.2011.00088
Received: 11 November 2011; Paper pending published: 23 November 2011;
Accepted: 16 December 2011; Published online: 03 January 2012.
Edited by:
Laurent Brossay, Brown University, USAReviewed by:
Scott H. Robbins, Immune Design Corporation, USAFrancisco Borrego, Food and Drug Administration, USA
Luis J. Sigal, Fox Chase Cancer Center, US Minor Outlying Islands
Copyright: © 2012 Horowitz, Stegmann and Riley. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Eleanor M. Riley, Department of Immunology and Infection, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, UK. e-mail: eleanor.riley@lshtm.ac.uk
†Present address: Amir Horowitz, Departments of Structural Biology, and Microbiology and Immunology, Stanford University Medical School, Fairchild Building D-157, 299 Campus Drive West, Stanford, CA 94305-5126, USA.