 Yan Zhao
Yan Zhao Lina Song1†
Lina Song1† Shan Shan
Shan Shan Hua Jin
Hua Jin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 15 October 2024
Sec. Molecular Cytogenetics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1486974
Background: The clinical phenotypes of 16p13.11 microduplication syndrome have been extensively reported in previous studies, mostly about adults and children, with limited information available on fetal cases. This study aims to explore the genotype-phenotype correlation of fetuses with 16p13.11 microduplication syndrome and analyze the characteristics of prenatal diagnosis indications and provide clinical information for prenatal and postnatal genetic counseling.
Methods: We conducted a retrospective analysis of 3,451 pregnant women who underwent invasive prenatal diagnosis for SNP array between January 2018 and December 2022 at the Jinan Maternal and Child Health Hospital. Descriptive statistical analysis was performed on the prenatal diagnosis indications, pedigree analysis, pregnancy outcomes and postnatal follow-up of 15 fetuses with 16p13.11 microduplication syndrome.
Results: SNP array revealed that 15 fetuses had duplications in the 16p13.11 region with varying prenatal diagnosis indications. Among the cases, 6/15 exhibited ultrasound abnormalities, 5/15 had abnormal chromosomal copy number variations as indicated by non-invasive prenatal testing (NIPT), one case involved advanced maternal age, and 3/15 had other abnormalities. 16p13.11 microduplication syndrome was closely related to ultrasound abnormalities, especially structural abnormalities and soft marker anomalies (abnormal ultrasonic soft indicators), while the indication of NIPT could improve the detection rate of copy number variations (CNVs) in this region. Only 7/15 fetuses underwent pedigree verification, with one case of de novo 16p13.11 microduplication, and the others inherited from one parent. Pregnancy was terminated in 2/15 cases and the outcome of one case is unknown due to loss to follow-up. Among the remaining cases, only one case exhibited a ventricular septal defect, while another presented with omphalocele. No other obvious abnormalities were reported postnatally.
Conclusion: The prenatal phenotypes of fetuses with 16p13.11 microduplication were highly associated with ultrasound abnormalities but lacked specificity. Comprehensive genetic tracing, outcome analysis, and follow-up are essential for providing accurate prenatal and postnatal genetic counseling.
16p13.11 microduplication is a rare genetic disorder with incomplete penetrance and variable expressivity, associated with a range of neurodevelopmental and congenital disorders such as developmental delay, intellectual disability, language deficits, behavioral abnormalities, dysmorphic facial features, attention deficit hyperactivity disorder (ADHD), autism, and cardiovascular anomalies (Hamad et al., 2023). The 16p13.11 microduplication is a susceptibility factor for various neurodevelopmental disorders, though its molecular mechanisms, and candidate genes remain unclear (Watson et al., 2014). Chromosome 16 is rich in segmental duplications (SDs), also known as low copy repeats (LCRs), which account for more than 10% of its euchromatic content (Loftus et al., 1999; Martin et al., 2004; Ramalingam et al., 2011). These chromosome rearrangements are an important cause leading to neurodevelopmental disorders (Ingason et al., 2011).
The pathogenicity of 16p13.11 microdeletion has been well-established, yet the clinical significance of 16p13.11 microduplication remains controversial (Allach El Khattabi et al., 2020). Ullmann (Ullmann et al., 2007) firstly reported the pathogenicity of 16p13.1 microduplication, primarily linking it to intellectual disability and autism spectrum disorders. While Hannes (Hannes et al., 2009) identified recurrent reciprocal duplication of 16p13.1 was a common population variant (5/1682, 0.29%) suggesting that it may be a rare benign variation. In recent years, numerous copy number variations (CNVs) have been detected resulting from the widespread clinical application of chromosome microarray analysis (CMA). It is now broadly accepted that 16p13.1 microduplication may be associated with cognitive impairment, developmental delay, behavioral abnormalities, delayed language development, schizophrenia, and a variety of congenital anomalies, including cardiovascular abnormalities, skeletal abnormalities, aortic disease, renal and urinary system malformations (Kirov et al., 2009; Gumuslu et al., 2015; Erhart et al., 2018; Calderoni et al., 2020). However, individuals with normal phenotypes and unaffected relatives have also been identified, that complicating the interpretation of clinical significance of 16p13.11 microduplications in the context of prenatal genetic counseling, particularly.
Notably, most previous literatures on 16p13.11 microduplications have focused on adults and children, but limited data available on prenatal fetal phenotypes. In this study, 15 fetuses with recurrent 16p13.11 microduplication were identified using single-nucleotide polymorphism microarray (SNP array) technology. We retrospectively analyzed the prenatal diagnosis indications, parental origin, pregnancy outcome and follow-up results of fetus with 16p13.11 microduplication syndrome to further explore the genotype-phenotype correlations and elucidate the underlying genetic mechanisms of 16p13.11 microduplication syndrome.
This retrospective analysis was conducted on data from 3,451 pregnant women who underwent invasive prenatal diagnosis by SNP array testing at the Prenatal Diagnosis Center of Jinan Maternal and Child Health Hospital between January 2018 and December 2022. Only 15 cases with 16p13.11 microduplication and no other pathogenic CNVs were included in this study. The mean age of the pregnant women was 30.5 years (range: 21–42 years), and the mean gestational age was 21 weeks (range: 18–27 weeks). All participants underwent transabdominal amniocentesis after signing informed consent and received genetic counseling from professional genetic doctor. This study was approved by the Medical Ethics Committee of Jinan Maternal and Child Health Hospital.
Amniotic fluid samples from the pregnant women or peripheral blood from the couples were collected between 18 and 24 weeks of gestation following informed consent. DNA extraction was performed using the Tiangen (Beijing) Whole Blood/Tissue Genomic DNA Extraction Kit (Catalog No: DP304) and adhering strictly to the kit’s protocol. Genomic DNA was quantified using the NanoDrop 2000 spectrophotometer (Thermo Scientific) and stored at −20°C.
Whole-genome DNA were processed through digestion, ligation, polymerase chain reaction (PCR), purification, fragmentation, labeling and hybridization steps using the Affymetrix CytoScan750K array. Data calculation and interpretation were carried out using Chromosome Analysis Suite (ChAS) version 4.0.
The pathogenicity of the identified CNVs was further evaluated based on relevant databases. The following databases were mainly referred to: International public Database of Genomic Variants (DGV) (http://dgv.tcag.ca/dgv/app/home), DECIPHER Pathological Variation Database (http://decipher.sanger.ac.uk/browser), the International Standards for Cytogenomic Arrays Consortium (ISCA) (https://www.iscaconsortium.org/), the Online Mendelian Inheritance in Man (OMIM) (http://www.omim.org) and the Clinical Genome Resource (GlinGen) (http://www.clinicalgenome.org). Additionally, GENE, PubMed, and other relevant sources were consulted for further insights.
The variation classification of CNVs pathogenicity followed the guidelines set forth by the American College of Medical Genetics and Genomics (ACMG, 2019). The CNVs were scored according to the following criteria:
①: Pathogenic: 0.99 points or higher, ②Likely Pathogenic: 0.90 to 0.98 points, ③Uncertain Significance: −0.89 to 0.89 points, ④Likely Benign: −0.90 to −0.98 points, ⑤Benign: −0.99 points or lower.
All fetuses underwent regular prenatal examinations with dynamic monitoring of fetal growth and development via ultrasound. Pregnancy outcomes and neonatal health status were obtained through telephone follow-up. For live births, regular telephone follow-ups were conducted to record the child’s growth, development milestones and language development. The most recent follow-up was completed in May 2023.
SNP array analysis identified duplications in the 16p13.11 region in 15 fetuses (Table 1). The size of fragments ranged from 0.79 to 2.82 Mb, involved 7 to 13 OMIM genes, five of which were morbid genes of clinical relevant diseases (Table 2), The duplications either overlapped or flanked the typical 1.65 Mb 16p13.11 microduplication (chr16:14986684-1648668, GRCh37). These duplications spanned different regions within the single-copy sequence interval ((I, II, III). Out of 15 cases, 9 exhibited typical duplications covering intervals I and II, while cases 7 and 15 showed atypical duplications encompassing intervals II and III. Notably, Interval II was present in all cases (Figure 1).
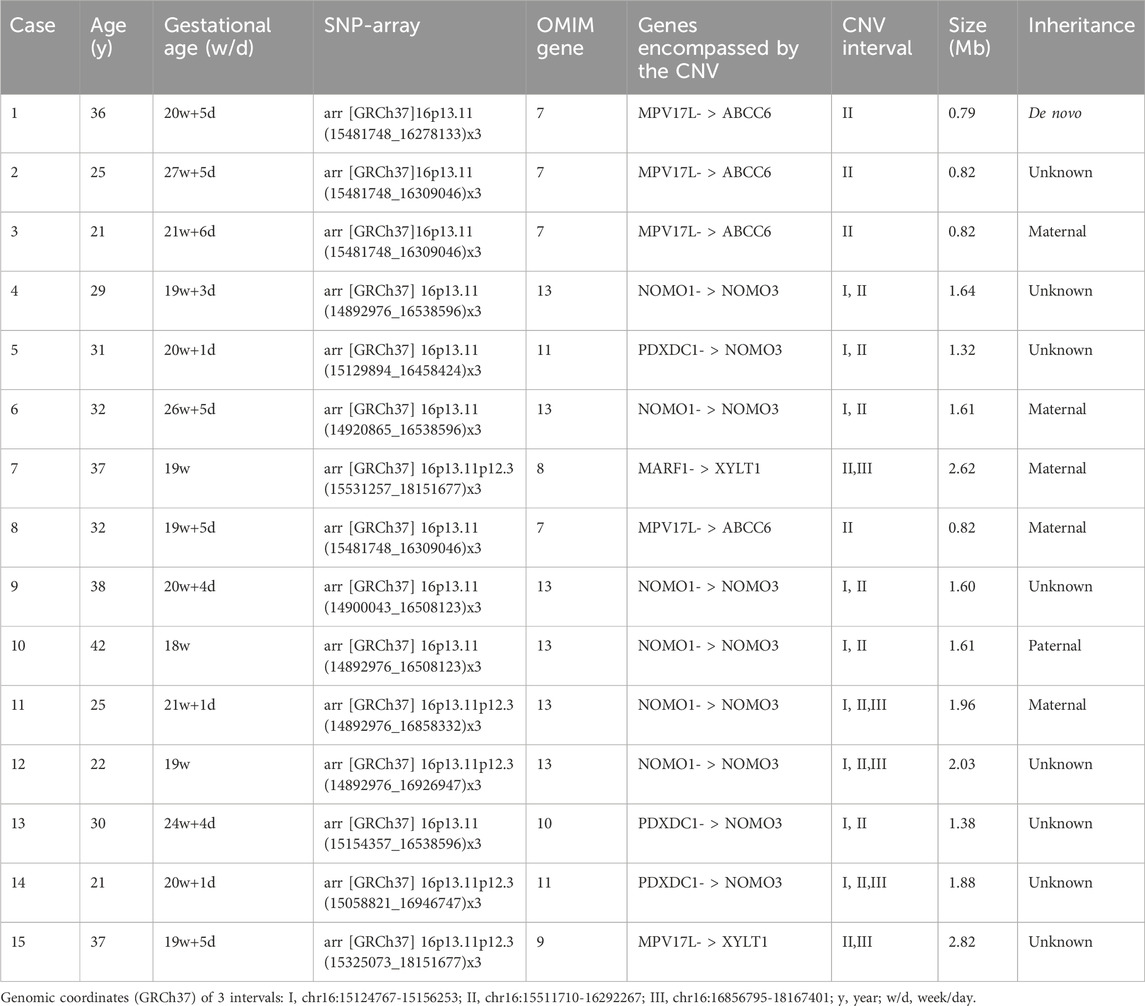
Table 1. SNP array of 15 fetuses with 16p13.11 microduplication.
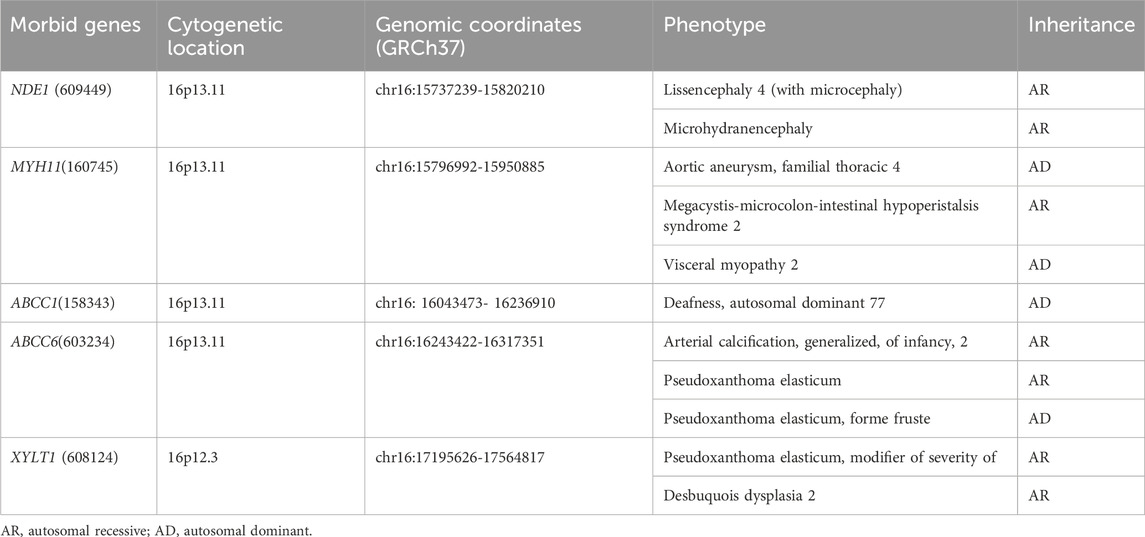
Table 2. Clinical relevant OMIM Morbid genes of 15 fetuses with 16p13.11 microduplication.
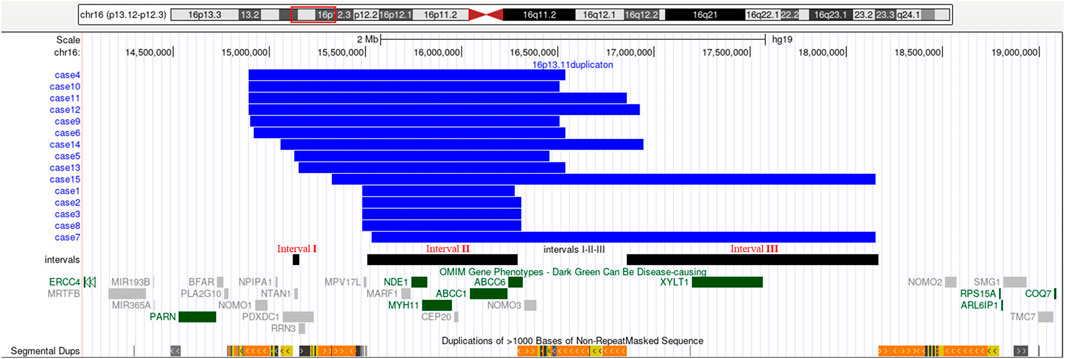
Figure 1. 16p13.11 microduplication detected using SNP-array. SNP-array revealed 16p13.11 microduplication in fetus case1∼case15. Refer to the intervals I, II and III marked in the figure to determine the location of the 16p13.11 duplication breakpoints in 15 fetuses. Case 1, 2, 3, 8 have smaller typical duplicatons encompassed interval II, while Case 7, 15 have larger atypical duplicatons encompassed interval II and III. The breakpoints of case 4, 5 ,6, 9, 10, 13 encompassed interval I and II, and the breakpoints of case 11, 12, 14 encompassed interval I, II and III. Note: The red rectangle in the chromosome diagram marks the location of 16p13.11. The blue bars indicate the location of duplications in the 16p13.11 region for 15 fetuses. The black bars, from left to right, represent intervals I, II and III respectively. OMIM genes are also labeled, dark green indicates clinical relevant OMIM Morbid genes, and gray indicates other OMIM genes.
The copy number variations (CNVs) in the 16p13.11 region of 15 fetuses were analyzed using public databases. No similar variation reports were included in the DGV database. The DECIPHER database contains multiple cases classified as pathogenic and likely pathogenic, as well as reports of similar variants whose clinical significance remains unclear. According to the ClinGen database, the duplication covers nearly the entire area of the 16p13.11 recurrent region (BP2-BP3, includes MYH11), with a triplosensitivity score of 2, indicating some evidence of pathogenicity. Previous literatures have identified this region as a susceptibility locus for neurocognitive disorders. Duplications in this region have been associated with autism, intellectual developmental delay, and various congenital developmental abnormalities (Ullmann et al., 2007; Mefford et al., 2009). Clinical presentations are generally nonspecific such as developmental delay, intellectual disability, learning difficulties, though asymptomatic carriers have also been reported (Kendall et al., 2019; Allach El Khattabi et al., 2020). The ClinVar database also reports pathogenic and uncertain variants associated with duplications in this region. Based on the ACMG Variant Classification Guidelines (2019), pathogenicity scores ranged from −0.89 to 0.89 points indicating that the clinical significance of these duplications remains uncertain.
Unfortunately, the parents of 8 fetuses declined pedigree verification. In the remaining 7 fetuses that underwent family validation, one case was de novo (1/7), and the remaining 6 cases were inherited from their parents (6/7). Of these, 5 were maternally inherited (5/7), one case was paternally inherited (1/7) with all parents being asymptomatic carriers (Table 1).
Among the prenatal diagnosis indications for fetuses carrying 16p13.11 microduplication, the most common was ultrasound abnormalities which accounted for 6 of the 15 cases. These abnormalities included structural anomalies (3/15) and soft marker anomalies/abnormal ultrasonic soft indicators (3/15). Structural abnormalities included 1 case of ventricular septal defect, 1 case of omphalocele and 1 case of right kidney agenesis. The soft marker anomalies included 1 case of bilateral choroid plexus cysts combined with a single umbilical artery, 1 case of single umbilical artery and 1 case of unilateral choroid plexus cyst combined with advanced maternal age. NIPT suggested the presence of chromosomal abnormalities in 5 of the 15 cases, with 3 cases showing microduplication of chromosome 16 and 2 cases indicating other chromosomal abnormalities. It is worth noting that advanced maternal age, as a significant indication for prenatal diagnosis, account for 5 of the 15 fetuses carrying 16p13.11 microduplications (Table 3).
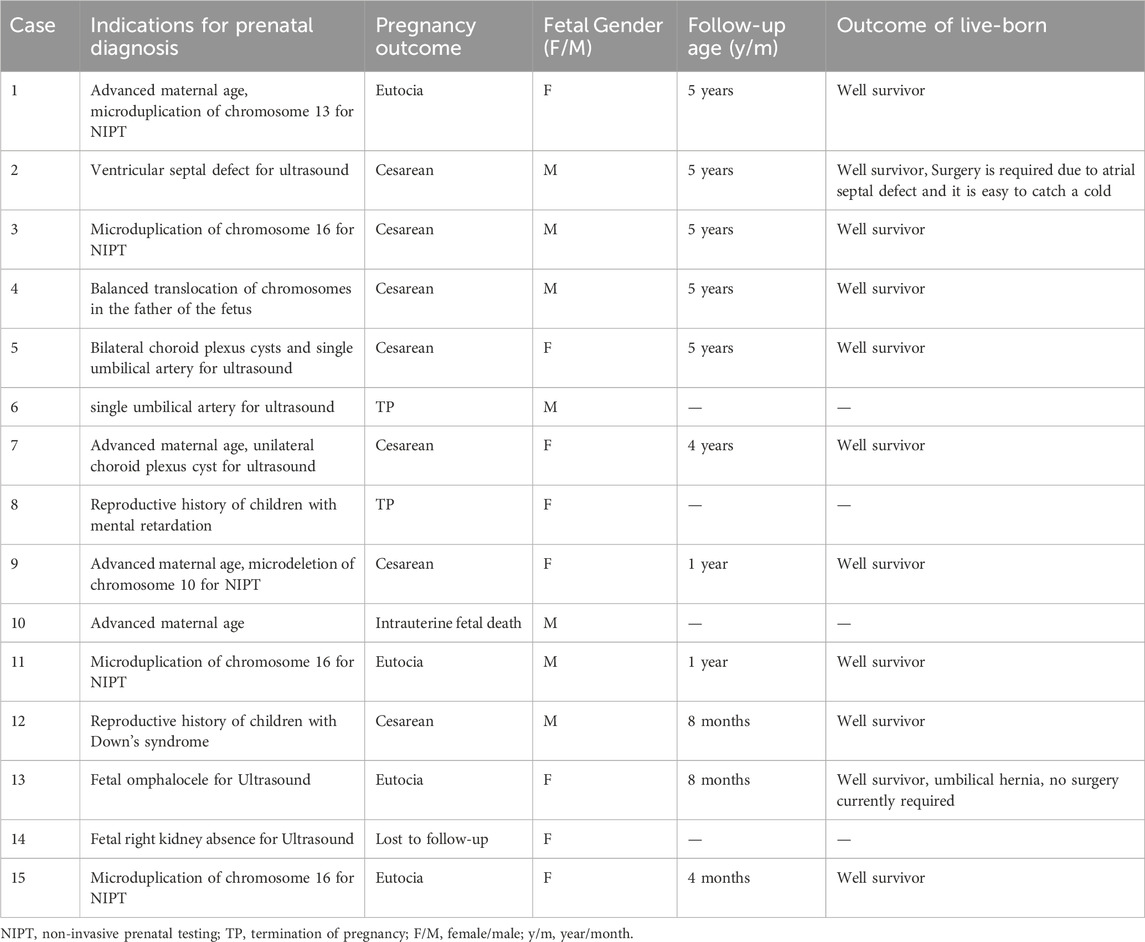
Table 3. Clinical information of 15 fetuses with 16p13.11 microduplication.
Of the 15 fetuses with 16p13.11 microduplications, except for one fetus was lost to follow-up, the pregnancies of an additional 3 fetuses were terminated. The remaining 11 fetuses carried to term following thorough adequate genetic counseling regarding the associated risks (Table 3). The age of the 11 surviving children ranged from 4 months to 5 years till the time of publication. One child with a ventricular septal defect was last followed up at 5 years of age. The child displayed normal intelligence, motor skills and language, which were confirmed by his patents. But they noted a susceptibility to colds, as the child had not undergone heart surgery. Another child with omphalocele was last followed up at 8 months and was reported to be able to roll over and sit independently, with a present lack of need for surgery. The other 9 children exhibited no notable abnormalities based on information come from their parents.
The short arm of chromosome 16 (16p) is a hotspot region for chromosomal copy number variations (CNVs), particularly the 16p13.11 region (Ciaccio et al., 2017; Redaelli et al., 2019; Atli et al., 2022). This region consists of three single-copy sequence intervals (I, II, III), flanked by low copy repeats (LCRs) (Ingason et al., 2011), which predispose to chromosomal rearrangements through non-allelic homologous recombination (NAHR). Intervals I and II are critical for copy number variation (Nagamani et al., 2011). In this study, SNP array analysis of amniotic fluid of 3,451 pregnant women revealing 15 fetuses with varying sizes of microduplications in the 16p13.11 region, ranging from 0.79 to 2.82 Mb. The breakpoints of 15 cases with 16p13.11 microduplications covered different regions across the three single-copy sequence intervals (I, II, III). Notably, There were 9 cases whose breakpoints encompassed intervals I and II, and all cases involved interval II. The concentration of breakpoints in intervals I and II suggests that these two regions may harbor key genes associated with clinical phenotype, consistent with previous findings (Nagamani et al., 2011). Interval I located between 15.12 and 15.15 Mb, contains 3 OMIM genes, including NTAN1, which encodes an asparagine-specific N-terminal amidase, which is an enzyme involved in regulating the half-life of proteins in vivo. Inactivation of the Ntan1 in mice is associated with abnormal neural activities, such as changes in social behavior, impaired memory, impairment of spatial and non-spatial learning abilities (Kwon et al., 2000; Balogh et al., 2001). Interval II located between 15.51 and 16.29 Mb, contains 7 OMIM genes, including NDE1, which is highly expressed in the brain and plays a pivotal role in the growth and development of cerebral cortex (Luttik et al., 1998; Nagamani et al., 2011). Knockout of Nde1 in mice leads to abnormalities in the cerebral cortex and microcephaly (Feng and Walsh, 2004). Moreover, NDE1 gene is regarded as a strong candidate gene associated with phenotypes of neural development (Soto-Perez et al., 2020). Interval II identified as a dosage-sensitive region, represents the core pathogenic locus of 16p13.11 (Tropeano et al., 2013), which consistents with our findings that all cases encompassed interval II.
The 16p13.11 microduplication is recognized as a neurodevelopmental susceptibility locus with incomplete penetrance and variable expression, which is an important impact on developmental delay, learning difficulties and behavioral abnormalities (Coe et al., 2014; Nuttle et al., 2016). The phenotype in adults and children has been well described in the previous literatures, such as cognitive impairment, behavioral abnormalities, cardiac and aortic malformations, skeletal abnormalities and other organ abnormalities (Balogh et al., 2001; Hamad et al., 2023).
However, there are few reports on the prenatal phenotype associated with this microduplication. Redaelli (Redaelli et al., 2019) detected 366 prenatal specimens using comparative genomic hybridization (aCGH) and found that one fetus with prenatal indications of non-immune edema carried 16p13.11 microduplications. Dabkowska (Dabkowska et al., 2020) identified a 16p13.11 microduplication in a fetus with encephalocele. Cai (Cai et al., 2022) found 15 fetuses with 16p13.11 microduplication by SNP array detected from prenatal diagnosis of 9,000 pregnant women, which indicated the most relevant prenatal indicators were structural or soft marker anomalies on ultrasound. Similarly, in this study, ultrasound abnormalities were the most common indication for prenatal diagnosis observed in 6 out of 15 cases, including 3 with structural abnormalities and 3 with soft marker anomalies. Hamad (Hamad et al., 2023) noted congenital cardiac anomalies in 16 of 206 patients with 16p13.11 microduplication, with the most common feature of which was ventricular septal defect. Houcinat (Houcinat et al., 2015) described a family with congenital renal and urinary system anomalies associated with 16p13.11 microduplication, in which the father had unilateral kidney dysplasia and the son suffered from chronic kidney disease and pelvic ureteral obstruction. Shi (Shi et al., 2021) reported that the rate of chromosomal abnormalities was as high as 29.6% on fetal omphalocele. These findings highlight the association between ultrasound abnormalities or soft marker anomalies and chromosomal rearrangements involving 16p13.11 region (Lan et al., 2020; Su et al., 2021). In our cohort, NIPT suggested chromosomal abnormalities in 5 out of 15 cases, 3 of which involved microduplication of chromosome 16. With the widespread clinical application of NIPT, the detection rate of CNVs involving 16p13.11 microduplications has been greatly improved (Du et al., 2018). Advanced maternal age, a known high-risk factor for chromosomal abnormalities (Wang et al., 2019), was the prenatal diagnosis indication in 5 out of 15 cases carrying 16p13.11 microduplications, possibly contributing to the higher detection rate in our study. The specific relationship between advanced age factors and the occurrence of 16p13.11 microduplications is still unclear.
Initially speculated a “benign variant” due to its presence in phenotypically normal parents, however, the pathogenicity of 16p13.11 microduplication is gradually recognized as more patients detected. (Hannes et al., 2009; Nagamani et al., 2011). The penetrance is estimated at around 7%–10.6% (Kendall et al., 2019; Kirov et al., 2014), and the duplication may be inherited from asymptomatic parents or occur de novo (Redaelli et al., 2019). It has been reported that approximately 93.6% of CNVs inherited from unaffected parents (Nagamani et al., 2011; Hamad et al., 2023). In our study, 7 cases underwent pedigree verification, and one case was de novo (1/7), while the remaining 6 were inherited (6/7), accounting for about 85.7% inherited from asymptomatic parents, consistent with previous literature. Notably, 5 cases were inherited from the mother (5/7), while one case had paternal inheritance (1/7), indicating a higher maternal inheritance rate. Tropeano (Tropeano et al., 2013) found a male bias in the 16p13.11 region, but there was no gender bias in our samples (7 males and 8 females), likely due to the small sample size.
Our follow-up revealed a 15.4% penetrance rate via telephone follow-up, which is higher than the levels reported in the literatures (Kendall et al., 2019; Kirov et al., 2014; Martin et al., 2020). There are two children exhibiting clinical phenotypes: one with ventricular septal defect and the other with omphalocele. This discrepancy may be affected by the limited sample size and the nature of telephone follow-up of our study, which may influence parental reports Additionally, genetic factors beyond 16p13.11 CNVs, such as other undetected mutations, could contribute to the phenotypes, but further testing was declined by their parents. Children with 16p13.11 microduplications might lack of specific phenotypes and the abnormalities may progressively emerge during growth or relatively mild. Although no abnormalities are evident in the neonatal period, affected individuals may show varying degrees of cognitive impairments and behavioral abnormalities with increasing age (Delicado et al., 2014). Therefore, ongoing monitoring of the growth and development of individuals with 16p13.11 microduplications is crucial.
In conclusion, when CNVs in the 16p13.11 region are detected during fetal development, clinical decisions should be based on a comprehensive analysis of fetal ultrasound findings, familial history and detailed genetic counseling instead of terminating the pregnancy solely. The potential for varying phenotypic expression within the same family underscores the importance of considering the origin in the parental generation and recurrence risks in comprehensive genetic counseling.
This study has several limitations. First, the sample size was small, with only 15 cases of fetal 16p13.11 microduplications detected. Second, the point mutations of genes were not detected by SNP array used in this study, but the next-generation sequencing was not employed. Third, the longest follow-up case in our study was tracked up to 5 years and clinical phenotypes associated with 16p13.11 microduplications are mainly observed in adults and children. So it is essential to conduct long-term follow-ups of these children in future studies to observe their growth and development. In future, research with larger sample size and longer follow-up period are necessary to better understand the genotype-phenotype correlation and provide more comprehensive genetic counseling.
The use of SNP array is an effective tool for diagnose variations in the 16p13.11 region. Our findings indicate that microduplication of 16p13.11 is strongly associated structural and soft marker anomalies detected via prenatal ultrasound. The application of NIPT (non-invasive prenatal testing) has enhanced the detection rate of prenatal 16p13.1 microduplications. Simultaneously, comprehensive prenatal phenotypic analyses, follow-ups on assessments of pregnancy outcomes and postnatal follow-ups for fetuses with 16p13.11 microduplications were conducted in this study. The majority of these fetuses were found to be in good health after birth, offering valuable insights for clinical genetic counseling.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
The studies involving humans were approved by Medicine, Jinan Maternal and Child Ethics Committee Health Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
YZ: Writing–original draft, Writing–review and editing. LS: Data curation, Investigation, Writing–review and editing. SZ: Data curation, Writing–review and editing. FH: Data curation, Writing–review and editing. SS: Data curation, Writing–review and editing. HJ: Funding acquisition, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is supported by Shandong Province Medical and Health Science and Technology Plan (202105030122), Technology Bureau Clinical Medical Science and Technology Innovation Plan (202328018) and Jinan Municipal Health Commission Science and Technology Development Plan Project (2024303006).
This work was strongly supported by the Prenatal Diagnosis Center and Ultrasound Department of Jinan Maternal and Child Health Hospital. We thank all patients who participated in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Allach El Khattabi, L., Heide, S., Caberg, J. H., Andrieux, J., Doco Fenzy, M., Vincent-Delorme, C., et al. (2020). 16p13.11 microduplication in 45 new patients: refined clinical significance and genotype-phenotype correlations. J. Med. Genet. 57 (5), 301–307. doi:10.1136/jmedgenet-2018-105389
Atli, E. I., Yalcintepe, S., Atli, E., Demir, S., Mail, C., and Gurkan, H. (2022). Clinical implications of chromosome 16 copy number variation. Mol. Syndromol. 13 (3), 184–192. doi:10.1159/000517762
Balogh, S. A., McDowell, C. S., Tae Kwon, Y., and Denenberg, V. H. (2001). Facilitated stimulus-response associative learning and long-term memory in mice lacking the NTAN1 amidase of the N-end rule pathway. Brain. Res. 892 (2), 336–343. doi:10.1016/s0006-8993(00)03268-6
Cai, M., Que, Y., Chen, X., Chen, Y., Liang, B., Huang, H., et al. (2022). 16p13.11 microdeletion/microduplication in fetuses: investigation of associated ultrasound phenotypes, genetic anomalies, and pregnancy outcome follow-up. Bmc. Pregnancy. Childbirth. 22 (1), 913. doi:10.1186/s12884-022-05267-w
Calderoni, S., Ricca, I., Balboni, G., Cagiano, R., Cassandrini, D., Doccini, S., et al. (2020). Evaluation of chromosome microarray analysis in a large cohort of females with autism spectrum disorders: a single center Italian study. J. Pers. Med. 10 (4), 160. doi:10.3390/jpm10040160
Ciaccio, C., Tucci, A., Scuvera, G., Estienne, M., Esposito, S., and Milani, D. (2017). 16p13 microduplication without CREBBP involvement: moving toward a phenotype delineation. Eur. J. Med. .Genet. 60 (3), 159–162. doi:10.1016/j.ejmg.2016.12.006
Coe, B. P., Witherspoon, K., Rosenfeld, J. A., van Bon, B. W., Vulto-van Silfhout, A. T., Bosco, P., et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 46 (10), 1063–1071. doi:10.1038/ng.3092
Dabkowska, S., Kucinska-Chahwan, A., Beneturska, A., Ilnicka, A., Nowakowska, B., Panek, G., et al. (2020). Prenatal diagnosis and clinical significance of cephalocele-A single institution experience and literature review. Prenat. Diagn. 40 (5), 612–617. doi:10.1002/pd.5654
Delicado, A., Fernandez, L., de Torres, M. L., Nevado, J., Garcia-Santiago, F. A., Rodriguez, R., et al. (2014). Familial imbalance in 16p13.11 leads to a dosage compensation rearrangement in an unaffected carrier. Bmc. Med. Genet. 15, 116. doi:10.1186/s12881-014-0116-3
Du, Y., Lin, J., Lan, L., Dong, Y., Zhu, J., Jiang, W., et al. (2018). Detection of chromosome abnormalities using current noninvasive prenatal testing: a multi-center comparative study. Biosci. .Trends. 12 (3), 317–324. doi:10.5582/bst.2018.01044
Erhart, P., Brandt, T., Straub, B. K., Hausser, I., Hentze, S., Bockler, D., et al. (2018). Familial aortic disease and a large duplication in chromosome 16p13.1. Med 6 (3), 441–445. doi:10.1002/mgg3.371
Feng, Y., and Walsh, C. A. (2004). Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 44 (2), 279–293. doi:10.1016/j.neuron.2004.09.023
Gumuslu, K. E., Savli, H., Sunnetci, D., Cine, N., Kara, B., Eren Keskin, S., et al. (2015). A CGH array study in nonsyndromic (primary) autism patients: deletions on 16p13.11, 16p11.2, 1q21.1, 2q21.1q21.2, and 8p23.1. Turk J.Med. Sci. 45 (2), 313–319. doi:10.3906/sag-1310-81
Hamad, A., Sherlaw-Sturrock, C. A., Glover, K., Salmon, R., Low, K., Nair, R., et al. (2023). Expanding the phenotypic spectrum of Chromosome 16p13.11 microduplication: a multicentric analysis of 206 patients. Eur. J. Med. Genet. 66 (4), 104714. doi:10.1016/j.ejmg.2023.104714
Hannes, F. D., Sharp, A. J., Mefford, H. C., de Ravel, T., Ruivenkamp, C. A., Breuning, M. H., et al. (2009). Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J. Med. Genet. 46 (4), 223–232. doi:10.1136/jmg.2007.055202
Houcinat, N., Llanas, B., Moutton, S., Toutain, J., Cailley, D., Arveiler, B., et al. (2015). Homozygous 16p13.11 duplication associated with mild intellectual disability and urinary tract malformations in two siblings born from consanguineous parents. Am. J. Med. Genet. A 167A (11), 2714–2719. doi:10.1002/ajmg.a.37212
Ingason, A., Rujescu, D., Cichon, S., Sigurdsson, E., Sigmundsson, T., Pietilainen, O. P., et al. (2011). Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol. Psychiatry. 16 (1), 17–25. doi:10.1038/mp.2009.101
Kendall, K. M., Bracher-Smith, M., Fitzpatrick, H., Lynham, A., Rees, E., Escott-Price, V., et al. (2019). Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK Biobank. Br. J. Psychiatry 214 (5), 297–304. doi:10.1192/bjp.2018.301
Kirov, G., Grozeva, D., Norton, N., Ivanov, D., Mantripragada, K. K., Holmans, P., et al. (2009). Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 18 (8), 1497–1503. doi:10.1093/hmg/ddp043
Kirov, G., Rees, E., Walters, J. T., Escott-Price, V., Georgieva, L., Richards, A. L., et al. (2014). The penetrance of copy number variations for schizophrenia and developmental delay. Biol. Psychiatry. 75 (5), 378–385. doi:10.1016/j.biopsych.2013.07.022
Kwon, Y. T., Balogh, S. A., Davydov, I. V., Kashina, A. S., Yoon, J. K., Xie, Y., et al. (2000). Altered activity, social behavior, and spatial memory in mice lacking the NTAN1p amidase and the asparagine branch of the N-end rule pathway. Mol. Cell. Biol. 20 (11), 4135–4148. doi:10.1128/MCB.20.11.4135-4148.2000
Lan, L., Wu, H., She, L., Zhang, B., He, Y., Luo, D., et al. (2020). Analysis of copy number variation by sequencing in fetuses with nuchal translucency thickening. J. Clin. Lab. Anal. 34 (8), e23347. doi:10.1002/jcla.23347
Loftus, B. J., Kim, U. J., Sneddon, V. P., Kalush, F., Brandon, R., Fuhrmann, J., et al. (1999). Genome duplications and other features in 12 Mb of DNA sequence from human chromosome 16p and 16q. Genomics 60 (3), 295–308. doi:10.1006/geno.1999.5927
Luttik, M. A., Overkamp, K. M., Kotter, P., de Vries, S., van Dijken, J. P., and Pronk, J. T. (1998). The Saccharomyces cerevisiae NDE1 and NDE2 genes encode separate mitochondrial NADH dehydrogenases catalyzing the oxidation of cytosolic NADH. J. Biol. Chem. 273 (38), 24529–24534. doi:10.1074/jbc.273.38.24529
Martin, C. L., Wain, K. E., Oetjens, M. T., Tolwinski, K., Palen, E., Hare-Harris, A., et al. (2020). Identification of neuropsychiatric copy number variants in a health Care system population. JAMA. Psychiatry. 77 (12), 1276–1285. doi:10.1001/jamapsychiatry.2020.2159
Martin, J., Han, C., Gordon, L. A., Terry, A., Prabhakar, S., She, X., et al. (2004). The sequence and analysis of duplication-rich human chromosome 16. Nature 432 (7020), 988–994. doi:10.1038/nature03187
Mefford, H. C., Cooper, G. M., Zerr, T., Smith, J. D., Baker, C., Shafer, N., et al. (2009). A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Res. 19 (9), 1579–1585. doi:10.1101/gr.094987.109
Nagamani, S. C., Erez, A., Bader, P., Lalani, S. R., Scott, D. A., Scaglia, F., et al. (2011). Phenotypic manifestations of copy number variation in chromosome 16p13.11. Eur. J. Hum. Genet. 19 (3), 280–286. doi:10.1038/ejhg.2010.184
Nuttle, X., Giannuzzi, G., Duyzend, M. H., Schraiber, J. G., Narvaiza, I., Sudmant, P. H., et al. (2016). Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature 536 (7615), 205–209. doi:10.1038/nature19075
Ramalingam, A., Zhou, X. G., Fiedler, S. D., Brawner, S. J., Joyce, J. M., Liu, H. Y., et al. (2011). 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J. Hum. Genet. 56 (7), 541–544. doi:10.1038/jhg.2011.42
Redaelli, S., Maitz, S., Crosti, F., Sala, E., Villa, N., Spaccini, L., et al. (2019). Refining the phenotype of recurrent rearrangements of chromosome 16. Int. J. Mol. Sci. 20 (5), 1095. doi:10.3390/ijms20051095
Shi, X., Tang, H., Lu, J., Yang, X., Ding, H., and Wu, J. (2021). Prenatal genetic diagnosis of omphalocele by karyotyping, chromosomal microarray analysis and exome sequencing. Ann. Med. 53 (1), 1285–1291. doi:10.1080/07853890.2021.1962966
Soto-Perez, J., Baumgartner, M., and Kanadia, R. N. (2020). Role of NDE1 in the development and evolution of the gyrified cortex. Front. Neurosci. 14, 617513. doi:10.3389/fnins.2020.617513
Su, L., Wu, X., Lin, N., Xie, X., Cai, M., Wang, M., et al. (2021). Different cutoff values for increased nuchal translucency in first-trimester screening to predict fetal chromosomal abnormalities. Int. J. Gen. Med. 14, 8437–8443. doi:10.2147/IJGM.S330960
Tropeano, M., Ahn, J. W., Dobson, R. J., Breen, G., Rucker, J., Dixit, A., et al. (2013). Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders. PloS. One. 8 (4), e61365. doi:10.1371/journal.pone.0061365
Ullmann, R., Turner, G., Kirchhoff, M., Chen, W., Tonge, B., Rosenberg, C., et al. (2007). Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum. Mutat. 28 (7), 674–682. doi:10.1002/humu.20546
Wang, J., Chen, L., Zhou, C., Wang, L., Xie, H., Xiao, Y., et al. (2019). Application of copy number variation sequencing for prenatal diagnosis in women at an advanced maternal age. 36 (6): 533–537. doi:10.3760/cma.j.issn.1003-9406.2019.06.001
Keywords: 16p13.11, SNP-array, microduplication, CNVs, prenatal diagnosis
Citation: Zhao Y, Song L, Zhang S, Hou F, Shan S and Jin H (2024) Prenatal diagnosis and postnatal follow-up of 15 fetuses with 16p13.11 microduplication syndrome. Front. Genet. 15:1486974. doi: 10.3389/fgene.2024.1486974
Received: 27 August 2024; Accepted: 30 September 2024;
Published: 15 October 2024.
Edited by:
Thomas Liehr, Friedrich Schiller University Jena, GermanyReviewed by:
Anja Weise, University Hospital Jena, GermanyCopyright © 2024 Zhao, Song, Zhang, Hou, Shan and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shan Shan, MTA1NzMzNjU4MEBxcS5jb20=; Hua Jin, dG9ueXNoaXJseUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.