
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 27 February 2025
Sec. Genetics of Common and Rare Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1472907
 Wenjing Wang1,2,3†
Wenjing Wang1,2,3† Tielun Yin1,2,3†Xinyu Zhang1,2,3
Tielun Yin1,2,3†Xinyu Zhang1,2,3 Zhaoxia Wang4,5
Zhaoxia Wang4,5 Tianyun Wang1,6,7Shuo Zhang1,2,3
Tianyun Wang1,6,7Shuo Zhang1,2,3 Yingshuang Zhang1,2,3
Yingshuang Zhang1,2,3 Dongsheng Fan1,2,3*
Dongsheng Fan1,2,3*Background: Oculopharyngodistal myopathy (OPDM) is a rare hereditary muscle disease characterized by progressive ptosis, ophthalmoplegia, dysphagia, dysarthria, and distal muscle weakness. The genetic basis was identified in 2019 with CGG repeat expansions in the noncoding region of LRP12. Similar expansions in GIPC1, NOTCH2NLC, and RILPL1 were later linked to OPDM, classifying the disease into OPDM1-4. OPDM4, associated with RILPL1, was discovered in 2022 with a few confirmed cases worldwide, leaving its clinical features and pathogenic mechanisms largely unexplored.
Case presentation: We present a patient with OPDM4 who had suffered progressive ptosis, external ophthalmoplegia, pharyngeal weakness, facial muscle weakness, and distal limb weakness over the past 20 years. Electromyography (EMG) revealed myogenic damage and normal H-reflex latency. A biopsy of the left biceps brachii revealed myogenic changes with atypical rimmed vacuoles in some muscle fibers. Screening for extra-muscular system involvement revealed no obvious involvement of the heart or central nervous system. Genetic analysis confirmed 126 CGG repeat expansions in RILPL1 and excluded abnormal CGG repeat expansions in LRP12, GIPC1, and NOTCH2NLC.
Conclusion: This case broadens the spectrum of CGG repeat numbers in the RILPL1 gene associated with OPDM4. In addition, systematic medical examinations revealed several new characteristics of OPDM4, which have not been reported previously, such as normal H reflex, potential mild cognitive impairment, etc. These findings expand our knowledge of the phenotypic spectrum of diseases caused by repeat CGG expansions in RILPL1.
Oculopharyngodistal myopathy (OPDM) is a rare hereditary muscle disease that is clinically defined by progressive symptoms including ptosis, ophthalmoplegia, dysphagia, dysarthria, and distal muscle weakness. Satoyoshi and Kinoshita first described OPDM in four Japanese families in 1977 (Satoyoshi and Kinoshita, 1977). Since then, more than 300 cases of OPDM across diverse ethnicities have been documented. Despite OPDM being a long-standing disease, its genetic basis remained unknown until recently. In 2019, CGG repeat expansions in the noncoding region of low-density lipoprotein receptor-related protein 12 (LRP12) were identified as causative for OPDM (Ishiura et al., 2019). Subsequently, similar expansions in the noncoding regions of GAIP C-terminus-interacting-protein 1 (GIPC1), NOTCH2 N-terminal-like protein C (NOTCH2NLC), and Rab interacting lysosomal protein like 1 (RILPL1) were subsequently linked to the OPDM phenotype (Deng et al., 2020; Ogasawara et al., 2020; Xi et al., 2021; Yu et al., 2021a; An et al., 2022; Yu et al., 2022; Zeng et al., 2022; Yang et al., 2023; YU et al., 2023; Eura et al., 2024; Ma et al., 2024). The OPDM subtypes associated with these genes can be classified into four types: OPDM1 to OPDM4. The four subtypes of OPDM have been reported to exhibit an autosomal dominant mode of inheritance (Ishiura et al., 2019; Deng et al., 2020; Ogasawara et al., 2020; Fukuda et al., 2021; Kumutpongpanich et al., 2021; Xi et al., 2021; Yu et al., 2021a; Yu et al., 2022; Zeng et al., 2022). Even though certain OPDM patients appear sporadic or autosomal recessive, pedigree analysis revealed that such individuals have asymptomatic fathers with extremely long CGG repeat expansions (>200 repeats) (Deng et al., 2020; Kumutpongpanich et al., 2021; Yu et al., 2021a). This phenomenon is likely attributable to a hypermethylation mechanism, which silences the transcription of the host genes in the expanded allele, whereas the normal allele compensates for the gene function, thereby producing an asymptomatic phenotype (Yu et al., 2021a). The causative genes for OPDM are still being discovered, such as ABCD3 and LOC642361/NUTM2B-AS1 (Cortese et al., 2024; Gu et al., 2024; Pongpakdee et al., 2024). Among these genes, the causative gene RILPL1 for OPDM4 was only discovered very recently in 2022 (Yu et al., 2022). Currently, only approximately 20 patients are genetically confirmed to have OPDM4. These OPDM4 cases have been reported only in mainland China (Yu et al., 2022; Zeng et al., 2022; Yang et al., 2023; Eura et al., 2024). Given the rarity of reported OPDM4 cases, our understanding of its clinical features and pathogenic mechanisms remains highly limited. In this case report, we described a patient with a genetically confirmed diagnosis of OPDM4. This report aims to contribute to the limited body of literature on OPDM4, deepening our understanding of the characteristics of OPDM4.
A 50-year-old male was admitted to Peking University Third Hospital on 18 February 2024, due to progressive ptosis, pharyngeal weakness, and facial muscle weakness during the past 20 years. Initially, he experienced eye movement difficulties, accompanied by bilateral exophthalmos and incomplete eyelid closure. Five years ago, he developed voice changes, slow swallowing, and marked drooping of the lower lip (Figure 1; Table 1). He did not experience subjective limb weakness, sensory abnormalities, or other neurological symptoms. He was previously in good health, with no history of hypertension, diabetes, or coronary heart disease. He smoked half a pack of cigarettes and drank one bottle of beer daily for over 20 years but quit them 15 years ago. No other individuals in his family had similar symptoms. Neurological examination revealed dysarthria, external ophthalmoplegia, facial muscle weakness and pharyngeal weakness (Figure 1A). Muscle strength was Grade IV for distal muscles of upper limbs, Grade IV+ for distal muscles of lower limbs, and Grade V for proximal muscles of both limbs. Tendon reflexes were absent with negative pathological signs in both limbs (Table 1).
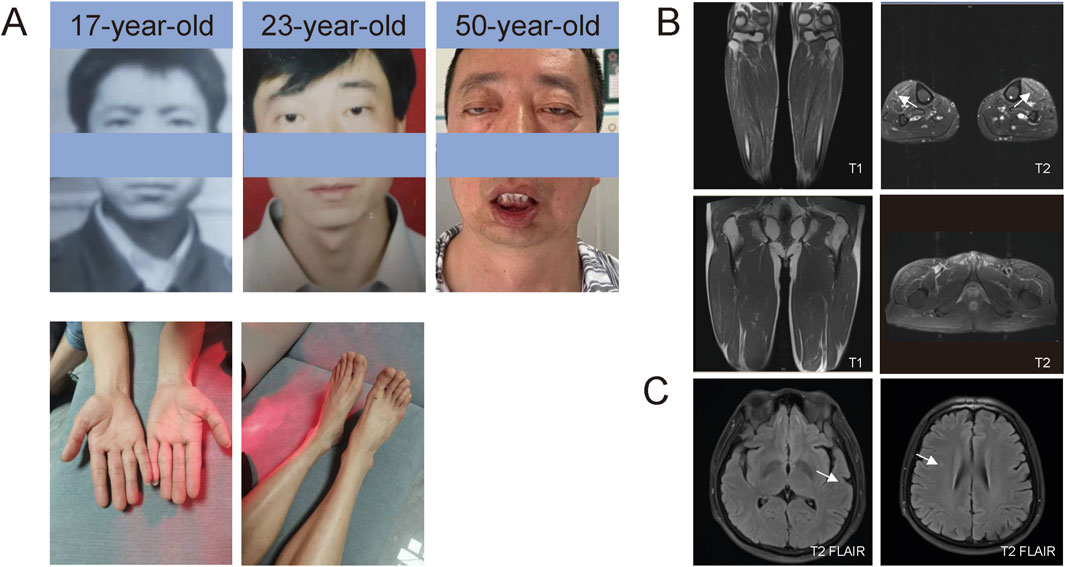
Figure 1. Photos of patients and MRI. (A) Facial photos at different ages and of the distal limbs. (B) T1- and T2-weighted MRI sequences of the legs. Muscle MRI showed T2 hyperintensities, suggesting edema of the lower legs (tibialis anterior). (C) T2/FLAIR sequences of the brain MRI. Brain MRI revealed T2/FLAIR hyperintensities, suggesting mild white matter demyelination.
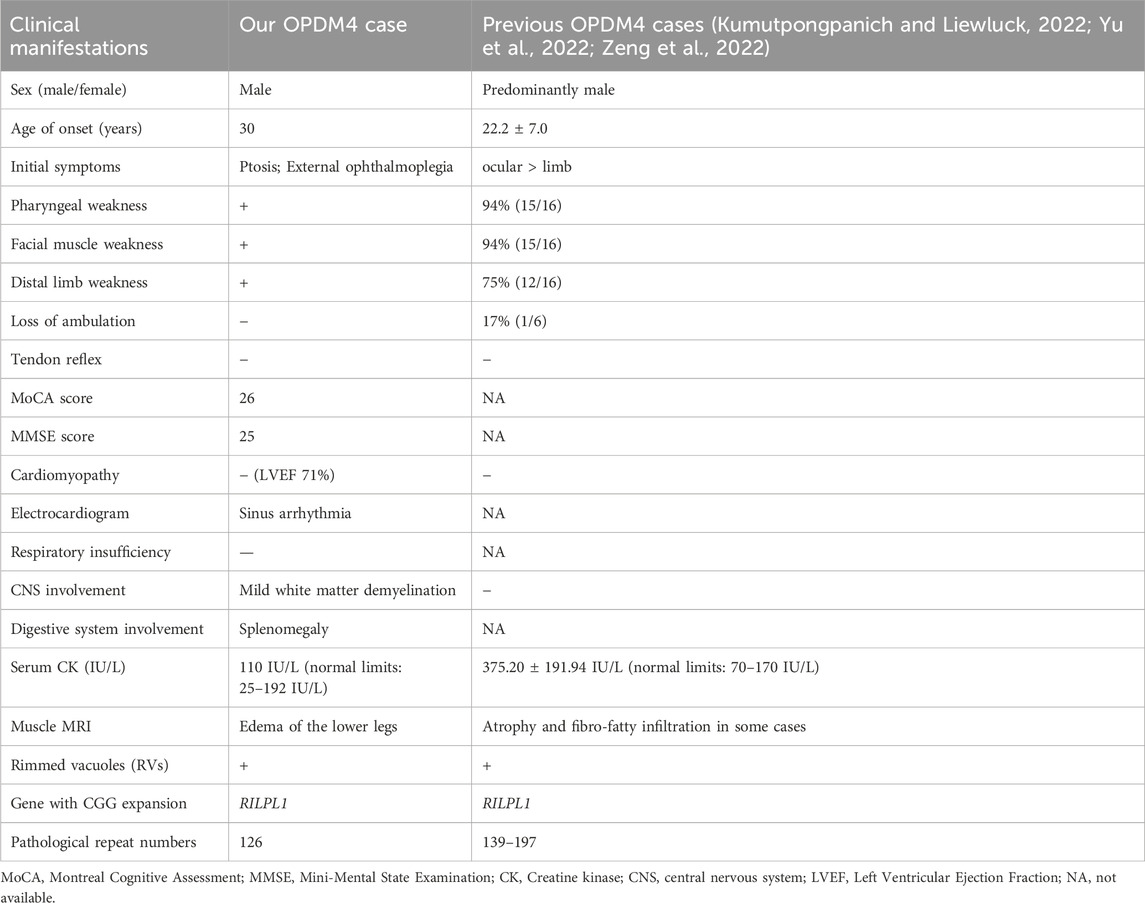
Table 1. Summary of clinical manifestations.
Comprehensive auxiliary examinations were performed. Laboratory tests, including Routine blood, urine, and stool tests, along with biochemical tests, coagulation function, thyroid function, and autoantibodies, were all within normal ranges, and creatine kinase was 110 IU/L (normal limits: 25–192 IU/L). Electrophysiological examinations showed normal motor and sensory nerve conduction in all four limbs, with normal F-wave latency and occurrence rates of 50%–100% in both the median and ulnar nerves, and normal H-reflex latency in both tibial nerves. Electromyography (EMG) revealed myotonic discharges and myogenic damage in the left first interosseous muscle, right gastrocnemius, left biceps brachii, and left quadriceps. Muscle MRI revealed mild muscle edema in the lower legs (Figure 1B), and brain MRI revealed mild white matter demyelination and bilateral mastoiditis (Figure 1C). A biopsy of the left biceps brachii revealed myogenic changes. The muscle fibers exhibited mild size variability, with a few small round atrophic and degenerative fibers, and individual muscle fibers containing vacuoles. We also conducted cognitive function tests on the patient who had a middle school education level. The patient scored 26 points on the Montreal Cognitive Assessment (MoCA) and 25 points on the Mini-Mental State Examination (MMSE). An electrocardiogram revealed sinus arrhythmia. Echocardiography revealed no significant abnormalities. Color Doppler ultrasound of the digestive system indicated splenomegaly (Tables 1, 2). Genetic testing, including dynamic mutation analysis of the four causative genes for OPDM (LRP12, GIPC1, NOTCH2NLC, and RILPL1) revealed that the patient had CGG repeat numbers of 7 and >78 in the OPDM4 causative gene RILPL1, with one allele exceeding the normal range. Fluorescence amplicon length analysis PCR (AL-PCR) technology revealed that RILPL1 contains 126 CGG repeats, in which the primers RILPL1-F (5′-Fam-CCATTGAGCGCAACTCGGATC-3′) and RILPL1-AL-R (5′-GGGTGTGCGGGCCCGG-3′) were used. Repeat numbers in the LRP12, GIPC1, and NOTCH2NLC loci were within the normal range. Additionally, testing for dynamic mutations in the poly-adenine-binding protein nuclear 1 (PABPN1) gene, which is associated with oculopharyngeal muscular dystrophy (OPMD) (Malerba et al., 2017), revealed normal GCN repeat numbers. Whole exome sequencing and mitochondrial gene testing did not reveal any pathogenic or likely pathogenic variants explaining the patient’s phenotype (Figure 2).
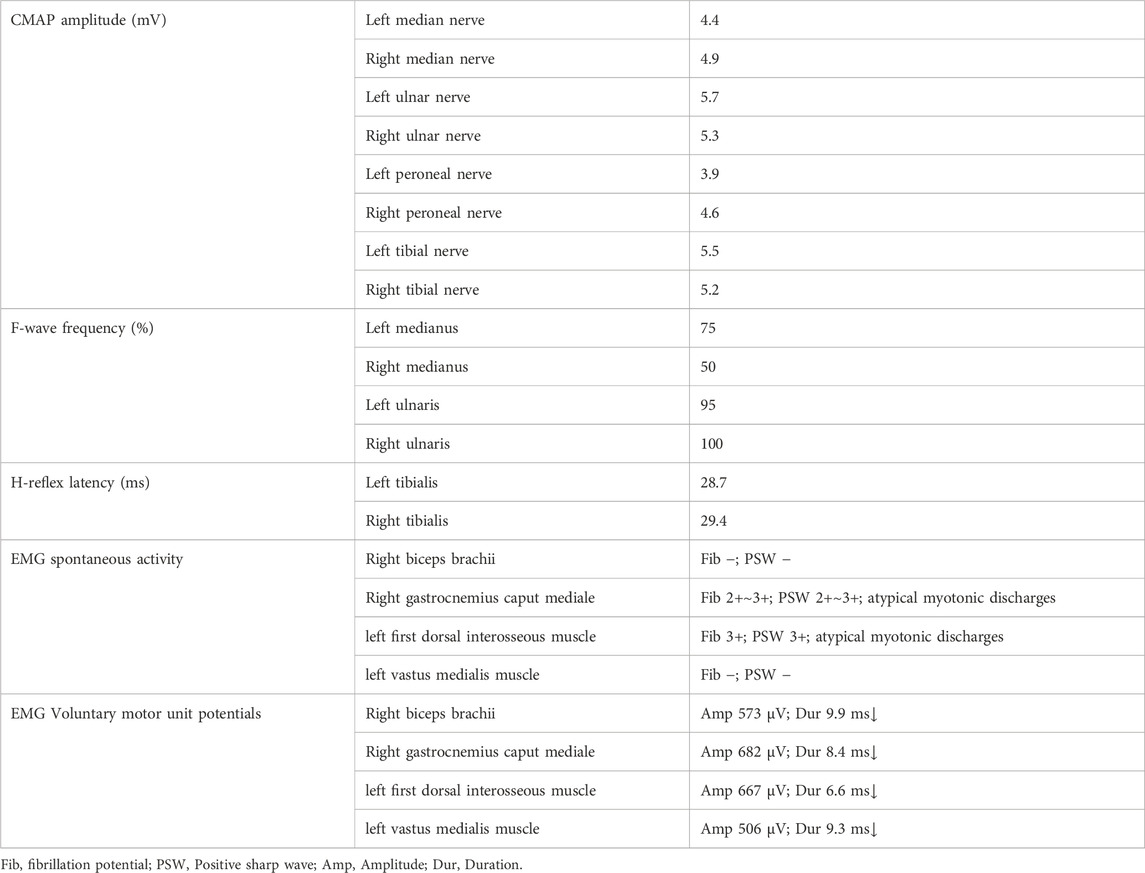
Table 2. Summary of electrophysiological data.
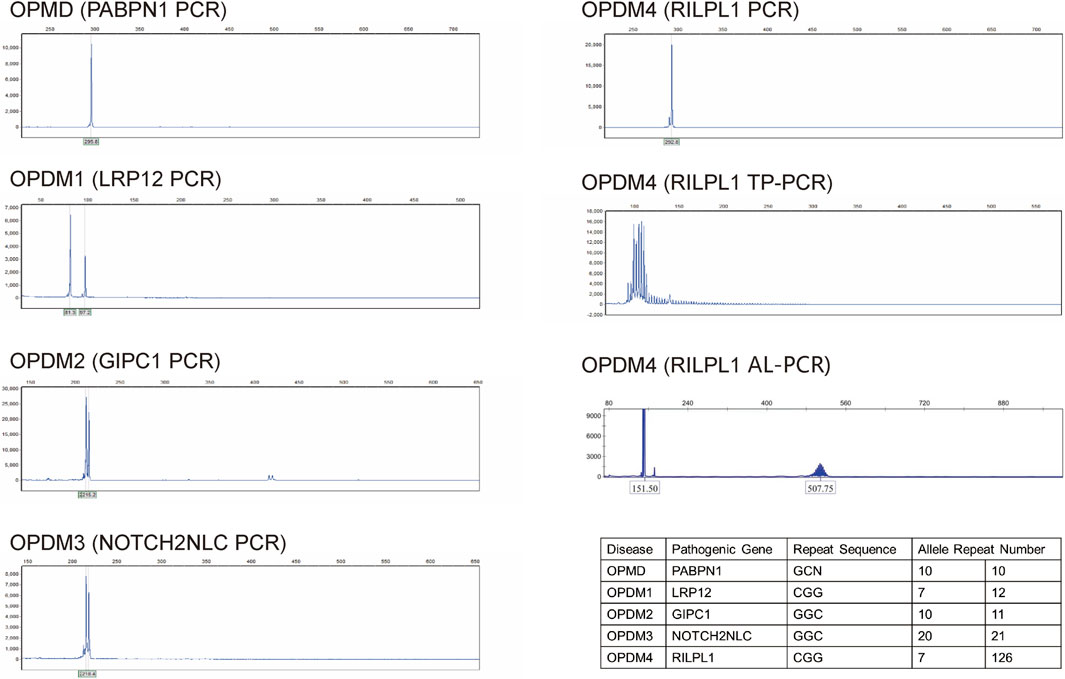
Figure 2. Dynamic mutation analysis of the four causative genes for OPDM (LRP12, GIPC1, NOTCH2NLC, and RILPL1) and OPMD (PABPN1) showed that repeat numbers in the LRP12, GIPC1, NOTCH2NLC and PABPN1 loci were within the normal range. Triplet repeat-primed PCR (TP-PCR) revealed that the patient had CGG repeat numbers of 7 and >78 in the OPDM4 causative gene RILPL1, with one allele exceeding the normal range. Fluorescence amplicon length analysis PCR (AL-PCR) technology detected 126 CGG repeats in RILPL1. Repeat numbers in the LRP12, GIPC1, NOTCH2NLC, and PABPN1 loci were within normal ranges.
OPDM4 is an exceptionally rare disorder (Yu et al., 2022; Zeng et al., 2022; Yang et al., 2023; Eura et al., 2024). It is urgently needed to further accumulate cases to deepen our understanding of the clinical features and pathogenic mechanisms of OPDM4. This case report presents comprehensive clinical manifestations, diagnostic approaches, and genetic findings, with the goal of enhancing and expanding our knowledge of this rare disorder. We found that the CGG repeat count in the RILPL1 gene was 126, which was smaller than the previously reported minimum pathogenic repeat count of 139. Through systematic medical examinations, we also discovered some new characteristics of OPDM4, which have not been reported previously, such as a normal H reflex, and potential mild cognitive impairment.
In our case, we broadened the spectrum of CGG repeat numbers in the RILPL1 gene associated with OPDM4. Among a large Chinese cohort, 11 out of 51 OPDM patients (21.6%) were diagnosed with OPDM4. The average age of onset was approximately 24 years, with repeat sizes ranging from 139 to 197 (Yu et al., 2022). In this case, the patient had 126 CGG repeat expansions in RILPL1, which was less than 139. Therefore, it is reasonable to deduce that CGG repeat expansions ranging from 126 to 197 are pathological.
With the progress of research, OPDM1-3 have been found to affect the extramuscular system. Some OPDM1 patients have been reported to develop respiratory insufficiency, cardiomyopathy of undetermined cause (atrial fibrillation, heart failure, second-degree atrioventricular block, and dilated cardiomyopathy), and neurologic abnormalities (dementia, idiopathic Parkinson disease and mild cognitive impairment) (Saito et al., 2020; Kumutpongpanich et al., 2021; Shimizu et al., 2022; Hobara et al., 2025). GGC repeat expansions in OPDM2 causative gene GIPC1 have been found to be associated with movement disorders (Fan et al., 2022; Pan et al., 2022; Zhou et al., 2022; Murayama et al., 2024). OPDM3 and neuronal intranuclear inclusion disease (NIID) share the same cause, GGC repeat expansions in NOTCH2NLC. The pathological process of OPDM3 extends beyond the oculopharyngeal muscles, and involves various extraskeletal muscle organs, including the central and peripheral nervous systems, as well as cardiac, respiratory, and gastrointestinal systems (Ishiura et al., 2019; Tian et al., 2019; Ogasawara et al., 2020; Cao et al., 2021; Yu et al., 2021a; Yu et al., 2021b; Boivin and Charlet-Berguerand, 2022; Huang et al., 2022; Ma et al., 2024). However, there is currently insufficient research on extramuscular system involvement in OPDM4 patients. Therefore, screening for extramuscular system involvement is also crucial for OPDM4 patients, as muscular symptoms may be only a part of the phenotypic spectrum caused by CGG repeat expansions in RILPL1. In this case, we thoroughly screened for extramuscular system involvement, such as the central nervous system, heart, and digestive system. In this patient, brain MRI revealed mild white matter demyelination. He scored 26 points on the MoCA and 25 points on the MMSE, which indicates potential mild cognitive impairment. Besides a brief history of smoking and alcohol use, he has no vascular risk factors such as hypertension or diabetes. There is a possibility that the mild white matter demyelination and potential mild cognitive impairment were related to OPDM4. Echocardiography and EEG showed no evidence of abnormalities in heart structure or cardiac functions. He had no breathing difficulty until now. Doppler ultrasound of the digestive system indicated splenomegaly, but it is unclear whether this is related to the disease. These findings expand our knowledge of the phenotypic spectrum of OPDM4 caused by repeat CGG expansions in RILPL1.
We conducted comprehensive electrophysiological examinations on this patient, which provided insights into the affected muscle sub-areas. Notably, the patient’s H-reflex was normal, despite the complete absence of tendon reflexes. This discrepancy is intriguing because the reflex arcs for the H-reflex and tendon reflexes are similar, and involve the afferent pathway, central processing, efferent pathway, and effector. The key distinction lies in the fact that the H-reflex is triggered by electrical stimulation, whereas the tendon reflex is elicited by mechanical stimulation, such as tapping the tendon. The H-reflex primarily involves type Ia sensory fibers activated by electrical stimulation of sensory nerves, whereas the tendon reflex is triggered by muscle stretch, stimulating sensory nerves within muscle spindles. Given that the patient’s nerve conduction was normal, it is reasonable to infer that the affected muscle sub-area in the patient was muscle spindles. This finding suggested that muscle spindles could be susceptible or affected early in OPDM4 patients.
Our case also provides valuable evidence for differentiating OPDM4 from OPMD and other OPDM subtypes. OPDM and OPMD are similar and even believed to be indistinguishable in terms of their myopathological features. However, OPMD is caused by an alanine expansion mutation in PABPN1 gene (Brais et al., 1998). Clinically, OPMD is characterized by oculopharyngeal muscle involvement and rimmed vacuolar pathology, with proximal limb muscle weakness being a typical feature, rather than distal weakness (Victor et al., 1962; Tomé et al., 1997). Our patient exhibited distal limb weakness, and genetic testing revealed no abnormalities in the PABPN1 gene. In addition, this case confirms the differences in clinical features between OPDM4 and OPDM1-3, consistent with previous studies. It has been reported that 90% (9/10) of OPDM4 patients initially present with ptosis or dysarthria, and 60% do not exhibit distal muscle weakness until 10–20 years after these initial symptoms (Yu et al., 2022). On the contrary, distal-limb weakness was the most common initial symptom in OPDM1 (3 out of 4, 75%), OPDM2 (14 out of 19, 73.68%), and OPDM3 (8 out of 9, 87.5%) (Deng et al., 2020; Ogasawara et al., 2020; Xi et al., 2021; Yu et al., 2021a; Yu et al. 2021b; Yu et al. 2022; Zeng et al., 2022; Yang et al., 2023; Eura et al., 2024; Ma et al., 2024). In this case, 20 years ago, the patient experienced initial symptoms of ptosis and external ophthalmoplegia. To date, the patient has not reported subjective limb weakness, although neurological examination revealed a mild decrease in distal muscle strength. His clinical presentation was consistent with the distinct characteristics previously reported.
All OPDM subtypes exhibit autosomal dominant inheritance (Ishiura et al., 2019; Deng et al., 2020; Ogasawara et al., 2020; Kumutpongpanich et al., 2021; Xi et al., 2021; Yu et al., 2021a; Yu et al., 2022). Interestingly, this patient had no relevant family history. Due to the rarity of reported OPDM4 cases, it was difficult to deduce the exact mode of inheritance. However, some findings have been reported for other OPDM subtypes. In OPDM1-3, the CGG repeat has been observed to expand or contract over generations in some families. Currently, asymptomatic carriers with large expansions (>200–300) are reported to be fathers of affected offspring with shorter expansions, indicating a bias towards the male germline (Deng et al., 2020; Fukuda et al., 2021; Kumutpongpanich et al., 2021; Yu et al., 2021a). Similar repeat contractions in affected offspring have been reported in asymptomatic parents, typically fathers, with fragile X messenger ribonucleoprotein 1 (FMR1) CGG repeats in Fragile X syndrome (Nolin et al., 2019). One possible explanation is that an expansion beyond a certain length could lead to hypermethylation in the promoter region, thus silencing gene transcription and preventing the production of toxic mRNA (Kumutpongpanich et al., 2021). In our case, the patient’s mother is currently 81 years of age and in very good health, without any symptoms similar to those of this patient. His father passed away at the age of 73 years because of myocardial infarction. The father did not exhibit clinical manifestations similar to those of the patient throughout life. Based on the patterns observed in other OPDM subtypes, we speculate that the CGG repeat in RILPL1 was inherited from the patient’s asymptomatic father.
Our study has several limitations. Although muscle pathology revealed rimmed vacuoles (RVs), a hallmark of OPDM, we were unable to detect another key marker, p62-positive nuclear inclusions. This limitation arose because the biopsy was performed years ago at another hospital, and the patient declined a repeat procedure. Furthermore, conducting genetic testing on the patient’s mother would strengthen our hypothesis that the CGG repeat expansion in RILPL1 was inherited from his asymptomatic father, but consent was not obtained.
In conclusion, this case broadens the genetic and phenotypic spectrum of CGG repeat numbers in the RILPL1 gene associated with OPDM4. In addition, systematic medical examinations revealed some new characteristics of OPDM4. For OPDM4, we still need to accumulate a large number of cases to summarize its clinical features and causative mechanisms. With more OPDM4 cases and muscle-related examinations reported, enhancing our understanding of the relationship between genotype and phenotype is meaningful. In terms of physical examination and auxiliary tests, we should not limit ourselves to muscle-related examinations, which will enhance our understanding of the phenotypic spectrum caused by CGG repeat expansions in RILPL1.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
WW: Funding acquisition, Investigation, Writing–original draft. TY: Investigation, Writing–original draft. XZ: Investigation, Writing–original draft. ZW: Investigation, Methodology, Writing–review and editing. TW: Investigation, Methodology, Writing–review and editing. SZ: Investigation, Methodology, Writing–review and editing. YZ: Investigation, Methodology, Writing–review and editing. DF: Conceptualization, Funding acquisition, Investigation, Writing–review and editing, Supervision.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Natural Science Foundation of China (82101489, 81873784, 82071426) and Young Elite Scientists Sponsorship Program by BAST (BYESS2023317).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
An, R., Chen, H., Gu, W., and Xu, Y. (2022). Oculopharyngodistal myopathy with CGG repeat expansions in GIPC1: the first report from southwestern China. Neurol. Sci. 43, 3989–3993. doi:10.1007/s10072-022-06005-y
Boivin, M., and Charlet-Berguerand, N. (2022). Trinucleotide CGG repeat diseases: an expanding field of polyglycine proteins? Front. Genet. 13, 843014. doi:10.3389/fgene.2022.843014
Brais, B., Bouchard, J.-P., Xie, Y.-G., Rochefort, D. L., Chrétien, N., Tomé, F. M., et al. (1998). Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 18, 164–167. doi:10.1038/ng0298-164
Cao, L., Yan, Y., and Zhao, G. (2021). NOTCH2NLC-related repeat expansion disorders: an expanding group of neurodegenerative disorders. Neurol. Sci. 42, 4055–4062. doi:10.1007/s10072-021-05498-3
Cortese, A., Beecroft, S. J., Facchini, S., Curro, R., Cabrera-Serrano, M., Stevanovski, I., et al. (2024). A CCG expansion in ABCD3 causes oculopharyngodistal myopathy in individuals of European ancestry. Nat. Commun. 15, 6327. doi:10.1038/s41467-024-49950-2
Deng, J., Yu, J., Li, P., Luan, X., Cao, L., Zhao, J., et al. (2020). Expansion of GGC repeat in GIPC1 is associated with oculopharyngodistal myopathy. Am. J. Hum. Genet. 106, 793–804. doi:10.1016/j.ajhg.2020.04.011
Eura, N., Ogasawara, M., and Nishino, I. (2024). Recent topics of oculopharyngodistal myopathy. Neurology Clin. Neurosci. 12, 10–15. doi:10.1111/ncn3.12680
Fan, Y., Shen, S., Yang, J., Yao, D., Li, M., Mao, C., et al. (2022). GIPC1 CGG repeat expansion is associated with movement disorders. Ann. neurology 91, 704–715. doi:10.1002/ana.26325
Fukuda, H., Yamaguchi, D., Nyquist, K., Yabuki, Y., Miyatake, S., Uchiyama, Y., et al. (2021). Father-to-offspring transmission of extremely long NOTCH2NLC repeat expansions with contractions: genetic and epigenetic profiling with long-read sequencing. Clin. Epigenetics 13, 204–217. doi:10.1186/s13148-021-01192-5
Gu, X., Yu, J., Jiao, K., Deng, J., Xia, X., Qiao, K., et al. (2024). Non-coding CGG repeat expansion in LOC642361/NUTM2B-AS1 is associated with a phenotype of oculopharyngodistal myopathy. J. Med. Genet. 61, 340–346. doi:10.1136/jmg-2023-109345
Hobara, T., Ando, M., Higuchi, Y., Yuan, J. H., Yoshimura, A., Kojima, F., et al. (2025). Linking LRP12 CGG repeat expansion to inherited peripheral neuropathy. J. Neurology, Neurosurg. and Psychiatry 96, 140–149. doi:10.1136/jnnp-2024-333403
Huang, X.-R., Tang, B.-S., Jin, P., and Guo, J. F. (2022). The phenotypes and mechanisms of NOTCH2NLC-related GGC repeat expansion disorders: a comprehensive review. Mol. Neurobiol. 59, 523–534. doi:10.1007/s12035-021-02616-2
Ishiura, H., Shibata, S., Yoshimura, J., Suzuki, Y., Qu, W., Doi, K., et al. (2019). Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 51, 1222–1232. doi:10.1038/s41588-019-0458-z
Kumutpongpanich, T., and Liewluck, T. (2022). Oculopharyngodistal myopathy: the recent discovery of an old disease. Muscle and Nerve 66 (6), 650–652. doi:10.1002/mus.27735
Kumutpongpanich, T., Ogasawara, M., Ozaki, A., Ishiura, H., Tsuji, S., Minami, N., et al. (2021). Clinicopathologic features of oculopharyngodistal myopathy with LRP12 CGG repeat expansions compared with other oculopharyngodistal myopathy subtypes. JAMA neurol. 78, 853–863. doi:10.1001/jamaneurol.2021.1509
Ma, J., Zhang, H., Meng, B., Qin, J., Liu, H., Pang, X., et al. (2024). CGG repeat expansion in NOTCH2NLC causing overlapping oculopharyngodistal myopathy and neuronal intranuclear inclusion disease with diffusion weighted imaging abnormality in the cerebellum. J. Clin. Neurology (Seoul, Korea) 20, 580–590. doi:10.3988/jcn.2023.0486
Malerba, A., Klein, P., Bachtarzi, H., Jarmin, S. A., Cordova, G., Ferry, A., et al. (2017). PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun. 8, 14848. doi:10.1038/ncomms14848
Murayama, A., Nagaoka, U., Sugaya, K., Shimazaki, R., Miyamoto, K., Matsubara, S., et al. (2024). Sequential development of parkinsonism in two patients with oculopharyngodistal type myopathy in GIPC1-related repeat expansion disorder. Neuromuscul. Disord. 44, 104465. doi:10.1016/j.nmd.2024.104465
Nolin, S. L., Glicksman, A., Tortora, N., Allen, E., Macpherson, J., Mila, M., et al. (2019). Expansions and contractions of the FMR1 CGG repeat in 5,508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. Part A 179, 1148–1156. doi:10.1002/ajmg.a.61165
Ogasawara, M., Iida, A., Kumutpongpanich, T., Ozaki, A., Oya, Y., Konishi, H., et al. (2020). CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol. Commun. 8, 204–208. doi:10.1186/s40478-020-01084-4
Pan, Y., Xue, J., Chen, J., Zhang, X., Tu, T., Xiao, Q., et al. (2022). Assessment of GGC repeat expansion in GIPC1 in patients with Parkinson's disease. Mov. Disord. 37, 1557–1559. doi:10.1002/mds.29041
Pongpakdee, S., Apiwattanakul, M., Termglinchan, T., Witoonpanich, R., Dejthevaporn, C., Lee, T., et al. (2024). CGG/CCG repeat expansions in LOC642361/NUTM2B-AS1 in Thai patients with oculopharyngodistal myopathy. Neurol. Genet. 10, e200170. doi:10.1212/NXG.0000000000200170
Saito, R., Shimizu, H., Miura, T., Hara, N., Mezaki, N., Higuchi, Y., et al. (2020). Oculopharyngodistal myopathy with coexisting histology of systemic neuronal intranuclear inclusion disease: clinicopathologic features of an autopsied patient harboring CGG repeat expansions in LRP12. Acta Neuropathol. Commun. 8, 75–5. doi:10.1186/s40478-020-00945-2
Satoyoshi, E., and Kinoshita, M. (1977). Oculopharyngodistal myopathy: report of four families. Archives Neurology 34, 89–92. doi:10.1001/archneur.1977.00500140043007
Shimizu, T., Ishiura, H., Hara, M., Shibata, S., Unuma, A., Kubota, A., et al. (2022). Expanded clinical spectrum of oculopharyngodistal myopathy type 1. Muscle and Nerve 66, 679–685. doi:10.1002/mus.27717
Tian, Y., Wang, J.-L., Huang, W., Zeng, S., Jiao, B., Liu, Z., et al. (2019). Expansion of human-specific GGC repeat in neuronal intranuclear inclusion disease-related disorders. Am. J. Hum. Genet. 105, 166–176. doi:10.1016/j.ajhg.2019.05.013
Tomé, F. M., Chateau, D., Helbling-Leclerc, A., and Fardeau, M. (1997). Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul. Disord. 7, S63–S69. doi:10.1016/s0960-8966(97)00085-0
Victor, M., Hayes, R., and Adams, R. D. (1962). Oculopharyngeal muscular dystrophy: a familial disease of late life characterized by dysphagia and progressive ptosis of the eyelids. N. Engl. J. Med. 267, 1267–1272. doi:10.1056/NEJM196212202672501
Xi, J., Wang, X., Yue, D., Dou, T., Wu, Q., Lu, J., et al. (2021). 5′ UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain 144, 601–614. doi:10.1093/brain/awaa426
Yang, X., Zhang, D., Shen, S., Li, P., Li, M., Niu, J., et al. (2023). A large pedigree study confirmed the CGG repeat expansion of RILPL1 Is associated with oculopharyngodistal myopathy. BMC Med. Genomics 16, 253. doi:10.1186/s12920-023-01586-9
Yu, J., Deng, J., Guo, X., Shan, J., Luan, X., Cao, L., et al. (2021a). The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain 144, 1819–1832. doi:10.1093/brain/awab077
Yu, J., Luan, X., Yu, M., Zhang, W., Lv, H., Cao, L., et al. (2021b). GGC repeat expansions in NOTCH2NLC causing a phenotype of distal motor neuropathy and myopathy. Ann. Clin. Transl. Neurology 8, 1330–1342. doi:10.1002/acn3.51371
Yu, J., Shan, J., Yu, M., Di, L., Xie, Z., Zhang, W., et al. (2022). The CGG repeat expansion in RILPL1 is associated with oculopharyngodistal myopathy type 4. Am. J. Hum. Genet. 109, 533–541. doi:10.1016/j.ajhg.2022.01.012
Yu, J.-x., Yu, M., and Zhang, W. (2023). Oculopharyngodistal myopathy caused by CGG repeat expansion in 5'untranslated region of LRP12 gene: four cases report. Chin. J. Contemp. Neurology Neurosurg. 23, 798.
Zeng, Y. H., Yang, K., Du, G. Q., Chen, Y. K., Cao, C. Y., Qiu, Y. S., et al. (2022). GGC repeat expansion of RILPL1 is associated with oculopharyngodistal myopathy. Ann. Neurology 92, 512–526. doi:10.1002/ana.26436
Keywords: oculopharyngodistal myopathy, RILPL1, CGG repeat, OPDM4, myopathy
Citation: Wang W, Yin T, Zhang X, Wang Z, Wang T, Zhang S, Zhang Y and Fan D (2025) A case report of oculopharyngodistal myopathy with 126 CGG repeat expansions in RILPL1. Front. Genet. 16:1472907. doi: 10.3389/fgene.2025.1472907
Received: 02 August 2024; Accepted: 05 February 2025;
Published: 27 February 2025.
Edited by:
Changhe Shi, First Affiliated Hospital of Zhengzhou University, ChinaReviewed by:
Jun Mitsui, The University of Tokyo, JapanCopyright © 2025 Wang, Yin, Zhang, Wang, Wang, Zhang, Zhang and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongsheng Fan, ZHNmYW4yMDEwQGFsaXl1bi5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.