 Enrico Capobianco
Enrico Capobianco- University of Miami, Center for Computational Science, Miami, FL, United States
Big data are expected to exert profound impacts on medicine. High-throughput technologies, electronic medical records, high-resolution imaging, multiplexed omics, these are examples of fields that are progressing at a fast pace. Because they all yield complex heterogeneous data types, managing such variety and volumes is a challenge. While the computation power required to analyze them is available, the main difficulty consists in interpreting the results. In light of the emerging precision medicine paradigm, oncology is influenced by digital phenotypes characterizing disease expression, In particular, digital biomarkers could become critical for the evaluation of clinical endpoints. Currently, integrative approaches are conceived for the analysis of multi-evidenced data, i.e., data generated from multiple sources, such as cells, organs, individual lifestyle and social habits, environment, population dynamics, etc. The granularity, the scales of measurement, the model prediction accuracy, these are factors justifying an ongoing progressive differentiation from evidence-based medicine, typically based on a relatively small and unique scale of the experiments, thus well assimilated by a mathematical or statistical model. A premise of precision medicine is the N-of-1 paradigm, inspired by a focus on individualization. However, diversity, amount, and complexity of input data points that are needed for individual assessments, suggest centrality of systems inference principles. In turn, a revised paradigm is acquiring relevance, say (N-of-1)c, where the exponent c indicates connectivity. What makes connectivity such a key factor? For instance, the synergy embedded but often latent in the data layers, namely signatures, profiles, etc., which can lead to many stratified directions. Reference then goes to the biological and medical insights due to data integration, here discussed in view of the current oncological trends.
Trends in Health
Digital health is legitimating what was described about 30 years ago by Dawkins (1982), when presenting the extended phenotype principle. Phenotypic features, which include biological and genetic ones, are subject to continuous change as a result of all types of environmental interactions. Technology is clearly a game changer in most environments, especially those lived by humans, and is driving the transition toward digital phenotypes (Jain et al., 2015; Beckmann and Lew, 2016). An example is provided by digital biomarkers (Meister et al., 2016). These represent structured and unstructured information re-organized qualitatively and quantified by measures, scores, and predictions. Medical imaging at the technological side, and lifestyle and social dynamics at the environmental and behavioral sides, represent possible sources of digital biomarkers influencing precision oncology. The ultimate aim is to identify patient groups with certain risk factors or presenting different response to therapy compared to other groups (Capobianco, 2016; Dominietto and Capobianco, 2016; O’Connor et al., 2016). Hence, finding new digital phenotypes augments the ability to diagnose and cure a variety of oncological conditions, beyond early detection, and leading to new disease classification, treatment, and management.
In cancer, which is manifestly heterotypic, early diagnosis is crucial. During treatment, the management of the disease is a priority and issues to be considered refer to both prevention and therapy. Naturally enough, patient engagement is also being revolutionized, particularly under the influence of drivers like social influences and m-health (Teare et al., 2017). Finally, following the primary goal of precision medicine of bringing individualized solutions in terms of therapies, new models beyond the P4 paradigm are destined to emerge (Hood and Flores, 2012; Schellekens et al., 2017). Multiple factors need to be considered for proper assessment of cancer therapeutic solutions. For instance, with reference to multimodal drug combinations designed to special drug regimens targeting cancer cells, among the involved factors, a role in the molecular context is played by monitoring and assessing the immune response, measuring feedback signals from the microenvironment, considering the lifestyle and diet influences, etc. These complement more specific factors, those characterizing complexity inherent to sub-clonality, resistance, recurrence. For example, clones are especially important to explain phenotypic heterogeneity, while offering insights in tumorigenesis and treatment response. These properties refer to the fact that sub-populations may be identified across a spectrum of differentiated cancer characteristics, like proliferation and growth rates, which complicate the assessment of the effects of the metastatic power (Tabassum and Polyak, 2015).
Concerning sub-clonality, the utility of sequential approaches to precision oncology is acquiring relevance, as from the recent interest in temporal collateral sensitivity (Pritchard et al., 2013; Zhao et al., 2016). In particular, as sub-clones compete for dominance, some may prevail when they are resistant to treatment, thus becoming drug-induced sub-clones. Then, they can mutually or synergistically cooperate in communities, thus favoring (even just transiently) the metastatic process through cross-seeding dynamics, i.e., when the sub-clones that are present at a site have originated at other sites (Gundem et al., 2015; Hong et al., 2015; Shen, 2015; Tabassum and Polyak, 2015).
Combinations of drugs or targeted therapies are considered a priority in clinical contexts, but the current trends also assign centrality to the understanding of a milieu of data layers informing on stromal cell–cell interactions and environmental factors (Dry et al., 2016). These layers include genomic profiles, epigenomic marks, risk factors, marker indicators of metastatic conditions, which can also be aligned with precursor and premalignant conditions. In such integrated context, the exposome (see examples of ongoing activities in http://humanexposomeproject.com/; http://www.projecthelix.eu/; http://www.exposomicsproject.eu/; http://emoryhercules.com/) is highly relevant.
Cancer cells communicate locally and globally with the microenvironment, due to systemic interactions in response to inflammation, immune responses, vasculature, and metabolism signals coming from the stroma. A worthwhile attempt is to enable reprogramming by inducing invasion even against the environmental constraints prone to prevent or block the metastatic development of cancer cells (Axelrod et al., 2006). Clearly enough, the possible definitions of a cancer system are highly dependent on the understanding of the systemic environmental influences that regulate progression and determine the metastatic process.
Cancer Heterogeneity
Many factors contribute to one of the most challenging features of cancer, heterogeneity, which is the main factor determining the observed variation in cancer phenotypes. In particular, the clonal variety with its distinct genetic profiles implies an inherent differentiation in molecular evolution due to multiple resistance mechanisms and consequently drug responsiveness. Intra-tumor heterogeneity implicates that patients present differences in relation to the same cancer type, and that the same patient presents differences between tumor sites. Consequently, there are substantial difficulties in predicting tumor dynamics and the associated outcomes. Note that spatiotemporal heterogeneity allows adaptation to varying microenvironment constraints (Keats et al., 2012; Fisher et al., 2013).
Furthermore, similar mutations may result in differentiated phenotypes, while dissimilar mutations can converge to the same phenotype (De Sousa e Melo et al., 2013). This complexity is of course destined to complicate tremendously every clinical phase, particularly the one called to establish therapeutic paths. An example is offered by the Molecular Analysis for Therapy Choice (NCI-MATCH), which is based on patients’ genetic features (Mullard, 2015). Another example is offered by the Molecular Profiling-based Assignment of Cancer Therapy (NCI-MPACT), for which the goal is targeting oncogenic driver mutations in main pathways (DNA repair, PI3-K/mTOR, Ras/Raf/MEK) of unresponsive patients (Do et al., 2015). These examples indicate the need of systematically organized clinical trials aimed to assess signaling pathway-driven therapies. In turn, new strategies for off-label targeted therapies might follow in the realm of particularly resistant cases and rare cancers (Xue and Wilcox, 2016). In such cases, the emerging paradigm is that of the basket-trial design, aimed to conduct several independent and parallel phase II trials due to simultaneous screening over genetic aberrations detected by NGS-derived gene panels, thus enabling the assessment of a multitude of treatments in a large number of clinical trials (Redig and Jänne, 2015).
Back to sub-clonality, the role of clone-specific mutations is emphasized. Such alterations cover a wide variety of cases and suggest that clone-driven inference should become central from both a systems and a dynamic perspective. In particular, the latter involves slow- and fast-growing clones, likewise early- and late-emerging clones (Wang et al., 2013a,b). Therefore, Figure 1 illustrates the point that due to the role of the microenvironment and the imposed constraints, it is crucial to refer novel phenotypes by clone–clone and clone–stroma interactions to study disease progression. Due to the lack of experimental models (Marusyk et al., 2014), the role of computational models needs to acquiring further value, especially models centered on clonal networks. One type of complexity comes from cancer drivers that combine in many possible ways, while triggering state transitions. The focus of next generation computational algorithms should mainly be the identification of such transitions, and the prediction of tipping points or early warning signals useful to indicate closeness to the state switches. A primary aim remains the mitigation of the effects of such changes by taking preventive interventions. In principle, the identification may also extend to attractor states, those in which stationarity would be restored (Brock et al., 2015; Taherian Fard and Ragan, 2017). When considering possible sources of variation in cancer systems, i.e., genetic, epigenetic, and other, the attention shifts to their translation into systems perturbations, like in the case of treatments targeted to control growth and proliferation. Drug sensitivity represents a crucial factor, one enabling selectivity between sub-clones, suggesting that different data will be needed to inform on phenotypes and differentiated cancer-inducing agents.
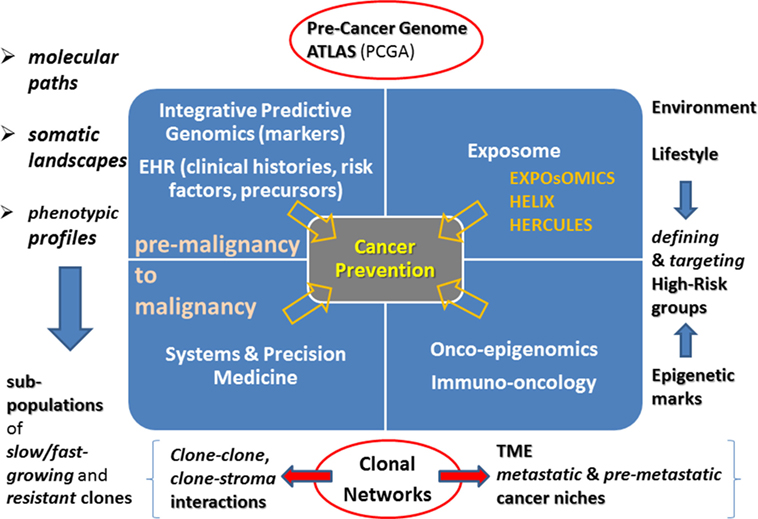
Figure 1. Cancer prevention landmarks. Integrative predictive genomics and EHR will be the drivers of novel systems and precision medicine approaches targeted to deciphering paths from premalignancy to malignancy. Identification of clonal subpopulations and their interactome are challenges ahead. Another rich and highly unexplored source is the Exposome together with its links with the Epigenome. A current challenge is re-defining onco-epigenomics and immune-oncology research. Clonal networks will be increasingly characterized in terms of tumor microenvironment and centered on epigenetic marks.
However, multiple phenotypes could span complexities that could still be governed by a relatively limited number of principles. In such regard, computational algorithms will be determinant for estimating the needed reduction of dimensionality able to approximate the distance between data bulks (or ranks). Thus, while finding signatures may continue to be a goal (Wang et al., 2015), these indicators will step from simply molecular ones (genes, proteins) to network ones (gene or protein modules), with the connectivity between nodes capturing even the higher dependencies and synergies in the system.
It is, therefore, likely that precision medicine, while focusing on the N-of-1 paradigm (Brannon and Sawyers, 2013), will induce a revision of the paradigm, namely (N-of-1)c empowered with the “c” or connectivity factor. This would be primarily acting as a driver of synergy between complex variables embedded in multilayered data, and as a result, spanning multiple stratifications. Consequently, the crucial identifications to be faced involve novel sensors of systems phase transitions that might inform on the insurgence of metastasis, tumor recurrence, drug selection, therapy assessment. Then, further identifications would consider diversification into categories ranging from clone specific to clone-pervasive evidences. Clearly enough, learning from each patient should make the above context actionable toward individualized solutions, but learning from few samples can be a limiting factor due to reduced statistical power for evaluating the uncertainty of predictions and the replicability of the results. Finally, higher rates of false positives call for adjustment by multiple testing of control for type 1 error rate, which needs to be rebalanced with the benefit of the novel identification types (Jain et al., 2015; Grody, 2016).
Role of High-Throughput Genomics
Next generation sequencing generates massively parallel amounts of data through the sequences of genomic regions explored in shotgun mode. The applications are expected to reach many possible directions, especially aimed at selecting patients for specific therapies. In cancer experiments, the malignant phenotypes represent a variety of possible detections obtainable in multiple ways. Hundreds of genes are currently targeted, but only a few are actionable, i.e., effectively and/or successfully “drugged” according to approved drugs (Alyass et al., 2015; Dietel et al., 2015). Then, different marker types will become central to future analyses, from prognostic markers (focused on disease outcomes) to predictive markers (focused on identifying patient sub-groups responsive to specific treatments).
However, it is well known that spotting cancer phenotypes is a cumbersome task from the computational standpoint. Many possible variants (in the order of several thousand hits for whole exome sequencing and several million hits for whole genome sequencing) reveal only limited causative examples. Also, only a relatively small number of variants can lead to either drugged or druggable targets. Currently, the detection of mutations is mostly left to targeted panels covering hundreds or even thousands of target loci (Malapelle et al., 2015; Marrone et al., 2015). This way, the detection of low-level mosaicism referred to mutations common across tumors can be ensured. At the same time, access to genetic information can be enabled at an individual basis, and therapy can be targeted to specific mutational signatures.
A couple of limitations must be mentioned: (a) variants present in more than 1% of normal human chromosomes are most likely not clinically relevant in cancer (Strom, 2016); (b) variability among patients remains an issue in the response to treatments. Given different types of heterogeneities, cellular one in which a mix of tumor and stroma cells is found, as well as molecular one due to sub-clonality, the path forward in the study of cancer genomes implies integrative approaches to cover methylation profiles and variation from non-coding genomic regions in view of a deeper assessment of differential expression. From a methodological side, the relationships among profiles or data layers should include cross-interactions, aimed to find possible multilayered latent structures underlying intra-layer dynamics (Kim et al., 2016; Tordini et al., 2016; Hasin et al., 2017; Huang et al., 2017). From a biomedical perspective, new markers will aim to measure drug response, as with pharmacodynamic ones, i.e., molecular indicators of drug effect on a target and useful to associate drug regimen, target effect, and biological tumor response (https://next.cancer.gov/developmentresources/pd_biomarker.htm), and also those neoantigens arising from cancer-specific mutations, and requiring genomics-proteomics pairing (Snyder et al., 2014).
Differential Measures Beyond Expression
Cancer networks have nowadays become quite popular. They are not simply descriptive or explorative tools, but they are generators of hypotheses under very complex conditions that they can represent quite straightforwardly. Among the systems-level insights that networks provide, the most promising implications refer to identification of novel drug targets (Ishitsuka et al., 2016; Vinayagam et al., 2016; Sharma et al., 2017). Only a few examples of such applications are currently available, but their promising ideas depend on known properties:
(a) Networks are modular structures (Newman, 2012; Bonnet et al., 2015; Fortunato, 2016). Due to the properties of modules, or communities, which embed more significant dependence among their components than with the external entities, the focus can be put on nodes treated as single targets but also on modules representing multiple connected targets. The relevance of therapeutic targets is of course high, as some of them cannot be directly used, but an indirect monitoring through their interactors could be allowed by tracking the significant connectivity paths surrounding them (Mora et al., 2014, 2016)
(b) Topological measures establish a series of measurable features that can be used to characterize network nodes, say target genes or proteins, and this might improve the discrimination power of algorithms in identifying with significance. Therefore, for each available sample not only genes (1…..N) or proteins (1…..P) could be put into associative (correlative or causal) or interactive communication, but also features (1…..F) can be embedded at each of such entities (nodes, links), thus expanding the dimensionality of the informative array exploitable for inference. To bridge with the earlier introduced digital biomarkers, it is tempting to enrich the network with therapy-related variables, such as response, follow-up, drug effects, etc. (Dominietto et al., 2015; Dominietto and Capobianco, 2016)
(c) Dependence in networks is naturally imported through the inherent metric based on the node connectivity patterns. This metric might be extended to cover highly complex network configurations (De Domenico et al., 2013, 2016). Network metrics go beyond the ones generally adopted in clustering, and as a result, more confidence can be assigned to the various associations between nodes, especially when comparing normal to disease networks, or temporal trajectories of disease networks across disease stages (say, pre- and posttreatment). Clearly enough, there is some level of uncertainty to consider, which requires statistical evaluations of significance, but also elucidation of the role of network entropy in cancer. Notably, the so-called fluctuation theorem of dynamical systems was proposed to establish criteria of assessment of cancer system hallmarks, through a correlation between systems entropy (or uncertainty) and resilience (robustness). Cancer cells are shown to be characterized by increased network entropy, while differential gene expressions are anticorrelated with local network entropy changes. This is important for possible identifications of novel drug targets and for the role of gene essentiality in knock-down experiments (West et al., 2012).
Cancer Prevention
Together with the concept of clonal evolution, and following the developments of the cancer stem cell hypothesis, the differentiation status of cancer cells is a determinant of their functional properties (Shackleton et al., 2009). Consequently, phenotypic heterogeneity in cancer reflects also non-genetic diversity occurring in normal cells (Marusyk et al., 2012). For instance, a driver of cancer development takes into consideration phenotypic states represented by the so-called epigenetic landscapes, together with their inter-attractor dynamics.
Another important consideration is that mutations are observed in healthy or pre- or even non-malignant cells too, thus, beyond the acquired mutations that may be selected and result clinically relevant, particularly at a later stage of cancer development. While establishing the clonal structure of tumors is very important for predicting its evolution (Rosenthal et al., 2017), the detection of very early cancer stages will depend on the examination of healthy samples and will allow the definition of molecular hallmarks of premalignant conditions (Ryan and Faupel-Badger, 2016), and even the foreseen achievement of a precancer genome atlas (Campbell et al., 2016; Kensler et al., 2016). Examples are already available, for instance with a series of recent studies centered on melanoma: one in which mutations were examined from precursor to invasive lesions (Shain et al., 2015), another in which single-cell RNA-Seq was employed to profile a multitude of cell types (Tirosh et al., 2016), and then one focusing on subtypes from multiple sites (Hayward et al., 2017).
To conclude, a couple of considerations are dedicated to non-primary cancer prevention, i.e., to other actions requiring preventive and timely interventions. An example is provided by dormancy, in which the metastatic process is inactive for years, and offers a therapeutic window against metastasis especially by targeting pathways that may help preserving the dormant state in cells. A more intermediate strategy to tackle the metastasis cascade process at stages of still limited and treatable lesions would require the colonization process to be locally interrupted by targeting at specific sites pre-metastatic niches (Steeg, 2016; Peinado et al., 2017). Figure 1 sketches some relationships that call for future in-depth (re-)examination, characterization, and further interoperable and actionable development in view of adapting and refining the cancer prevention map.
Author Contributions
The paper is conceived and written by the author.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MW and handling Editor declared their shared affiliation.
Acknowledgments
The author thanks two anonymous reviewers for constructive comments.
References
Alyass, A., Turcotte, M., and Meyre, D. (2015). From big data analysis to personalized medicine for all: challenges and opportunities. BMC Med. Genomics 8:33. doi: 10.1186/s12920-015-0108-y
Axelrod, R., Axelrod, D. E., and Pienta, K. J. (2006). Evolution of cooperation among tumor cells. Proc. Natl. Acad. Sci. U.S.A. 103, 13474–13479. doi:10.1073/pnas.0606053103
Beckmann, J. S., and Lew, D. (2016). Reconciling evidence-based medicine and precision medicine in the era of big data: challenges and opportunities. Genome Med. 8, 134. doi:10.1186/s13073-016-0388-7
Bonnet, E., Calzone, L., and Michoel, T. (2015). Integrative multi-omics module network inference with lemon-tree. PLoS Comput. Biol. 11:e1003983. doi:10.1371/journal.pcbi.1003983
Brannon, A. R., and Sawyers, C. L. (2013). “N of 1” case reports in the era of whole-genome sequencing. J. Clin. Invest. 123, 4568–4570. doi:10.1172/JCI70935
Brock, A., Krause, S., and Ingber, D. E. (2015). Control of cancer formation by intrinsic genetic noise and microenvironmental cues. Nat. Rev. Cancer 15, 499–509. doi:10.1038/nrc3959
Campbell, J. D., Mazzilli, S. A., Reid, M. E., Dhillon, S. S., Platero, S., Beane, J., et al. (2016). The case for a pre-cancer genome atlas (PCGA). Cancer Prev. Res. (Phila) 9, 119–124. doi:10.1158/1940-6207.CAPR-16-0024
Capobianco, E. (2016). Imaging-driven digital biomarkers. Cancer Transl. Med. 2, 61–63. doi:10.4103/2395-3977.181440
De Domenico, M., Granell, C., Porter, M. A., and Arenas, A. (2016). The physics of spreading processes in multilayer networks. Nat. Phys. 12, 901–906. doi:10.1038/nphys3865
De Domenico, M., Sole’-Ribalta, A., Cozzo, E., Kivela, M., Moreno, Y., Porter, M. A., et al. (2013). Mathematical formulation of multilayer networks. Phys. Rev. X3, 041022. doi:10.1103/PhysRevX.3.041022
De Sousa e Melo, F., Vermeulen, L., Fessler, E., and Medema, J. P. (2013). Cancer heterogeneity – a multifaceted view. EMBO Rep. 14, 686–695. doi:10.1038/embor.2013.92
Dietel, M., Johrens, K., Laffert, M. V., Hummel, M., Blaker, H., Pfitzner, B. M., et al. (2015). A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: a review focusing on clinical relevance. Cancer Gene Ther. 22, 417–430. doi:10.1038/cgt.2015.39
Do, K., O’Sullivan Coyne, G., and Chen, A. P. (2015). An overview of the NCI precision medicine trials – NCI-MATCH and MPACT. Chin. Clin. Oncol. 4, 31. doi:10.3978/j.issn.2304-3865.2015.08.01
Dominietto, M., Tsinoremas, N., and Capobianco, E. (2015). Integrative analysis of cancer imaging readouts by networks. Mol. Oncol. 9, 1–16. doi:10.1016/j.molonc.2014.08.013
Dominietto, M. D., and Capobianco, E. (2016). Expected impacts of connected multimodal imaging in precision oncology. Front. Pharmacol. 7:451. doi:10.3389/fphar.2016.00451
Dry, J. R., Yang, M., and Saez-Rodriguez, J. (2016). Looking beyond the cancer cell for effective drug combinations. Genome Med. 8, 125. doi:10.1186/s13073-016-0379-8
Fisher, R., Pusztai, L., and Swanton, C. (2013). Cancer heterogeneity: implications for targeted therapeutics. Br. J. Cancer 108, 479–485. doi:10.1038/bjc2012.581
Fortunato, S. (2016). Community detection in networks: a user guide. Phys. Rep. 659, 1–44. doi:10.1016/j.physrep.2016.09.002
Grody, W. W. (2016). The next generation of cancer management. Cancer Biol. Med. 13, 1–2. doi:10.20892/j.issn.2095-3941.2016.0027
Gundem, G., Van Loo, P., Kremeyer, B., Alexandrov, L. B., Tubio, J. M. C., Papaemmanuil, E., et al. (2015). The evolutionary history of lethal metastatic prostate cancer. Nature 520, 353–357. doi:10.1038/nature14347
Hasin, Y., Seldin, M., and Lusis, A. (2017). Multi-omics approaches to disease. Genome Biol. 18, 83. doi:10.1186/s13059-017-1215-1
Hayward, N. K., Wilmott, J. S., Waddell, N., Johansson, P. A., Field, M. A., Nones, K., et al. (2017). Whole-genome landscapes of major melanoma subtypes. Nature 545, 175–180. doi:10.1038/nature22071
Hong, M. K. H., Macintyre, G., Wedge, D. C., Van Loo, P., Patel, K., Lunke, S., et al. (2015). Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat. Commun. 6, 1–12. doi:10.1038/ncomms7605
Hood, L., and Flores, M. (2012). A personal view on systems medicine and the emergence of proactive P4 medicine: predictive, preventive, personalized and participatory. Nat. Biotechnol. 29, 613–624. doi:10.1016/j.nbt.2012.03.004
Huang, S., Chaudhary, K., and Garmire, L. X. (2017). More is better: recent progress in multi-omics data integration methods. Front. Genet. 8:84. doi:10.3389/fgene.2017.00084
Ishitsuka, M., Akutsu, T., and Nacher, J. C. (2016). Critical controllability in proteome-wide protein interaction network integrating transcriptome. Sci. Rep. 6, 23541. doi:10.1038/srep23541
Jain, S. H., Powers, B. W., Hawkins, J. B., and Brownstein, J. S. (2015). The digital phenotype. Nat. Biotechnol. 33, 462–463. doi:10.1038/nbt.3223
Keats, J. J., Chesi, M., Egan, J. B., Garbitt, V. M., Palmer, S. E., Braggio, E., et al. (2012). Clonal competition with alternating dominance in multiple myeloma. Blood 120, 1067–1076. doi:10.1182/blood-2012-01-405985
Kensler, T. W., Spira, A., Garber, J. E., Szabo, E., Lee, J. J., Dong, Z., et al. (2016). Transforming cancer prevention through precision medicine and immune-oncology. Cancer Prev. Res. (Phila) 9, 2–10. doi:10.1158/1940-6207.CAPR-15-0406
Kim, M., Rai, N., Zorraquino, V., and Tagkopoulos, I. (2016). Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat. Commun. 7, 13090. doi:10.1038/ncomms13090
Malapelle, U., De Stefano, A., Carlomagno, C., Bellevicine, C., and Troncone, G. (2015). Next-generation sequencing in the genomic profiling of synchronous colonic carcinomas: comment on Li et al (2015). J. Clin. Pathol. 68, 946–947. doi:10.1136/jclinpath-2015-203205
Marrone, M., Potosky, A. L., Penson, D., and Freedman, A. N. (2015). A 22 gene-expression assay, decipher® (GenomeDx biosciences) to predict five-year risk of metastatic prostate cancer in men treated with radical prostatectomy. PLoS Curr. 7. doi:10.1371/currents.eogt.761b81608129ed61b0b48d42c04f92a4
Marusyk, A., Almendro, V., and Polyak, K. (2012). Intra-tumor heterogeneity: a looking glass for cancer? Nat. Rev. Cancer 12, 323–334. doi:10.1038/nrc3261
Marusyk, A., Tabassum, D. P., Altrock, P. M., Almendro, V., Michor, F., and Polyak, K. (2014). Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514, 54–58. doi:10.1038/nature13556
Meister, S., Deiters, W., and Becker, S. (2016). Digital health and digital biomarkers – enabling value chains on health data. Curr. Dir. Biomed. Eng. 2, 577–581. doi:10.1515/cdbme-2016-0128
Mora, A., Taranta, M., Zaki, N., Badidi, E., Cinti, C., and Capobianco, E. (2014). Ensemble inference by integrative cancer networks. Front. Genet. 5:59. doi:10.3389/fgene.2014.00059
Mora, A., Taranta, M., Zaki, N., Cinti, C., and Capobianco, E. (2016). Epigenetically driven network cooperativity: meta-analysis in multi-drug resistant osteosarcoma. J. Complex Netw. 4, 296–317. doi:10.1093/comnet/cnv017
Mullard, A. (2015). NCI-MATCH trial pushes cancer umbrella trial paradigm. Nat. Rev. Drug Discov. 14, 513–515. doi:10.1038/nrd4694
Newman, M. E. J. (2012). Communities, modules and large-scale structure in networks. Nat. Phys. 8, 25–31. doi:10.1038/nphys2162
O’Connor, J. P. B., Aboagye, E. O., Adams, J. E., Aerts, H. J. W. L., Barrington, S. F., Beer, A. J., et al. (2016). Imaging biomarker roadmap for cancer studies. Nat. Rev. Clin. Oncol. 14, 169–196. doi:10.1038/nrclinonc.2016.162
Peinado, H., Zhang, H., Matei, I. R., Costa-Silva, B., Hoshino, A., Rodrigues, G., et al. (2017). Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317. doi:10.1038/nrc.2017.6
Pritchard, J. R., Bruno, P. M., Gilbert, L. A., Capron, K. L., Lauffenburger, D. A., and Hemann, M. T. (2013). Defining principles of combination drug mechanisms of action. Proc. Natl. Acad. Sci. U.S.A. 110, E170–E179. doi:10.1073/pnas.1210419110
Redig, A. J., and Jänne, P. A. (2015). Basket trials and the evolution of clinical trial design in an era of genomic medicine. J. Clin. Oncol. 33, 975–977. doi:10.1200/JCO.2014.59.8433
Rosenthal, R., McGranahan, N., Herrero, J., and Swanton, C. (2017). Deciphering genetic intratumor heterogeneity and its impact on cancer evolution. Annu. Rev. Cancer Biol. 1, 223–240. doi:10.1146/annurev-cancerbio-042516-011348
Ryan, B. M., and Faupel-Badger, M. (2016). The hallmarks of premalignant conditions: a molecular basis for cancer prevention. Semin. Oncol. 43, 22–35. doi:10.1053/j.seminoncol.2015.09.007
Schellekens, H., Aldosari, M., Talsma, H., and Mastrobattista, E. (2017). Making individualized drugs a reality. Nat. Biotechnol. 35, 507–513. doi:10.1038/nbt.3888
Shackleton, M., Quintana, E., FEaron, E. R., and Morrison, S. J. (2009). Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 138, 822–829. doi:10.1016/j.cell.2009.08.017
Shain, A. H., Yeh, I., Kovalyshyn, I., Sriharan, A., Talevich, E., Gagnon, A., et al. (2015). The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 373, 1926–1936. doi:10.1056/NEJMoa1502583
Sharma, A., Cinti, C., and Capobianco, E. (2017). Multitype network-guided target controllability in phenotyically characterized osteosarcoma: role of tumor microenvironment. Front. Immunol. 8:918. doi:10.3389/fimmu.2017.00918
Shen, M. M. (2015). Cancer: the complex seeds of metastasis. Nature 520, 298–299. doi:10.1038/nature14377
Snyder, A., Makarov, V., Merghoub, D., Yuan, J., Zaretsky, J. M., Desrichard, A., et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199. doi:10.1056/NEJMoa1406498
Strom, S. P. (2016). Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med. 13, 3–11. doi:10.28092/j.issn.2095-3941.2016.0004
Tabassum, D. P., and Polyak, K. (2015). Tumorigenesis: it takes a village. Nat. Rev. Cancer 15, 473–483. doi:10.1038/nrc3971
Taherian Fard, A., and Ragan, M. A. (2017). Modeling the attractor landscape of disease progression: a network-based approach. Front. Genet. 8:48. doi:10.3389/fgene.2017.00048
Teare, H. J. A., Hogg, J., Kaye, J., Luqmani, R., Rush, E., Turner, A., et al. (2017). The RUDY study: using digital technologies to enable a research partnership. Eur. J. Hum. Genet. 25, 816–822. doi:10.1038/ejhg.2017.57
Tirosh, I., Izar, B., Prakadan, S. M., Wadsworth, M. H. II, Treacy, D., Trombetta, J. J., et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. doi:10.1126/science.aad0501
Tordini, F., Aldinucci, M., Milanesi, L., Lio’, P., and Merelli, I. (2016). The genome conformation as an integrator of multi-omic data: the example of damage spreading in cancer. Front. Genet. 7:194. doi:10.3389/fgene.2016.00194
Vinayagam, A., Gibson, T. E., Lee, H. J., Yilmazel, B., Roesel, C., Hu, Y., et al. (2016). Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc. Natl. Acad. Sci. U.S.A. 113, 4976–4981. doi:10.1073/pnas.1603992113
Wang, E., Zaman, N., Mcgee, S., Milanese, J. S., Masoudi-Nejad, A., and O’Connor-McCourt, M. (2015). Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin. Cancer Biol. 30, 4–12. doi:10.1016/j.semcancer.2014.04.002
Wang, E., Zou, J., Zaman, N., Beitel, L. K., Trifiro, M., and Paliouras, M. (2013a). Cancer systems biology in the genome sequencing era: part 1, dissecting and modeling of tumor clones and their networks. Semin. Cancer Biol. 23, 279–285. doi:10.1016/j.semcancer.2013.06.002
Wang, E., Zou, J., Zaman, N., Beitel, L. K., Trifiro, M., and Paliouras, M. (2013b). Cancer systems biology in the genome sequencing era: part 2, evolutionary dynamics of tumor clonal networks and drug resistance. Semin. Cancer Biol. 23, 286–292. doi:10.1016/j.semcancer.2013.06.001
West, J., Bianconi, G., Severini, S., and Teschendorff, A. E. (2012). Differential network entropy reveals cancer system hallmarks. Sci. Rep. 2, 802. doi:10.1038/srep00802
Xue, Y., and Wilcox, W. R. (2016). Changing paradigm of cancer therapy: precision medicine by next-generation sequencing. Cancer Biol. Med. 13, 12–18. doi:10.28092/j.issn.2095-3941.2016.0003
Keywords: precision medicine, big data, digital health, genomics, cancer networks
Citation: Capobianco E (2017) Precision Oncology: The Promise of Big Data and the Legacy of Small Data. Front. ICT 4:22. doi: 10.3389/fict.2017.00022
Received: 13 June 2017; Accepted: 11 August 2017;
Published: 29 August 2017
Edited by:
Taha Yasseri, University of Oxford, United KingdomReviewed by:
Matt Willis, University of Oxford, United KingdomManlio De Domenico, Universidad Rovira i Virgili, Spain
Copyright: © 2017 Capobianco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enrico Capobianco, ZWNhcG9iaWFuY28mI3gwMDA0MDttZWQubWlhbWkuZWR1