 Hua He
Hua He Lijuan Long2
Lijuan Long2 Manling Tang
Manling Tang Jia Chen
Jia Chen- 1Laboratory Medicine Center, Zhuzhou Hospital Affiliated to Xiangya School of Medicine, Central South University, Zhuzhou, China
- 2Department of Critical Care Medicine, Zhuzhou Hospital Affiliated to Xiangya School of Medicine, Central South University, Zhuzhou, China
- 3Reproductive Medicine Center, Jiangxi Maternal and Child Health Hospital, Nanchang, China
- 4Department of Radiology, Zhuzhou Hospital Affiliated to Xiangya School of Medicine, Central South University, Zhuzhou, China
- 5Central Laboratory, Jiangxi Maternal and Child Health Hospital, Nanchang, China
Introduction: Biallelic loss-of-function variants in the SLC13A5 (solute carrier family 13, member 5) gene are responsible for autosomal recessive developmental and epileptic encephalopathy 25 with amelogenesis imperfecta (DEE25). Until now, no pathogenic variants of SLC13A5 has been reported among the Chinese population.
Methods: A Chinese Han pediatric patient with epilepsy and global developmental delay was described in this study. Trio-whole exome sequencing (WES) including the patient and her parents was performed to determine the genetic basis of the phenotype. Potential pathogenic variants were subsequently confirmed by Sanger sequencing. Additionally, we conducted an extensive review of the literature regarding SLC13A5 variants to analyze their associated phenotypic characteristics.
Results: Trio-WES revealed a novel homozygous variant c.1705T>G in SLC13A5 associated with clinical manifestations in the proband. The variant was also detected in her parents and unaffected sister, who were both heterozygous carriers. The variant is a nonstop substitution that is predicted to extend the SLC13A5 protein by 174 amino acids (p.569Gluext174). Analysis of previously published cases indicated that SLC13A5 patient in our study exhibited overlapping symptoms.
Discussion: We identified a novel homozygous nonstop mutation in the SLC13A5 gene of a Chinese patient with DEE25. This study expands the mutation spectrum of SLC13A5 and will have significant implications for the proband’s family in terms of medical management and genetic counseling.
1 Introduction
Developmental and epileptic encephalopathies (DEEs) are a group of clinically and genetically heterogeneous neurological disorders characterized by frequent epileptic activity and developmental impairment (Guerrini et al., 2023). DEEs represent the most severe forms of epilepsy, typically manifesting in neonates or children (Scheffer et al., 2021). Genetic factors play a significant role in the development of DEEs. To date, over 100 genes have been associated with DEEs due to rapid advances in diagnostic tools, significantly enhancing our understanding of their molecular basis (McTague et al., 2016; Guerrini et al., 2023).
Autosomal recessive variants of the SLC13A5 gene (MIM*608305) have been reported to cause developmental and epileptic encephalopathy 25 with amelogenesis imperfecta (DEE25, MIM #615905) in early infancy (Matricardi et al., 2020; Milosavljevic et al., 2022). The SLC13A5 gene is located on chromosome 17p13.1 and consists of 12 exons (Inoue et al., 2002a). It encodes a plasma membrane sodium-dependent citrate carrier known as the Na + -coupled citrate (NaCT) transporter, which is highly expressed in the brain, teeth, liver, and testis (Inoue et al., 2002b). Neurons are incapable of de novo synthesis of tricarboxylic acid cycle intermediates, therefore, the uptake of intermediates like citrate is crucial for maintaining energy status and neurotransmitter production (Hardies et al., 2015; Henke et al., 2020). Biallelic mutations, whether homozygous or compound heterozygous, in the SLC13A5 gene can impair citrate uptake, disrupting tricarboxylic acid cycle metabolism and resulting in neonatal epilepsy (Goodspeed et al., 2022). SLC13A5 was first identified as a causative gene of DEE25 in 2014 (Thevenon et al., 2014). To date, over 40 pathogenic variants have been reported in the SLC13A5 gene (Brown et al., 2021; Beltran, A.S., 2024). However, none of these variants have been reported in the Chinese population.
In our study, we identified a novel nonstop mutation in the SLC13A5 gene in a Chinese family affected by infant epilepsy and developmental delay. The patient exhibited a range of clinical features, including dental anomalies, intellectual disability, motor difficulties, and speech impairments, alongside abnormalities detected through neuroimaging. We conducted a thorough literature review on SLC13A5 variants to analyze their associated phenotypic characteristics.
2 Materials and methods
2.1 Whole-exome sequencing and analysis
Ethical approval for this study was granted by the Ethics Committee of Jiangxi Maternal and Child Health Hospital. Written informed consent was obtained from the parents of the patient. Genomic DNA was extracted from peripheral blood samples using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The main steps of whole exome sequencing (WES) analysis were performed as previously described (Guo et al., 2018; Chen et al., 2020). Exomes were captured using the SureSelect XT Human All Exon V6 kit (Agilent Technologies, Santa Clara, CA, United States) and sequenced on an Illumina HiSeq sequencer (Illumina, San Diego, CA, United States). After quality control assessment with FastQC, reads were mapped to the UCSC RefSeq database hg19 human reference genome using BWA to remove duplications. Variant calling and annotation were conducted using GATK and ANNOVAR. Candidate mutations identified by WES were confirmed by Sanger sequencing. The pathogenicity of these candidate variants was assessed following the 2015 ACMG/AMP classification guidelines (Richards et al., 2015).
2.2 Protein predictions
The protein sequence of human SLC13A5 was retrieved from the UniProt database (https://www.uniprot.org/), and AlphaFold2, an online protein structure prediction tool, was utilized for modeling SLC13A5 structure. All structural figures were generated using PyMol.
3 Results
3.1 Clinical features of the patient
The proband was a 5-year-old girl born to physically healthy via cesarean section at term. The parents were non-consanguineous and from different cities in Jiangxi province. The proband experienced a multifocal status epilepticus characterized by long-lasting focal seizures the day after birth. During her first year, seizures were occurred approximately every 3–4 months. Following treatment with antiseizure medications valproic acid and levetiracetam, seizure frequency significantly decreased. She exhibited profound intellectual disability and severe motor impairment, presenting with autistic-like behavior characterized by lack of eye contact, inability to speak, and disinterest in surroundings. She required support to stand or sit due to severe muscle tone, particularly in both upper limbs (Figure 1A). Dental hypoplasia and amelogenesis imperfecta were noted (Figure 1B). Brain MRI revealed demyelination in the white matter (Figure 1C), enlargement of the temporal horn of the lateral ventricle, and atrophy of the hippocampus and parahippocampal gyrus (Figures 1D, E). The patient had a healthy 12-year-old sister and another sister who exhibited similar symptoms and died from seizures at 3 months of age, neither had undergone genetic testing.
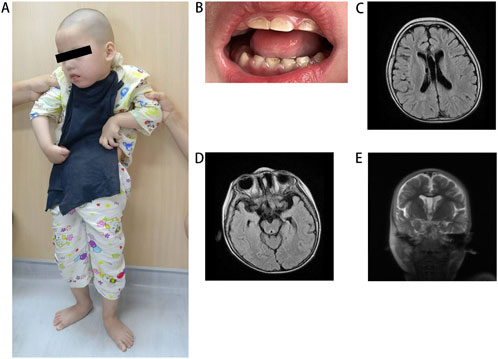
Figure 1. Clinical features of the patient. (A) The patient at the standing position. (B) Illustrative picture of teeth hypodontia and amelogenesis imperfecta in patient. (C) MRI shows white matter demyelination, (D) enlargement of the temporal horn of the lateral ventricle and (D, E) atrophy of the hippocampus and parahippocampal gyrus. MRI, magnetic resonance imaging.
3.2 Identification of a nonstop SLC13A5 variant
Whole-exome sequencing analyses were conducted on the patient and her parents to identify the genetic basis of her condition. A homozygous variant (c.1705T>G) in the SLC13A5 gene (NM_177,550.5) was identified as contributing to the patient’s symptoms. Sanger sequencing confirmed the homozygous mutation in the patient and revealed that her parents and unaffected sister were heterozygous carriers of the c.1705T>G (p.569Gluext174) mutation (Figure 2A, B). This variant has not been previously reported in the literature and is absent from the gnomAD, HGMD, and ClinVar databases, suggesting it is a novel causative variation for DEE25. According to the 2015 ACMG guidelines [15], the pathogenicity of the c.1705T>G variant was classified based on criteria PM2_Supporting + PM4_Very strong, indicating it is likely pathogenic. No additional variants in genes associated with epilepsy and/or dental abnormalities were identified in the proband.
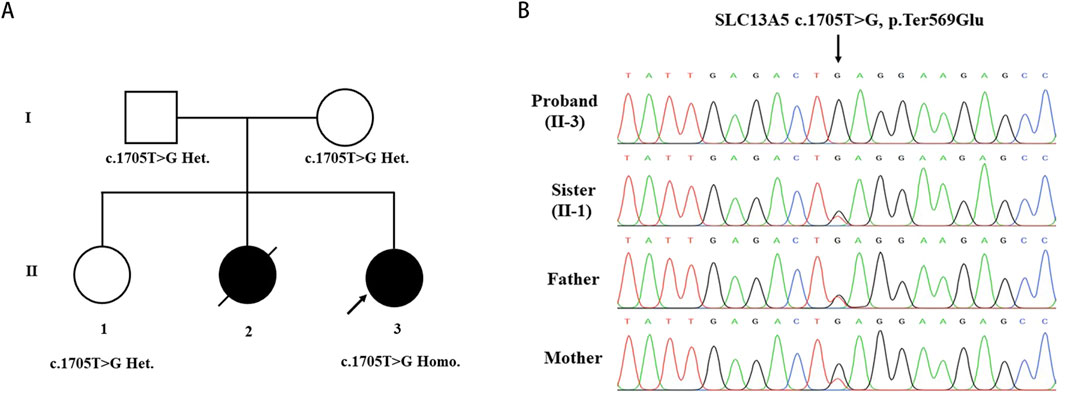
Figure 2. Identification of the SLC13A5 Variant. (A) Pedigree of the investigated family. (B) Sanger sequencing show homozygous mutation in the proband(II-3), heterozygous mutation in sister (II-1) and heterozygous mutation in parents. Arrow indicated the nonstop mutation changing the TAG stop codon in GAG.
3.3 Structural modelling of the SLC13A5 Variant
Sequence analysis revealed that the c.1705T>G variant identified in this study changes the TAG stop codon of the SLC13A5 gene to a glutamic acid-encoding GAG codon. Downstream at position c.*520_522, there is another TGA stop codon, resulting in the expression of a chimeric protein composed of the regular 568 SLC13A5 residues and an additional 174-residue C-terminal extension (p.569Gluext174) (Figure 3A). Structural modeling indicated that the substitution of the stop codon to glutamic acid created an α-helical domain and random coils that were absent in the wild-type protein (Figure 3B).
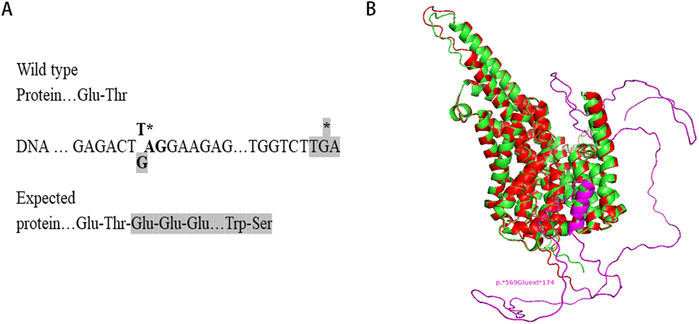
Figure 3. Structural modelling of the mutant SLC13A5. (A) Protein expressed from the mutant allele is C-terminally extended. (B) Structural modeling of SLC13A5 protein by AlphaFold2. The p. Ter569Glu Variant, wild type, and extended portion are shown in red, green, and purple, respectively. Models are visualized in PyMOL.
4 Discussion
Biallelic loss-of-function mutations in the SLC13A5 gene typically lead to DEE25, characterized by neurological manifestations such as epilepsy and psychomotor developmental delay (Alsemari et al., 2024). In this study, we identified a novel SLC13A5 nonstop variant (c.1705T>G) in a Chinese girl who began experiencing seizures shortly after birth. The phenotypic spectrum observed in our patient overlaps with previously reported cases, including early onset seizures, profound intellectual disability, global developmental delay, and dental abnormalities (Supplementary Table 1). Additionally, our patient exhibited auxiliary features such as brain MRI abnormalities and hypertonia. According to the literature, neuroimaging abnormalities are present in approximately one-third of SLC13A5-related DEE cases, with white matter abnormalities being the most common (Whitney et al., 2023). The affected patients may initially present with infantile hypotonia and later develop increased muscle tone, pyramidal signs, spasticity, dystonia, and/or ataxia. This range of symptoms reflects the clinical heterogeneity of the disease (Arvio and Lähdetie, 2020).
From the literature review (Supplementary Table 1), most SLC13A5 variants are missense mutations, though a smaller number of frameshift, splicing, insertion, and intronic mutations have also been reported. These variants are typically either homozygous or compound heterozygous and follow an autosomal recessive inheritance pattern. Patients with these mutations exhibit developmental impairments of varying severity, often accompanied by a range of neurological signs and symptoms, including axial hypotonia, peripheral hypertonia, dystonia, dyskinesia, spasticity, ataxia, choreoathetosis, and microcephaly. Current clinical and genetic data do not show consistent genotype-phenotype correlations (Matricardi et al., 2020). Even within families with more than one affected child, there is significant phenotype heterogeneity among siblings with identical genotypes (Anselm et al., 2017; Arvio and Lähdetie, 2020; Matricardi et al., 2020).
The human SLC13A5 protein consists of 568 amino acids and is located at the plasma membrane, where it mediates the transport of citrate from extracellular environment into the cell (Inoue et al., 2002a). Citrate is crucial for maintaining cellular metabolic homeostasis, particularly in energy production and neurotransmitter synthesis within the central nervous system (CNS) (Li and Wang, 2021). Secondary structure predictions indicate that SLC13A5 is predominantly α-helical, comprising approximately 65.3% α-helices, including 11 transmembrane α-helices, with 33% in loops and less than 2% in β-strands (Jaramillo-Martinez et al., 2021). The protein forms a homodimer, with each protomer containing a scaffold domain and a transport domain (Sauer et al., 2021). Mutations in SLC13A5 can disrupt citrate transport directly or affect protein expression and localization, depending on the position of the mutated residue (Jaramillo-Martinez et al., 2021; Beltran, 2024). Our case study presents a novel nonstop mutation that results in a C-terminally extended protein with an additional 174 residues (p.569Gluext174). Structural modeling revealed that this mutation introduces an α-helical domain and random coils not present in the wild-type protein. Functional research, such as citrate uptake assays, is needed to elucidate the underlying mechanisms of the mutation’s defects in SLC13A5-related epilepsy.
Nonstop variants, which convert stop codons into sense codons, are exceedingly rare mutations reported in various diseases (Hamby et al., 2011; Vaché et al., 2023), such as autosomal dominant Charcot-Marie-Tooth disease type 2 (Bock et al., 2018) and excessive hemorrhaging due to mutations in coagulation factor X (Ameri et al., 2007). When an alternative in-frame stop codon exists in the 3′untranslated region (UTR), translation can continue normally, resulting in the production of a C-terminally extended protein. The pathogenicity of these extra polypeptides has been investigated in a few cases. Studies have shown that these mutations can lead to altered subcellular localization in some instances (Sun et al., 2017) and accumulation of mutant proteins in others (Bock et al., 2018). These findings suggest that C-terminally extended proteins can confer a toxic gain-of-function, which contrasts with the loss-of-function effects typically seen in nonstop decay pathways. Further research is needed to fully understand the genetic mechanisms underlying C-terminal elongation in SLC13A5 and its implications for disease pathogenesis.
In summary, we identified the first SLC13A5 mutation (c.1705T>G, p.569Gluext174) in a Chinese population. This nonstop substitution is rare and broadens the mutational spectrum of the SLC13A5 gene. The discovery of this pathogenic variant will have significant implications for the proband’s family in terms of medical management and genetic counseling.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://figshare.com/, https://doi.org/10.6084/m9.figshare.28795916.v1.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Jiangxi Maternal and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
HH: Data curation, Funding acquisition, Writing – original draft, Writing – review and editing. LL: Investigation, Methodology, Writing – review and editing. MT: Investigation, Methodology, Writing – review and editing. QX: Formal Analysis, Resources, Writing – review and editing. SD: Formal Analysis, Resources, Writing – review and editing. GC: Formal Analysis, Resources, Writing – review and editing. YZ: Data curation, Validation, Writing – review and editing. QW: Data curation, Validation, Writing – review and editing. JC: Conceptualization, Funding acquisition, Writing – review and editing, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the grants from the National Natural Science Foundation of China (82160317), Natural Science Foundation of Hunan Province (2024JJ9557 and 2024JJ7648), and Jiangxi Provincial Natural Science Foundation (20224BAB206018 and 20232BAB216097).
Acknowledgments
We thank the patient and relatives for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1474390/full#supplementary-material
References
Alsemari, A., Guzmán-Vega, F. J., Meyer, B. F., and Arold, S. T. (2024). Novel homozygous variants of SLC13A5 expand the functional heterogeneity of a Homogeneous syndrome of early infantile epileptic encephalopathy. Pediatr. Neurol. 151, 68–72. doi:10.1016/j.pediatrneurol.2023.10.005
Ameri, A., Machiah, D. K., Tran, T. T., Channell, C., Crenshaw, V., Fernstrom, K., et al. (2007). A nonstop mutation in the factor (F)X gene of a severely haemorrhagic patient with complete absence of coagulation FX. Thromb. Haemost. 98 (6), 1165–1169. doi:10.1160/th07-02-0125
Anselm, I., MacCuaig, M., Prabhu, S. B., and Berry, G. T. (2017). Disease heterogeneity in Na(+)/Citrate Cotransporter Deficiency. JIMD Rep. 31, 107–111. doi:10.1007/8904_2016_577
Arvio, M., and Lähdetie, J. (2020). Adult phenotype of the homozygous missense mutation c.655G>A, p.Gly219Arg in SLC13A5: a case report. Am. J. Med. Genet. A 182 (11), 2671–2674. doi:10.1002/ajmg.a.61802
Beltran, A. S. (2024). Novel Approaches to studying SLC13A5 disease. Metabolites 14 (2), 84. doi:10.3390/metabo14020084
Bock, A. S., Günther, S., Mohr, J., Goldberg, L. V., Jahic, A., Klisch, C., et al. (2018). A nonstop variant in REEP1 causes peripheral neuropathy by unmasking a 3'UTR-encoded, aggregation-inducing motif. Hum. Mutat. 39 (2), 193–196. doi:10.1002/humu.23369
Brown, T. L., Nye, K. L., and Porter, B. E. (2021). Growth and Overall health of patients with SLC13A5 citrate transporter disorder. Metabolites 11 (11), 746. doi:10.3390/metabo11110746
Chen, J., Yuan, H., Xie, K., Wang, X., Tan, L., Zou, Y., et al. (2020). A novel TAB2 nonsense mutation (p.S149X) causing autosomal dominant congenital heart defects: a case report of a Chinese family. BMC Cardiovasc Disord. 20 (1), 27. doi:10.1186/s12872-019-01322-1
Goodspeed, K., Liu, J. S., Nye, K. L., Prasad, S., Sadhu, C., Tavakkoli, F., et al. (2022). SLC13A5 Deficiency disorder: from genetics to gene Therapy. Genes (Basel) 13 (9), 1655. doi:10.3390/genes13091655
Guerrini, R., Conti, V., Mantegazza, M., Balestrini, S., Galanopoulou, A. S., and Benfenati, F. (2023). Developmental and epileptic encephalopathies: from genetic heterogeneity to phenotypic continuum. Physiol. Rev. 103 (1), 433–513. doi:10.1152/physrev.00063.2021
Guo, J. F., Zhang, L., Li, K., Mei, J. P., Xue, J., Chen, J., et al. (2018). Coding mutations in NUS1 contribute to Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 115 (45), 11567–11572. doi:10.1073/pnas.1809969115
Hamby, S. E., Thomas, N. S., Cooper, D. N., and Chuzhanova, N. (2011). A meta-analysis of single base-pair substitutions in translational termination codons ('nonstop' mutations) that cause human inherited disease. Hum. Genomics 5 (4), 241–264. doi:10.1186/1479-7364-5-4-241
Hardies, K., de Kovel, C. G., Weckhuysen, S., Asselbergh, B., Geuens, T., Deconinck, T., et al. (2015). Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 138 (Pt 11), 3238–3250. doi:10.1093/brain/awv263
Henke, C., Töllner, K., van Dijk, R. M., Miljanovic, N., Cordes, T., Twele, F., et al. (2020). Disruption of the sodium-dependent citrate transporter SLC13A5 in mice causes alterations in brain citrate levels and neuronal network excitability in the hippocampus. Neurobiol. Dis. 143, 105018. doi:10.1016/j.nbd.2020.105018
Inoue, K., Zhuang, L., and Ganapathy, V. (2002a). Human Na+ -coupled citrate transporter: primary structure, genomic organization, and transport function. Biochem. Biophys. Res. Commun. 299 (3), 465–471. doi:10.1016/s0006-291x(02)02669-4
Inoue, K., Zhuang, L., Maddox, D. M., Smith, S. B., and Ganapathy, V. (2002b). Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 277 (42), 39469–39476. doi:10.1074/jbc.M207072200
Jaramillo-Martinez, V., Urbatsch, I. L., and Ganapathy, V. (2021). Functional Distinction between human and Mouse sodium-coupled citrate transporters and its Biologic significance: an Attempt for structural basis using a Homology modeling Approach. Chem. Rev. 121 (9), 5359–5377. doi:10.1021/acs.chemrev.0c00529
Li, Z., and Wang, H. (2021). Molecular mechanisms of the SLC13A5 gene Transcription. Metabolites 11 (10), 706. doi:10.3390/metabo11100706
Matricardi, S., De Liso, P., Freri, E., Costa, P., Castellotti, B., Magri, S., et al. (2020). Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia 61 (11), 2474–2485. doi:10.1111/epi.16699
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A., and Scheffer, I. E. (2016). The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 15 (3), 304–316. doi:10.1016/S1474-4422(15)00250-1
Milosavljevic, S., Glinton, K. E., Li, X., Medeiros, C., Gillespie, P., Seavitt, J. R., et al. (2022). Untargeted Metabolomics of Slc13a5 Deficiency reveal Critical liver-brain Axis for Lipid homeostasis. Metabolites 12 (4), 351. doi:10.3390/metabo12040351
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Sauer, D. B., Song, J., Wang, B., Hilton, J. K., Karpowich, N. K., Mindell, J. A., et al. (2021). Structure and inhibition mechanism of the human citrate transporter NaCT. Nature 591 (7848), 157–161. doi:10.1038/s41586-021-03230-x
Scheffer, I. E., Hulihan, J., Messenheimer, J., Ali, S., Keenan, N., Griesser, J., et al. (2021). Safety and Tolerability of Transdermal Cannabidiol Gel in children with developmental and epileptic encephalopathies: a Nonrandomized Controlled trial. JAMA Netw. Open 4 (9), e2123930. doi:10.1001/jamanetworkopen.2021.23930
Sun, J., Hao, Z., Luo, H., He, C., Mei, L., Liu, Y., et al. (2017). Functional analysis of a nonstop mutation in MITF gene identified in a patient with Waardenburg syndrome type 2. J. Hum. Genet. 62 (7), 703–709. doi:10.1038/jhg.2017.30
Thevenon, J., Milh, M., Feillet, F., St-Onge, J., Duffourd, Y., Jugé, C., et al. (2014). Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 95 (1), 113–120. doi:10.1016/j.ajhg.2014.06.006
Vaché, C., Cubedo, N., Mansard, L., Sarniguet, J., Baux, D., Faugère, V., et al. (2023). Identification and in vivo functional investigation of a HOMER2 nonstop variant causing hearing loss. Eur. J. Hum. Genet. 31 (7), 834–840. doi:10.1038/s41431-023-01374-0
Keywords: developmental and epileptic encephalopathy, SLC13A5, whole exome sequencing, homozygous, nonstop mutation
Citation: He H, Long L, Tang M, Xu Q, Duan S, Chen G, Zhao Y, Wu Q and Chen J (2025) Identification of a novel homozygous SLC13A5 nonstop mutation in a Chinese family with epileptic encephalopathy and developmental delay. Front. Genet. 16:1474390. doi: 10.3389/fgene.2025.1474390
Received: 01 August 2024; Accepted: 02 April 2025;
Published: 17 April 2025.
Edited by:
Edoardo Monfrini, University of Milan, ItalyReviewed by:
Antonio Gennaro Nicotera, University of Messina, ItalyVidal Yahya, University of Milan, Italy
Copyright © 2025 He, Long, Tang, Xu, Duan, Chen, Zhao, Wu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia Chen, ZHJlYW1odW50ZXJfY2pAMTYzLmNvbQ==