 Davide Vecchio1*
Davide Vecchio1* Marina Macchiaiolo1Michaela V. Gonfiantini1
Marina Macchiaiolo1Michaela V. Gonfiantini1 Filippo M. Panfili1
Filippo M. Panfili1 Francesco Petrizzelli2Niccolò Liorni2,3
Francesco Petrizzelli2Niccolò Liorni2,3 Fabiana Cortellessa1
Fabiana Cortellessa1 Lorenzo Sinibaldi1Ippolita Rana1
Lorenzo Sinibaldi1Ippolita Rana1 Emanuele Agolini4
Emanuele Agolini4 Dario Cocciadiferro4
Dario Cocciadiferro4 Nicole Colantoni5
Nicole Colantoni5 Michela Semeraro6Cristiano Rizzo6
Michela Semeraro6Cristiano Rizzo6 Annalisa Deodati5,7
Annalisa Deodati5,7 Nicola Cotugno5,8
Nicola Cotugno5,8 Serena Caggiano9
Serena Caggiano9 Elisabetta Verrillo9
Elisabetta Verrillo9 Carlotta G. Nucci10Serpil Alkan11Jorge M. Saraiva12,13,14Joaquim De Sá12Pedro M. Almeida12Jayanth Krishna15
Carlotta G. Nucci10Serpil Alkan11Jorge M. Saraiva12,13,14Joaquim De Sá12Pedro M. Almeida12Jayanth Krishna15 Paola S. Buonuomo1
Paola S. Buonuomo1 Diego Martinelli6Carlo Dionisi Vici6
Diego Martinelli6Carlo Dionisi Vici6 Viviana Caputo3Andrea Bartuli1
Viviana Caputo3Andrea Bartuli1 Antonio Novelli4Tommaso Mazza2,4
Antonio Novelli4Tommaso Mazza2,4- 1Rare Diseases and Medical Genetics Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 2Bioinformatics Unit, Fondazione IRCCS Casa Sollievo Della Sofferenza, San Giovanni Rotondo, Italy
- 3Department of Experimental Medicine, Sapienza University of Rome, Rome, Italy
- 4Translational Cytogenomics Research Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 5Department of Systems Medicine, University of Rome Tor Vergata, Rome, Italy
- 6Division of Metabolic Diseases, Bambino Gesù Children’s Hospital IRCCS, Rome, Italy
- 7Diabetology and Growth Disorders Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 8Research Unit of Clinical Immunology and Vaccinology, IRCCS Bambino Gesù Children’s Hospital, Rome, Italy
- 9Pediatric Pulmonology and Cystic Fibrosis Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 10Neurosurgery Unit, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 11Department of Pediatrics, Centre Hospitalier Universitaire, CHU, Liège, Belgium
- 12Medical Genetics Department, Hospital Pediátrico de Coimbra, Unidade Local de Saúde de Coimbra, Coimbra, Portugal
- 13University Clinic of Pediatrics, Faculty of Medicine, University of Coimbra, Coimbra, Portugal
- 14Clinical Academic Center of Coimbra, Hospital Pediátrico de Coimbra, Unidade Local de Saúde de Coimbra, Coimbra, Portugal
- 15Krishna Institute of Medical Sciences (KIMS Hospital), Hyderabad, India
Introduction: Infantile hypotonia with psychomotor retardation and characteristic facies-1 (IHPRF1, MIM#615419) is a rare, birth onset, autosomal recessive disorder caused by homozygous or compound heterozygous truncating variants in NALCN gene (MIM#611549) resulting in a loss-of-function effect.
Methods: We enrolled a new IHPRF1 patients’ cohort in the framework of an international multicentric collaboration study. Using specialized in silico pathogenicity predictors and ad hoc structural analyses, we assessed the mechanistic consequences of the deleterious variants retrieved on NALCN structure and function.
Results: To date 38 different NALCN variants have been retrieved from 33 different families, 26 from unrelated and 22 from related patients. We report on five new IHPRF1 patients from four different families, harboring four newly identified and one previously retrieved variant that exhibited a markedly significant functional impact, thereby compromising the functionality of the protein complex.
Discussion: By widening the functional spectrum of biallelic variants affecting the NALCN gene, this article broadens the IHPRF1 syndrome’s genotype-phenotype correlation and gives new insight into its pathogenic mechanism, diagnosis, and clinical management.
1 Introduction
Infantile hypotonia with psychomotor retardation and characteristic facies-1 (IHPRF1, MIM#615419) is a rare autosomal recessive disorder with an onset at birth or early in infancy, with a typical severe course. Individuals affected by IHPRF1 are generally characterized by moderate-severe hypotonia, global developmental delay, and dysmorphic features (Al-Sayed et al., 2013; Bramswig et al., 2018; Karimi, et al., 2020) IHPRF1 is caused by homozygous truncating variants in NALCN gene (MIM#611549) on chromosome 13q33 resulting in a loss-of-function (LoF) effect (Bramswig et al., 2018; Bend et al., 2016) NALCN encodes for sodium leak channel, non-selective (NALCN) protein, a voltage-independent, nonselective cation channel that belongs to a family of voltage-gated sodium and calcium channels regulating the resting membrane potential and excitability of neurons with passive, intracellular movement of sodium ions (National Library of Medicine: NCBI, 2024). Novel evidence suggests that NALCN plays a role also in other pathways such as pain sensation (Zhang and Wei, 2023). NALCN LoF variants lead to reduced sodium leak causing hyperpolarization of the resting membrane potential explaining the hypoexcitability phenotype that characterizes IHPRF1 disease (Bramswig et al., 2018; Bend et al., 2016). Interestingly, heterozygous pathogenic variants in NALCN gene are associated, with another disorder predominantly characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay (CLIFAHDD, MIM#616266) that owns a significant phenotypic overlap degree with IHPRF1, but also shows distal arthrogryposis (Chong et al., 2015).
Other important IHPRF1 features are seizures, brain anomalies (cortical atrophy and/or thin corpus callosum, e.g.,), muscle wasting, strabismus and/or nystagmus, scoliosis, feeding difficulties, constipation, sleep disturbance (secondary to altered circadian rhythm) and breathing abnormalities. Dysmorphic features frequently reported in IHPRF1 are prominent forehead with triangular face, microcephaly, micrognathia, smooth philtrum, thin upper lip, low-set and large ears, slender nose, and strabismus (Al-Sayed et al., 2013; Liu et al., 2013; Bramswig et al., 2018). Periodic breathing and sleep apnea, that usually increase in severity during sleep, are frequently reported both in patients with IHPRF1 and CLIFAHDD (Lozic et al., 2016; Gal et al., 2016; Bourque et al., 2018; Campbell et al., 2018; Maselli et al., 2022; Winczewska-Wiktor et al., 2022) and are generally alleviated by oxygen supplementation or in some cases with support of Non-Invasive Ventilation (NIV) (Lozic et al., 2016).
To date 48 patients have been reported with IHPRF1 (Al-Sayed et al., 2013; Köroğlu et al., 2013; Farwell et al., 2015; Gal et al., 2016; Angius et al., 2018; Bourque et al., 2018; Bramswig et al., 2018; Campbell et al., 2018; Carneiro et al., 2018; Takenouchi et al., 2018; Karimi, et al., 2020; Ope et al., 2020; Maselli et al., 2022; Khan et al., 2022; Abul-Husn et al., 2023; Tehrani Fateh et al., 2023; Susgun et al., 2024) with 38 different NALCN variants from 26 unrelated and 22 related patients (33 different families) in homozygosity or compound heterozygosity. Here, we report on five new IHPRF1 patients enrolled in the framework of an international multicentric collaboration study, four with homozygous variants and one with compound heterozygous deleterious variants in NALCN gene, in order to: I. further delineate the IHPRF1 genotype-phenotype correlation and its clinical management, II: broaden the spectrum of biallelic variants in NALCN gene, and III: assess the mechanistic consequences of these variants on the protein structure and function by resorting to specialized in silico pathogenicity predictors and ad hoc structural analyses.
2 Materials and methods
2.1 Study population and data collection
This study enrolled five pediatric male patients with NALCN deleterious variants. All patients’ data were collected during clinical assessments, after obtaining informed consent and recorded on an anonymized dataset. Patients’ inclusion spanned from May 2023 to March 2024. An extensive literature review on IHPRF1 was conducted by using Embase® and MEDLINE® databases. Data collection included: 1) general information: gender, age, inheritance, nationality; 2) phenotypic description collected by clinical evaluation; 3) laboratory and instrumental investigations: blood tests, EEG, brain MRI, polysomnography. In addition, the main clinical features of the patients with recurrent pathogenic variants were annotated according to the human phenotype ontology (HPO) (Köhler et al., 2021) (Figure 1).
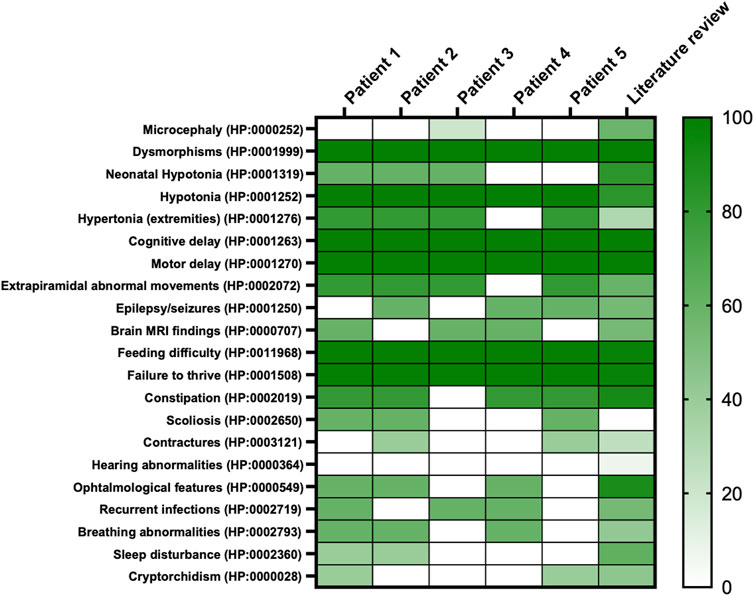
Figure 1. Heatmap of main clinical features frequencies of present cohort compared to literature. On the right, the colored scale depicts patients’ features frequency; from absence and/or very low frequency: light green to very high frequency: deep green. On the left, features retrieved in the whole cohort reported per HPO codes.
2.2 Exome sequencing
Whole Exome Sequencing (WES) was performed on proband and parents’ DNA using the Twist Human Core Exome Kit (Twist Bioscience), according to the manufacturer’s protocol, and sequenced on the Illumina NovaSeq 6000 platform. The BaseSpace pipeline and the Geneyx software LifeMap Sciences were respectively used for the variant calling and annotating variants. Sequencing data were aligned to the hg19 human reference genome.
Variants were reported according to NM_052867.4 and NP_443099.1.
Global minor allele frequency (GMAF) for analyzed variants was reported according to the Genome Aggregation Database (gnomAD) v4.1.0. Based on the guidelines of the American College of Medical Genetics and Genomics (ACMG), a minimum coverage depth of 30X was considered suitable for analysis. Variants were also examined for Qscore and visualized by the Integrative Genome Viewer (IGV) v2.17.4. Clinical interpretation of variants was reported according to the ACMG/AMP 2015 guidelines using the Intervar tool (Li and Wang, 2017).
2.3 Biochemical analysis
The biochemical analysis of urinary oligosaccharides both on fresh urine and dried urine spots (DUS), was performed with a UHPLC/MS-MS (Ultra-high performance liquid chromatography tandem mass spectrometry) method. The pre-analytical procedure was obtained without derivatization and using maltoheptaose as internal standard. Urine Samples were ultra-filtered and normalized to a creatinine concentration of 1 mM. The spectrometric analysis was performed in multiple reaction monitoring (MRM) acquisition both in positive and negative mode for all target oligosaccharides (OS) transitions, including the glucose tetrasaccharide Glc4. For each MRM, the median (50th percentile) of several control samples was calculated and results were expressed as MoM. The chromatographic run was 28 min (Heiner-Fokkema et al., 2020).
2.4 In-silico assessment of variant pathogenicity
The pathogenic potential of each variant retrieved in our cohort was assessed in silico with a pool of variant type-specific software predictors and structural biology methods.
2.4.1 Variant type-specific software predictors
The frameshift variant, p.Tyr1431Leufs*27, causing a premature termination codon (PTC) was evaluated with MutationTasting 2021 (Steinhaus et al., 2021), CADD (Schubach et al., 2024), SIFT-Indel (Hu and Ng, 2013), FATHMM-indel (Ferlaino et al., 2017), and CAPICE (Li et al., 2020). The effect of the nonsense variants, p. (Arg842Ter) and p.Arg855Ter, was evaluated using CADD, DANN (Quang et al., 2015), Eigen (Ionita-Laza et al., 2016), GERP++ (Davydov et al., 2010), LRT (Chun and Fay, 2009), MutationTaster (Schwarz et al., 2010), VEST4 (Carter et al., 2013), MPA (Yauy et al., 2018), and fathmm-MKL (Shihab et al., 2015). In addition, we queried the MetaDome web server (Wiel et al., 2019) to inspect the NALCN tolerance to these variants. The splicing variant, c.2889 + 2T > A, was evaluated using MutationTaster 2021, CADD, Eigen, and MPA, as well as splicing-specific predictors, such as SpliceAI (Jaganathan et al., 2019), Pangolin (Zeng and Li, 2022), MaxEntScan (Yeo and Burge, 2004), and SPiP (Leman et al., 2022). Finally, being the putative pathogenic effect of the missense variant p. (Cys1417Tyr) less straightforward than all the previous variants, it was studied by resorting to both 24 third-party pathogenicity predictors, i.e., CADD, Eigen, MPA, SIFT (Ng and Henikoff, 2003), SIFT4G (Vaser et al., 2016), PolyPhen2 (Adzhubei et al., 2013), FATHMM (Shihab et al., 2013), AlphaMissense (Cheng et al., 2023), REVEL (Ioannidis et al., 2016), ClinPred (Alirezaie et al., 2018), Meta SVM (Kim et al., 2017), Meta LR (Chen et al., 2022), Mistic (Chennen, et al., 2020), DEOGEN2 (Raimondi et al., 2017), DANN, GERP++, LRT, M-CAP (Jagadeesh et al., 2016), MutationTaster (Schwarz et al., 2010), MutationAssessor, MetaLR, PROVEAN (Choi et al., 2012), VEST4, fathmm-MKL, and structural biology methods.
Evolutionary conservation was assessed using GERP++, phyloP, phastCons, and SiPhy tools.
2.4.2 Structural biology
Atomic coordinates of the NALCN protein were retrieved from the AlphaFold Protein Structure Database (Ittisoponpisan et al., 2019), where a high-quality model of the protein was available, with a confidence score (pLDDT) ranging from very high (>90) for the intermembrane ion transport domain to low/very low (<50) for the cytoplasmic α-helical turns and loops. Then, structural damages caused by the missense variant p. (Cys1417Tyr) were investigated using the Missense3D web tool (Schymkowitz et al., 2005). Finally, the stability of the mutant structure was investigated thermodynamically through the BuildModel function implemented in the FoldX algorithm (10.1093/nar/gki387), which was run with default parameters as in Castellana et al. (2021) and Frustaci et al. (2018).
3 Results
3.1 Clinical cohort description
Main clinical features in the present cohort and their frequencies were compared to the previous literature (Table 1). Available clinical features of patients 1 and 4 are depicted (Figure 2). The main reported features in our cohort were compared with literature (Figure 1), while IHPRF1 patients’ clinical features retrieved are cumulatively reported (Supplementary Table S1). The location of the IHPRF1, both from literature review and those from the present study, are shown along the NALCN protein (Figure 3).
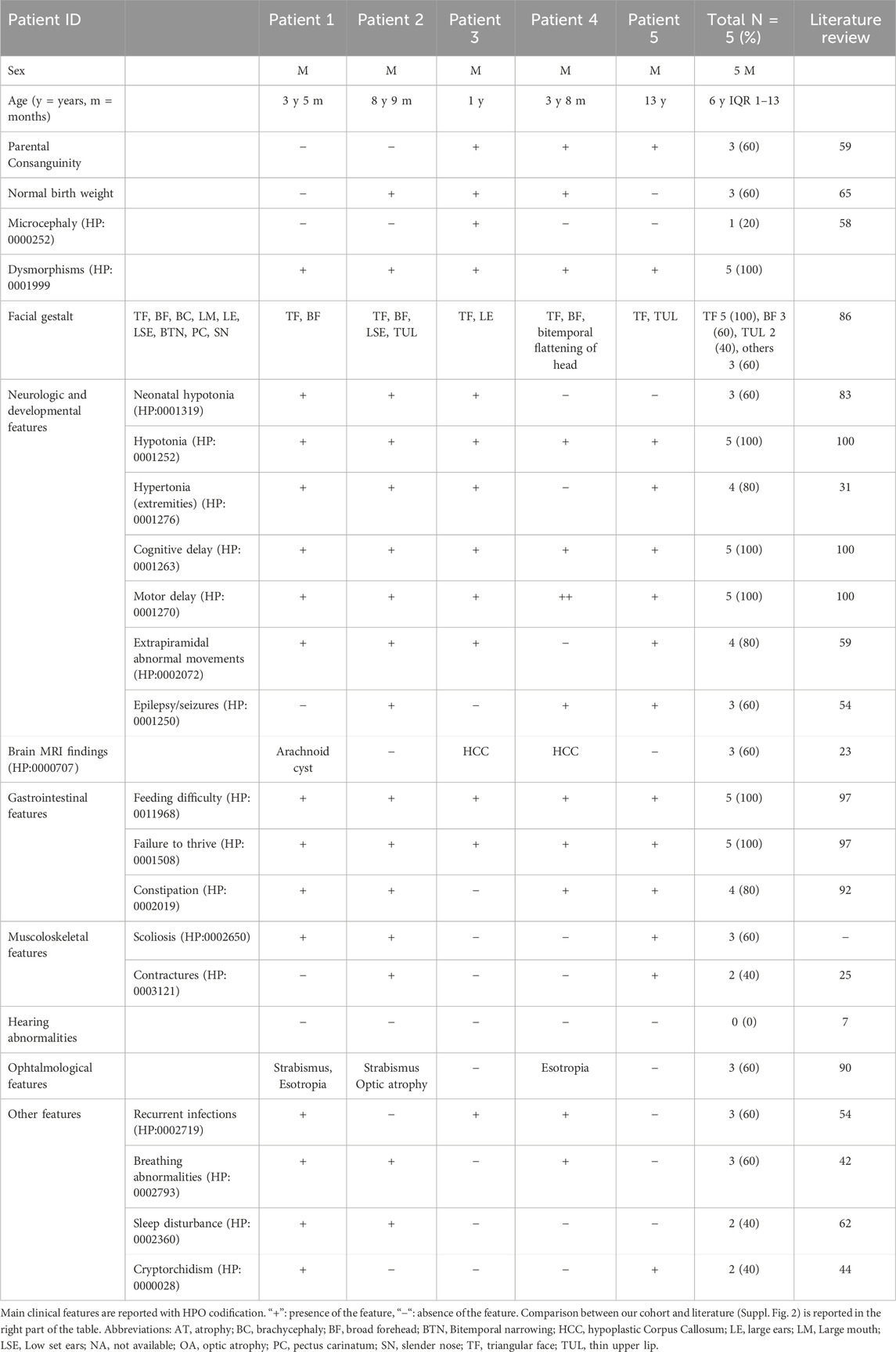
Table 1. Main clinical features in IHPRF1 patients reported in present cohort and literature review.
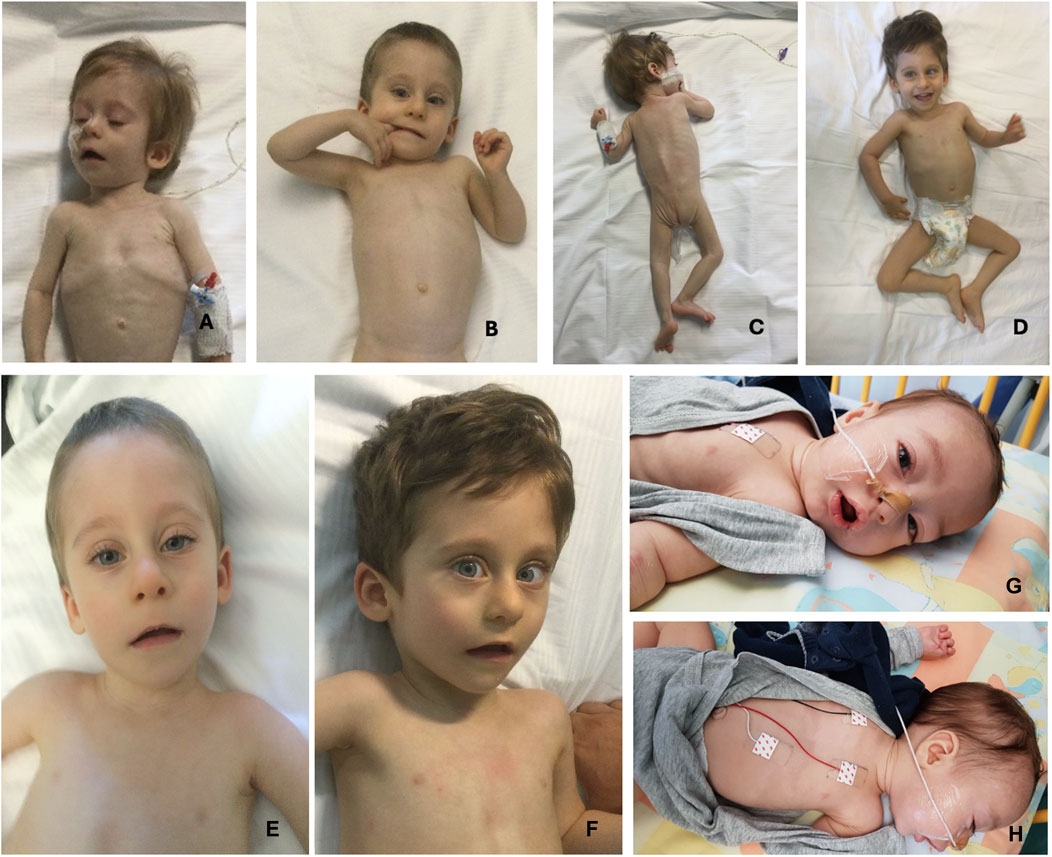
Figure 2. Pictures of two patients from the present cohort. From (A–D), pictures of patient 1 (from 9 months of age to 2 years and 7 months). (A, B) pictures depict severe emaciation of the patient and hypotonia whereas before neurosurgery. Pictures (C, D) show patient 1’s clinical improvement and weight gain after surgery. (E, F) (respectively taken at 18 months and 2 years and 7 months of age) depict the patient 1’s dysmorphic features: triangular face, high forehead, large ears, and mild convergent strabismus. (G, H) show patient 4 facial gestalt: triangular face, broad forehead, bitemporal narrowing and the presence of NG tube for enteral feeding.
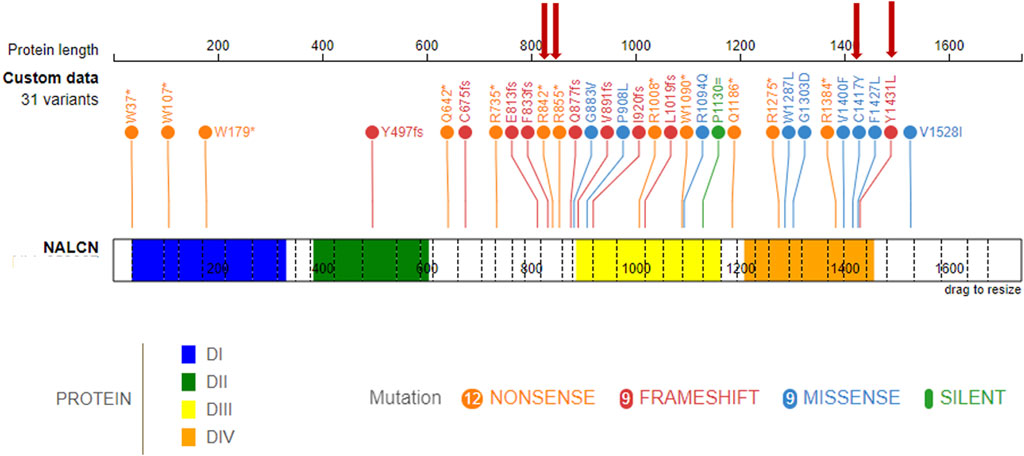
Figure 3. IHPRF1-associated LoF known variants along the NALCN protein. (top) known NALCN variants colored by protein effect (five splicing variants not included). The protein is made up of four homologous domains: DI (blue, 36–331), DII (green, 384–604), DIII (yellow, 887–1163), DIV (orange, 1209–1457), each with six transmembrane segments. Red arrows indicate variants retrieved in the present study.
Patient Report 1: Three-year and five-month-old male, second child of healthy not-consanguineous parents. Family history was not informative for any genetic diseases, his older sister is in good clinical condition. He was born after a regular pregnancy at 38 weeks of gestation from caesarean delivery for previous caesarean section. Anthropometric parameters at birth: weight 2,300 g (-2.5 SD) indicative for a small for gestational age child, length 46 cm (-2.5 SD), and occipitofrontal circumference (OFC) 35 cm (+0.82 SD). APGAR score 9/9. At birth, he showed neonatal hypotonia and dysmorphic somatic features triangular face, high forehead, large ears, and mild convergent strabismus. During follow-up at neurological evaluation were noted: severe psychomotor retardation, predominantly axial hypotonia (not reached the sitting position), hypertonia of the extremities and paroxysmal episodes and rotatory eye movements. The patient came to our observation at the age of 9 months and was admitted to our hospital for the history of failure to thrive (FTT) and finding of a suprasellar cystic lesion detected at brain MRI in another hospital. He presented linear growth during the first 6 months of his life, followed by progressive poor weight gain (Supplementary Figure S1). In consideration of the severe FTT an extensive work-up was performed. To exclude an inborn error of metabolism several examinations were performed resulting normal except for the analysis of urinary oligosaccharides on fresh urine which revealed increased levels of glucose tetrasaccharide (Glc4) in several samples analyzed (Glc4 from 6 to 15 multiple of the medians, MoM, RR < 5). Endocrinology analysis showed reduced levels of IGF1 and IGFBP3 (-2 SD), with normal basal GH. Leptin levels appeared also reduced on multiple measurements (0.57 and 0.06, RR 2.05–5.63 ng/mL). Brain MRI documented the presence of a cyst (diameter 32 mm × 18 mm) which displaced the surrounding structures (pituitary stalk, cerebral peduncles, pons and basilar artery) and modest increase in the size of the lateral ventricles and periencephalic spaces in fronto-temporal regions bilaterally (Figure 4A). Thus, clinical suspicion of Diencephalic Syndrome (DS) was assessed. Endoscopic third ventriculostomy surgery was performed (Figure 4B) with a progressive slight increase in the growth rate and the anatomopathological analysis confirmed the diagnosis of arachnoid cyst, a benign lesion. After surgery, the patient developed iatrogenic hypoadrenalism requiring replacement therapy with hydrocortisone. In the suspicion of sleep apnea, a nocturnal polysomnography examination was performed revealing the presence of periodic breathing, associated with desaturations and normal CO2 value. Supplemental oxygen during sleep was prescribed with a progressive amelioration of the periodic breathing (Supplementary Figure S2). Interestingly a slight increase of homovanillic acid and homovanillic/vanilmandelic acid ratio (HVR) values was detected during the investigation for the cyst, showing a progressive normalization after O2 supplementation (Supplementary Figure S3). Because of the history of recurrent pneumonia, immunological screening was performed. Partial functional defects of T and B lymphocytes were reported after in vitro stimulation with PHA, OKT3, and CpG and a reduction of NK activity was found. He underwent orchidopexy surgery for cryptorchidism at 2 years of age. For the presence of scoliosis, he was prescribed with Cheneau brace. Last auxological parameters at 2-year and 6-month of age: height 85.5 cm (−2 SD), weight 8.5 Kg (−3 SD), OFC 48 cm (−1 SD). Echocardiogram, urinary tract ultrasound, audiological screening and electroencephalography (EEG) resulted normal. WES in trio was then performed revealing the nonsense variant c.2563C > T (p.Arg855Ter) and the splicing variant c.2889 + 2T > A (p.?) in the NALCN gene in compound heterozygosity.
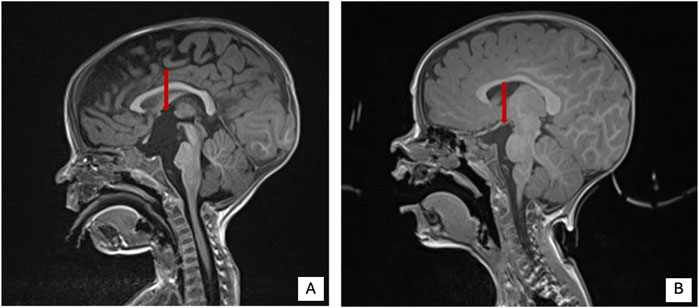
Figure 4. Brain MRI pre and post-surgery in patient 1 of present cohort. (A) Brain MRI pre surgery. (B) Brain MRI post-surgery. Red arrow shows the cystic lesion.
Patient Report 2: Eight-year and 9-month-old male, with a prenatal history of intrauterine growth restriction. At birth, he presented with hypotonia, plagiocephaly, feeding difficulties and poor weight gain. From a neurological standpoint, he has severe psychomotor development delay and epilepsy with an onset at around 6 months. EEG showed initially burst-suppression pattern and later a pre-hypsarrhythmic pattern. He currently has drug-resistant epilepsy, with sporadic seizures. At neurological evaluation was noted severe irritability and involuntary movements with choreoathetosis in the upper limbs and hypertrophy of the thigh muscles, axial hypotonia and hyperreflexia of the limbs. No gait or language acquisition. He is completely dependent on activities of daily living. For the presence of FTT he required nasogastric tube (NG tube) feeding between 6 months and 2 years of age. He also had a dislocated right hip and severe scoliosis (43° Cobb), currently treated with a brace. Surgery is being considered for the future. From a respiratory point of view, at the age of 3 years and 9 months he underwent a polygraph sleep study which showed severe central hypoventilation characterized by constant periodic breathing with central apnea. He underwent non-invasive ventilation at the age of 4 years and 5 months, not tolerate. Parents opted not to continue with non-invasive ventilation. He maintained respiratory stability with no significant infectious complications, but he keeps his habitual irregular breathing pattern and apnea. He also has visual impairment (strabismus and optic nerve hypoplasia) and complaints of constipation. Current auxological parameters: height −2.5 SD, OFC −1.85 SD; weight −3 SD. The analysis of urinary oligosaccharides performed on DUS was normal (Glc4 1.3 MoM, RR < 15). Brain MRI which did not identify any relevant alterations. Finally, WES detected the homozygous nonsense variant c.2524C > T (p.Arg842Ter) in NALCN gene.
Patient Report 3: One-year-old male, born from a third-degree consanguineous marriage with an history of delayed achievement of milestones, feeding issues, poor weight gain, and several episodes of respiratory tract infections (in two cases needing hospitalization for Pneumonia). Perinatal and family history were not contributive. On examination, dysmorphisms were noted (triangular face, prominent ears with smooth pinna and hypodontia). Neurological examination showed limb hypotonia. The rest of the systemic examination was not significant. Brain MRI showed hypoplastic corpus callosum. EEG resulted normal. WES performed on the proband and his parents revealed the homozygous missense variant c.4250G > A (p.Cys1417Tyr) in NALCN gene. Symptomatic treatment with physiotherapy and occupational therapy was advised to address the concerns in developmental delay.
Patient Report 4: Three-years and eight-month-old male born at 37 weeks of gestational age with a weight of 2,670 g, from a consanguineous family from Morocco with other two children without medical complaints. One episode of hypoglycemia in the neonatal period. Dysmorphic features: bitemporal flattening of the head, wide forehead, triangular face. He presented with a history of feeding difficulties with FTT requiring NG tube and subsequently percutaneous gastric tube (G tube), severe hypotonia, global amyotrophy, severe constipation with frequent fecal impactions. Brain MRI showed a thin corpus callosum. At 8 months, for the presence of spasms he underwent long-term EEG that showed generalized waves-spikes. Valproate and vigabatrin was initiated gradually. Clonazepam was added because of refractory seizures. Patient is now controlled by these three drugs. When he was 2-year-old, he was diagnosed with sleeping apnea syndrome with mild desaturation bur without alveolar hypoventilation. At ophtalmological evaluation finding of esotropia and absence of pursuit movements of the eyes. WES performed on the proband and his parents revealed the homozygous frameshift variant c.4291dupT (p.Tyr1431Leufs*27) variant in NALCN gene.
Patient Report 5: Thirteen-year-old male born at term without neonatal complications with a weight of 2,450 g. Dysmorphic features: triangular face, thin upper lip. He presented with a history of severe hypotonia with lower limbs hypertonia and choreic movements. Diagnosis of epileptic encephalopathy initially treated with valproate, phenobarbital and clonazepam, then phenobarbital was switched to lamotrigine because of persistent absence and atonic crisis. Brain MRI was normal. He underwent bilateral inguinal hernia and bilateral cryptorchidism surgical correction at 2 years old. To date he shows good eye contact and interaction with peers, no language acquisition or gait needing a wheelchair. At ophthalmological evaluation finding of strabismus. He presented early with feeding difficulties and gastro-esophageal reflux needing percutaneous gastrostomy. G tube was removed at 2 years of age for parents willing. Currently, he is eating only blended food for difficulties in mastication and swallowing. Over the time he also showed pica eating disorder, scoliosis and severe constipation. WES performed on the proband and his parents revealed the homozygous frameshift variant c.4291dupT (p.Tyr1431Leufs*27) variant in NALCN gene.
3.2 Biochemical analysis
The urinary oligosaccharides analysis was performed on fresh urine for patient 1 and DUS for patient 2. Two negative MRM transitions of the tetrasaccharide Glc4 and its isomer maltotetraose (M4) were selected as the most characteristic and the Glc4 transition was used for the quantification. For patient 1, we extensively analyzed 7 urine samples at different collection times (2021-2023) obtaining Glc4 increased values of MoM ranging between a minimum of 6 and a maximum of 15 (6, 7.4, 11.8, 13.1, 13.7, 15). Only the last sample analyzed had a normal Glc4 value (4 MoM). The reference value for Glc4 in urine was <5 MoM. For patient 2, we analyzed only 1 DUS sample obtaining the normal value of 1.3 MoM for Glc4. The reference value for Glc4 in DUS was <15 MoM.
3.3 In-silico assessment of the pathogenicity of variants
The pathogenicity of all variants under examination was evaluated in silico using an array of variant type-specific software predictors and structural biology methods. p. (Tyr1431LeufsTer27) caused the formation of a premature termination codon (PTC) 27 residues after the Tyr1431 site. The PTC caused, in turn, the abolishment of a significant portion of the cytoplasmic α-helical turns and loops of the protein and the loss of part of the DIV-S6 transmembrane helix, with a putative disruption of the fundamental activation gate (Figure 5B). While we could not predict whether the protein undergoes degradation upon this mutation, we reported full agreement on its pathogenic effect among five distinct in silico indel-specific predictors (Table 2). This variant was not annotated in gnomAD, and was classified as “Likely pathogenic” according to the ACMG/AMP 2015 guidelines. The two nonsense variants p.(Arg842Ter) and p. (Arg855Ter) were considered pathogenic by seven on eight queried predictors and to be phylogenetically highly conserved through 100 vertebrates according to all computed conservation scores (Table 2). Structurally, both variants determine the complete loss of 2 out of 4 transmembrane domains, with dramatic consequences on the protein stability. Variants were reported in gnomAD with different allele frequencies (0,000007435 and 0,0000254, respectively) and also in Clinvar (as “Likely pathogenic” and “Pathogenic,” respectively). Both variants were classified as “Pathogenic” according to the ACMG/AMP 2015 guidelines (Li and Wang, 2017). The splicing variant c.2889 + 2T > A (p.?) is predicted to have a pathogenic effect by all six queried tools that could handle intronic variants (Table 2). Functionally, all splicing-specific queried software packages, i.e., Splice AI, Pangolin, MaxEntScan, and SPiP, predict it to affect the splicing machinery regarding the exon 25. Two splicing events are envisaged. The one less likely (Splice AI score = 0.32) is the loss of the acceptor site of exon 25 located −133 bp from the variant position, thereby causing the exon to skip. The more probable event is the loss of the exon 25 donor site located 2 bp upstream of the variant site (Splice AI score = 1.00), which causes transcription elongation of 24 intronic nucleotides, i.e., GTAAGTCTTTGCTTATTGCCTAAATGA, before forming a PTC. Structurally, the skipping of exon 25 would affect the voltage-sensor domain 3 (VSD3), with putative consequences on the protein functionality. On the contrary, a so early protein truncation would have consequences on the protein stability like those described above for the nonsense variants in the unlikely case the protein was actually translated (Figure 5B). Variant was not annotated in gnomAD and was classified as “Pathogenic” according to the ACMG/AMP 2015 guidelines (Li and Wang, 2017). Finally, the missense variant p.(Cys1417Tyr) is deemed pathogenic by all queried tools (Table 3). The site is highly conserved through vertebrates and exhibits a MetaDome tolerance score of 0.67, which is compatible with a slightly intolerant site to mutations.
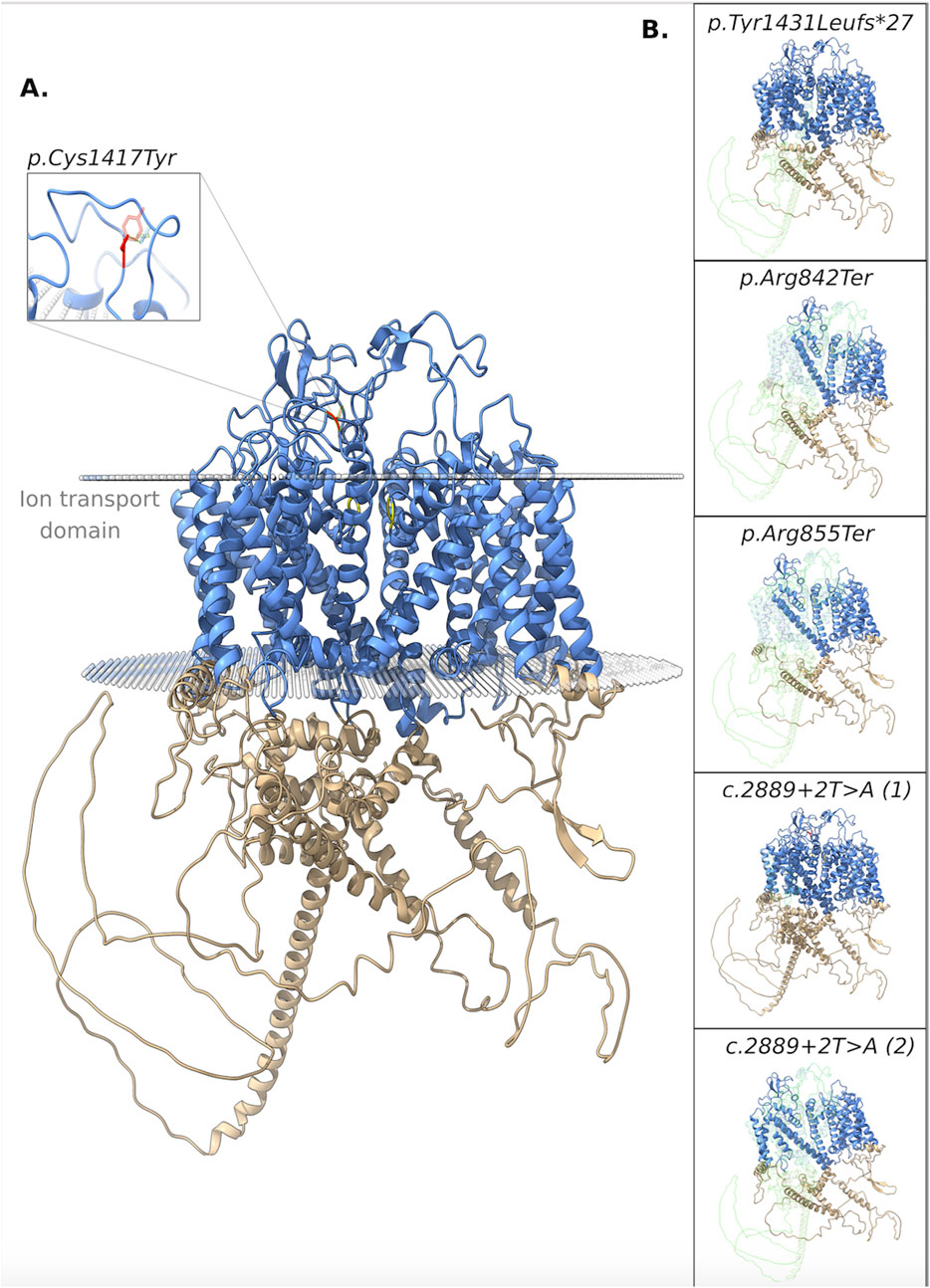
Figure 5. 3D structure of the NALCN protein. (A) Functional domains: ion transport domain and the selectivity filter highlighted in cyan and yellow, respectively. Focus on the breakage of a disulphide bond. (B) Each box shows the impact of a different variant on the protein structure. In particular, the portions of the native protein that are lost due to the frameshift, nonsense, and splicing (the two putative consequences) variants are shown.
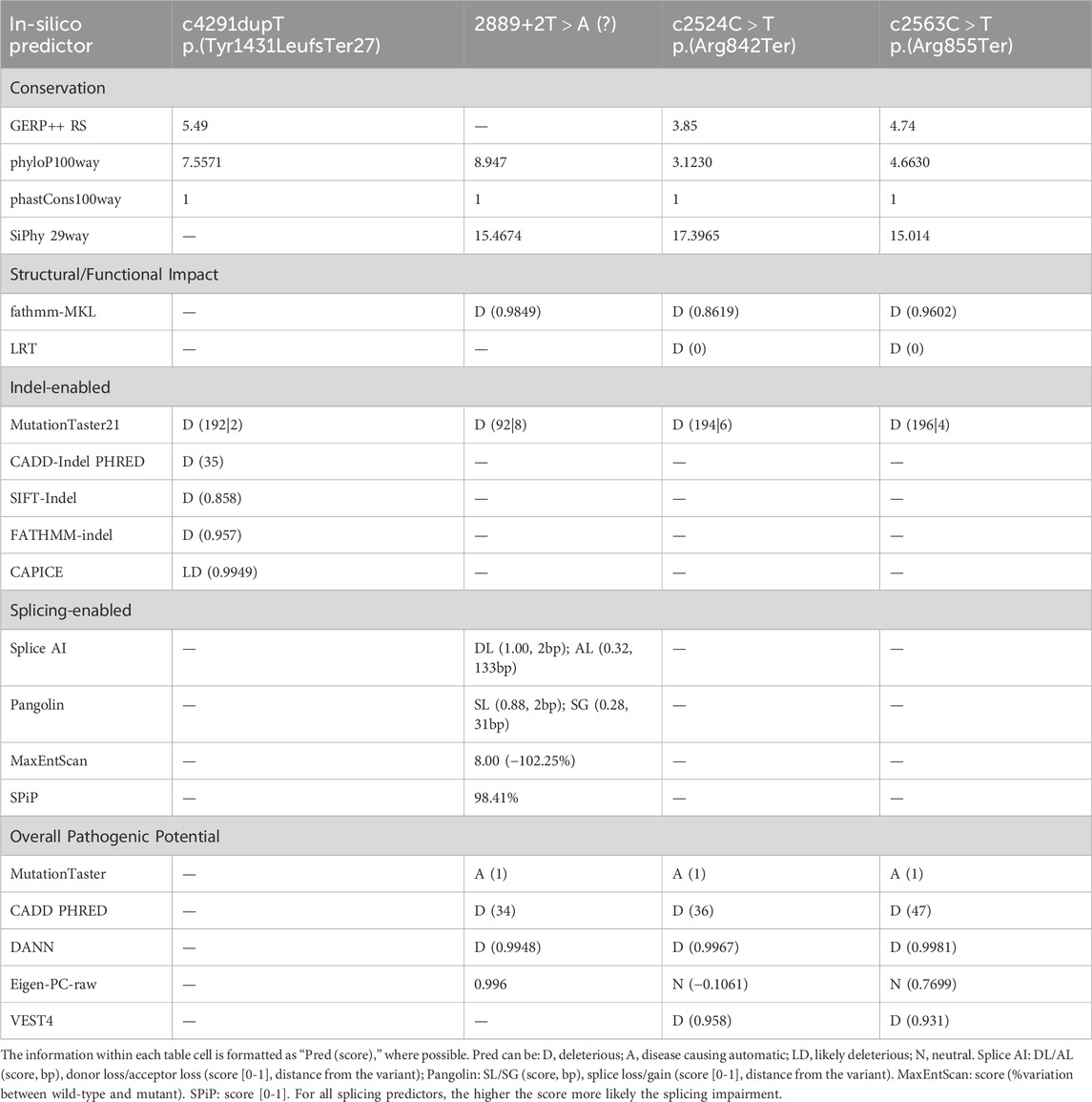
Table 2. Table summarizing in silico pathogenicity predictions for identified nonsense and splicing genetic variants. Each column represents a specific variant, and each row represents a different in silico pathogenicity prediction tool.
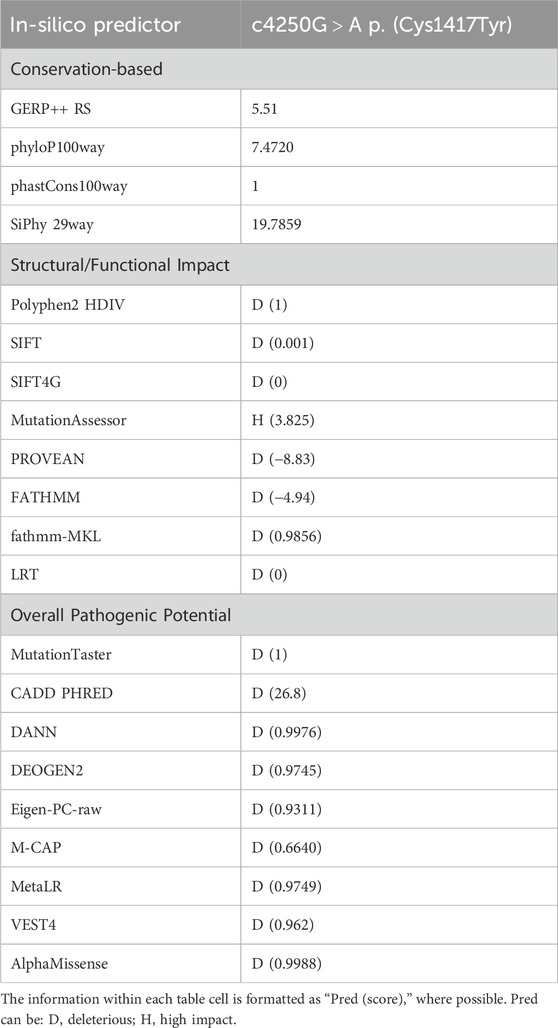
Table 3. Table summarizing in silico pathogenicity predictions for the identified missense variant. Each row represents a different in silico pathogenicity prediction tool.
From a structural standpoint and considering that the NALCN is a four-domain (domains DI to DIV, Figure 3) ion channel characterized by a transmembrane region (in blue in Figure 5A), where each domain includes six transmembrane segments, and two soluble regions that protrude in both extracellular and cytoplasmic sides (Kschonsak et al., 2020), the missense p. (Cys1417Tyr) variant is located into one of these extracellular soluble loops (Figure 5A), where an elaborate network of disulfide-bond from each domain creates an appendage above the pore selectivity filter. As reported by Missense3D, this residue substitution can be classified as damaging due to the disruption of one of these fundamental disulphide bonds, in particular between the Cys1417 and the Cys1405. We assessed the protein stability using the FoldX algorithm to further confirm its thermodynamic impact. Following free energy calculation, we estimated a clear ΔΔG increase (ΔΔGp.(Cys1417Tyr) = 10.9587 + 0.005 kcal/mol), compatible with a highly destabilizing effect. This variant, which was reported in gnomAD with a global AF of 0,000003424, was classified as a Variant of Uncertain Significance (VUS) according to the ACMG/AMP 2015 guidelines (Li and Wang, 2017).
4 Discussion
Pathogenetic variants of the NALCN gene, in homozygosity or compound heterozygosity with a putative LoF role, are associated with IHPRF1, that is inherited in an autosomal recessive manner, in contrast to heterozygous pathogenic variants in NALCN with a GoF effects, that are associated with CLIFAHDD. According to Monteil et al. (2024), variants reported in CLIFAHDD are mostly missense and are concentrated within the pore-forming S5 and S6 helices, while IHPRF1 variants are broadly distributed and the majority are truncating variants (nonsense, frameshift or deletions) (Monteil et al., 2024). LoF variants in IHPRF1 seem to cause hyperpolarization of the resting membrane potential explaining the hypoexcitability phenotype that characterizes IHPRF1 disease with profound hypotonia (Bend et al., 2016; Bramswig et al., 2018). On the other hand, most of the literature seems to indicate that the putative mechanism underlying CLIFAHDD is the gain of function of NALCN explaining the presence of arthrogryposis caused by hyperexcitability of the motor unit, secondary to the depolarization of the resting membrane potential, resulting in a hypercontracted phenotype (Bramswig et al., 2018; Bouasse et al., 2019). However, according to Bend et al. (2016), some variants reported in CLIFAHDD, seem to have a loss of function mechanism acting as dominant negative in functional assays on C. elegans (CLIFAHDD-dominant Antimorphic) (Bramswig et al., 2018).
In this study, we reported five male patients from four different families of whom four carried homozygous variants (patient 2, 3, 4, and 5) and one (patient 1) inherited a compound heterozygosity in the NALCN gene. Only the nonsense variant c.2563C > T (p.Arg855Ter) retrieved in patient 1 was already previously reported by Karimi et al. (2020), the others (1 splicing, 1 missense, 1 frameshift, and 1 nonsense) are retrieved for the first time. To date, 36 different variants have been reported in the literature in homozygosis or compound heterozygosis associated to IHPRF1, and among them, 12 nonsense, 9 missense, 9 frameshift, 5 splicing, and 1 synonymous variant (Figure 3; Supplementary Table S2). Frequencies of the main clinical features are comparable with those previously reported in literature (Table 1; Figure 1). Considering the clinical phenotype of our patients is it possible to observe that the whole cohort experienced feeding difficulties (needing in some cases NG or G tube) and FTT. This finding is consistent with the literature on IHPRF1 (Al-Sayed et al., 2013; Bramswig et al., 2018; Campbell et al., 2018). Interestingly, patient 1, after 6 months of linear growth, presented with severe FTT, in the presence of adequate caloric intake. Finding at brain MRI of a large arachnoid cyst in the suprasellar region needing surgical corrections led to the clinical diagnosis of DS. DS is a rare cause of FTT in infants associated with central nervous system tumors in the hypothalamic region. Clinical presentation includes severe emaciation, lipodystrophy, normal linear growth, and histories of poor weight gain starting at about 6 months of age despite having normal caloric intake (Burr et al., 1976; Trapani et al., 2022). Other features described but more inconstant are hyperalertness, hyperkinesia, hydrocephalus, nystagmus, visual field defects, and vomiting. The DS is characterized by the absence of specific laboratory findings related to hypothalamic dysfunction and it is often a diagnosis of exclusion after a long differential diagnostic work-up (Burr et al., 1976; Fleischman et al., 2005). MRI is considered the gold standard for the diagnosis of a cerebral mass causing DS (Hoffmann et al., 2014; Trapani et al., 2022). Treatment includes surgical resection, radiation therapy, and chemotherapy, and it is often followed by weight gain (Kim, 2012; Tosur et al., 2017). Significantly, during the diagnostic workup for patient 1 an elevation of urinary oligosaccharides with increased levels of glucose tetrasaccharide (Glc4) was reported. Glc4 (Glca1-6Glca1-4Glca1-4Glc), is a glycogen-derived dextrin that correlates with the extent of glycogen accumulation in skeletal muscle (Young et al., 2009). Glc4 is described in the literature as the most sensitive biochemical biomarker of Glycogen storage diseases (GSDs) type II (acid α-glucosidase deficiency, Pompe disease, PD, OMIM #232300) a metabolic disorder affecting mainly the liver and muscle. It was described in patients performing a whole-body muscle magnetic resonance imaging a correlation between muscle strength, the amount of fatty infiltration in muscle, and urinary Glc4 (Khan et al., 2020). The degree of elevation appears to correlate with the severity of the clinical phenotype and the stage of Pompe disease (Piraud et al., 2020; Heiner-Fokkema et al., 2020), but it can be elevated in other conditions associated with increased glycogen storage, including certain leukemias and sarcomas, Duchenne muscular dystrophy and acute pancreatitis (Young et al., 2009). Glc4 levels were markedly increased also in three disorders belonging to the autophagy machinery: Vici syndrome, Yunis-Varon syndrome, and Danon disease, all sharing a cardiomuscular involvement with increased glycogen storage and variable vacuoles accumulation detectable on light microscopy at the level of skeletal and cardiac muscle (Semeraro et al., 2021; Mastrogiorgio et al., 2021). Since muscular wasting and severe hypotonia are frequently reported in IHPRF1, we hypothesized that Glc4 could be a marker of the neuromuscular damage in this condition, so we measured Glc4 also in patient 2 but the examination tested negative. Interestingly, the urinary glucosaccharides exam was repeated in patient 1 after neurosurgery and amelioration of the growth pattern, with Glc4 levels normalization. This finding led us to hypothesize that Glc4 elevations may be considered as a DS biomarker which could be associated likewise retrieved in patient 1, with severe muscular wasting and FTT. Of note leptin levels in our patient were suppressed in several determinations, with a slight increase after surgery and weight gain. Leptin is a pleiotropic hormone primarily secreted in adipose tissue by adipocytes and is implicated in a wide range of biological functions that control different processes such as the regulation of body weight and energy expenditure, stimulates thermogenesis, and reduces appetite, reproductive function, immune response, and bone metabolism (Greco et al., 2021). In recent years, leptin has been widely studied in obesity and anorexia nervosa, and its plasma levels are related to BMI (Miller, 2011). In literature, DS is associated in large case series with low levels of leptin and concomitant increase of ghrelin (Brauner et al., 2006; Trapani et al., 2022). In some cases, reductions in leptin levels have been reported after surgical lesions’ excision probably due to a direct secretion by the tumor mass (Velasco et al., 2014). In humans, decreasing leptin concentrations in response to food deprivation are responsible for the starvation-induced suppression of the hypothalamic-pituitary-gonadal axes (Sainsbury et al., 2002). Low levels of leptin have been observed also in patients with FTT (Shaoul et al., 2003). According to Monteil et al. (2024) leptin seems to act as a positive regulator of NALCN. Recent evidence demonstrated how leptin is a potent respiratory stimulant, by activating NALCN to depolarize neurons type 1 of the solitary tract (NTS) that express the long form of leptin receptor (LepRn), providing a glutamatergic pathway that relays excitation to premotor breathing control areas in the ventral respiratory group (rVRG) which harbors bulbospinal inspiratory premotor neurons (Do et al., 2020). Do et al. (2020) also showed that the leptin-induced depolarization in type-1 cells was abolished in NALCN-cKO mice (selective deletion of NALCN in LepRb neurons), with an increase of breathing irregularities, concluding that NTS LepRb neurons contribute to the stability of the respiratory pattern and reduce the occurrence of both central and obstructive apneas. Moreover, previous studies demonstrated that injection of leptin into NTS of rats stimulates respiratory output (Inyushkina et al., 2010). This observation led us to speculate on the hypothesis that low levels of leptin in the context of IHPRF1 or CLIFAHDD can worsen the clinical picture of these patients. In this perspective, good management of FTT and weight loss could ameliorate the phenotype of the patients with IHPRF1, in particular for periodic breathing. Patient 1 presented a progressive increase of leptin values after surgery and weight gain (from 0.06 ng/mL to 1.18 ng/mL, even if still under reference value) and this was accompanied by improvement of clinical condition and periodic breathing. The patient had already started oxygen supplementation so it is difficult to assess if the increase in leptin levels may have improved this aspect. We suggest that further studies on leptin levels in a larger cohort of patients with IHPRF1 and CLIFAHDD with FTT are needed to better assess the role of this hormone in the variability of clinical features.
Breathing abnormalities are frequently reported in both patients with IHPRF1 (Campbell et al., 2018; Maselli et al., 2022) and CLIFAHDD (Gal et al., 2016; Lozic et al., 2016; Bourque et al., 2018; Winczewska-Wiktor et al., 2022). NALCN mutant mice have a severely disrupted respiratory rhythm interrupted by periods of apnea and die within 24 h of birth. Breathing pattern in NALCN null mice is characterized by apnea for 5 s followed by a burst of breathing for 5 s at a rate of 5 episodes of apnea per minute imputable to a complete loss of electrical activity from the fourth cervical root (C4) that innervates the diaphragm with rhythmic electrical signals (Lu et al., 2007). Respiratory rhythms are generated by inspiratory neurons in the pre-Bötzinger complex (preBötC) network of the medulla generating the rhythmic output to cranial and spinal motor neurons. This network is modulated by Substance P that exerts excitatory effects in preBötC neurons by modulating a nonselective cation channel with the properties of NALCN (Lozic et al., 2016; Monteil et al., 2024). Moreover, NALCN is expressed in CO2/H (+)-sensitive neurons of retrotrapezoid nucleus (RTN) that regulate breathing. In vivo, RTN-specific knockdown of NALCN reduced CO2-evoked neuronal activation and breathing (Shi et al., 2016). The main treatment for periodic breathing and apnea in the reported cases is based on oxygen supplementation at night (Lozic et al., 2016; Campbell et al., 2018), in some cases requiring NIV (Maselli et al., 2022). Tracheostomy is rarely required (Lozic et al., 2016; Vivero et al., 2017). In our cohort, we reported three patients with breathing abnormalities, patient 1 with periodic breathing and severe central apneas requiring oxygen supplementation, patient 2 with periodic breathing and severe central apneas requiring NIV (discontinued on parent’s decision) and patient 4 with mild central apneas not requiring medical treatment. Interestingly, an increase in urinary homovanillic acid, vanilmandelic acid and NSE values was observed over time in patient 1 during the workup for the presence of brain cysts (before detecting a benign lesion at anatomopathological evaluation) that did not normalize after surgery. Conversely, after the introduction of oxygen supplementation, a decrease in the respective values was observed, with normalization of the homovanillic/vanilmandelic acid ratio (Supplementary Figure S3). Thus, it is possible to hypothesize that the progressive normalization of the urinary homovanillic acid, vanilmandelic acid and their ratio, could be associated to the amelioration of periodic breath, after the low flow oxygen therapy. In literature, several studies suggested an alteration of the sympathetic nervous system with an increase in urinary norepinephrine levels in apneic patients (Dimsdale et al., 1995). To date, most of the studies evaluated this relation in obstructive sleep apnea (OSA). In a recent pediatric systematic review, it was reported that catecholamine levels can be pointed out as a marker for sympathetic nervous system excitability in OSA, and correlate with specific clinical, cardiovascular, neurobehavioral, and metabolic parameters, contributing to increased morbidity (Sica et al., 2021). It is shown that continuous positive airway pressure (CPAP) treatment in patients with OSA reduces catecholamine levels and blood pressure (Green et al., 2021). Sympathetic nervous system activation was observed also in patients with central sleep apnea (McLaren et al., 2019). Recurrent infections, in particular respiratory tract infections, are another important clinical finding among IHPRF1 and CLIFAHDD patients (Bramswig et al., 2018; Karimi, et al., 2020; Tehrani Fateh et al., 2023). In our cohort, we detected two patients with recurrent infections, patient 1 with frequent respiratory infections requiring antibiotic prophylaxis, and patient 3 with multiple episodes of pneumonia. According to Bramswig et al. (2018), recurrent infections in the cohort were not associated with immunological alterations (specific information on test performed was not reported) and were correlated with FTT. Recently, Tehrani Fateh et al. (2023) described a IHPRF1 patient with recurrent urinary tract infections hypothesizing that abnormal contraction of detrusor muscle due to NALCN LoF could lead to incomplete emptying of the bladder with an increased risk of infections. However, patient 1 of the present cohort was found with partial functional defects of T and B lymphocytes that were reported after in vitro stimulation with PHA, OKT3, and CpG and a reduction of NK activity. According to Karimi et al. (2020), male individuals seem to display a more severe phenotype when compared to female individuals, even in the same family. Interestingly, estrogen and progesterone play a role in the regulation of NALCN expression in human myometrial muscle cells suggesting a sex-driven regulation for NALCN. All patients in the present cohort are males and some features such as epilepsy and breathing abnormalities are slightly higher compared to the literature review (which includes both male and female individuals), confirming that the male sex worsens the phenotype in this disease.
Finally, all variants cause a markedly significant functional impact, thereby compromising the functionality of the protein complex, as indicated by in silico and structural analyses. Variants introducing premature stop codon may act at different levels, mutant transcripts might undergo nonsense-mediated mRNA decay, or, alternatively, the proteins could be produced but may be severely affected in terms of function and interaction with other elements of the complex. When considering patients harboring nonsense truncating variants in codons 842 (p.Arg842Ter) and 855 (p.Arg855Ter) in the present cohort, namely, patient 1 and patient 2 and the two siblings described by Karimi et al. (2020), it is interesting to observe that all of these four patients seems to display breathing abnormalities that in our two cases are severe, needing in one case oxygen supplementation (patient 1) and NIV (patient 2). We also reported a new missense variant in homozygosity, p.Cys1417Tyr, located into an extracellular soluble loop of the protein. To date, missense variants are less frequently reported as pathogenic, and, only one missense variant c.3860G > T, (p.Trp1287Leu) was reported in homozygosity in three patients from the same family localizing on a site close to the outer region of the channel, with likely milder functional consequences (Al-Sayed et al., 2013). Close to the missense homozygous variant p.Cys1417Tyr described in the present work, a missense variant c.4281C > A, p.Phe1427Leu in compound heterozygosity with a splicing variant (c.4281C > A and c.4103 + 2T > C) in two siblings located in the predicted S6 pore forming segment of domain IV was reported (Bramswig et al., 2018). Of interest, the phenotype of these patients, along with the clinical picture of patient 3 from the present cohort harboring the missense homozygous variant c.4250G>A p.Cys1417Tyr seems to be milder compared with patients harboring truncating variants in NALCN. Further studies and functional in vitro analyses will be necessary to characterize the molecular effects that truncating and missense variants have at the molecular level to enable more accurate genotype-phenotype correlations.
5 Conclusion
In this study, we focused on WES, in silico pathogenicity predictors, and structural analyses to examine genetic variants retrieved from a new IHPRF1 patients’ cohort. We described five new IHPRF1 patients from four different families, harboring four newly identified and one previously retrieved variant whose deep investigation exhibited a markedly significant functional impact, thereby compromising the functionality of the NALCN complex. The present findings, taken together, widen the pathogenic mechanism and functional spectrum of biallelic variants affecting the NALCN gene, as well as the IHPRF1 syndrome diagnosis and genotype-phenotype correlation.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the study involving human samples in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
DV: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–original draft, Writing–review and editing, Resources. MM: Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing–review and editing. MG: Data curation, Formal Analysis, Investigation, Validation, Writing–review and editing. FMP: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing–original draft, Writing–review and editing. FP: Formal Analysis, Methodology, Software, Validation, Visualization, Writing–original draft, Resources. NL: Formal Analysis, Methodology, Software, Validation, Visualization, Writing–original draft, Resources. FC: Data curation, Formal Analysis, Investigation, Validation, Writing–review and editing. LS: Data curation, Formal Analysis, Investigation, Validation, Writing–review and editing. IR: Data curation, Formal Analysis, Investigation, Validation, Writing–review and editing. EA: Formal Analysis, Investigation, Methodology, Software, Validation, Writing–review and editing. DC: Formal Analysis, Investigation, Methodology, Software, Validation, Writing–review and editing. NeC: Data curation, Formal Analysis, Investigation, Validation, Writing–review and editing. MS: Formal Analysis, Investigation, Methodology, Validation, Writing–original draft. CR: Formal Analysis, Investigation, Methodology, Validation, Writing–original draft. AD: Data curation, Investigation, Validation, Writing–review and editing. NaC: Data curation, Investigation, Validation, Writing–review and editing. SC: Investigation, Validation, Writing–review and editing. EV: Investigation, Validation, Writing–review and editing. CN: Investigation, Validation, Writing–review and editing. SA: Data curation, Investigation, Validation, Writing–original draft. JS: Data curation, Investigation, Validation, Writing–original draft. JD: Investigation, Validation, Writing–original draft. PA: Investigation, Validation, Writing–review and editing. JK: Investigation, Validation, Writing–original draft. PB: Formal Analysis, Writing–original draft, Methodology, Data curation, Investigation, Validation, Writing–review and editing. DM: Writing–review and editing, Resources, Methodology, Investigation. CD: Validation, Writing–review and editing, Methodology, Investigation. VC: Data curation, Writing–original draft, Resources, Methodology, Formal Analysis, Visualization, Writing–review and editing, Software. AB: Writing–review and editing, Investigation, Funding acquisition, Methodology, Data curation, Validation, Formal Analysis, Conceptualization, Supervision. AN: Validation, Writing–review and editing, Supervision, Methodology, Data curation, Formal Analysis, Investigation. TM: Writing–review and editing, Methodology, Supervision, Conceptualization, Software, Writing–original draft, Formal Analysis, Project administration, Visualization, Data curation, Resources, Validation.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Italian Ministry of Health with Current Research funds.
Acknowledgments
This study has been generated within the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN-ITHACA) [EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516]. The authors thank patients and their families for their generosity in taking part in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1477940/full#supplementary-material
References
Abul-Husn, N. S., Marathe, P. N., Kelly, N. R., Bonini, K. E., Sebastin, M., Odgis, J. A., et al. (2023). Molecular diagnostic yield of genome sequencing versus targeted gene panel testing in racially and ethnically diverse pediatric patients. Genet. Med. 25, 100880. doi:10.1016/j.gim.2023.100880
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). “7.20.1-7.20.41. Predicting functional effect of human missense mutations using PolyPhen-2,”. Hoboken, NJ: Current Protocols in Human Genetics. Chapter, Unit7.20. doi:10.1002/0471142905.hg0720s76
Alirezaie, N., Kernohan, K. D., Hartley, T., Majewski, J., and Hocking, T. D. (2018). ClinPred: prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. Am. J. Hum. Genet. 103, 474–483. doi:10.1016/j.ajhg.2018.08.005
Al-Sayed, M. D., Al-Zaidan, H., Albakheet, A., Hakami, H., Kenana, R., Al-Yafee, Y., et al. (2013). Mutations in NALCN cause an autosomal-recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am. J. Hum. Genet. 93, 721–726. doi:10.1016/j.ajhg.2013.08.001
Angius, A., Cossu, S., Uva, P., Oppo, M., Onano, S., Persico, I., et al. (2018). Novel NALCN biallelic truncating mutations in siblings with IHPRF1 syndrome. Clin. Genet. 93, 1245–1247. doi:10.1111/cge.13162
Bend, E. G., Si, Y., Stevenson, D. A., Bayrak-Toydemir, P., Newcomb, T. M., Jorgensen, E. M., et al. (2016). NALCN channelopathies: distinguishing gain-of-function and loss-of-function mutations. Neurology 87, 1131–1139. doi:10.1212/WNL.0000000000003095
Bouasse, M., Impheng, H., Servant, Z., Lory, P., and Monteil, A. (2019). Functional expression of CLIFAHDD and IHPRF pathogenic variants of the NALCN channel in neuronal cells reveals both gain- and loss-of-function properties. Sci. Rep. 9, 11791. doi:10.1038/s41598-019-48071-x
Bourque, D. K., Dyment, D. A., MacLusky, I., Kernohan, K. D., and Care and McMillan, H. J. (2018). Periodic breathing in patients with NALCN mutations. J. Hum. Genet. 63, 1093–1096. doi:10.1038/s10038-018-0484-1
Bramswig, N. C., Bertoli-Avella, A. M., Albrecht, B., Al Aqeel, A. I., Alhashem, A., Al-Sannaa, N., et al. (2018). Genetic variants in components of the NALCN–unc80–unc79 ion channel complex cause a broad clinical phenotype (NALCN channelopathies). Hum. Genet. 137, 753–768. doi:10.1007/s00439-018-1929-5
Brauner, R., Trivin, C., Zerah, M., Souberbielle, J. C., Doz, F., Kalifa, C., et al. (2006). Diencephalic syndrome due to hypothalamic tumor: a model of the relationship between weight and puberty onset. J. Clin. Endocrinol. Metabolism 91, 2467–2473. doi:10.1210/jc.2006-0322
Burr, I. M., Slonim, A. E., Danish, R. K., Gadoth, N., and Butler, I. J. (1976). Diencephalic syndrome revisited. J. Pediatr. 88, 439–444. doi:10.1016/s0022-3476(76)80260-0
Campbell, J., FitzPatrick, D. R., Azam, T., Gibson, N. A., Somerville, L., Joss, S. K., et al. (2018). NALCN dysfunction as a cause of disordered respiratory rhythm with central apnea, 141, S485–S490. doi:10.1542/peds.2017-0026
Carneiro, T. N., Krepischi, A. C., Costa, S. S., Tojal da Silva, I., Vianna-Morgante, A. M., Valieris, R., et al. (2018). Utility of trio-based exome sequencing in the elucidation of the genetic basis of isolated syndromic intellectual disability: illustrative cases. Appl. Clin. Genet. 11, 93–98. doi:10.2147/TACG.S165799
Carter, H., Douville, C., Stenson, P. D., Cooper, D. N., and Karchin, R. (2013). Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14 (Suppl. 3), S3. doi:10.1186/1471-2164-14-S3-S3
Castellana, S., Biagini, T., Petrizzelli, F., Parca, L., Panzironi, N., Caputo, V., et al. (2021). MitImpact 3: modeling the residue interaction network of the Respiratory Chain subunits. Nucleic Acids Res. 49, D1282–D1288. doi:10.1093/nar/gkaa1032
Chen, Y., Liu, L., Li, J., Jiang, H., Ding, C., and Zhou, Z. (2022). MetaLR: meta-tuning of learning rates for transfer learning in medical imaging. arXiv:2206.01408v2. [cs.CV]. doi:10.48550/arXiv.2206.01408
Cheng, J., Novati, G., Pan, J., Bycroft, C., Žemgulytė, A., Applebaum, T., et al. (2023). Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492. doi:10.1126/science.adg7492
Chennen, K., Weber, T., Lornage, X., Kress, A., Böhm, J., Thompson, J., et al. (2020). MISTIC: a prediction tool to reveal disease-relevant deleterious missense variants. PLOS ONE 15, e0236962. PubMed: 32735577. doi:10.1371/journal.pone.0236962
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLOS ONE 7, e46688. doi:10.1371/journal.pone.0046688
Chong, J. X., McMillin, M. J., Shively, K. M., Beck, A. E., Marvin, C. T., Armenteros, J. R., et al. (2015). De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am. J. Hum. Genet. 96, 462–473. doi:10.1016/j.ajhg.2015.01.003
Chun, S., and Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. doi:10.1101/gr.092619.109
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLOS Comput. Biol. 6, e1001025. doi:10.1371/journal.pcbi.1001025
Dimsdale, J. E., Coy, T., Ziegler, M. G., Ancoli-Israel, S., and Clausen, J. (1995). The effect of sleep apnea on plasma and urinary catecholamines. Sleep 18, 377–381. PubMed: 7676172.
Do, J., Chang, Z., Sekerková, G., McCrimmon, D. R., and Martina, M. (2020). A leptin-mediated neural mechanism linking breathing to metabolism. Cell. Rep. 33, 108358. doi:10.1016/j.celrep.2020.108358
Farwell, K. D., Shahmirzadi, L., El-Khechen, D., Powis, Z., Chao, E. C., Tippin Davis, B., et al. (2015). Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 17, 578–586. doi:10.1038/gim.2014.154
Ferlaino, M., Rogers, M. F., Shihab, H. A., Mort, M., Cooper, D. N., Gaunt, T. R., et al. (2017). An integrative approach to predicting the functional effects of small indels in non-coding regions of the human genome. BMC Bioinforma. 18, 442. doi:10.1186/s12859-017-1862-y
Fleischman, A., Brue, C., Poussaint, T. Y., Kieran, M., Pomeroy, S. L., Goumnerova, L., et al. (2005). Diencephalic syndrome: a cause of failure to thrive and a model of partial growth hormone resistance. Pediatrics 115, e742–e748. doi:10.1542/peds.2004-2237
Frustaci, A., De Luca, A., Guida, V., Biagini, T., Mazza, T., Gaudio, C., et al. (2018). Novel α-actin gene mutation p.(Ala21Val) causing familial hypertrophic cardiomyopathy, myocardial noncompaction, and transmural crypts. Clinical-pathologic correlation. J. Am. Heart Assoc. 7 (Ala21Val), e008068. doi:10.1161/JAHA.117.008068
Gal, M., Magen, D., Zahran, Y., Ravid, S., Eran, A., Khayat, M., et al. (2016). A novel homozygous splice site mutation in NALCN identified in siblings with cachexia, strabismus, severe intellectual disability, epilepsy and abnormal respiratory rhythm. Eur. J. Med. Genet. 59, 204–209. doi:10.1016/j.ejmg.2016.02.007
Greco, M., De Santo, M., Comandè, A., Belsito, E. L., Andò, S., Liguori, A., et al. (2021). Leptin-activity modulators and their potential pharmaceutical applications. Biomolecules 11, 1045. doi:10.3390/biom11071045
Green, M., Ken-Dror, G., Fluck, D., Sada, C., Sharma, P., Fry, C. H., et al. (2021). Meta-analysis of changes in the levels of catecholamines and blood pressure with continuous positive airway pressure therapy in obstructive sleep apnea. J. Clin. Hypertens. 23, 12–20. doi:10.1111/jch.14061
Heiner-Fokkema, M. R., van der Krogt, J., de Boer, F., Fokkert-Wilts, M. J., Maatman, R. G. H. J., Hoogeveen, I. J., et al. (2020). The multiple faces of urinary glucose tetrasaccharide as biomarker for patients with hepatic glycogen storage diseases. Genet. Med. 22, 1915–1916. doi:10.1038/s41436-020-0878-2
Hoffmann, A., Gebhardt, U., Sterkenburg, A. S., Warmuth-Metz, M., and Müller, H. L. (2014). Diencephalic syndrome in childhood craniopharyngioma—results of German multicenter studies on 485 long-term survivors of childhood craniopharyngioma. J. Clin. Endocrinol. Metabolism 99, 3972–3977. doi:10.1210/jc.2014-1680
Hu, J., and Ng, P. C. (2013). SIFT indel: predictions for the functional effects of amino acid insertions/deletions in proteins. PLOS ONE 8, e77940. doi:10.1371/journal.pone.0077940
Inyushkina, E. M., Merkulova, N. A., and Inyushkin, A. N. (2010). Mechanisms of the respiratory activity of leptin at the level of the solitary tract nucleus. Neurosci. Behav. Physiology 40, 707–713. doi:10.1007/s11055-010-9316-2
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885. doi:10.1016/j.ajhg.2016.08.016
Ionita-Laza, I., McCallum, K., Xu, B., and Buxbaum, J. D. (2016). A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 48, 214–220. doi:10.1038/ng.3477
Ittisoponpisan, S., Islam, S. A., Khanna, T., Alhuzimi, E., David, A., and Sternberg, M. J. E. (2019). Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J. Mol. Biol. 431, 2197–2212. doi:10.1016/j.jmb.2019.04.009
Jagadeesh, K. A., Wenger, A. M., Berger, M. J., Guturu, H., Stenson, P. D., Cooper, D. N., et al. (2016). M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48, 1581–1586. doi:10.1038/ng.3703
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting splicing from primary sequence with deep learning. Cell. 176, 535–548. doi:10.1016/j.cell.2018.12.015
Kang, Y., and Chen, L. (2022). Structure and mechanism of NALCN-FAM155A-UNC79-UNC80 channel complex. Nat. Commun. 13, 2639. doi:10.1038/s41467-022-30403-7
Karimi, A. H., Karimi, M. R., Farnia, P., Parvini, F., and Foroutan, M. (2020). A homozygous truncating mutation in NALCN causing IHPRF1: detailed clinical manifestations and a review of literature. Appl. Clin. Genet. 13, 151–157. doi:10.2147/TACG.S261781
Khan, A., Tian, S., Tariq, M., Khan, S., Safeer, M., Ullah, N., et al. (2022). NGS-driven molecular diagnosis of heterogeneous hereditary neurological disorders reveals novel and known variants in disease-causing genes. Mol. Genet. Genomics 297, 1601–1613. doi:10.1007/s00438-022-01945-8
Khan, A. A., Boggs, T., Bowling, M., Austin, S., Stefanescu, M., Case, L., et al. (2020). Whole-body magnetic resonance imaging in late-onset Pompe disease: clinical utility and correlation with functional measures. J. Inherit. Metabolic Dis. 43, 549–557. doi:10.1002/jimd.12190
Kim, E. (2012). Symptomatic Rathke cleft cyst: clinical features and surgical outcomes. World Neurosurg. 78, 527–534. doi:10.1016/j.wneu.2011.12.091
Kim, S., Jhong, J. H., Lee, J., and Koo, J. Y. (2017). Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 10, 2. doi:10.1186/s13040-017-0126-8
Köhler, S., Gargano, M., Matentzoglu, N., Carmody, L. C., Lewis-Smith, D., Vasilevsky, N. A., et al. (2021). The human phenotype ontology in 2021. Nucleic Acids Res. 49, D1207–D1217. doi:10.1093/nar/gkaa1043
Köroğlu, Ç., Seven, M., and Tolun, A. (2013). Recessive truncating NALCN mutation in infantile neuroaxonal dystrophy with facial dysmorphism. J. Med. Genet. 50, 515–520. doi:10.1136/jmedgenet-2013-101634
Kschonsak, M., Chua, H. C., Noland, C. L., Weidling, C., Clairfeuille, T., Bahlke, O. Ø., et al. (2020). Structure of the human sodium leak channel NALCN. Nature 587, 313–318. doi:10.1038/s41586-020-2570-8
Kschonsak, M., Chua, H. C., Weidling, C., Chakouri, N., Noland, C. L., Schott, K., et al. (2022). Structural architecture of the human NALCN channelosome. Nature 603, 180–186. doi:10.1038/s41586-021-04313-5
Leman, R., Parfait, B., Vidaud, D., Girodon, E., Pacot, L., Le Gac, G., et al. (2022). SPiP: splicing Prediction Pipeline, a machine learning tool for massive detection of exonic and intronic variant effects on mRNA splicing. Hum. Mutat. 43, 2308–2323. doi:10.1002/humu.24491
Li, Q., and Wang, K. (2017). InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 100, 267–280. doi:10.1016/j.ajhg.2017.01.004
Li, S., van der Velde, K. J., de Ridder, D., van Dijk, A. D. J., Soudis, D., Zwerwer, L. R., et al. (2020). CAPICE: a computational method for Consequence-Agnostic Pathogenicity Interpretation of Clinical exome variations. Genome Med. 12, 75. doi:10.1186/s13073-020-00775-w
Liu, Y., Yan, X., and Zhou, T. (2013). TBCK influences cell proliferation, cell size and mTOR signaling pathway. PLOS ONE 8, e71349. doi:10.1371/journal.pone.0071349
Lozic, B., Johansson, S., Lovric Kojundzic, S., Markic, J., Knappskog, P. M., Hahn, A. F., et al. (2016). Novel NALCN variant: altered respiratory and circadian rhythm, anesthetic sensitivity. Ann. Clin. Transl. Neurology 3, 876–883. doi:10.1002/acn3.362
Lu, B., Su, Y., Das, S., Liu, J., Xia, J., and Ren, D. (2007). The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. 129, 371–383. doi:10.1016/j.cell.2007.02.041
Maselli, K., Park, H., Breilyn, M. S., and Arens, R. (2022). Severe central sleep apnea in a child with biallelic variants in NALCN. J. Clin. Sleep Med. 18, 2507–2513. doi:10.5664/jcsm.10146
Mastrogiorgio, G., Macchiaiolo, M., Buonuomo, P. S., Bellacchio, E., Bordi, M., Vecchio, D., et al. (2021). Clinical and molecular characterization of patients with adenylosuccinate lyase deficiency. Orphanet J. rare Dis. 16 (1), 112. doi:10.1186/s13023-021-01731-6
McLaren, A. T., Bin-Hasan, S., and Narang, I. (2019). Diagnosis, management and pathophysiology of central sleep apnea in children. Paediatr. Respir. Rev. 30, 49–57. doi:10.1016/j.prrv.2018.07.005
Miller, K. K. (2011). Endocrine dysregulation in anorexia nervosa update. J. Clin. Endocrinol. Metabolism 96, 2939–2949. doi:10.1210/jc.2011-1222
Monteil, A., Guérineau, N. C., Gil-Nagel, A., Parra-Diaz, P., Lory, P., and Senatore, A. (2024). New insights into the physiology and pathophysiology of the atypical sodium leak channel NALCN. Physiol. Rev. 104, 399–472. doi:10.1152/physrev.00014.2022
National library of medicine NALCN sodium leak channel, nonselective Homo sapiens human. (2024). Accessed August 07, 2024. Bethesda, MD: National Library of Medicine. Available at: https://www.ncbi.nlm.nih.gov/gene/259232.
Ng, P. C., and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi:10.1093/nar/gkg509
Ope, O., Bhoj, E. J., Nelson, B., Li, D., Hakonarson, H., and Sobering, A. K. (2020). A homozygous truncating NALCN variant in two Afro-Caribbean siblings with hypotonia and dolichocephaly. Am. J. Med. Genet. Part A 182, 1877–1880. doi:10.1002/ajmg.a.61744
Piraud, M., Pettazzoni, M., de Antonio, M., Vianey-Saban, C., Froissart, R., Chabrol, B., et al. (2020). Urine glucose tetrasaccharide: a good biomarker for glycogenosis type II and III? A study of the French cohort. Mol. Genet. Metabolism Rep. 23, 100583. PubMed: 32382504. doi:10.1016/j.ymgmr.2020.100583
Quang, D., Chen, Y., and Xie, X. X. (2015). DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761–763. doi:10.1093/bioinformatics/btu703
Raimondi, D., Tanyalcin, I., Ferté, J., Gazzo, A., Orlando, G., Lenaerts, T., et al. (2017). DEOGEN2: prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. 45, W201-W206–W206. doi:10.1093/nar/gkx390
Sainsbury, A., Cooney, G. J., and Herzog, H. (2002). Hypothalamic regulation of energy homeostasis. Best Pract. and Res. Clin. Endocrinol. and Metabolism 16, 623–637. doi:10.1053/beem.2002.0230
Schubach, M., Maass, T., Nazaretyan, L., Röner, S., and Kircher, M. (2024). CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 52, D1143–D1154. doi:10.1093/nar/gkad989
Schwarz, J. M., Rödelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi:10.1038/nmeth0810-575
Schymkowitz, J., Borg, J., Stricher, F., Nys, R., Rousseau, F., and Serrano, L. (2005). The FoldX web server: an online force field. Nucleic Acids Res. 33 (Web Server Issue), W382–W388. doi:10.1093/nar/gki387
Semeraro, M., Sacchetti, E., Deodato, F., Coşkun, T., Lay, I., Catesini, G., et al. (2021). A new UHPLC-MS/MS method for the screening of urinary oligosaccharides expands the detection of storage disorders. Orphanet J. Rare Dis. 16, 24. doi:10.1186/s13023-020-01662-8
Shamseldin, H. E., Faqeih, E., Alasmari, A., Zaki, M. S., Gleeson, J. G., and Alkuraya, F. S. (2016). Mutations in UNC80, encoding part of the UNC79-UNC80-NALCN channel complex, cause autosomal-recessive severe infantile encephalopathy. Am. J. Hum. Genet. 98, 210–215. doi:10.1016/j.ajhg.2015.11.013
Shaoul, R., Kessel, A., Toubi, E., Lanir, A., Glazer, O., and Jaffe, M. (2003). Leptin and cytokines levels in children with failure to thrive. J. Pediatr. Gastroenterology Nutr. 37, 487–491. doi:10.1097/00005176-200310000-00016
Shi, Y., Abe, C., Holloway, B. B., Shu, S., Kumar, N. N., Weaver, J. L., et al. (2016). Nalcn is a ‘leak’ sodium channel that regulates excitability of brainstem chemosensory neurons and breathing. J. Neurosci. 36, 8174–8187. doi:10.1523/JNEUROSCI.1096-16.2016
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L. A., Edwards, K. J., et al. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65. doi:10.1002/humu.22225
Shihab, H. A., Rogers, M. F., Gough, J., Mort, M., Cooper, D. N., Day, I. N. M., et al. (2015). An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543. doi:10.1093/bioinformatics/btv009
Sica, E., De Bernardi, F., Nosetti, L., Martini, S., Cosentino, M., Castelnuovo, P., et al. (2021). Catecholamines and children obstructive sleep apnea: a systematic review. Sleep. Med. 87, 227–232. doi:10.1016/j.sleep.2021.09.007
Steinhaus, R., Proft, S., Schuelke, M., Cooper, D. N., Schwarz, J. M., and Seelow, D. (2021). MutationTaster2021. Nucleic Acids Res. 49, W446–W451. doi:10.1093/nar/gkab266
Stray-Pedersen, A., Cobben, J. M., Prescott, T. E., Lee, S., Cang, C., Aranda, K., et al. (2016). Biallelic mutations in UNC80 cause persistent hypotonia, encephalopathy, growth retardation, and severe intellectual disability. Am. J. Hum. Genet. 98, 202–209. doi:10.1016/j.ajhg.2015.11.004
Susgun, S., Yucesan, E., Goncu, B., Hasanoglu Sayin, S., Kina, U. Y., Ozgul, C., et al. (2024). Two rare autosomal recessive neurological disorders identified by combined genetic approaches in a single consanguineous family with multiple offspring. Neurol. Sci. 45, 2271–2277. doi:10.1007/s10072-023-07211-y
Takenouchi, T., Inaba, M., Uehara, T., Takahashi, T., Kosaki, K., and Mizuno, S. (2018). Biallelic mutations in NALCN: expanding the genotypic and phenotypic spectra of IHPRF1. Am. J. Med. Genet. Part A 176, 431–437. doi:10.1002/ajmg.a.38543
Tehrani Fateh, S., Bagheri, S., Sadeghi, H., Salehpour, S., Fazeli Bavandpour, F., Sadeghi, B., et al. (2023). Extending and outlining the genotypic and phenotypic spectrum of novel mutations of NALCN gene in IHPRF1 syndrome: identifying recurrent urinary tract infection. Neurol. Sci. 44, 4491–4498. doi:10.1007/s10072-023-06960-0
Tosur, M., Tomsa, A., and Paul, D. L. (2017). Diencephalic syndrome: a rare cause of failure to thrive. BMJ Case Rep. 2017, bcr2017220171. doi:10.1136/bcr-2017-220171
Trapani, S., Bortone, B., Bianconi, M., Rubino, C., Sardi, I., Lionetti, P., et al. (2022). Diencephalic syndrome in childhood, a challenging cause of failure to thrive: miniseries and literature review. Italian J. Pediatr. 48, 147. doi:10.1186/s13052-022-01316-4
Urquhart, D. S. (2018). NALCN dysfunction as a cause of disordered respiratory rhythm with central apnea. Pediatrics 141, S485–S490. doi:10.1542/peds.2017-0026
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M., and Ng, P. C. (2016). SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9. doi:10.1038/nprot.2015.123
Velasco, P., Clemente, M., Lorite, R., Ventura, M. C., Gros, L., Sanchez de Toledo, J., et al. (2014). The role of leptin in diencephalic syndrome. Pediatrics 133, e263–e266. doi:10.1542/peds.2012-3196
Vivero, M., Cho, M. T., Begtrup, A., Wentzensen, I. M., Walsh, L., Payne, K., et al. (2017). Additional de novo missense genetic variants in NALCN associated with CLIFAHDD syndrome. Clin. Genet. 91, 929–931. doi:10.1111/cge.12899
Wiel, L., Baakman, C., Gilissen, D., Veltman, J. A., Vriend, G., and Gilissen, C. (2019). MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat. 40, 1030–1038. doi:10.1002/humu.23798
Winczewska-Wiktor, A., Hirschfeld, A. S., Badura-Stronka, M., Wojsyk-Banaszak, I., Sobkowiak, P., Bartkowska-Śniatkowska, A., et al. (2022). Central apneas due to the CLIFAHDD syndrome successfully treated with pyridostigmine. Int. J. Environ. Res. Public Health 19, 775. doi:10.3390/ijerph19020775
Wu, J., Li, Q., Li, Y., Lin, J., Yang, D., Zhu, G., et al. (2014). A long type of TBCK is a novel cytoplasmic and mitotic apparatus-associated protein likely suppressing cell proliferation. J. Genet. Genomics 41, 69–72. doi:10.1016/j.jgg.2013.12.006
Yauy, K., Baux, D., Pegeot, H., Van Goethem, C., Mathieu, C., Guignard, T., et al. (2018). MoBiDiC prioritization algorithm, a free, accessible, and efficient pipeline for single-nucleotide variant annotation and prioritization for next-generation sequencing routine molecular diagnosis. J. Mol. Diagnostics 20, 465–473. doi:10.1016/j.jmoldx.2018.03.009
Yeo, G., and Burge, C. B. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394. doi:10.1089/1066527041410418
Young, S. P., Zhang, H., Corzo, D., Thurberg, B. L., Bali, D., Kishnani, P. S., et al. (2009). Long-term monitoring of patients with infantile-onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet. Med. 11, 536–541. doi:10.1097/GIM.0b013e3181a87867
Zeng, T., and Li, Y. I. (2022). Predicting RNA splicing from DNA sequence using Pangolin. Genome Biol. 23, 103. doi:10.1186/s13059-022-02664-4
Keywords: NALCN, IHPRF1, CLIFAHDD, channelosome complex, genotype-phenotype correlation, rhythmic behaviors, structural biology
Citation: Vecchio D, Macchiaiolo M, Gonfiantini MV, Panfili FM, Petrizzelli F, Liorni N, Cortellessa F, Sinibaldi L, Rana I, Agolini E, Cocciadiferro D, Colantoni N, Semeraro M, Rizzo C, Deodati A, Cotugno N, Caggiano S, Verrillo E, Nucci CG, Alkan S, Saraiva JM, De Sá J, Almeida PM, Krishna J, Buonuomo PS, Martinelli D, Dionisi Vici C, Caputo V, Bartuli A, Novelli A and Mazza T (2024) Widening the infantile hypotonia with psychomotor retardation and characteristic Facies-1 Syndrome’s clinical and molecular spectrum through NALCN in-silico structural analysis. Front. Genet. 15:1477940. doi: 10.3389/fgene.2024.1477940
Received: 08 August 2024; Accepted: 27 November 2024;
Published: 11 December 2024.
Edited by:
Xiaonan Zhao, Baylor College of Medicine, United StatesReviewed by:
Ewelina Bukowska-Olech, Poznan University of Medical Sciences, PolandPietro Hiram Guzzi, Magna Græcia University, Italy
Fatih Kelesoglu, Istanbul University, Türkiye
Copyright © 2024 Vecchio, Macchiaiolo, Gonfiantini, Panfili, Petrizzelli, Liorni, Cortellessa, Sinibaldi, Rana, Agolini, Cocciadiferro, Colantoni, Semeraro, Rizzo, Deodati, Cotugno, Caggiano, Verrillo, Nucci, Alkan, Saraiva, De Sá, Almeida, Krishna, Buonuomo, Martinelli, Dionisi Vici, Caputo, Bartuli, Novelli and Mazza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Vecchio, ZGF2aWRlLnZlY2NoaW9Ab3BiZy5uZXQ=