
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CLASSIFICATION article
Front. Immunol. , 08 November 2011
Sec. Primary Immunodeficiencies
volume 2 - 2011 | https://doi.org/10.3389/fimmu.2011.00054
 Waleed Al-Herz1,2
Waleed Al-Herz1,2 Aziz Bousfiha3
Aziz Bousfiha3 Jean-Laurent Casanova4,5 Helen Chapel6 Mary Ellen Conley7,8* Charlotte Cunningham-Rundles9
Jean-Laurent Casanova4,5 Helen Chapel6 Mary Ellen Conley7,8* Charlotte Cunningham-Rundles9 Amos Etzioni10 Alain Fischer11
Amos Etzioni10 Alain Fischer11 Jose Luis Franco12 Raif S. Geha13 Lennart Hammarström14
Jose Luis Franco12 Raif S. Geha13 Lennart Hammarström14 Shigeaki Nonoyama15
Shigeaki Nonoyama15 Luigi Daniele Notarangelo13,16*
Luigi Daniele Notarangelo13,16* Hans Dieter Ochs17
Hans Dieter Ochs17 Jennifer M. Puck18 Chaim M. Roifman19 Reinhard Seger20 and Mimi L. K. Tang21,22,23
Jennifer M. Puck18 Chaim M. Roifman19 Reinhard Seger20 and Mimi L. K. Tang21,22,23
We report the updated classification of primary immunodeficiency diseases, compiled by the ad hoc Expert Committee of the International Union of Immunological Societies. As compared to the previous edition, more than 15 novel disease entities have been added in the updated version. For each disorders, the key clinical and laboratory features are provided. This updated classification is meant to help in the diagnostic approach to patients with these diseases.
The International Union of Immunological Societies (IUIS) Expert Committee on Primary Immunodeficiency met in New York City, May 31–June 1, 2011 to update the classification of human primary immunodeficiencies (PIDs). Novel developments in gene discovery and increased knowledge in the mechanisms that govern immune system development and function have resulted in the identification of several novel PIDs in the last 2 years.
The classification of primary immunodeficiencies (PIDs) provides a framework to help in the diagnostic approach to patients. As in recent classifications, eight major groups of PIDs have been included in the Tables; however the order of the Tables has been changed with Table 2 now describing the “Well-defined syndromes with immunodeficiency” (previously Table 3) to reflect the immunological similarity between the disorders included in this Table and those in Table 1, “Combined immunodeficiencies.”
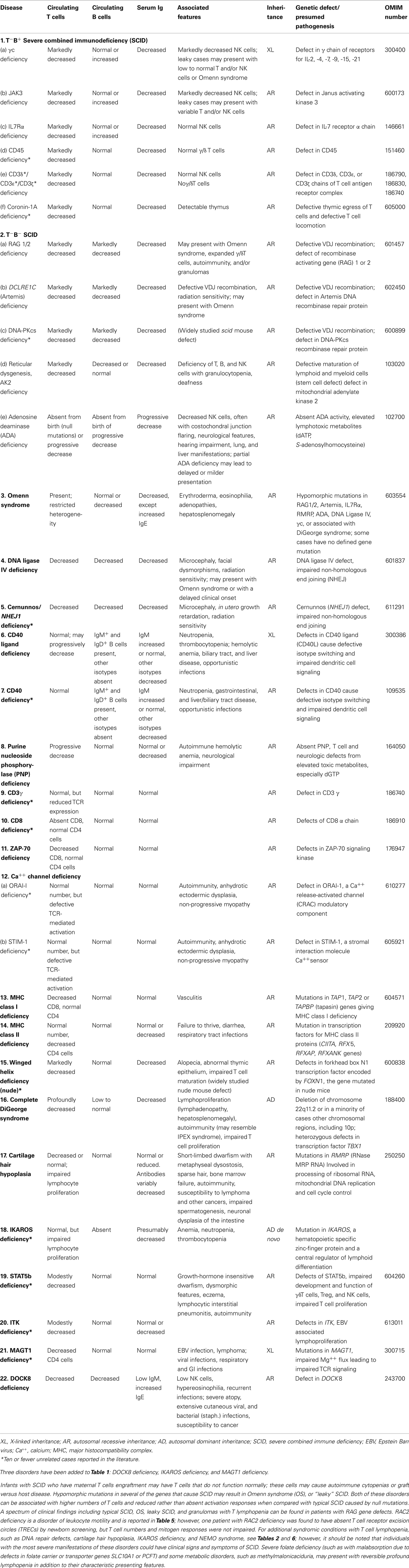
Table 1. Combined immunodeficiencies.
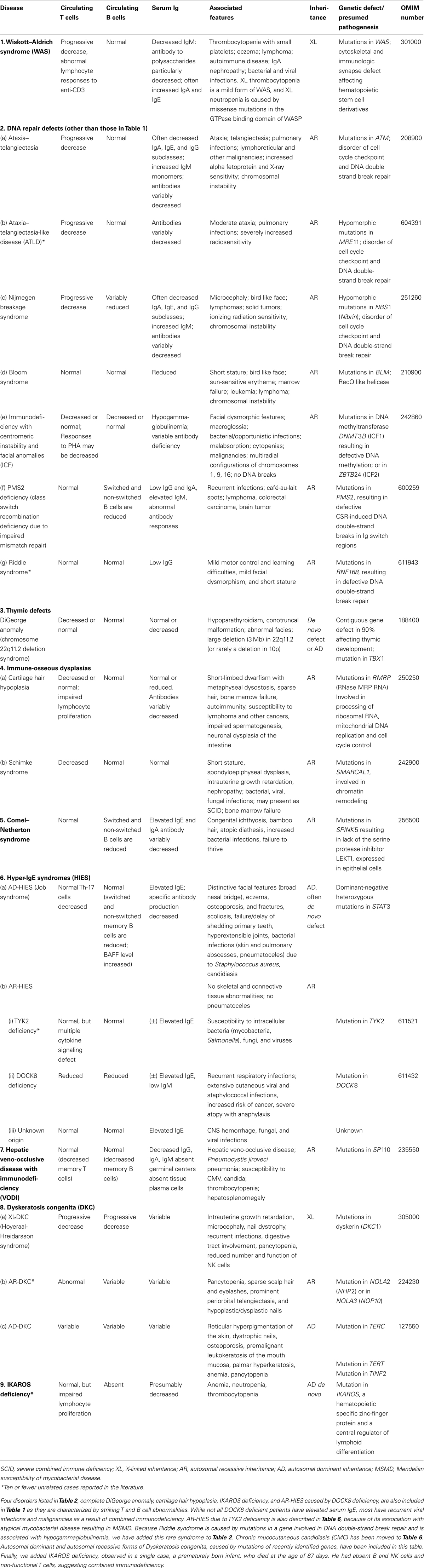
Table 2. Well-defined syndromes with immunodeficiency.
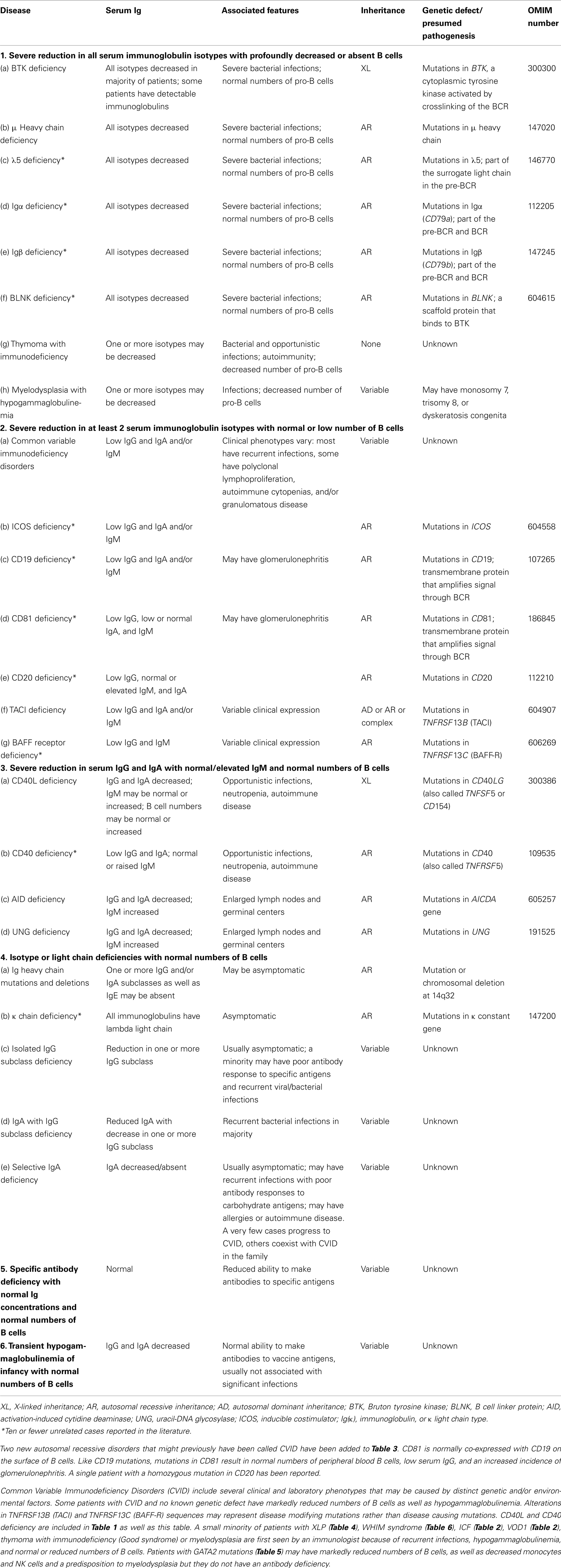
Table 3. Predominantly antibody deficiencies.
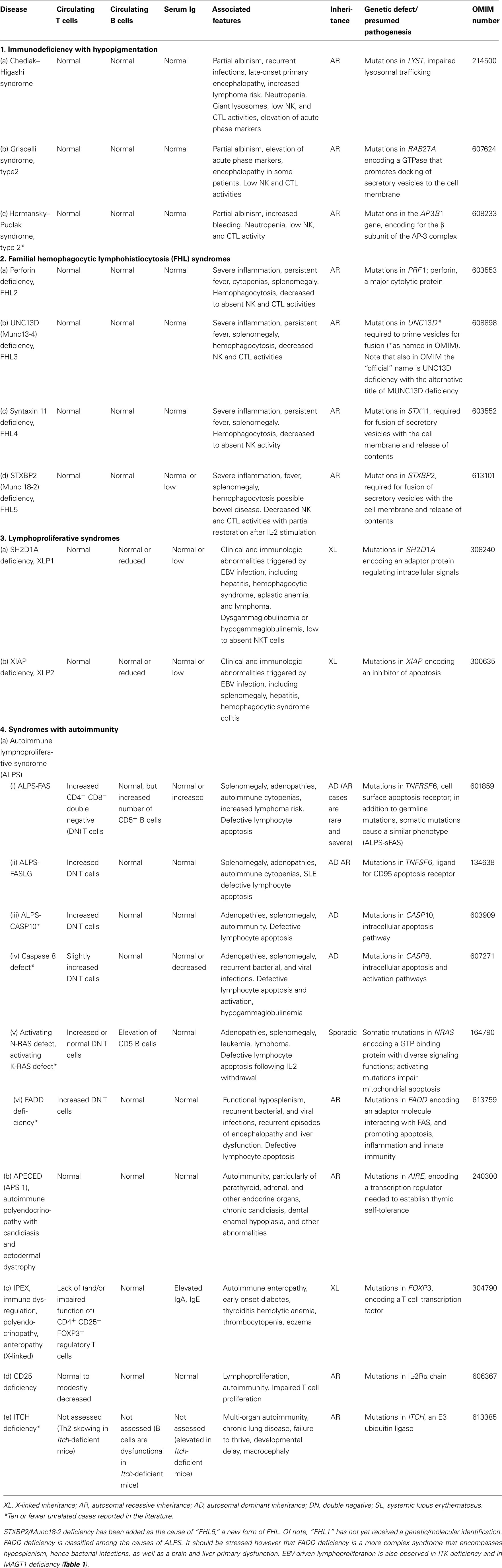
Table 4. Diseases of immune dysregulation.
Any classification of human disorders is somewhat arbitrary, and the classification of PIDs is no exception. Some disorders might well belong to more than one group. CD40 ligand deficiency, for example, is reported both in Tables 1 and 3 (“Predominantly antibody deficiencies”), to reflect the facts that failed B cell isotype switching was historically the most prominent feature of this condition (originally named Hyper-IgM syndrome) and that some patients survive into adulthood without significant opportunistic infections and do well with only immunoglobulin replacement therapy. Explanatory notes provided after each Table offer additional information (particularly where a condition appears in more than one Table) and indicate which new disorders have been added to that Table.
Although this updated classification reports on the most typical immunological findings and associated clinical and genetic features for the various PIDs, there is extensive clinical, immunological, and molecular heterogeneity that can not be easily recapitulated in a brief summary. To facilitate a more rigorous analysis of each disease, a column has been added on the right with a hyperlink to refer to its catalog number in the Online Mendelian Inheritance in Man (OMIM) publicly accessible database (www.omim.org) of human genetic disorders. It is suggested that the reader consult this regularly updated and fully referenced resource.
The prevalence of the various PIDs varies in different countries. For this reason, in this new classification, we have elected to avoid giving a comment on the relative frequency of PID disorders. However, an asterisk has been placed in the first column, after the disease name, to identify disorders for which fewer than 10 unrelated cases have been reported in the literature. Some of these forms of PID can be considered extremely rare. Others have only recently been identified and it may be that more patients will be detected over time.
Finally, it is increasingly recognized that different mutations in the same gene may result in different phenotypes and may be associated with different patterns of inheritance. This concept of clinical, immunological, and genetic heterogeneity is assuming foremost importance. Notes in the text or in the footnotes identify such heterogeneity, when known.
The scope of the IUIS Expert Committee on Primary Immunodeficiency is to increase awareness, facilitate recognition, and promote optimal treatment for patients with Primary Immunodeficiency disorders worldwide. For this reason, in addition to periodically revising the Classification of PIDs, the Expert Committee is also actively involved in the development of diagnostic criteria and in providing, upon request, advice with regard to therapeutic guidelines.
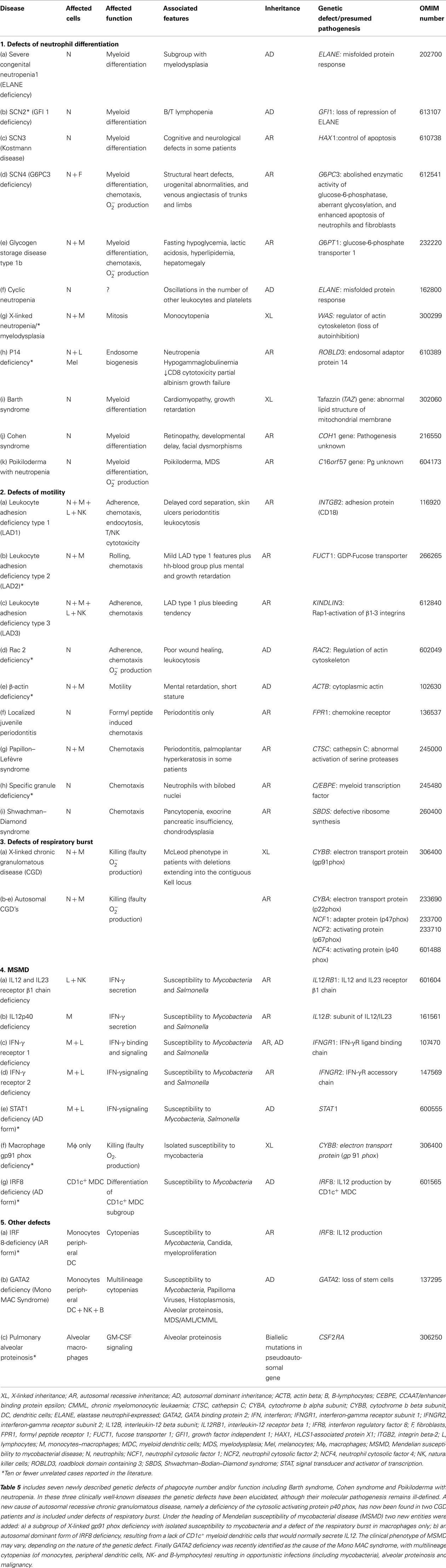
Table 5. Congenital defects of phagocyte number, function, or both.
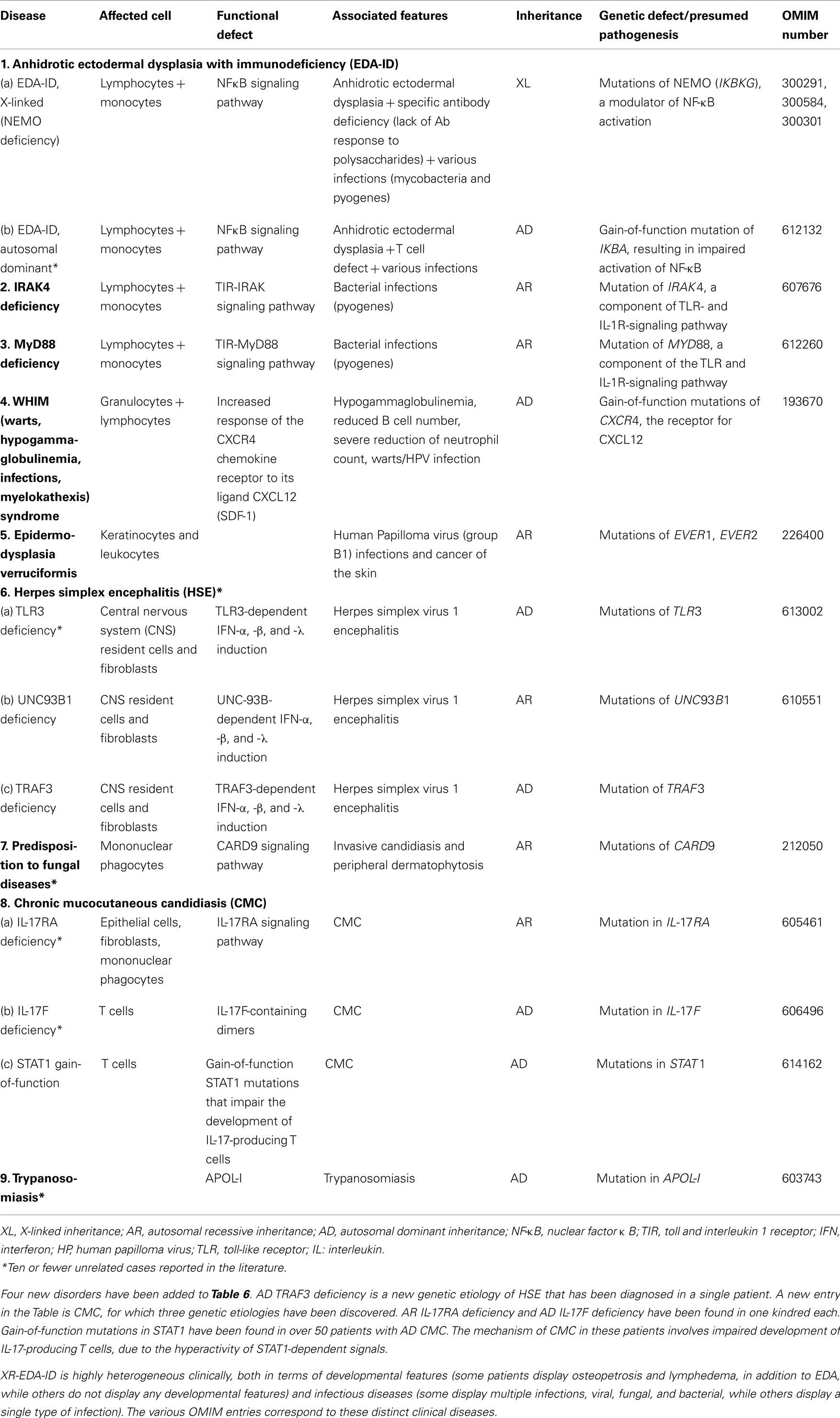
Table 6. Defects in innate immunity.
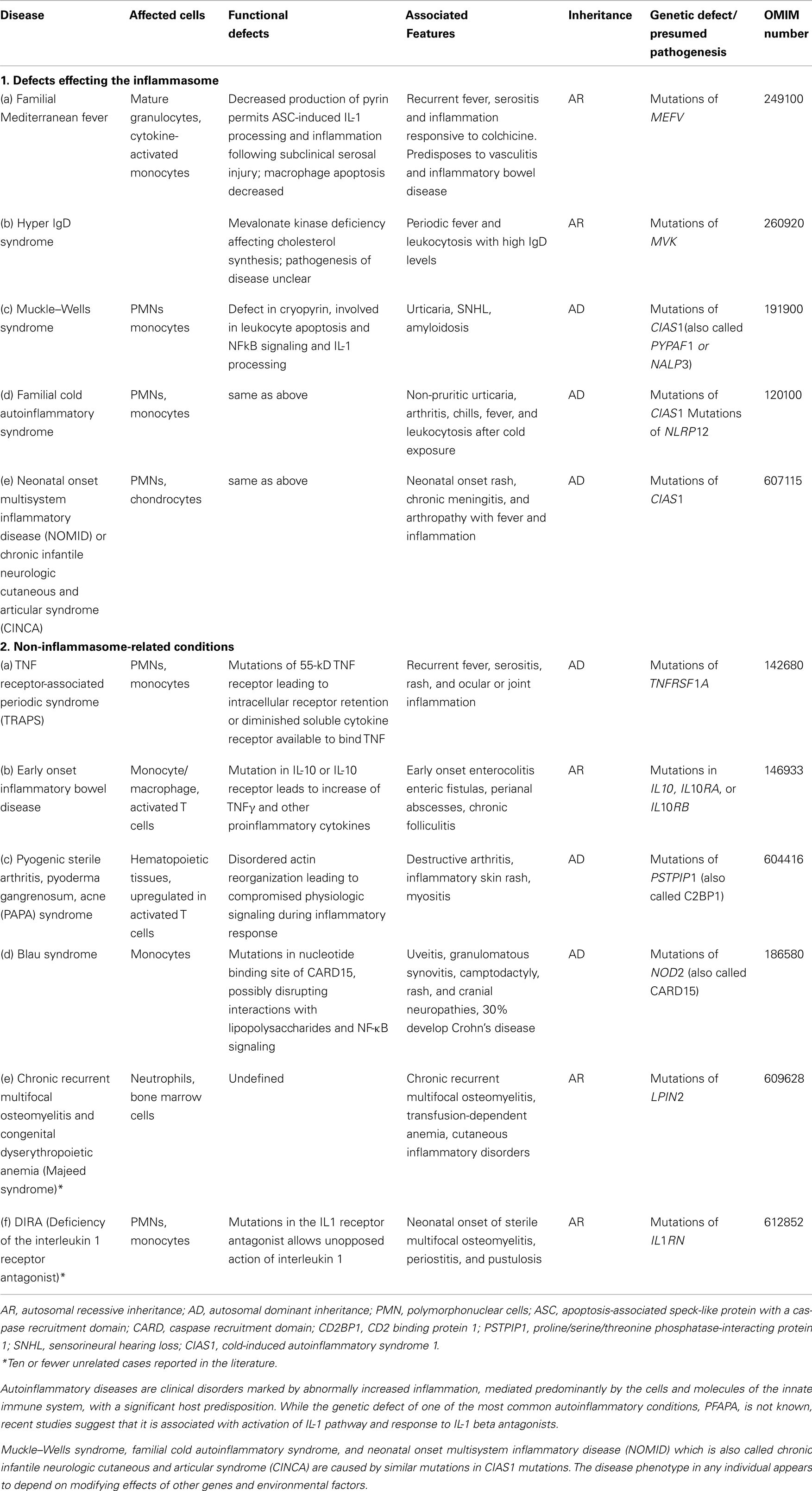
Table 7. Autoinflammatory disorders.
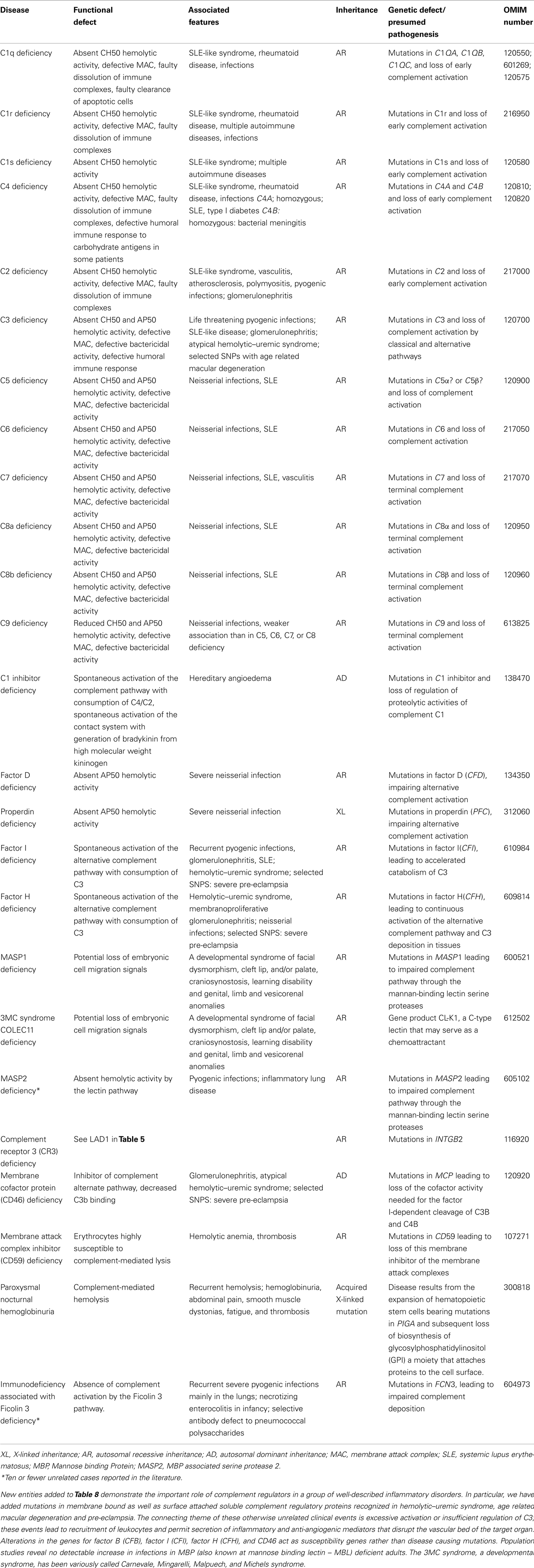
Table 8. Complement deficiencies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Keywords: primary immunodeficiency diseases
Citation: Al-Herz W, Bousfiha A, Casanova J-L, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Fischer A, Franco JL, Geha RS, Hammarström L, Nonoyama S, Notarangelo LD, Ochs HD, Puck JM, Roifman CM, Seger R and Tang ML (2011) Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front. Immun. 2:54. doi: 10.3389/fimmu.2011.00054
Received: 25 August 2011; Accepted: 20 September 2011;
Published online: 08 November 2011.
Edited by:
Eric Meffre, Yale University School of Medicine, USAReviewed by:
Eric Meffre, Yale University School of Medicine, USACopyright: © 2011 Al-Herz, Bousfiha, Casanova, Chapel, Conley, Cunningham-Rundles, Etzioni, Fischer, Franco, Geha, Hammarström, Nonoyama, Notarangelo, Ochs, Puck, Roifman, Seger and Tang. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Mary Ellen Conley, Department of Pediatrics, University of Tennessee College of Medicine, Memphis, TN, USA; Department of Immunology MS 351, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105, USA. e-mail:bWFyeWVsbGVuLmNvbmxleUBzdGp1ZGUub3Jn; Luigi Daniele Notarangelo, Division of Immunology and The Manton Center for Orphan Disease Research, Children’s Hospital Boston, Karp Research Building, Room 20217, 1 Blackfan Circle, Boston, MA 02115, USA. e-mail:bHVpZ2kubm90YXJhbmdlbG9AY2hpbGRyZW5zLmhhcnZhcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.