 Wangdong Deng1,2,3
Wangdong Deng1,2,3 Xinyi Cai4*
Xinyi Cai4*- 1Department of Urology, Longgang District Central Hospital of Shenzhen, Shenzhen, China
- 2Shenzhen Clinical School of Medicine, Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, China
- 3College of Medicine, Shantou University, Shantou, China
- 4Department of Pathology, Shantou University Medical College, Shantou, China
Introduction: Ficus hirta, known as Wuzhimaotao in China, is a dioecious plant species within the Moraceae family, highly regarded for its medicinal and ecological roles.
Methods: Utilizing a hybrid assembly methodology, combining Nanopore and Illumina sequencing, we achieved a detailed mitochondrial genomic architecture.
Results: The genome exhibits single circular structures, spans 486,226 base pairs with 45.21% GC content, and encompasses 31 distinct protein-coding genes.Our analysis extends to gene content, codon usage, intergenomic transfers, phylogenetic relationships, and RNA editing patterns. Notably, the mitochondrial and chloroplast genomes share 15 homologous fragments, underpinning intercellular gene exchange. Phylogenetic positioning confirms Ficus hirta within Moraceae, closely allied with Morus notabilis.
Discussion: This comprehensive mitochondrial genome elucidation not only augments the biological understanding of Ficus hirta but also enriches genomic resources for future research.
1 Introduction
Ficus hirta, known as Wuzhimaotao in China, is a dioecious plant species within the Moraceae family, highly regarded for its medicinal and ecological roles (Wu et al., 2013; Yu et al., 2008; Sun et al., 2016). It has been extensively used in traditional herbal medicine, particularly in the Lingnan regions of China, with its dry root, Radix Fici Hirtae, being utilized for therapeutic purposes. Key bioactive compounds have been identified as the main active ingredients contributing to its anti-inflammatory and antioxidant effects (Cheng et al., 2017; Chen et al., 2020).
Ecologically, F. hirta is integral to its ecosystem, participating in intricate mutualisms. It depends on species-specific fig wasps for pollination, while vertebrates, including birds and bats, facilitate seed dispersal, emphasizing its role in promoting local biodiversity (Yu et al., 2010). Due to overharvesting driven by market demand for its medicinal properties, wild populations have significantly declined, raising concerns for its long-term survival. Despite this, genetic studies have revealed high genetic diversity and low population differentiation, indicating that F. hirta populations still have the resilience needed for conservation efforts and potential sustainable breeding programs (Lu et al., 2022).
Although much is known about the medicinal properties and chloroplast genome of F. hirta, its mitochondrial genome remains largely unexplored (Liu et al., 2019). Mitochondrial genomes are essential for understanding key aspects of plant energy metabolism, phylogenetic relationships, and genomic architecture. Recent advancements in sequencing technologies, particularly hybrid assembly methods combining long-read and short-read data, have enabled comprehensive analyses of plant mitochondrial genomes. This opens new avenues to investigate gene content, codon usage, intergenomic transfers, and evolutionary dynamics, making the study of F. hirta’s mitochondrial genome particularly significant in expanding our understanding of the species at the genomic level.
This study presents the first complete mitochondrial genome of F. hirta, alongside a comparative analysis with other species in the Rosales order. The study sheds light on intergenomic transfers between mitochondrial and chloroplast genomes, offering valuable insights into their evolutionary interactions. By analyzing phylogenetic relationships within the Moraceae family, this research enhances our understanding of F. hirta’s genomic framework, contributing to broader knowledge about the evolutionary divergence of species within the Rosales order. The findings serve as an important resource for future conservation strategies and genomic studies of this ecologically and pharmacologically important species.
2 Materials and methods
2.1 Plant material and DNA extraction
Fresh leaves of F. hirta were collected from multiple locations in the Lingnan region of China. Total genomic DNA was extracted from approximately 100 mg of fresh leaf tissue using the cetyltrimethylammonium bromide (CTAB) method (Doyle and Doyle, 1987). DNA quantity and quality were assessed using a NanoDrop spectrophotometer and agarose gel electrophoresis (Wilfinger et al., 1997). High-quality DNA samples were selected for sequencing.
2.2 Sequencing and genome assembly
We employed a hybrid sequencing strategy using Illumina NovaSeq (150-bp paired-end reads, ∼5 Gb) and Nanopore PromethION (∼10 Gb of long-read data). Long-read assembly was first performed using Flye (v2.9) with default parameters, generating a graph-based assembly in GFA format. To identify mitochondrial contigs, we built a local BLAST database using makeblastdb (BLAST + v2.11.0) and conducted a BLASTn search against conserved mitochondrial genes from Arabidopsis thaliana, using parameters: evalue 1e-5 -outfmt 6 -max_hsps 10 -word_size 7 -task blastn-short. The identified mitochondrial contigs were visualized using Bandage (v0.8.1) for manual selection. Different mapping software and parameters for short-read and long-read sequencing data were used. For the short-read sequencing data, the paired-end sequencing reads were aligned to the reference genome using a multi-step mapping pipeline. First, the BWA-MEM algorithm (v0.7.17) was employed to generate genome indices with the command “bwa index”. Subsequent read alignment was performed using “bwa mem” with 10 computational threads (-t 10), processing both forward and reverse read files. The SAM output was piped to samtools (v1.15.1) for binary conversion (-bS) while filtering out unmapped reads (-F 12) using eight parallel threads (-@ 8). Reads were then name-sorted (sort -n) to maintain proper pairing information before being converted back to FASTQ format. The final aligned read pairs were output through samtools’ fastq module with thread-optimized processing (-@ 8). For the long-read sequencing data, Long-read sequencing data processing was performed through an optimized alignment pipeline. The reference genome was initially indexed using minimap2 (v2.24) with the command “minimap2 -d ref1.mmi”. Sequence alignment was executed using the Nanopore-preset parameters (-ax map-ont) with eight computational threads (-t 8), processing compressed long-read data (ont.fastq.gz) through “minimap2 -ax map-pb -t 8 ref1.mmi ont.fastq.gz > out.sam”. The resulting SAM file was converted to BAM format using samtools (v1.15.1) with unmapped read filtering (-F 4) and 8-thread parallelization (-@ 8). Final FASTQ generation was accomplished through bedtools’ (v2.30.0) bamtofastq module, producing the processed long-read dataset. The final hybrid assembly was performed using Unicycler (v0.4.8) with default parameters, producing a circular mitochondrial genome, which was confirmed and visualized again using Bandage (v0.8.1). The final mitochondrial genome assembly was polished using Illumina short-read data with Pilon (v1.23) to improve assembly accuracy.
2.3 Genome annotation and analysis
The annotation of the mitochondrial genome was performed using GeSeq (v1.85), with A. thaliana and Liriodendron tulipifera mitochondrial genomes used as reference sequences. We chose A. thaliana and Liriodendron tulipifera as references for gene annotation because they are well-annotated model plants with high-quality mitochondrial annotation covering typical angiosperm gene sets, which widely used for mitochondrial reference. tRNAscan-SE (v2.0) was employed for the identification of tRNA genes, while BLASTN (v2.11.0) was used for rRNA annotation. The annotations were manually curated using the Apollo software (v2.6.6) to ensure accuracy. The complete circular genome map was visualized using OGDRAW (v1.3.1) to represent gene locations and orientations.
2.4 Codon usage and repeat sequence analysis
Relative synonymous codon usage (RSCU) for all protein-coding genes (PCGs) was calculated using MEGA (v7.0.26). To assess repeat structures within the genome, MISA-web (v2.1) was used to identify simple sequence repeats (SSRs), and REPuter (v2.74) was employed to detect dispersed repeats, including forward, palindromic, and complementary repeats. Tandem repeat sequences were identified using Tandem Repeat Finder (TRF, v4.09) 81% as matching-score threshold for tandem repeats. This threshold is a commonly used default in Tandem Repeat Finder for moderate stringency, balancing sensitivity and specificity. And all repeat sequence results were visualized using Circos (v0.69–9).
2.5 Intergenomic transfer and phylogenetic analysis
Homologous regions between the mitochondrial and chloroplast genomes were identified using BLASTN (v2.11.0) with a minimum alignment length of 100 bp and identity greater than 90%. The sequence transfer between the two genomes was further analyzed and visualized using Circos (v0.69-9). The chloroplast genome was assembled using GetOrganelle software (v1.7.7.0) (Jin et al., 2020), annotated using CPGAVAS2 software (Shi et al., 2019), and then corrected using CPGView software (Liu et al., 2023). The rrn16S, rrn23S, rrn4.5S, and rrn5S genes were identified based on sequence homology and conserved domain searches. Subsequently, we masked or removed the corresponding regions from further analyses to prevent potential biases introduced by highly conserved sequences. This methodological approach aligns with previous studies emphasizing the necessity of removing highly conserved organellar genes to improve the accuracy of horizontal gene transfer detection (Mower et al., 2010; Timmis et al., 2004). Phylogenetic analysis was performed using MAFFT (v7.475) for multiple sequence alignment and IQ-TREE (v2.1.4) for maximum likelihood analysis. Morus notabilis and other closely related species in the Moraceae family were included for comparative analysis The phylogenetic tree included 31 species across multiple families. Rosaceae species included Pyrus × bretschneideri (NC_065218.1), Pyrus betulifolia (NC_054332.1), Pyrus communis (NC_065229.1), Eriobotrya japonica (NC_045228.1), Sorbus aucuparia (NC_052880.1), Malus sylvestris (NC_065226.1), Malus sieversii (NC_065225.1), Chaenomeles speciosa (OL450370.1), Torminalis glaberrima (MT610102.1), Photinia serratifolia (NC_065220.1), Malus baccata (NC_065224.1), Prunus armeniaca (NC_065228.1), Prunus mira (NC_065231.1), Prunus kanzakura (NC_065230.1), Fragaria iturupensis (NC_062833.1), Fragaria gracilis (NC_062834.1), Fragaria vesca (NC_065239.1), Potentilla anserina (ON478170.1), Potentilla micrantha (NC_062588.1), Rosa rugosa (NC_065237.1), Rosa chinensis (NC_065236.1), Rubus chingii (NC_065238.1), and Geum urbanum (NC_065221.1). Moraceae species included F. hirta (This Research) and Morus notabilis (NC_041177.1). Cannabaceae species included Cannabis sativa (NC_029855.1) and Humulus lupulus (MW413894.1). Rhamnaceae species included Ziziphus mauritiana (NC_068745.1) and Ziziphus jujuba (NC_029809.1). Fabaceae species included Glycine max (NC_020455.1), Senna tora (NC_038053.1).
2.6 RNA editing prediction
RNA editing sites in the mitochondrial genome of F. hirta were predicted using the PREP-Mt tool (v0.9), which is specifically designed to identify RNA editing events in plant mitochondrial genomes. The tool predicts C-to-U conversions, the most common form of RNA editing in plant mitochondria. The sequences of all 31 protein-coding genes from the F. hirta mitochondrial genome were input into the program. The cutoff value for editing prediction was set to 0.9, following standard parameters for accurate detection of potential RNA editing sites in plant mitochondrial genomes.
2.7 Synteny analysis
Synteny analysis was performed to compare the F. hirta mitochondrial genome with those of related species. Homologous regions between the mitochondrial genomes of F. hirta and other species were identified using the BLASTN tool (v2.11.0), with a minimum alignment length of 500 bp and sequence identity greater than 90%. MCScanX (v1.0) was used to detect and analyze syntenic blocks, including regions of conservation and rearrangement. The syntenic relationships were visualized using Circos (v0.69-9), providing a comprehensive view of genome structure variation among the species.
3 Results
3.1 Complete mitogenome sequence of Ficus hirta
The mitochondrial genome of F. hirta is organized as a single circular molecule (Figure 1). The genome spans 486,226 base pairs (bp) with a GC content of 45.21%, which is comparable to other plant mitochondrial genomes within the Rosales order (Zhang et al., 2022). Annotation of the F. hirta mitochondrial genome identified 31 protein-coding genes, 19 tRNA genes, and three rRNA genes, with the tRNA and rRNA genes containing multi-copy variants, as shown in Supplementary Table S1. The protein-coding genes include 24 core mitochondrial genes and seven non-core genes, following the conserved gene set typically observed in plant mitochondrial genomes. Comparative analysis with A. thaliana and Liriodendron tulipifera confirmed the presence of all core genes, indicating a high level of conservation. Additionally, no novel protein-coding genes specific to F. hirta were identified, further supporting the evolutionary stability of plant mitochondrial genomes. The core genes include 5 ATP synthase genes (atp1, atp4, atp6, atp8, and atp9); 9 NADH dehydrogenase genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9); four cytochrome c biogenesis genes (ccmB, ccmC, ccmFC, and ccmFN); three cytochrome c oxidase genes (cox1, cox2, and cox3); one protein transport subunit gene (mttB); one maturases gene (matR); and one cytochrome b gene (cob). The non-core genes include one ribosomal protein large subunit gene (rpl16); four ribosomal protein small subunit genes (rps4, rps7, rps12, rps13); and two succinate dehydrogenase genes (sdh3, sdh4). Details of genome assembly and gene annotations could be found in Supplementary Data Sheet 1.
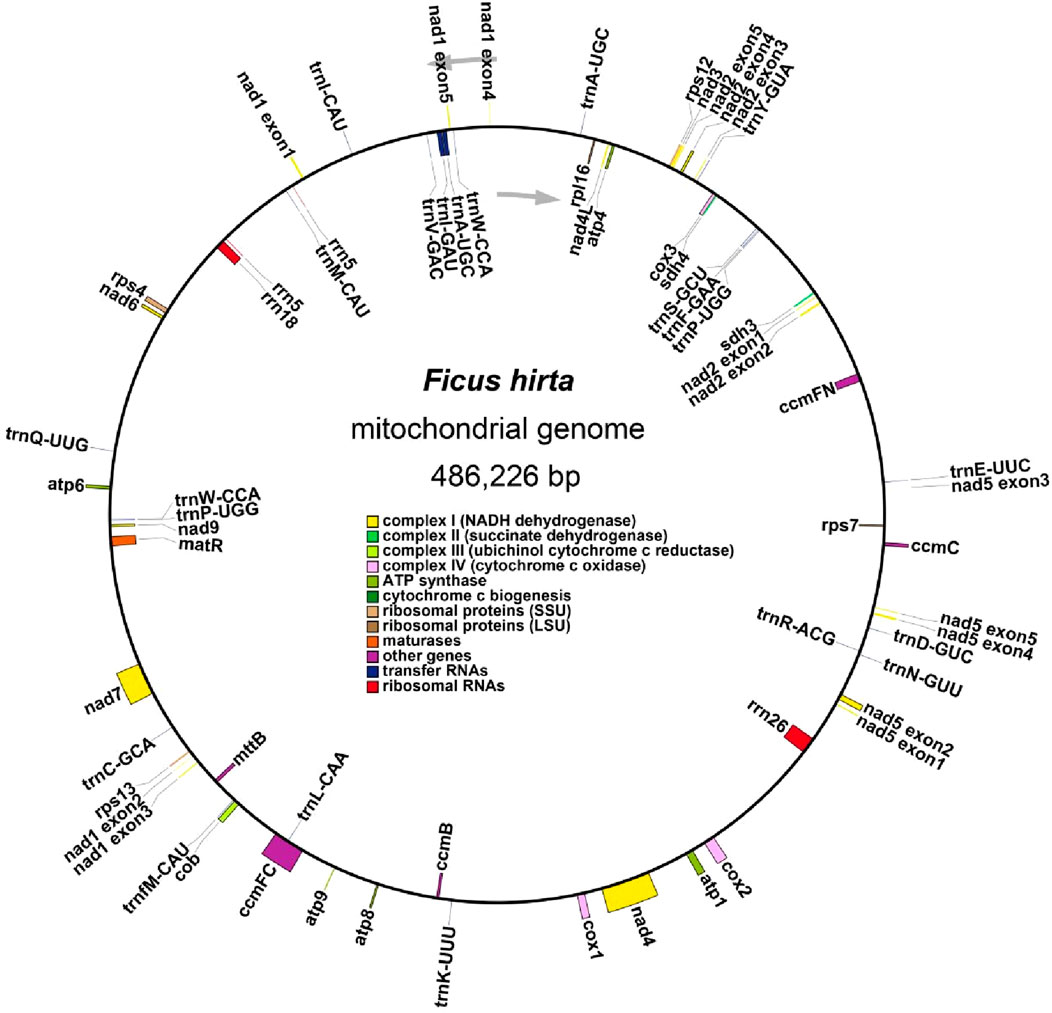
Figure 1. Detailed mitogenome map of Ficus hirta Colored boxes denote conserved mitochondrial genes categorized according to their functional products. Genes located inside the circle are transcribed in a clockwise direction, whereas those positioned outside the circle are transcribed in a counterclockwise direction. Genes associated with various functional categories are designated by distinct colors.
3.2 Codon usage analysis of protein-coding genes (PCGs) in Ficus hirtas
Codon usage analysis was conducted on the 31 unique PCGs of the Ficus hirtas mitochondrial genome (Figure 2; Supplementary Table S2). The codon usage for each amino acid is presented in Supplementary Table S2. Codons with a relative synonymous codon usage (RSCU) greater than one are considered preferentially used by amino acids. As shown in Figure 2, apart from the start codon AUG and tryptophan (UGG), both of which have an RSCU value of 1, the PCGs of the mitochondrial genome exhibit a general codon usage bias. For example, alanine (Ala) shows a strong preference for GCU, with the highest RSCU value of 1.63 among mitochondrial PCGs, followed by proline (Pro), which preferentially uses CCU, with an RSCU value of 1.58. Notably, phenylalanine (Phe) has a maximum RSCU value of less than 1.2, indicating a relatively weak codon usage bias.
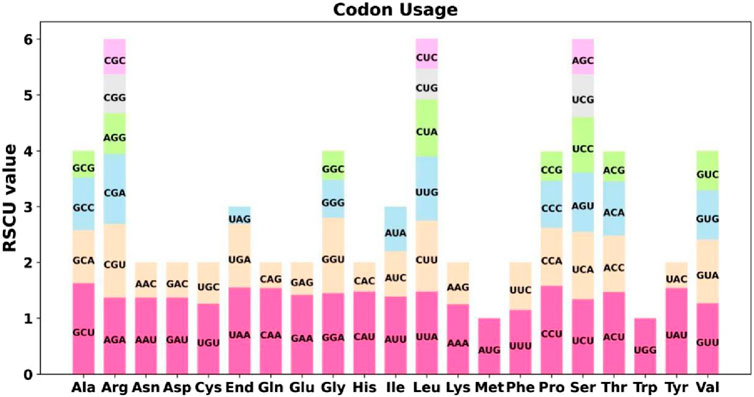
Figure 2. Codon Usage Analysis of the Ficus hirta Mitochondrial Genome. X-axis, codon families; Y-axis, the RSCU value.
3.3 Repeat sequence analysis on the Ficus hirta mitochondrial genome
A repeat sequence analysis was performed on the F. hirta mitochondrial genome (Figure 3; Supplementary Tables S3–S5). A total of 153 SSRs were detected, with mononucleotide and dinucleotide SSRs accounting for 58.82% of the total SSRs, and mononucleotide adenine repeats making up 53.57% of mononucleotide SSRs. No hexanucleotide SSRs were observed. Tandem repeats, which are core repeat units ranging from 7 to 200 bp, were also identified, with 15 tandem repeats found in the F. hirta mitochondrial genome, exhibiting a matching score greater than 81% and lengths varying from 15 to 48 bp.
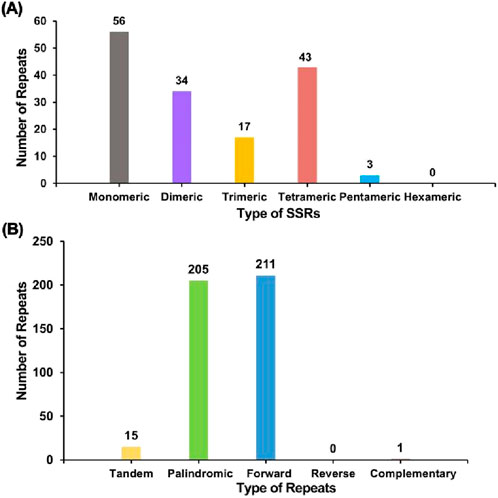
Figure 3. Repeat sequence analysis on the Ficus hirta mitochondrial genome. (A) The x-axis represents the types of SSRs, and the y-axis indicates the number of repeat fragments. The gray legend represents mononucleotide SSRs, the purple legend represents dinucleotide SSRs, the yellow legend represents trinucleotide SSRs, the red legend represents tetranucleotide SSRs, and the blue legend represents pentanucleotide SSRs. No hexanucleotide SSRs were detected in this mitochondrial genome. (B) The x-axis represents the types of repeat sequences, and the y-axis indicates the number of repeat fragments. The yellow legend represents tandem repeats, the green legend represents palindromic repeats, the blue legend represents forward repeats, and the red legend represents complementary repeats. No reverse repeats were detected in this mitochondrial genome.
Additionally, 417 dispersed repeats longer than 30 bp were detected, comprising 205 palindromic repeats, 211 forward repeats, and one complementary repeat, with no reverse repeats observed. The longest palindromic repeat extended 12,060 bp, while the longest forward repeat measured 521 bp. These repeat sequences were distributed across the mitochondrial genome, primarily occurring within intergenic regions, which is consistent with findings in other plant mitochondrial genomes (Fukasawa et al., 2024; Zhong et al., 2022; Wang et al., 2024).
3.4 Sequence transfer analysis of mitochondrial genome structure in Ficus hirta
Based on sequence similarity analysis, a total of 15 segments were identified as homologous between the mitochondrial and chloroplast genomes, with a combined length of 4,979 bp, accounting for 1.02% of the mitochondrial genome. The longest segment, MTPT7, spans 1,266 bp. Annotation of these homologous sequences revealed three complete genes, all of which are tRNA genes (trnD-GUC, trnI-CAU, trnM-CAU), located within the 15 homologous segments (Figure 4; Supplementary Table S6 and Supplementary Data Sheet 2).
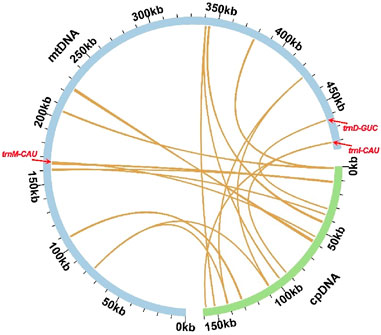
Figure 4. Sequence transfer analysis. Blue arcs represent the mitochondrial genome, while green arcs represent the chloroplast genome. The yellow lines between the arcs correspond to homologous genome segments. The homologous fragments containing complete tRNA genes (trnD-GUC, trnI-CAU, and trnM-CAU) were distinctly marked.
3.5 phylogenetic analysis of mitochondrial genome structure in Ficus hirta
Phylogenetic analysis was conducted based on the DNA sequences of 23 conserved mitochondrial protein-coding genes (PCGs) (Figure 5). A phylogenetic tree was constructed for 31 species across five families of angiosperms. The shared protein-coding genes include atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFC, ccmFN, cob, cox1, cox2, cox3, matR, mttB, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9. Two mitochondrial genomes from the Fabaceae family were used as outgroups. The phylogenetic topology based on mitochondrial DNA aligns with the most recent classification by the Angiosperm Phylogeny Group (APG) (Tap et al., 2016). Species F. hirta belongs to the order Rosales, family Moraceae, and is closely related to Morus notabilis (Chinese mulberry).
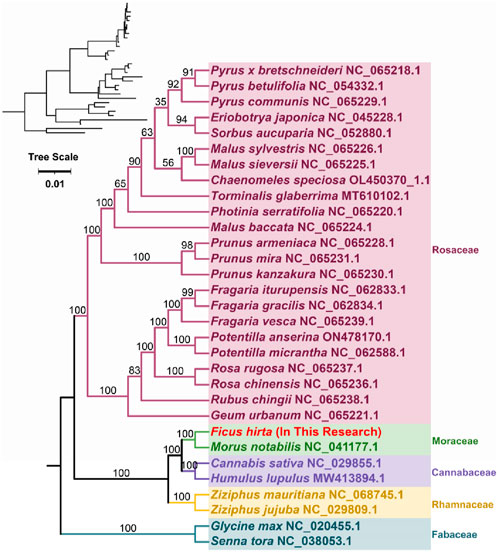
Figure 5. Phylogenetic analysis was conducted based on the DNA sequences of 23 conserved mitochondrial protein-coding genes (PCGs). The tree scale represents the number of nucleotide substitutions per site. The bootstrap values are indicated at the nodes.
3.6 Analysis of RNA editing events of mitochondrial genome structure in Ficus hirta
RNA editing events were identified for the 31 unique PCGs from the F. hirta mitochondrial genome (Figure 6). The cutoff value was set to 0.9. Under this criterion, a total of 419 potential RNA editing sites were computationally identified across the 31 mitochondrial PCGs, all of which involved C-to-U base editing. Among the mitochondrial genes, the nad4 gene had the highest number of RNA editing sites, with 47 identified edits. The second highest was the nad7 gene, with 36 RNA editing events. According to the annotation results, there are a total of three RNA editing events related to translation, including the start codon ACG of nad1 and the stop codons CAA of atp9 and CGA of ccmFC, which are likely generated by RNA editing. Through C-to-U editing, these codons are converted into the start codon AUG and the stop codons UAA/UGA, respectively (Figure 7; Supplementary Table S7).
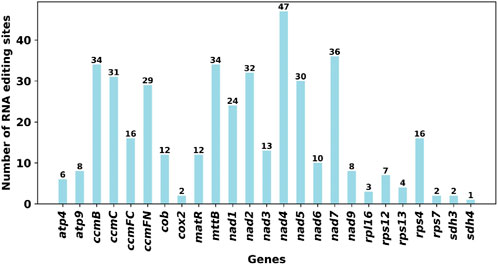
Figure 6. The Predicted Number of RNA Editing Sites in Mitochondrial PCGs, which focused on predicted C-to-U RNA editing sites.
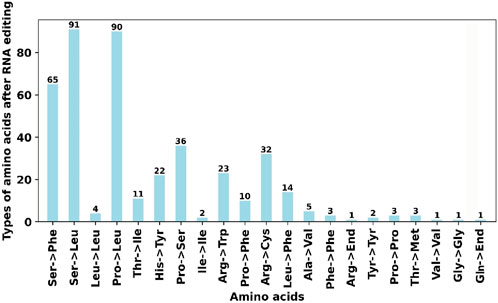
Figure 7. Types of amino acids after RNA editing.
3.7 Synteny analysis of mitochondrial genome structure in Ficus hirta
As shown in Figure 8, the red arc areas represent regions of inversion, while the gray areas indicate regions with high homology. To better present the results, collinear blocks shorter than 0.5 kb were not retained in the analysis. Homologous collinear blocks were detected between F. hirta species and closely related species within the Rosales order, but these blocks were relatively short. Additionally, some blank regions were identified, representing sequences unique to the F. hirta species with no homology to other species. The results indicate that the arrangement of collinear blocks among the mitochondrial genomes of these six species is inconsistent, and the mitochondrial genome of F. hirta species has undergone extensive genome rearrangement compared to closely related species. The mitochondrial genome sequences of the six Rosales species are highly non-conserved in their arrangement, having experienced extremely frequent genome recombination.
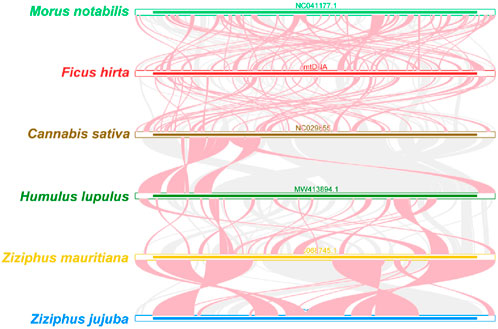
Figure 8. Synteny Analysis of Mitochondrial Genome Structure in Ficus hirta. The red arc areas represent regions of inversion, while the gray areas indicate regions with high homology.
4 Discussion
The assembly of the F. hirta mitochondrial genome provides valuable insights into its structure, gene content, and evolutionary conservation. The genome is organized as a circular molecule spanning 486,226 bp, with a GC content of 45.21%. The annotation identified 31 protein-coding genes, 19 tRNA genes, and three rRNA genes, all of which are highly conserved. Furthermore, annotation with A. thaliana and Liriodendron tulipifera confirms that F. hirta retains the core mitochondrial gene set, with no novel protein-coding genes identified.
One of the most intriguing findings is the presence of 15 homologous fragments shared between the mitochondrial and chloroplast genomes, suggesting significant interorganellar gene transfer events. These findings align with previous research in plants, where intracellular gene transfer between the chloroplast and mitochondrial genomes is a common phenomenon (Nguyen et al., 2020; Gui et al., 2016). The transfer of tRNA genes between organelles could be an adaptive mechanism that enhances genome plasticity. The study by Cui et al. (2021) supports the notion that chloroplast-to-mitochondrial transfers play a role in mitochondrial genome expansion. In species such as watermelon and melon, chloroplast-derived sequences account for 7.6% and 2.73% of the total mitochondrial genome, respectively. These findings highlight that mitochondria have a greater capacity to integrate foreign sequences, likely due to their more flexible DNA recombination mechanisms. Our findings in F. hirta are consistent with this trend, where small chloroplast-derived fragments were identified within the mitochondrial genome (Cui et al., 2021; Gao et al., 2020), supporting the hypothesis that such transfers contribute to the structural evolution of mitochondria (Cui et al., 2021).
The comparative phylogenetic analysis confirmed the close evolutionary relationship between F. hirta and Morus notabilis. However, notable differences in mitochondrial genome organization were observed between F. hirta and other members of the Rosales order, indicating frequent genome rearrangements within the group. Such rearrangements could be driven by the large repeat sequences identified in the genome, which may mediate recombination events.
Our codon usage bias analysis highlighted a preference for A/T-ending codons at the third position, a pattern commonly observed in plant mitochondrial genomes (Bi et al., 2020; Shidhi et al., 2021). From an evolutionary perspective, this bias is likely shaped by a combination of mutation pressure and natural selection (Ji et al., 2024; Cao et al., 2023). In terms of gene expression regulation, biased codon usage may impact mRNA stability and translation efficiency (Hia et al., 2019).
RNA editing in plant mitochondria is predominantly C-to-U conversions, which play a critical role in restoring conserved amino acid sequences that are functionally essential for mitochondrial proteins. This process helps correct mutations at the transcript level, ensuring proper protein functionality without requiring extensive changes in the DNA sequence (Ichinose and Sugita, 2016). The identification of 419 RNA editing sites, primarily involving C-to-U conversions, further underscores the complexity of gene expression in plant mitochondria. Such extensive RNA editing, especially in essential genes like nad4 and nad7, may be crucial for ensuring proper protein function. Our findings are based on predictions using PREP-Mt (v0.9), which is designed to detect C-to-U conversions, the most common form of RNA editing in plant mitochondria. Future research could leverage RNA-seq data to experimentally validate these predictions and explore the functional consequences of RNA editing.
5 Conclusion
In conclusion, the complete sequencing and annotation of the F. hirta mitochondrial genome provide valuable genomic resources that enhance our understanding of its biology and evolution. The intergenomic transfers, frequent genome rearrangements, and RNA editing events observed in F. hirta contribute to a broader understanding of plant mitochondrial genome dynamics. Future studies should focus on functional analyses of the transferred genes and their roles in mitochondrial function and adaptation.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI’s BankIt, under the following accession numbers: mitochondrial genome data 2944861, and the chloroplast genome 2945015.
Author contributions
WD: Formal analysis, Funding acquisition, Methodology, Software, Writing – original draft. XC: Conceptualization, Supervision, Validation, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study is supported by Key Medical Discipline in Longgang District, Research personnel training program of Longgang Central Hospital, Longgang Medical Discipline Construction Fund, and Key Medical Discipline Construction Site in Longgang District.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1530105/full#supplementary-material
SUPPLEMENTARY TABLE S1 | Annotated genes list in the mitogenome of Ficus hirta.
SUPPLEMENTARY TABLE S2 | RSCU of each amino acid in the mitochondrial genome of Ficus hirta.
SUPPLEMENTARY TABLE S3 | Tandem repeat sequences in the mitochondrial genome of Ficus hirta.
SUPPLEMENTARY TABLE S4 | SSRs in the mitochondrial genome of Ficus hirta.
SUPPLEMENTARY TABLE S5 | Dispersed repeat sequences in the mitochondrial genome of Ficus hirta.
SUPPLEMENTARY TABLE S6 | The homologous DNA fragment in the Ficus hirta mitochondrial genome.
SUPPLEMENTARY TABLE S7 | RNA editing.
References
Bi, C., Lu, N., Xu, Y., and Lu, Z. (2020). Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 21 (11), 3778. doi:10.3390/ijms21113778
Cao, J.-K., Lei, T., Gu, J.-J., Song, C., and Qi, X. (2023). Codon bias analysis of the mitochondrial genome reveals natural selection in the nonbiting midge Microtendipes umbrosus Freeman, 1955 (Diptera: chironomidae). 1955 (Diptera Chironomidae) 99, 217–225. doi:10.3956/2023-99.4.217
Chen, C., Peng, X., Chen, J., and Wan, C. (2020). Antioxidant, antifungal activities of ethnobotanical Ficus hirta vahl. And analysis of main constituents by HPLC-MS. Biomedicines 8 (1), 15. doi:10.3390/biomedicines8010015
Cheng, J., Yi, X., Wang, Y., Huang, X., and He, X. (2017). Phenolics from the roots of hairy fig (Ficus hirta Vahl.) exert prominent anti-inflammatory activity. J. Funct. Foods 31, 79–88. doi:10.1016/j.jff.2017.01.035
Cui, H., Ding, Z., Zhu, Q., Wu, Y., Qiu, B., and Gao, P. (2021). Comparative analysis of nuclear, chloroplast, and mitochondrial genomes of watermelon and melon provides evidence of gene transfer. Sci. Rep. 11, 1595. doi:10.1038/s41598-020-80149-9
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem. Bullet. 19, 11–15.
Fukasawa, Y., Driguez, P., Bougouffa, S., Carty, K., Putra, A., Cheung, M. S., et al. (2024). Plasticity of repetitive sequences demonstrated by the complete mitochondrial genome of Eucalyptus camaldulensis. Front. Plant Sci. 15, 1339594-. doi:10.3389/fpls.2024.1339594
Gao, C., Wu, C., Zhang, Q., Zhao, X., Wu, M., Chen, R., et al. (2020). Characterization of chloroplast genomes from two salvia medicinal plants and gene transfer among their mitochondrial and chloroplast genomes. Front. Genet. 11, 574962-. doi:10.3389/fgene.2020.574962
Gui, S., Wu, Z., Zhang, H., Zheng, Y., Zhu, Z., Liang, D., et al. (2016). The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci. Rep. 6, 30158. doi:10.1038/srep30158
Hia, F., Yang, S. F., Shichino, Y., Yoshinaga, M., Murakawa, Y., Vandenbon, A., et al. (2019). Codon bias confers stability to human mRNAs. EMBO Rep. 20 (11), e48220. doi:10.15252/embr.201948220
Ichinose, M., and Sugita, M. (2016). RNA editing and its molecular mechanism in plant organelles. Genes 8 (1), 5. doi:10.3390/genes8010005
Ji, H., Liu, J., Chen, Y., Yu, X., Luo, C., Sang, L., et al. (2024). Bioinformatic analysis of codon usage bias of HSP20 genes in four cruciferous species. Plants (Basel, Switz.) 13 (4), 468. doi:10.3390/plants13040468
Jin, J.-J., Yu, W.-B., Yang, J.-B., Song, Y., dePamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21 (1), 241. doi:10.1186/s13059-020-02154-5
Liu, S., Ni, Y., Li, J., Zhang, X., Yang, H., Chen, H., et al. (2023). CPGView: a package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 23 (3), 694–704. doi:10.1111/1755-0998.13729
Liu, Y., Chen, W., Li, F., Li, C., Xie, X., Chao, Z., et al. (2019). The complete chloroplast genome sequence of Ficus hirta (Moraceae). Resources 4 (2), 4041–4042. doi:10.1080/23802359.2019.1689867
Lu, Y., Chen, J., Chen, B., Liu, Q., Zhang, H., Yang, L., et al. (2022). High genetic diversity and low population differentiation of a medical plant Ficus hirta Vahl., uncovered by microsatellite loci: implications for conservation and breeding. BMC plant Biol. 22 (1), 334. doi:10.1186/s12870-022-03734-2
Mower, J. P., Stefanović, S., Hao, W., Gummow, J. S., Jain, K., Ahmed, D., et al. (2010). Horizontal acquisition of multiple mitochondrial genes from a parasitic plant followed by gene conversion with host mitochondrial genes. BMC Biol. 8, 150. doi:10.1186/1741-7007-8-150
Nguyen, V. B., Linh Giang, V. N., Waminal, N. E., Park, H. S., Kim, N. H., Jang, W., et al. (2020). Comprehensive comparative analysis of chloroplast genomes from seven Panax species and development of an authentication system based on species-unique single nucleotide polymorphism markers. J. ginseng Res. 44 (1), 135–144. doi:10.1016/j.jgr.2018.06.003
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47 (W1), W65–W73. doi:10.1093/nar/gkz345
Shidhi, P. R., Biju, V. C., Anu, S., Vipin, C. L., Deelip, K. R., and Achuthsankar, S. N. (2021). Genome characterization, comparison and phylogenetic analysis of complete mitochondrial genome of evolvulus alsinoides reveals highly rearranged gene order in solanales. Life (Basel, Switz.) 11 (8), 769. doi:10.3390/life11080769
Sun, Z. H., Liang, F. L., Chen, Y. C., Liu, H. X., and Zhang, W. M. (2016). Two new xyloketals from the endophytic fungus Endomelanconiopsis endophytica derived from medicinal plant Ficus hirta. J. Asian Nat. Prod. Res. 18 (11), 1036–1041. doi:10.1080/10286020.2016.1188084
The Angiosperm Phylogeny Group Chase, M. W., Christenhusz, M. J. M., Fay, M. F., Byng, J. W., Judd, W. S., et al. (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical J. Linn. Soc. 181 (1), 1–20. doi:10.1111/boj.12385
Timmis, J. N., Ayliffe, M. A., Huang, C. Y., and Martin, W. (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5 (2), 123–135. doi:10.1038/nrg1271
Wang, R., Luo, Y., Lan, Z., and Qiu, D. (2024). Insights into structure, codon usage, repeats, and RNA editing of the complete mitochondrial genome of Perilla frutescens (Lamiaceae). Sci. Rep. 14 (1), 13940. doi:10.1038/s41598-024-64509-3
Wilfinger, W. W., Mackey, K., and Chomczynski, P. J. B. (1997). Effect of pH and ionic strength on the spectrophotometric assessment of nucleic acid purity. Biotechniques 22 (3), 474–481. doi:10.2144/97223st01
Wu, W. K., Chen, F. H., Yan, Q. R., and Song, W. (2013). Study on folk medicine used by Hakka in Meizhou. Zhongguo Zhong Yao Za Zhi 38 (22), 3984–3987.
Yu, H., Nason, J. D., Ge, X., and Zeng, J. (2010). Slatkin's Paradox: when direct observation and realized gene flow disagree. A case study in Ficus. Mol. Ecol. 19 (20), 4441–4453. doi:10.1111/j.1365-294X.2010.04777.x
Yu, H., Zhao, N., Chen, Y., and Edward Allen, H. (2008). Male and female reproductive success in the dioecious fig, Ficus hirta Vahl. in Guangdong Province, China: implications for the relative stability of dioecy and monoecy. Symbiosis 45, 121–127.
Zhang, Y., Shen, Z., Meng, X., Zhang, L., Liu, Z., Liu, M., et al. (2022). Codon usage patterns across seven Rosales species. BMC Plant Biol. 22 (1), 65. doi:10.1186/s12870-022-03450-x
Keywords: mitochondrial genomes, Ficus hirta, plant genomics, ecosystem, comparative analysis
Citation: Deng W and Cai X (2025) Complete mitochondrial genome of Ficus hirta and its comparative analysis. Front. Genet. 16:1530105. doi: 10.3389/fgene.2025.1530105
Received: 18 November 2024; Accepted: 17 March 2025;
Published: 23 April 2025.
Edited by:
Juli Jing, Cornell University, United StatesReviewed by:
Qiuyue Chen, University of Wisconsin-Madison, United StatesMeng Zhang, Max Planck Institute for Plant Breeding Research, Germany
Copyright © 2025 Deng and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinyi Cai, MTZ4eWNhaUBhbHVtbmkuc3R1LmVkdS5jbg==