
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 10 December 2024
Sec. Genetics of Common and Rare Diseases
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1503048
This article is part of the Research Topic Insights in Genetics of Common and Rare Diseases 2024 View all 11 articles
 Qi Yang1,2†
Qi Yang1,2† Qiang Zhang1,2†
Qiang Zhang1,2† Xunzhao Zhou1,2Juntan Feng3Shujie Zhang1,2Li Lin1,2
Xunzhao Zhou1,2Juntan Feng3Shujie Zhang1,2Li Lin1,2 Shang Yi1,2Zailong Qin1,2
Shang Yi1,2Zailong Qin1,2 Jingsi Luo1,2,4*
Jingsi Luo1,2,4*UBAP2L-deficiency syndrome, also known as neurodevelopmental disorder with impaired language, behavioral abnormalities, and dysmorphic facies (NEDLBF, OMIM 620494), is an extremely rare autosomal dominant disorder. This condition is caused by heterozygous variant in the UBAP2L gene (NM_014847.4, MIM 616472), which encodes the ubiquitin-associated protein 2-like protein involved in the formation of stress granules (SGs). To date, only one report has documented 12 loss-of-function variants in UBAP2L, all of which were identified as de novo variants. In our study, we recruited a Chinese family with two patients exhibiting intellectual disability and seizures. Whole-exome sequencing was performed on the proband, revealing a novel heterozygous frameshift variant, UBAP2L (NM_014847.4):c.2453_2454del (p.Tyr818Trpfs*3). The variant was inherited from the affected mother, and confirmed in the proband and his parents by Sanger sequencing. This is the first familial report of a deleterious UBAP2L variant. The proband in this family presented a clinical phenotype similar to NEDLBF, which includes intellectual disability, developmental delay, speech delay, facial dysmorphism, seizures, and behavioral abnormalities. The affected mother presented only mild intellectual disability and mild language impairment. By clinical evaluation of our patients and previously reported cases with UBAP2L variants, we propose that intellectual disability, developmental delay (particularly in speech), infants’ feeding difficulties, behavioural abnormalities and seizures are the main clinical features of NEDLBF patients. Our study expands the genetic and phenotypic spectrum associated with NEDLBF.
UBAP2L-deficiency syndrome, also known as neurodevelopmental disorder with impaired language, behavioral abnormalities, and dysmorphic facies (NEDLBF, OMIM 620494), is caused by heterozygous loss-of-function variants in the UBAP2L gene, located at 1q21.3 (Jia et al., 2022). This gene contains 27 exons and encodes the ubiquitin-associated protein 2-like protein, which is involved in the formation of stress granules (SGs). SGs play a crucial role in cell survival under stress conditions and have been implicated in the pathogenesis of cancer, neurodegeneration, inflammatory diseases, and viral infections (Panas et al., 2016; Buchan et al., 2008; Arimoto et al., 2008; Takahara and Maeda, 2012). Moreover, UBAP2L is involved in the ubiquitination and degradation of RNA polymerase II (RNAPII) by recruiting Cullin-based ubiquitin complexes (Herlihy et al., 2022). Additionally, UBAP2L is a spindle-associated protein that plays a significant role in cellular mitosis by regulating the activity of the kinase PLK1 (Guerber et al., 2023). It has been identified as an oncogene associated with various cancers, including glioma, prostate cancer, hepatocellular carcinoma (HCC), breast cancer, and colorectal cancer (Li and Huang, 2014; Zhao et al., 2015; Ye et al., 2017; He et al., 2018; Chai et al., 2016). Furthermore, Lingerer, the Drosophila homolog of the human UBAP2L gene, regulates the proliferation of growing tissues by modulating the JAK/STAT signalling pathway (Baumgartner et al., 2013). A recent study found that variants in the UBAP2L gene were associated with developmental delay (DD), speech delay, mild-to-severe intellectual disability (ID), feeding difficulties in infants, seizures, motor delay, various behavioral abnormalities, hypotonia, skeletal anomalies, facial dysmorphism, and other variable clinical features (Jia et al., 2022) (Figure 1A).
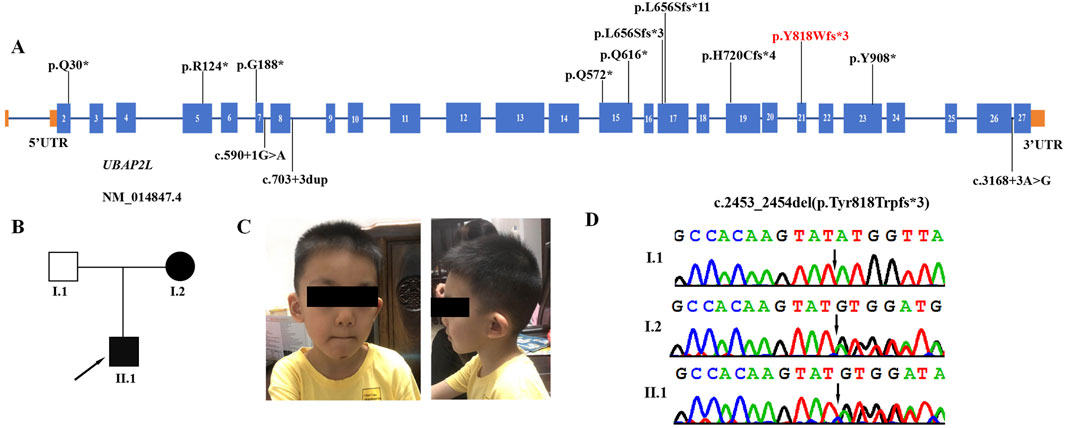
Figure 1. Clinical and genetic features. (A) The spectrum of UBAP2L pathogenic variants. The red variant is the novel variant identified in this study. (B) Family pedigree showing that both the mother and the proband are affected. (C) Facial appearance of the proband (II-1) at the age of 4 years old, showing cupped ear, long face and short nose. (D) DNA sequence chromatograms from Sanger sequencing of UBAP2L, showing a heterozygous frameshift variant UBAP2L (NM_014847.4: c.2453_2454del (p.Tyr818Trpfs*3)) in the proband. Sanger sequencing further revealed that his affected mother was heterozygous for the same variant and that his father was normal.
Here we report the first familial case of NEDLBF syndrome in a Chinese family with a novel pathogenic variant of UBAP2L (NM_014847.4):c.2453_2454del (p.Tyr818Trpfs*3) (Figure 1B). The mother carries this novel variant and passed it on to the proband. We provide a detailed description of the clinical phenotypes observed in both the proband and his mother. This report expands the spectrum of variants in the UBAP2L gene and provides additional molecular and clinical information insights to enhance our understanding of UBAP2L-deficiency syndrome.
The proband was initially referred to the Paediatric Endocrine Clinic of the Guangxi Maternal and Child Health Hospital due to behavioral issues and seizures. Written informed consents for publishing data and images were obtained from participants and the parents of the participant under the age of 16. The study was approved by the the Medical Ethics Committee of the Maternal and Child Health Hospital of Guangxi Autonomous Region. The patient was admitted following standard admission protocols, which included a comprehensive medical history, laboratory tests, physical examination, abdominal ultrasound, neurological examination, electroencephalogram, magnetic resonance imaging (MRI) of the brain, audiological examination, routine ophthalmological examination, and genetic testing.
We collected 5 mL peripheral blood samples from the proband and his parents for whole-exome sequencing. Genomic DNA was isolated from peripheral blood samples using the Lab-Aid DNA kit (Zeshan Biotechnology Co., Ltd., Xiamen, China), and its concentration and purity were assessed using a NanoDrop 1000 spectrophotometer (Thermo Scientific). Sequencing libraries were prepared using Agilent SureSelect Human Exon V6 kits (Agilent Technologies, Santa Clara, CA, United States) according to standard Illumina protocols and then sequenced with 100 bp paired-end reversible terminator technology on the Illumina Hiseq2000 platform at Huada Gene Technology Co. Ltd. (Shenzhen, China). Burrows-Wheeler Aligner software version 0.6.2 was used to map the clean reads to the the human genome assembly GRCh37. Annotation and classification of variants were performed using TGex software (LifeMap Sciences, Alameda, CA, United States). The variant analysis included those variants with a minor allele frequency of less than 1% in public databases (e.g., 1000Genomes Project, Genome Aggregation Database, Exome Sequencing Project, and ExAC) and in our in-house databases. The deleterious effect of the selected variants was evaluated using in silico prediction tools such as Variant Taster, CADD, SIFT, and PolyPhen2. Final interpretation and categorization of variants was performed using criteria developed by the American College of Medical Genetics (ACMG)/Association for Molecular Pathology (AMP) (Richards et al., 2015).
Sanger sequencing was used to validate the identified variants in the proband and his family members. Sanger primers flanking the candidate variant alleles were custom designed using Primer 3 software (https://bioinfo.ut.ee/primer3-0.4.0/primer3/) (forward primer: 5′-TCCGTGTCCTGTTCTAACCC-3′ and reverse primer: 5′- GCTGGGATGGGAAAGAGCTA-3′). PCR amplification was performed using Takara PrimeSTAR Max DNA Polymerase (Takara Biotechnology Co., Ltd., Dalian, China) under the following conditions:one cycle of initial denaturation at 95°C for 5 min; 35 cycles consisting of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C; followed by a final extension at 72°C for 5 min. The purified PCR fragments were then sequenced on an ABI 3500DX DNA analyzer (Applied Biosystems; Thermo FisherScientific, Inc.) using the Big Dye Terminator Cycle sequencing reaction kit (Perkin-Elmer, Applied Biosystems Division, Foster City, CA).
The proband was a Chinese boy and the first child of non-consanguineous parents. He was born prematurely at 34 weeks of gestation with a birth weight of 2,180 g (49.4th percentile), a birth length of 41.5 cm (50th percentile), a head circumference of 31 cm (48th percentile) and Apgar scores of 9, 9, and 10 at 1, 5, and 10 min, respectively. The proband was first admitted to Paediatric Department of the Guangxi Zhuang Autonomous Region Maternal and Child Health Hospital at the age of 4 years due to behavioral issues and seizure. He experienced mild developmental delay, achieving head control at 5 months, crawling at 10 months, and walking independently at 19 months. Speech development was delayed, with single words spoken at 20 months and complete sentences at 2 years and 4 months. Mild autistic traits were noted, including poor eye contact, repetitive behaviors, avoidance of group play, and unusual fixations and obsessions. At age 4, the Gesell Developmental Diagnostic Scale was used to assess his Developmental Quotient (DQ, DQ < 70 as low score): gross motor 80, fine motor 76, adaptive 58, language 57, and personal-social 56. A physical examination revealed that his height (102 cm, 25th percentile) and weight (15 kg, 25th percentile) were within normal ranges. Mild dysmorphic features included cupped ears, a long face, and a short nose (Figure 1C). The proband experienced his first seizure at 6 months of age during a fever, characterized as absence seizures occurring twice daily, each lasting about 5 s. Sleep electroencephalogram results showed spike, spike-slow, and polyspike-slow wave distributions in the right middle temporal region. Over the following years, he experienced several episodes of absence seizures, each coinciding with a very high fever (over 39°C) at onset. His seizures were controlled with valproate (VPA) treatment. The initial brain MRI at age 4 was normal.
Family history revealed that the proband’s mother experienced several absence seizures with high fever as a child which ceased after she began taking valproic acid (VPA). The mother, aged 34, has a normal height of 158 cm (30th percentile), and there was no sign of short stature during her childhood. She exhibits mild intellectual disability (IQ = 68) and mild language impairment. No behavioral abnormalities or dysmorphic features were observed. Unfortunately, the proband’s mother was unable to return for an EEG or brain MRI.
To determine the genetic etiology of the disease, whole-exome sequencing was performed on the proband. A total of 7.1 Gb of data was generated, with 99.4% of the target region covered and 99.0% of the targets covered more than 20X. There were 24,356 single nucleotide variants (SNVs) or insertion/deletion (indel) variants identified in the coding region and splice site (within 10 bp from the splice junction). After filtering out synonymous variants and those with a minor allele frequency (MAF) greater than 1% in public databases (e.g., 1000 Genomes Project, Genome Aggregation Database, Exome Sequencing Project and ExAC) as well as our in-house databases, a total of 758 variants were remained. Using TGex software (LifeMap Sciences, United States), 9 candidate variants in 9 genes (UBAP2L, CUX2, CACNA1I, SAMD12, HERC2, IQSEC1, NFASC, RERE, PLAA) were mapped to known phenotypes including seizures, intellectual disability, developmental delay, motor delay, language delay and autistic behaviour. The variants in the HERC2, IQSEC1, NFASC and PLAA genes were heterozygous (Supplementary Table S1). The disorders resulting from these genetic variants are autosomal recessive and have therefore been ruled out. The variants in the CUX2, CACNA1I, RERE and SAMD12 genes were transmitted from the unaffected father (Supplementary Table S1). Subsequently, a heterozygous frameshift variant located in exon 21 of UBAP2L (c.2453_2454del (p.Tyr818Trpfs*3)) was identified in the proband. Further Sanger sequencing determined that the variant was inherited from the probands’ mother. The frameshift variant was classified as likely pathogenic according to the ACMG/AMP guidelines. No pathogenic or likely pathogenic CNVs were detected in the WES data of the patient using the XHMM software and the depth of coverage methods.
UBAP2L, also known as NICE-4, is a highly conserved protein involved in various cellular biological processes, including spindle assembly, chromosome segregation, and regulation of cell proliferation and division (Guerber et al., 2023; He et al., 2018; Maeda et al., 2016). In a recent study by Jia et al., in 2022, loss-of-function variants in UBAP2L were associated with neurodevelopmental disorders (Jia et al., 2022). Their research demonstrated that Ubap2l knockout (KO) mice exhibited a lethal phenotype in most embryos, with a minority (2.6%) of surviving mice displaying significantly reduced size. Furthermore, heterozygous defective mice exhibited diminished preference for social novelty, cognitive deficits, and anxiety-like behavior, indicating a role for UBAP2L dysfunction in the neurodevelopmental deficits observed in affected individuals. In this report, we identified a novel heterozygous frameshift variant (c.2453_2454del (p.Tyr818Trpfs*3)) in the UBAP2L gene of a patient with a neurodevelopmental disorder (NDD) (Figure 1D). Sanger sequencing revealed that the affected mother was heterozygous for the same variant, while the unaffected father was not. This variant, located in exon 21 of the UBAP2L gene, introduces a premature termination codon that may activate the nonsense-mediated mRNA decay (NMD) pathway, resulting in a significant reduction in UBAP2L mRNA expression levels and subsequent loss of function. The variant is novel and is not present in the Human Gene variant Database (http://www. hgmd. cf.ac.uk/ac/), HPSD (http://liweilab.genetics. ac. cn/HPSD/), dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), ExAC and gnomAD (https://gnomad.broad institute. org/). Based on the ACMG/AMP classification guidelines, it is classified as a likely pathogenic variant (PVS1+PM2_supporting). This finding confirms that UBAP2L variants are likely responsible for the neurodevelopmental abnormalities in this family.
To date, only 14 affected individuals (including our patient) worldwide have been reported with UBAP2L variants (Jia et al., 2022). All identified variants (five frameshifts, three splicing, and six nonsense) generate null alleles (Figure 1D). The reported UBAP2L variants and the available clinical phenotypes for all cases are summarized in Table 1. Phenotypic analyses of affected individuals revealed significant heterogeneity in the characteristics observed in patients with UBAP2L loss-of-function variants; however, certain common characteristics (>50% of cases) could be identified. All cases exhibited neurodevelopmental abnormalities across multiple areas, ranging from mild to severe. Facial dysmorphic features were observed in almost all patients (10/11), but there was no consistency-some patients having only mild synophrys or epicanthus, while others having a variety of severe facial deformities. The speech domain appeared particularly vulnerable, with all patients (13/13) showing speech delays, and about half of them exhibiting marked delays, suggesting that language disorders are a typical symptom of UBAP2L deficiency syndrome. Intellectual disability was observed in 10 out of 12 patients, and motor delay was noted in 9 out of 13. Notably, the severity of these conditions was relatively mild, with only one patient exhibiting moderate to severe intellectual disability. Our patients also had mild intellectual disability and motor delay. Behavioral problems were reported in 10 of the 14 patients, including anxiety, ADHD, repetitive behaviors, obsessive behaviors, aggressive behaviors, self-injurious behaviors, emotional issues, and trichotillomania. Notably, the proband in this study exhibited autistic behaviors, such as repetitive actions, avoidance of group play, and unusual fixations and obsessions, whereas his mother did not display these issues. Multiple types of seizures were observed in most patients (9/14), with febrile seizures being the most common type. In the present study, both the proband and his mother had absence seizures during fever, which were controlled after treatment with valproic acid (VPA). Electroencephalography (EEG) abnormality were observed in four patients (4/7), including our proband. Most patients (8/12) experienced feeding difficulties in infancy, which improved with age. The proband in this study also had feeding difficulties during infancy. Skeletal and limb abnormalities were observed in six patients (6/13), including mild strephenopodia, clinodactyly of the V finger, lower limb skeletal anomalies (metatarsal adducts, femoral anteversion, tibial torsion), brachydactyly, joint stiffness (hands, elbows, knees, and feet), symphalangism of the thumb, scoliosis, kyphosis, nail hypoplasia, pointing finger joint contracture, broad halluces, proximally set thumbs, misalignment of the elbows and wrists, and prehensile/elongated halluces with varus deformity. Another area of concern is vision problems; more than half of the patients experienced issues in this domain, and early correction and treatment could benefit these children. Additionally, variable manifestations were observed among these patients. Several had failure to thrive (4/9), short stature (4/11), and microcephaly (4/11). Three patients had gastrointestinal problems, two had brain abnormalities, and two experienced sleep issues. Other dysmorphic features, such as hypertrichosis, café au lait spots, chronic bronchitis, recurrent urinary tract infections, asthma, and cutis marmorata, were also noted. In contrast, our patients did not have these variable dysmorphic features, including skeletal abnormalities, visual problems, failure to thrive, short stature, microcephaly, gastrointestinal problems, brain abnormalities, and other rare dysmorphic features.
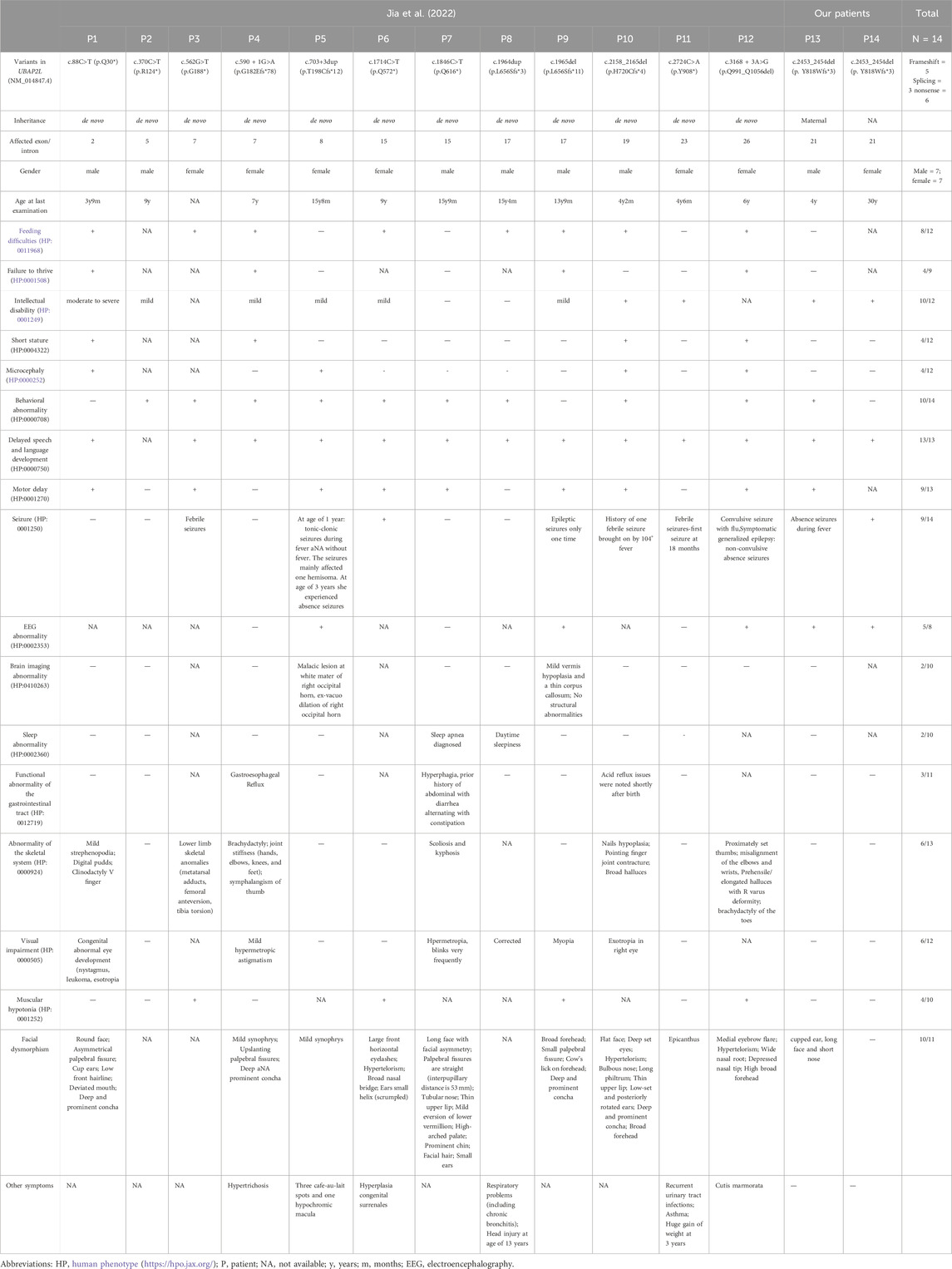
Table 1. Summary of the genetic and clinical features of the patients with UBAP2L-deficiency syndrome.
The mechanism by which UBAP2L variants cause developmental delay, speech delay, mild to severe intellectual disability (ID), seizures, hypotonia, behavioural abnormalities, hypotonia, skeletal and facial abnormalities, and other clinical symptoms remains unclear. There are multiple potential mechanisms by which UBAP2L may influence neurodevelopmental disorders. The UBAP2L protein consists of 483 amino acids and includes an N-terminal ubiquitin-associated (UBA) structural domain, which is involved in the ubiquitin-proteasome system and in the formation of aggregates induced by proteasome inhibitors (Maeda et al., 2016; Wilde et al., 2011). UBAP2L is a spindle-associated protein that plays a crucial role in cellular mitosis (Guerber et al., 2023). Previous studies have also reported that UBAP2L functions as a sperm protein that interacts with zona pellucida 3 in the human egg, playing an important role in the fertilization process (Naz and Dhandapani, 2010). Additionally, it is involved in the formation of complexes that regulate the activity of hematopoietic stem cells (Bordeleau et al., 2014). Furthermore, UBAP2L is implicated in the formation of stress granules (SGs), which are essential for cell survival under stress conditions and have been linked to the pathogenesis of cancer, neurodegeneration, inflammatory diseases, and viral infections (Panas et al., 2016; Buchan et al., 2008; Arimoto et al., 2008; Takahara and Maeda, 2012). Further functional studies are needed to enhance our understanding of the disorders associated with UBAP2L and the mechanisms by which they operate.
Among the identified variants, the SAMD12 variant (NM_207506.3, c.427C>T (p.Arg143*)) was a nonsense variant, present in the Genome Aggregation Database (gnomAD v.2.1.1) and dbSNP Database (rs764723335) with a minor allele frequency of 0.000007 (Supplementary Table S1). The variant is located in the last exon of the SAMD12 gene and results in a premature termination codon that produces a truncated protein, however, it remains unclear whether the variant causes nonsense-mediated decay (NMD). Variants in SAMD12 are associated with benign adult familial myoclonic epilepsy type 1 (BAFME1, OMIM 601068), a rare autosomal dominant disorder characterised by adult-onset cortical myoclonus with or without seizures (Ishiura et al., 2018; Cen et al., 2018; Lei et al., 2019). It has been reported to be associated with intronic TTTCA expansions. The molecular aetiology of the TTTCA repeat expansions in the pathogenesis of BAFME1 has not yet been elucidated. In this study, the proband’s seizure type is absence seizure with high fever. The father, who carries the same variant, has not yet shown any of these symptoms. Taken together, this variant is currently considered to be of uncertain clinical significance and therefore not responsible for the phenotype in the patients. However, as the onset of BAFME1 is usually in adulthood, long-term follow-up of these patients is necessary to assess possible disease progression.
In conclusion, we report the first familial cases of UBAP2L-deficiency syndrome with a novel loss-of-function variant UBAP2L identified in China. The proband in this family presented a clinical phenotype similar to NEDLBF, which includes intellectual disability, developmental delay, speech delay, facial dysmorphism, seizures, and behavioral abnormalities. The affected mother presented only mild intellectual disability and mild language impairment. The phenotypic variability associated with different UBAP2L variants suggests that further functional studies on the specific functions of these variants will enhance our understanding of the disease and its underlying mechanisms. The detailed genotype and phenotype information presented in this study is highly significant for the genetic diagnosis and genetic counselling of patients with UBAP2L-deficiency syndrome.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving humans were approved by The Institutional Review Board and Ethics Committee of Guangxi Maternal and Child Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
QY: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. QZ: Data curation, Methodology, Software, Writing–review and editing. XZ: Data curation, Formal Analysis, Software, Writing–review and editing. JF: Investigation, Resources, Software, Writing–review and editing. SZ: Data curation, Formal Analysis, Funding acquisition, Writing–review and editing. LL: Investigation, Methodology, Software, Validation, Writing–review and editing. SY: Data curation, Investigation, Methodology, Project administration, Software, Writing–review and editing. ZQ: Investigation, Methodology, Software, Validation, Writing–review and editing. JL: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing–original draft.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Health Department of Guangxi Province (Grant No. Z-A20220256, Z20190311 and Z-A20230305), the Guangxi Key Laboratory of reproductive health and birth defect prevention (21-220-22), Guangxi Key Laboratory of Birth Defects and Stem Cell Biobank (ZTJ2020002), the Guangxi Clinical Research Center for Pediatric Diseases (Guike AD22035121), the Guangxi Natural Science Foundation (Grant No. 2024GXNSFBA010072) and the Young Scientists Fund of the National Natural Science Foundation of China (No. 82201312).
We are grateful to the all the patients and their families participating in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1503048/full#supplementary-material
Arimoto, K., Fukuda, H., Imajoh-Ohmi, S., Saito, H., and Takekawa, M. (2008). Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 10 (11), 1324–1332. doi:10.1038/ncb1791
Baumgartner, R., Stocker, H., and Hafen, E. (2013). The RNA-binding proteins FMR1, rasputin and caprin act together with the UBA protein lingerer to restrict tissue growth in Drosophila melanogaster. PLoS Genet. 9, e1003598. doi:10.1371/journal.pgen.1003598
Bordeleau, M. E., Aucagne, R., Chagraoui, J., Girard, S., Mayotte, N., Bonneil, E., et al. (2014). UBAP2L is a novel BMI1-interacting protein essential for hematopoietic stem cell activity. Blood 124 (15), 2362–2369. doi:10.1182/blood-2014-01-548651
Buchan, J. R., Muhlrad, D., and Parker, R. (2008). P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 183 (3), 441–455. doi:10.1083/jcb.200807043
Cen, Z., Jiang, Z., Chen, Y., Zheng, X., Xie, F., Yang, X., et al. (2018). Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain 141, 2280–2288. doi:10.1093/brain/awy160
Chai, R., Yu, X., Tu, S., and Zheng, B. (2016). Depletion of UBA protein 2-like protein inhibits growth and induces apoptosis of human colorectal carcinoma cells. Tumour Biol. 37, 13225–13235. doi:10.1007/s13277-016-5159-y
Guerber, L., Vuidel, A., Liao, Y., Kleiss, C., Grandgirard, E., Sumara, I., et al. (2023). UBAP2L-dependent coupling of PLK1 localization and stability during mitosis. EMBO Rep. 24 (6), e56241. doi:10.15252/embr.202256241
He, J., Chen, Y., Cai, L., Li, Z., and Guo, X. (2018). UBAP2L silencing inhibits cell proliferation and G2/M phase transition in breast cancer. Breast Cancer 25 (2), 224–232. doi:10.1007/s12282-017-0820-x
Herlihy, A. E., Boeing, S., Weems, J. C., Walker, J., Dirac-Svejstrup, A. B., Lehner, M. H., et al. (2022). UBAP2/UBAP2L regulate UV-induced ubiquitylation of RNA polymerase II and are the human orthologues of yeast Def1. DNA Repair (Amst) 115, 103343. doi:10.1016/j.dnarep.2022.103343
Ishiura, H., Doi, K., Mitsui, J., Yoshimura, J., Matsukawa, M. K., Fujiyama, A., et al. (2018). Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 50, 581–590. doi:10.1038/s41588-018-0067-2
Jia, X., Zhang, S., Tan, S., Du, B., He, M., Qin, H., et al. (2022). De novo variants in genes regulating stress granule assembly associate with neurodevelopmental disorders. Sci. Adv. 8 (33), eabo7112. doi:10.1126/sciadv.abo7112
Lei, X. X., Liu, Q., Lu, Q., Huang, Y., Zhou, X. Q., Sun, H. Y., et al. (2019). TTTCA repeat expansion causes familial cortical myoclonic tremor with epilepsy. Eur. J. Neurol. 26, 513–518. doi:10.1111/ene.13848
Li, D., and Huang, Y. (2014). Knockdown of ubiquitin associated protein 2-like inhibits the growth and migration of prostate cancer cells. Oncol. Rep. 32, 1578–1584. doi:10.3892/or.2014.3360
Maeda, M., Hasegawa, H., Sugiyama, M., Hyodo, T., Ito, S., Chen, D., et al. (2016). Arginine methylation of ubiquitin-associated protein 2-like is required for the accurate distribution of chromosomes. FASEB J. 30 (1), 312–323. doi:10.1096/fj.14-268987
Naz, R. K., and Dhandapani, L. (2010). Identification of human sperm proteins that interact with human zona pellucida3 (ZP3) using yeast two-hybrid system. J. Reprod. Immunol. 84 (1), 24–31. doi:10.1016/j.jri.2009.10.006
Panas, M. D., Ivanov, P., and Anderson, P. (2016). Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 215, 313–323. doi:10.1083/jcb.201609081
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Takahara, T., and Maeda, T. (2012). Transient sequestration of TORC1 into stress granules during heat stress. Mol. Cell 47 (2), 242–252. doi:10.1016/j.molcel.2012.05.019
Wilde, I. B., Brack, M., Winget, J. M., and Mayor, T. (2011). Proteomic characterization of aggregating proteins after the inhibition of the ubiquitin proteasome system. J. Proteome Res. 10, 1062–1072. doi:10.1021/pr1008543
Ye, T., Xu, J., Du, L., Mo, W., Liang, Y., and Xia, J. (2017). Downregulation of UBAP2L inhibits the epithelial-mesenchymal transition via SNAIL1 regulation in hepatocellular carcinoma cells. Cell Physiol. Biochem. 41, 1584–1595. doi:10.1159/000470824
Keywords: UBAP2L-deficiency syndrome, UBAP2L, intellectual disability, developmental delay, Chinese
Citation: Yang Q, Zhang Q, Zhou X, Feng J, Zhang S, Lin L, Yi S, Qin Z and Luo J (2024) Whole-exome sequencing identified a novel heterozygous variant in UBAP2L in a Chinese family with neurodevelopmental disorder characterized by impaired language, behavioral abnormalities, and dysmorphic facies. Front. Genet. 15:1503048. doi: 10.3389/fgene.2024.1503048
Received: 28 September 2024; Accepted: 28 November 2024;
Published: 10 December 2024.
Edited by:
Mara Marongiu, National Research Council (CNR), ItalyReviewed by:
Laura Crisponi, National Research Council (CNR), ItalyCopyright © 2024 Yang, Zhang, Zhou, Feng, Zhang, Lin, Yi, Qin and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingsi Luo, eWFuZ3Fpc2tsbWcxMjZAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.