 Lilit Ghukasyan1†
Lilit Ghukasyan1† Gisane Khachatryan1,2†Tamara Sirunyan1,2†Arpine Minasyan1†Siras Hakobyan3,4Andranik Chavushyan1Varduhi Hayrapetyan1,2Hovsep Ghazaryan1
Gisane Khachatryan1,2†Tamara Sirunyan1,2†Arpine Minasyan1†Siras Hakobyan3,4Andranik Chavushyan1Varduhi Hayrapetyan1,2Hovsep Ghazaryan1 Gevorg Martirosyan1Gohar Mkrtchyan1
Gevorg Martirosyan1Gohar Mkrtchyan1 Valentina Vardanyan5,6Vahan Mukuchyan7Ashot Davidyants8
Valentina Vardanyan5,6Vahan Mukuchyan7Ashot Davidyants8 Roksana Zakharyan1,2*
Roksana Zakharyan1,2* Arsen Arakelyan1,2,3*
Arsen Arakelyan1,2,3*- 1Laboratory of Human Genetics, Institute of Molecular Biology NAS RA, Yerevan, Armenia
- 2Department of Bioengineering, Bioinformatics and Molecular Biology, Institute of Biomedicine and Pharmacy, Russian-Armenian University, Yerevan, Armenia
- 3Research Group of Bioinformatics, Institute of Molecular Biology NAS RA, Yerevan, Armenia
- 4Armenian Bioinformatics Institute, Yerevan, Armenia
- 5Department of Rheumatology, Yerevan State Medical University after Mkhitar Heratsi (YSMU), Yerevan, Armenia
- 6Department of Rheumatology, “Mikaelyan” Institute of Surgery, Yerevan, Armenia
- 7Department of Internal Medicine and Rheumatology, Nairi Medical Center, Yerevan, Armenia
- 8“Gisane” LLC, Davidyants Laboratories, Yerevan, Armenia
Familial Mediterranean Fever (FMF) is a genetic disorder with complex inheritance patterns and genotype-phenotype associations, and it is highly prevalent in Armenia. FMF typically follows an autosomal recessive inheritance pattern (OMIM: 249100), though it can occasionally display a rare dominant inheritance pattern with variable penetrance (OMIM։134610). The disease is caused by mutations in the MEFV gene, which encodes the pyrin protein. While the 26 most prevalent mutations account for nearly 99% of all FMF cases, more than 60 pathogenic mutations have been identified. In this study, we aimed to develop an affordable nanopore sequencing method for full-length MEFV gene mutation detection to aid in the diagnosis and screening of FMF. We employed a multiplex amplicon sequencing approach, allowing for the processing of up to 12 samples on both Flow cells and Flongle flow cells. The results demonstrated near-complete concordance between nanopore variant calling and qPCR genotypes. Moreover, nanopore sequencing identified additional variants, which were confirmed by whole exome sequencing. Additionally, intronic and UTR variants were detected. Our findings demonstrate the feasibility of full-gene nanopore sequencing for detecting FMF-associated pathogenic variants. The method is cost-effective, with costs comparable to those of the qPCR test, making it particularly suitable for settings with limited laboratory infrastructure. Further clinical validation using larger sample cohorts will be necessary.
1 Introduction
Familial Mediterranean fever (FMF) is a genetic autoinflammatory disease characterized by mutations within the MEFV (MEditerranean FeVer) gene that encodes the pyrin protein (French FMF Consortium, 1997; The International FMF Consortium, 1997; Papin et al., 2000).
Despite being considered a rare disease worldwide with an estimate of over 200,000 affected people (Gallego et al., 2023), it is common in populations of Mediterranean descent, including Armenians, Turks, Jews, Greeks, and Italians with a prevalence ranging from 10 to 1,500 per 100,000 population (Cerrito et al., 2015; Alparslan et al., 2020). Moreover, FMF prevalence (13–20/per 100,000) is notable in Japan (Migita et al., 2016), and recently it has also been reported in China (Wu et al., 2018).
As of today, 399 variants in the MEFV gene have been identified and their number is increasing with the development of sequencing approaches (Tufan and Lachmann, 2020). The majority of 63 known pathogenic/likely pathogenic variants are located on exon 10 which encodes the B30.2/SPRY domain, responsible for the activation of caspase-1 (source: Infevers database, https://infevers.umai-montpellier.fr/web/search.php, last accessed 15 August 2024). The most common variant of MEFV is M694V (c.2080A > G), which is also a dominant variant in the Armenian population (Hayrapetyan et al., 2016). Other frequent exon 10 variants are M694I (36.0%), V726A (29.5%), M680I (26.2%), R761H (4.9%). These variants constitute almost 75% of all FMF patients and are associated with more severe disease courses among Mediterranean populations. Up to two-thirds of registered variants distributed over the entire gene are either not classified or classified as variants of uncertain significance (VUS) due to their unknown clinical association (Accetturo et al., 2020).
FMF is primarily an autosomal recessive disorder, however, this pattern is not uniform across all cases, adding complexity to genetic counseling and risk assessment (Manukyan and Aminov, 2016). Many individuals with FMF are compound heterozygotes. Different combinations of mutations exhibit a wide range of FMF phenotypes diverse in their symptoms and intensity, indicating incomplete penetrance (Procopio et al., 2018).
Clinical and research laboratories in Armenia use PCR/qPCR-based approaches to test the presence of the most prevalent 12–26 pathogenic mutations, such as FMF StripAssay (12 mutations, ViennaLabs, Austria) or FMF Multiplex real-time PCR kit (26 mutations SNP Biotechnology, Turkey). The common disadvantages of these kits are the limited scope of mutations tested (12 or 26 of known 63), risks of interference for closely located mutations (for example, M680I (G/C-A), I692del, M694V, K695R) in available qPCR tests, and limited capacity for further multiplexing. These limitations can be overcome with Sanger and next-generation sequencing (NGS). Sanger sequencing is considered a gold standard for accuracy and offers opportunities for studying genetic variability on the scale of an exon or a full gene (Schmidt et al., 2022). However, it has been proven that the costs of Sanger sequencing per sample are very high due to the small target range (∼1,000 nucleotides in a single run) limiting its use in clinical settings (at least in low- and middle-income countries (LMICs), such as Armenia). Furthermore, sequencing a single gene like MEFV using high-throughput Illumina NGS platforms is both technically challenging and costly. The installation and operational costs of these instruments require significant investments in infrastructure and a large number of samples is required per run to make it cost-effective. In contrast, nanopore sequencing (NS) developed by Oxford Nanopore Technologies (ONT) offers a more cost-effective solution, with capital and consumable costs as low as approximately $1,000, making it particularly suitable for use in settings with limited laboratory infrastructure. NS is actively explored for the identification of pathogenic variants in various genetic disorders, such as Familial Hypercholesterolemia (Soufi et al., 2022), tandem repeat disorders (Stevanovski et al., 2022), Werner Syndrome (Miller et al., 2021), sickle cell disease (Christopher et al., 2021), and others (Minervini et al., 2020; Oehler et al., 2023).
In this study, we focused on developing an affordable nanopore-based full-gene sequencing test for detecting MEFV mutations to assist in the diagnosis of FMF.
2 Materials and methods
2.1 Clinical samples
Fourteen patients (5 males and 9 females) were recruited from the Mikaelyan Institute of Surgery and Nairi Medical Center. FMF was diagnosed based on the classification criteria for autoinflammatory recurrent fevers (Gattorno et al., 2019) and molecular genetic analysis of MEFV mutations using a commercially available qPCR kit for the detection of 26 common FMF mutations (FMF Multiplex real-time PCR kit, SNP Biotechnology, Turkey). Two control non-FMF subjects (females) were also recruited from the Institute of Molecular Biology NAS RA. The project was approved by the Ethics Committee of the Institute of Molecular Biology NAS RA (IRB 00004079, Protocol N3 from 23.08.2021).
Five milliliters of morning fasting blood was collected in K3-EDTA tubes. Genomic DNA from 200 μL blood was isolated using QIAamp DNA Blood Mini Kit (QIAGEN, MD, United States), according to the manufacturer’s instructions.
2.2 Primer design and PCR amplification
We used the NCBI RefSeq (LRG_190) locus sequence of the MEFV gene (accession: NG_007871) for primer design. Five pairs of multiplex primers each targeting approximately 4 kb region of the MEFV gene were selected using the primalscheme tool for multiplex primer selection (Quick et al., 2017). Each primer pair generated overlapping amplicons, together spanning approximately 99% of the MEFV gene. This size was chosen to minimize the number of multiplex or singleplex PCR reactions and ensure that common long-range polymerase can effectively amplify the fragments.
Validation qPCR primers were designed to check the presence of all fragments in pooled PCR reactions. Each set of primers targets a region within the MEFV fragment. Primers for qPCR with intercalating dyes were designed using the IDT PrimerQuest tool (Integrated DNA Technologies, Belgium) (Supplementary Table S1).
Amplicons were generated using Q5 Hot Start High-Fidelity 2X Master Mix (New England Biolabs, United States), LongAmp Hot Start Taq 2X Master Mix (New England Biolabs, United States), and BioMaster LR HS-PCR (2x) Mix (Biolabmix, Russia). qPCR validation of the obtained PCR amplicons was performed using HOT FIREPol EvaGreen qPCR Mix Plus (no ROX) (Solis BioDyne, Estonia). PCR reaction mix and cycling conditions are provided in Supplementary Table S2.
The integrity of the amplicons generated using PCR was checked using 1.5% agarose gel electrophoresis for 30 min with 1 Kb Plus DNA Ladder (BioFact, Seoul, Korea). Amplified fragments were visualized using ethidium bromide. Amplicon quantification was performed with a Nanodrop spectrophotometer (Thermo Fisher, MA, United States).
2.3 Sequencing library preparation and sequencing
For each sample, generated amplicons were purified with a 1x ratio of AMPure XP beads (Beckman Coulter, Brea, CA, United States) to exclude small nonspecific fragments, equilibrated by mass and pooled. Sequencing libraries were prepared according to the manufacturer’s (Oxford Nanopore Technologies, Oxford, United Kingdom) protocol 1) using native barcoding kits (EXP-NBD104, EXP-NBD114) in combination with a ligation sequencing kit (SQK-LSK109) or 2) Native Barcoding Kit 24 V14 (SQK-NBD114.24). The final sequencing library amount was 20–50 fmol.
Sequencing was performed on the MinION 1 Mkb device using R9.4 Flow cells or Flongle flow cells and R10.4 Flongle flow cells. The average run duration was 12–18 h.
2.4 Sequencing data analysis and variant calling
Raw sequencing data was basecalled and filtered with guppy basecaller (version 6.0.1) with a “high accuracy model” parameter. Basecalled data was then demultiplexed for downstream analysis. The alignment was done with a minimap2 aligner (version 2.24-r1122) the ONT genomic reads option on the reference sequence of the MEFV gene (accession: NG_007871) downloaded from the RefSeq (LRG_190) NCBI database (Li, 2018). BAM files were sorted and indexed for further exploration in integrative genomic viewer and variant calling analysis using samtools 1.13 (Li et al., 2009). Variant calling of the aligned file was done with longshot 1.0.0 software for variant detection from error-prone reads (Edge and Bansal, 2019). Another tool used for variant calling was P.E.P.P.E.R. (r.08) which uses recurrent neural networks for variant detection from long read data (Shafin et al., 2021). The resulting variants were annotated with the Open Custom Ranked Analysis of Variants Toolkit (openCRAVAT) (Pagel et al., 2020). The presence/absence of a variant was further confirmed by examination of BAM files using Integrated Genome Browser v2.12.3.
2.5 Whole exome sequencing
Whole exome sequencing (WES) of the same DNA samples was performed by a commercial sequencing provider (Macrogen, South Korea). Libraries were prepared with Agilent SureSelect V5 (Agilent Technologies, CA, United States) whole exome sequencing library preparation kit. Sequencing was performed on the Illumina Novaseq 6000 instrument (Illumina, SD, United States). Variant calling was performed with the Genome Analysis Toolkit v4.3 based on published best practices (Van der Auwera et al., 2013).
2.6 Performance analysis
Accuracy, sensitivity, and specificity analysis were performed as described elsewhere (Bartol, 2015) using the caret R package. qPCR results on mutation genotypes were considered as ground truth.
3 Results
A total of 14 patients (5 males and 9 females) and 2 healthy controls (2 females) underwent full MEFV gene nanopore sequencing (NS). The DNA samples were sequenced with library preparation using R9 or R10 chemistry on both Flow cells and Flongle flow cells. Additionally, whole genome sequencing data generated on the Illumina platform was available for some of the samples (see Table 1).
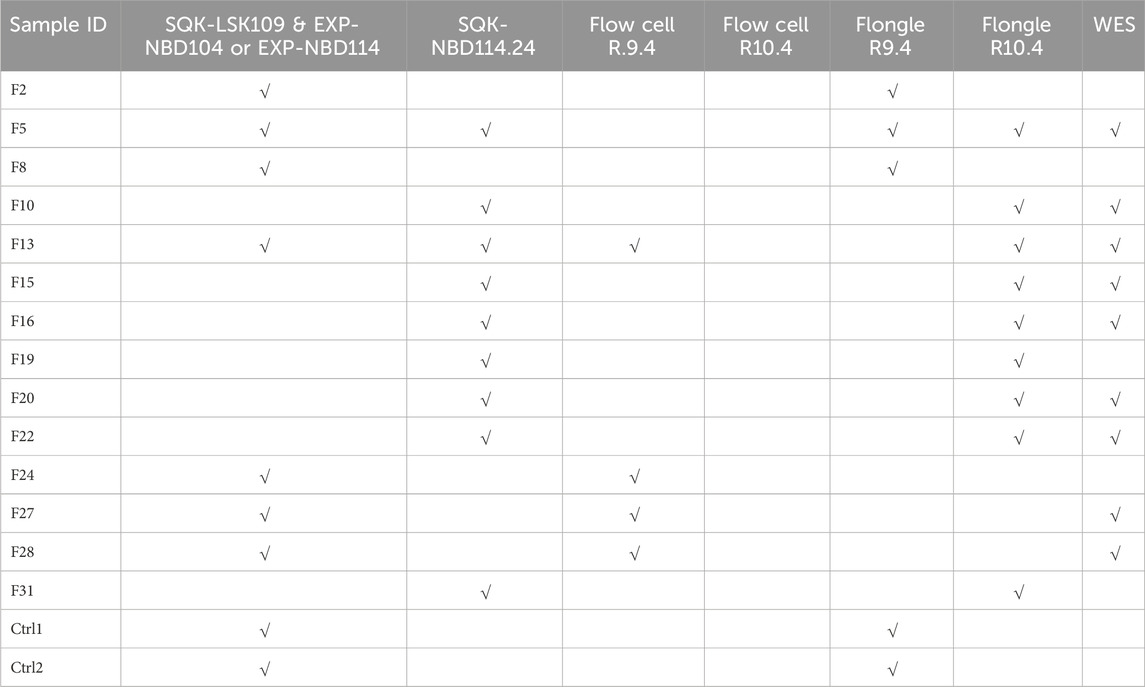
Table 1. Combinations of sample preps and flow cells for nanopore sequencing of full-length MEFV.
Nanopore sequencing statistics are shown in Figure 1. Using five tiling fragments, we achieved near-complete coverage of the full-length MEFV gene with balanced sequencing depth overall amplicons. The detailed statistics for individual runs are provided in Supplementary Data Sheet S1. The comparison of variants detected by NS and qPCR demonstrated a high level of concordance (Table 2). We performed a performance analysis of NS variant calling using qPCR results as the ground truth. Compared with qPCR the balanced accuracy of NS detection was 0.97 with a sensitivity of 0.93, specificity of 1.00, and F1 score of 0.97. From three NS replicates, two showed complete concordance of detected variants (samples F5 and F8). In one replicate of the F13 sample M694V was detected in a heterogeneous state (with V10 chemistry and R9.4 flow cell), while in the other replicate (with V14 chemistry and R10.4 flongle flow cell), the M694V variant was detected in a homozygous state concordant with qPCR results. Furthermore, in another sample (F19) additional pathogenic mutation V726A in a heterogeneous state was detected. Unfortunately, we did not have WES data for this sample to confirm the NS or qPCR results (Table 2).
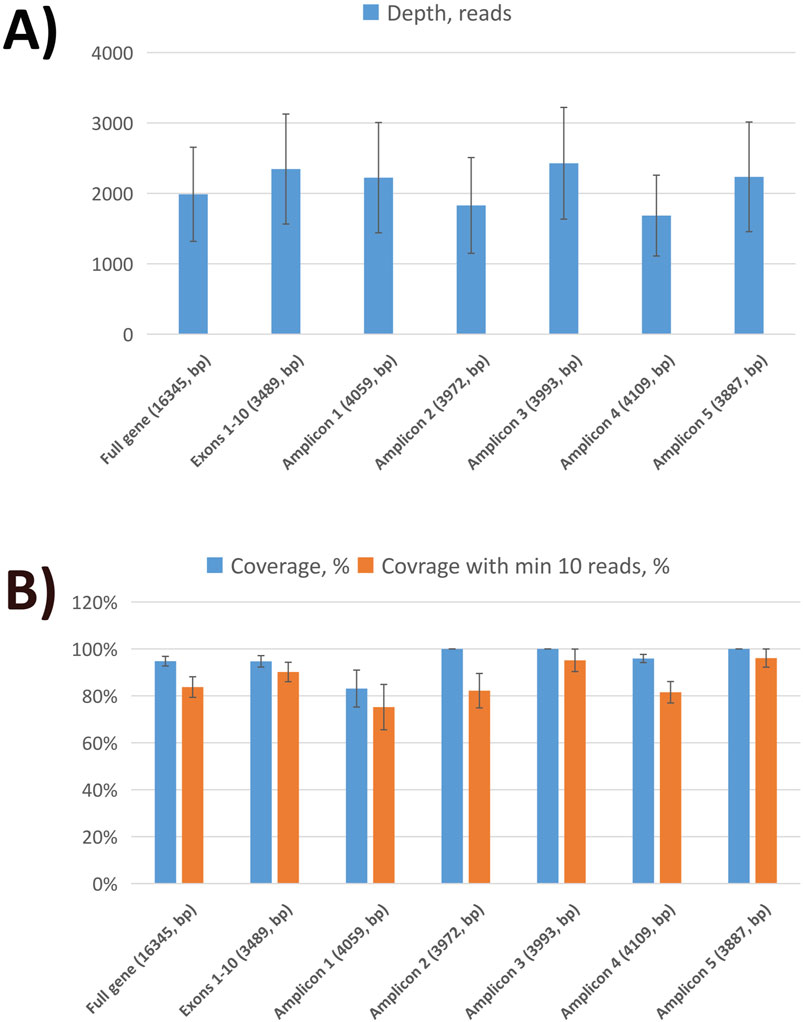
Figure 1. Sequencing statistics of MEFV gene using nanopore sequencing. (A) The average sequencing depth of full MEFV gene, exons, and individual amplicons. (B) Sequencing coverage of full MEFV gene, exons, and individual amplicons.
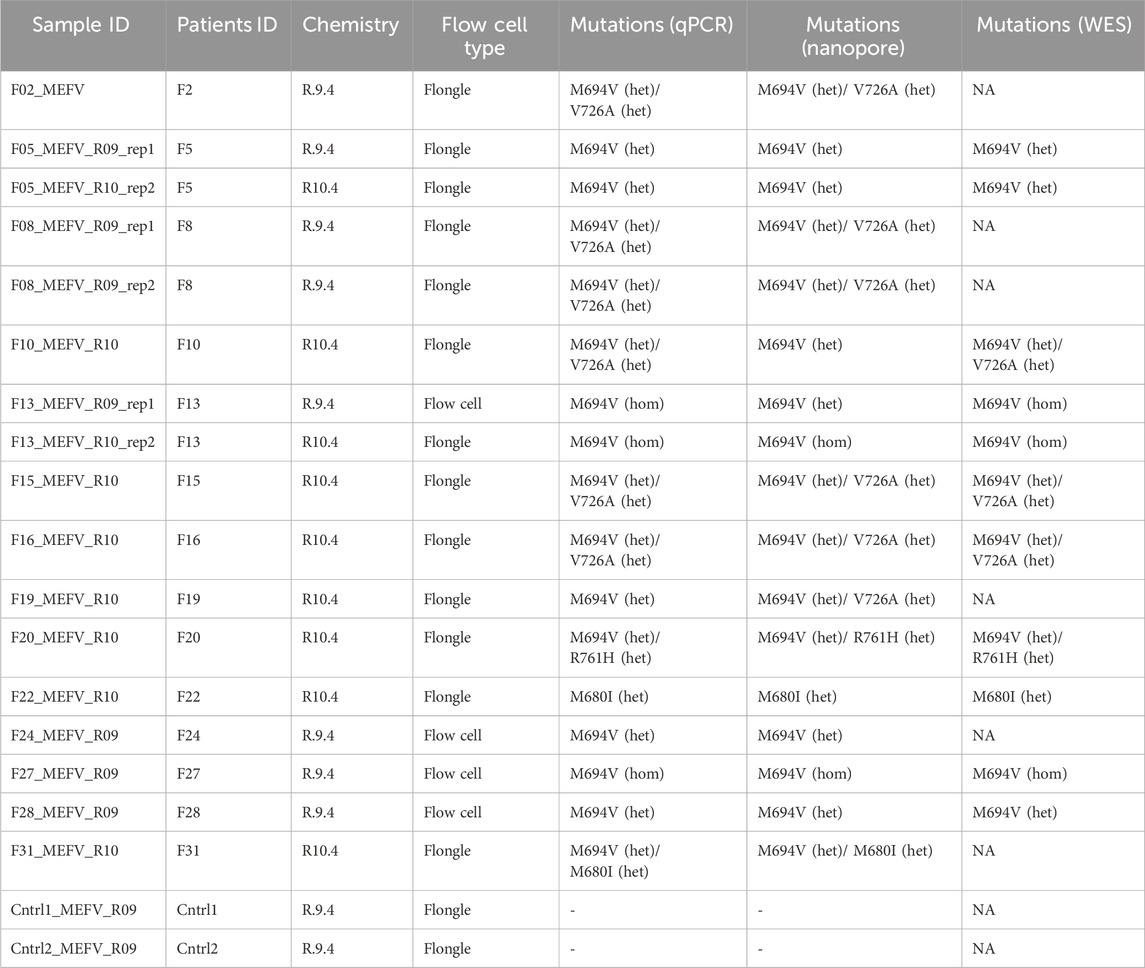
Table 2. Comparison of variant detection of full-gene nanopore sequencing, qPCR (ground truth), and WES.
Moreover, both NS and WES showed additional variants outside the scope of the PCR mutation panel (Supplementary Data Sheet S2). For example, both WES and NS identified the R202Q (Supplementary Figure S2). In addition, 19 intronic, 5′- and 3′- UTR variants were discovered with NS (Supplementary Data Sheet S2).
4 Discussion
We present a protocol for full MEFV gene sequencing with accuracy comparable to qPCR for detecting common variants. Unlike qPCR, our method is not restricted to a predefined set of variants or locations and can discover additional clinically relevant variants that qPCR will miss. In contrast to WES, it enables the detection of variants not only in exons but also in intronic and regulatory regions. Although all variants observed in noncoding variants in the 3′UTR and intronic regions were previously annotated in ClinVar and classified as benign (see Supplementary Data Sheet S2) this also shows a possibility to identify pathogenic mutations located in non coding regions of the gene such as rs773992396 G>C located in intron 6 (Balta et al., 2020). Furthermore, the sequencing protocol enables more in-depth studies to elucidate the role of coding variants in disease. For instance, while the R202Q variant is classified as benign, recent studies have highlighted its potential association with an inflammatory FMF phenotype (Kandur et al., 2022). Our protocol has also proven effective with new sequencing chemistry and Flongle flow cells. The latter, especially when used with 12-sample multiplexing, makes full-length MEFV gene sequencing both cost- and time-efficient, with results obtainable within 12–24 h including bioinformatics analysis and report generation. Additionally, tree samples were analyzed in replicates, and the variant calling results were consistent across different nanopore sequencing chemistries, aligning with the qPCR genotyping results. In one sample (F13), the correct genotype was detected using the new V14 but not the V10 chemistry suggesting that switching to the latest flow cells and library preparation kits may enhance genotyping accuracy. These observations may point out that, in clinical settings, it may be beneficial to conduct replicates during the initial phase to gather sufficient data for assessing genotyping consistency and intra-sample variability before deciding to switch to genotyping based on a single sample. Furthermore, the R9.4 flow cells and associated sample preparation reagents were discontinued in 2023-2024, leaving V14 chemistry and R10.4 flow cells as the only available option. The cost of full MEFV gene sequencing for 12 samples on a Flongle flow cell is approximately 30 USD, which is comparable to, and often less expensive than, the 26-mutation qPCR panel commonly used in clinical labs in Armenia. With 1 Gb output (2.5 Gb theoretical output according to the manufacturer), 12-sample multiplexing can yield up to 6,000x sequencing depth per sample. The SQK-NBD114.24 kit supports up to 24-sample multiplexing, which could theoretically provide around 3,000x depth per sample - still adequate for variant detection. Additionally, the SQK-NBD114.96 library preparation kit allows for 96-sample multiplexing, making it a viable option for large-scale screening. However, with smaller sample flows, using 24- or 96-sample multiplexing may significantly increase turnaround time. Finally, compared to the Illumina sequencing platform, our protocol offers greater flexibility in the number of samples required for cost-effective multiplexing.
Our method significantly improves upon the protocol proposed by Schmidt et al. (2022), which uses 8 pairs of primers targeting only the exons of the gene, against only 5 primer pairs to target the full gene. The accuracy of variant calling can be further enhanced by switching the basecalling mode to “super accurate”. Although this would require more computational resources and time, it has been shown to outperform default basecalling settings (Ni et al., 2023).
The small sample size is the main limitation of our study. Furthermore, the amount of DNA samples was not sufficient to run NS for all samples in replicates. Nonetheless, our findings, along with previous results (Schmidt et al., 2022), warrant the applicability of full-gene nanopore sequencing of MEFV in clinical settings. Furthermore, even with this small sample, we were able to collect patients with diverse genotype combinations (heterozygotes for a single allele, compound heterozygotes, and homozygotes), which could mitigate potential sampling bias.
In conclusion, we present a cost-effective nanopore sequencing-based protocol for MEFV gene sequencing, making it particularly suitable for settings with limited laboratory infrastructure. It can enable widespread and accurate detection of FMF-related mutations, thus supporting public health initiatives aimed at controlling the prevalence of FMF, particularly in LMICs of endemic regions. This protocol could also lead to the discovery of novel pathogenic variants within the MEFV gene, expanding our knowledge of FMF genetics.
Data availability statement
The data presented in the study are deposited in the NCBI Sequence Read Archive repository, accession number SRP544892.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Institute of Molecular Biology NAS RA (IRB 00004079). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
LG: Funding acquisition, Investigation, Validation, Writing–original draft. GK: Investigation, Validation, Writing–original draft. TS: Investigation, Validation, Writing–original draft. AM: Investigation, Validation, Writing–original draft. SH: Data curation, Formal Analysis, Software, Writing–original draft. AC: Investigation, Methodology, Supervision, Validation, Writing–original draft. VH: Investigation, Supervision, Validation, Writing–original draft. HG: Investigation, Supervision, Validation, Writing–original draft. GeM: Investigation, Supervision, Validation, Writing–original draft. GoM: Supervision, Writing–original draft, Investigation, Validation. VV: Resources, Writing–original draft. VM: Resources, Writing–original draft. AD: Writing–review and editing, Resources. RZ: Conceptualization, Funding acquisition, Project administration, Supervision, Writing–original draft, Writing–review and editing. AA: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Higher Education and Science Committee (former: Science Committee) of the MESCS of Armenia (21SCG-1F010 to RZ, 23EDP-1F009 to AA, 23AA-1F035 to LGh) and a research grant provided by the Armenian Engineers and Scientists of America (AESA) to RZ.
Acknowledgments
The author used OpenAI’s GPT-4.0 for revision of grammar and style of the parts of the text. Upon using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Conflict of interest
Author AD was employed by “Gisane” LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1493295/full#supplementary-material
References
Accetturo, M., D’Uggento, A. M., Portincasa, P., and Stella, A. (2020). Improvement of MEFV gene variants classification to aid treatment decision making in familial Mediterranean fever. Rheumatol. Oxf. Engl. 59, 754–761. doi:10.1093/rheumatology/kez332
Alparslan, Ö., Egeli, B. H., Demirel, Y., and Uğurlu, S. (2020). The prevalence of familial mediterranean fever and behçet’s disease: a cross-sectional study. Arch. Rheumatol. 35, 609–613. doi:10.46497/ArchRheumatol.2020.7769
Balta, B., Erdogan, M., Kiraz, A., Akalın, T., Baştug, F., and Bayram, A. (2020). A comprehensive molecular analysis and genotype-phenotype correlation in patients with familial mediterranean fever. Mol. Biol. Rep. 47, 1835–1843. doi:10.1007/s11033-020-05277-x
Bartol, T. (2015). Thoughtful use of diagnostic testing: making practical sense of sensitivity, specificity, and predictive value. Nurse Pract. 40, 10–12. doi:10.1097/01.NPR.0000470366.64804.35
Cerrito, L., Sicignano, L. L., Verrecchia, E., and Manna, R. (2015). “Epidemiology of FMF worldwide,” in Familial mediterranean fever. Editor M. Gattorno (Cham: Springer International Publishing), 81–90. doi:10.1007/978-3-319-14615-7_5
Christopher, H., Burns, A., Josephat, E., Makani, J., Schuh, A., and Nkya, S. (2021). Using DNA testing for the precise, definite, and low-cost diagnosis of sickle cell disease and other Haemoglobinopathies: findings from Tanzania. BMC Genomics 22, 902. doi:10.1186/s12864-021-08220-x
Edge, P., and Bansal, V. (2019). Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 10, 4660. doi:10.1038/s41467-019-12493-y
French FMF Consortium (1997). A candidate gene for familial Mediterranean fever. Nat. Genet. 17, 25–31. doi:10.1038/ng0997-25
Gallego, E., Arias-Merino, G., Sánchez-Díaz, G., Villaverde-Hueso, A., Posada de la Paz, M., and Alonso-Ferreira, V. (2023). Familial mediterranean fever in Spain: time trend and spatial distribution of the hospitalizations. Int. J. Environ. Res. Public. Health 20, 4374. doi:10.3390/ijerph20054374
Gattorno, M., Hofer, M., Federici, S., Vanoni, F., Bovis, F., Aksentijevich, I., et al. (2019). Classification criteria for autoinflammatory recurrent fevers. Ann. Rheum. Dis. 78 (8), 1025–1032. doi:10.1136/annrheumdis-2019-215048
Hayrapetyan, H. S., Sarkisian, T. F., and Atoyan, S. A. (2016). Risk of FMF development among heterozygous patients in Armenian population. Proc. YSU B Chem. Biol. Sci. 50, 48–52. doi:10.46991/PYSU:B/2016.50.3.048
Kandur, Y., Kocakap, D. B. S., Alpcan, A., and Tursun, S. (2022). Clinical significance of MEFV gene variation R202Q. Clin. Rheumatol. 41, 271–274. doi:10.1007/s10067-021-05906-1
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinforma. Oxf. Engl. 34, 3094–3100. doi:10.1093/bioinformatics/bty191
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinforma. Oxf. Engl. 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Manukyan, G., and Aminov, R. (2016). Update on pyrin functions and mechanisms of familial mediterranean fever. Front. Microbiol. 7, 456. doi:10.3389/fmicb.2016.00456
Migita, K., Izumi, Y., Jiuchi, Y., Iwanaga, N., Kawahara, C., Agematsu, K., et al. (2016). Familial Mediterranean fever is no longer a rare disease in Japan. Arthritis Res. Ther. 18, 175. doi:10.1186/s13075-016-1071-5
Miller, D. E., Sulovari, A., Wang, T., Loucks, H., Hoekzema, K., Munson, K. M., et al. (2021). Targeted long-read sequencing identifies missing disease-causing variation. Am. J. Hum. Genet. 108, 1436–1449. doi:10.1016/j.ajhg.2021.06.006
Minervini, C. F., Cumbo, C., Orsini, P., Anelli, L., Zagaria, A., Specchia, G., et al. (2020). Nanopore sequencing in blood diseases: a wide range of opportunities. Front. Genet. 11, 76. doi:10.3389/fgene.2020.00076
Ni, Y., Liu, X., Simeneh, Z. M., Yang, M., and Li, R. (2023). Benchmarking of Nanopore R10.4 and R9.4.1 flow cells in single-cell whole-genome amplification and whole-genome shotgun sequencing. Comput. Struct. Biotechnol. J. 21, 2352–2364. doi:10.1016/j.csbj.2023.03.038
Oehler, J. B., Wright, H., Stark, Z., Mallett, A. J., and Schmitz, U. (2023). The application of long-read sequencing in clinical settings. Hum. Genomics 17, 73. doi:10.1186/s40246-023-00522-3
Pagel, K. A., Kim, R., Moad, K., Busby, B., Zheng, L., Tokheim, C., et al. (2020). Integrated informatics analysis of cancer-related variants. JCO Clin. Cancer Inf. 4, 310–317. doi:10.1200/CCI.19.00132
Papin, S., Duquesnoy, P., Cazeneuve, C., Pantel, J., Coppey-Moisan, M., Dargemont, C., et al. (2000). Alternative splicing at the MEFV locus involved in familial Mediterranean fever regulates translocation of the marenostrin/pyrin protein to the nucleus. Hum. Mol. Genet. 9, 3001–3009. doi:10.1093/hmg/9.20.3001
Procopio, V., Manti, S., Bianco, G., Conti, G., Romeo, A., Maimone, F., et al. (2018). Genotype-phenotype correlation in FMF patients: a “non classic” recessive autosomal or “atypical” dominant autosomal inheritance? Gene 641, 279–286. doi:10.1016/j.gene.2017.10.068
Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith, A. D., Gangavarapu, K., et al. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276. doi:10.1038/nprot.2017.066
Schmidt, J., Berghaus, S., Blessing, F., Herbeck, H., Blessing, J., Schierack, P., et al. (2022). Genotyping of familial Mediterranean fever gene (MEFV)-Single nucleotide polymorphism-Comparison of Nanopore with conventional Sanger sequencing. PloS One 17, e0265622. doi:10.1371/journal.pone.0265622
Shafin, K., Pesout, T., Chang, P.-C., Nattestad, M., Kolesnikov, A., Goel, S., et al. (2021). Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat. Methods 18, 1322–1332. doi:10.1038/s41592-021-01299-w
Soufi, M., Bedenbender, S., Ruppert, V., Kurt, B., Schieffer, B., and Schaefer, J. R. (2022). Fast and easy nanopore sequencing workflow for rapid genetic testing of familial hypercholesterolemia. Front. Genet. 13, 836231. doi:10.3389/fgene.2022.836231
Stevanovski, I., Chintalaphani, S. R., Gamaarachchi, H., Ferguson, J. M., Pineda, S. S., Scriba, C. K., et al. (2022). Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing. Sci. Adv. 8, eabm5386. doi:10.1126/sciadv.abm5386
The International FMF Consortium (1997). Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell 90, 797–807. doi:10.1016/s0092-8674(00)80539-5
Tufan, A., and Lachmann, H. J. (2020). Familial Mediterranean fever, from pathogenesis to treatment: a contemporary review. Turk. J. Med. Sci. 50, 1591–1610. doi:10.3906/sag-2008-11
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.1–11. doi:10.1002/0471250953.bi1110s43
Keywords: familial mediterranean fever, FMF, MEFV, nanopore sequencing, variant detection, genetic testing
Citation: Ghukasyan L, Khachatryan G, Sirunyan T, Minasyan A, Hakobyan S, Chavushyan A, Hayrapetyan V, Ghazaryan H, Martirosyan G, Mkrtchyan G, Vardanyan V, Mukuchyan V, Davidyants A, Zakharyan R and Arakelyan A (2024) Genewise detection of variants in MEFV gene using nanopore sequencing. Front. Genet. 15:1493295. doi: 10.3389/fgene.2024.1493295
Received: 08 September 2024; Accepted: 11 November 2024;
Published: 29 November 2024.
Edited by:
Roberto Valli, University of Insubria, ItalyReviewed by:
Annalisa Frattini, National Research Council (CNR), ItalyArif Engin Cetin, Dokuz Eylul University, Türkiye
Copyright © 2024 Ghukasyan, Khachatryan, Sirunyan, Minasyan, Hakobyan, Chavushyan, Hayrapetyan, Ghazaryan, Martirosyan, Mkrtchyan, Vardanyan, Mukuchyan, Davidyants, Zakharyan and Arakelyan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arsen Arakelyan, YXJha2VseWFuYWFAc2NpLmFt; Roksana Zakharyan, cm9rc2FuYS56YWtoYXJ5YW5AcmF1LmFt
†These authors have contributed equally to this work and share first authorship