 Meiying Wei
Meiying Wei Bo Wang
Bo Wang Chaoqun Li
Chaoqun Li Xiaolan Li
Xiaolan Li Cai He
Cai He Yi Li
Yi Li- College of Forestry, Gansu Agricultural University, Lanzhou, China
Introduction: Nitraria tangutorum Bobr., a prominent xerophytic shrub, exhibits remarkable adaptability to harsh environment and plays a significant part in preventing desertification in northwest China owing to its exceptional drought and salinity tolerance.
Methods: To investigate the drought-resistant mechanism underlying N. tangutorum, we treated 8-week-old seedlings with polyethylene glycol (PEG)-6000 (20%, m/m) to induce drought stress. 27 samples from different tissues (leaves, roots and stems) of N. tangutorum at 0, 6 and 24 h after drought stress treatment were sequenced using PacBio single-molecule real-time (SMRT) sequencing and Illumina RNA sequencing to obtain a comprehensive transcriptome.
Results: The PacBio SMRT sequencing generated 44,829 non-redundant transcripts and provided valuable reference gene information. In leaves, roots and stems, we identified 1162, 2024 and 232 differentially expressed genes (DEGs), respectively. The Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that plant hormone signaling and mitogen-activated protein kinase (MAPK) cascade played a pivotal role in transmitting stress signals throughout the whole N. tangutorum plant following drought stress. The interconversion of starch and sucrose, as well as the biosynthesis of amino acid and lignin, may represent adaptive strategies employed by N. tangutorum to effectively cope with drought. Transcription factor analysis showed that AP2/ERF-ERF, WRKY, bHLH, NAC and MYB families were mainly involved in the regulation of drought response genes. Furthermore, eight physiological indexes, including content of proline, hydrogen peroxide (H2O2), malondialdehyde (MDA), total amino acid and soluble sugar, and activities of three antioxidant enzymes were all investigate after PEG treatment, elucidating the drought tolerance mechanism from physiological perspective. The weighted gene co-expression network analysis (WGCNA) identified several hub genes serve as key regulator in response to drought through hormone participation, ROS cleavage, glycolysis, TF regulation in N. tangutorum.
Discussion: These findings enlarge genomic resources and facilitate research in the discovery of novel genes research in N. tangutorum, thereby establishing a foundation for investigating the drought resistance mechanism of xerophyte.
1 Introduction
Drought is one of the most severe abiotic stresses worldwide that poses a significant threat to the survival and development of plants (Osmolovskaya et al., 2018). Due to the sessile nature, plants are subjected to endure drought stress and have evolved a sophisticated array of response mechanisms through long-term evolution. Once upon drought stress, plants perceive signals by various signaling transduction (Hirayama and Shinozaki, 2010; Takahashi et al., 2018). Plant hormones, including abscisic acid (ABA), auxin (IAA), cytokinin (CTK), gibberellin (GA), ethylene (ET), brassinosteroid (BR), jasmonic acid (JA) and salicylic acid (SA), have been identified for their involvement in the signaling networks as signal molecules (Lamaoui et al., 2018; Waadt, 2020). The mitogen-activated protein kinase (MAPK) cascade is also a stress-responsive signal transduction pathway that becomes activated in plants upon perceiving drought. MAPK cascade is involved in signal transduction between cells as well as within and outside the cell nucleus (Chen et al., 2023). Besides, due to the role as a second messenger in transducing extracellular stimuli into the intracellular environment, Ca2+ signaling also plays a pivotal role as a crucial signaling pathway in the drought response (Bartels and Sunkar, 2005). Along with signal transduction processes, different transcription factors (TF) like AP2/ERF, WRKY, NAC, bHLH, and MYB, are involved in the plant’s response to drought, and play essential roles through regulating the expression of downstream genes by binding with the cis-elements (Joshi et al., 2016). A series of drought-responsive genes will be activated, leading to the reprogramming the metabolism and growth of plant, resulting in various physiological and metabolic changes, such as the accumulation of reactive oxygen species (ROS) and activation of antioxidant enzymes (Li M. et al., 2022), dynamic interconversion between starch and sugars (Dong and Beckles, 2019), metabolism of amino acid and phenylpropanoid (Batista-Silva et al., 2019; Diniz et al., 2020; Dong and Lin, 2021). Ultimately, plants respond to drought at the whole-plant level through gene regulation and physiological adaptability. In conclusion, multiple signaling transduction pathways, TFs regulation and metabolic adjustments synergistically respond to drought stress in plants (Yang F. and Lv G., 2022).
Understanding the mechanisms of plant drought resistance is crucial for investigating strategies to mitigate the adverse effects of drought on plants and for cultivating genetically modified crops with enhanced drought tolerance through genetic technology (Ullah et al., 2017; Singh et al., 2015). The transcriptomic analysis has been extensively employed for elucidating the underlying mechanisms of plant drought resistance. The Illumina RNA-seq technique enables accurate quantification of genes alteration among tissues based on temporal and spatial expression profiling in many plants (Chen et al., 2021a). However, the effectiveness of reconstructing and annotating is hindered by the constraint imposed by short read. The SMRT sequencing provides the advantage of generating long and full-length transcripts, but it may be prone to inaccuracies (Li W. L. et al., 2021; Zhao et al., 2018). Therefore, the integration of Illumina RNA-seq with SMRT-seq can synergistically result in comprehensive transcriptional information for various plants species. The approach has been practiced in many studies, such as Haloxylon (Yang F. and Lv G. H., 2022), Populus ussuriensis (Li W. et al., 2021), Pinus massoniana L. (Mei et al., 2020) and Actinidia valvata (Li Z. et al., 2022).
Nitraria tangutorum Bobr. is a super-xerophytic shrub that falls under the Zygophyllaceae family, primarily found in the northwestern region of China (Zhu et al., 2021). As a desert plant, N. tangutorum possess easily produced branches, small leaves and extensive root system, showing strong adaptability to abiotic challenges like drought, high salinity, alkali condition, and sand burial in harsh environments. It is capable of thriving in arid and semiarid desert environments, where the average annual precipitation ranges from 140.9 to 302.2 mm (Zheng et al., 2014). These characteristics makes it an outstanding candidate species for investigating drought-stress tolerance mechanism. However, the research on N. tangutorum has mainly focused on its physiological responses to various stimuli (Gao et al., 2022; Didi et al., 2022; Yang et al., 2010a; Yang et al., 2010b), the identification of bioactive compounds in its fruit, leaves and seeds (Chen et al., 2021b; Hu et al., 2014; Jiang et al., 2021; Zhou et al., 2019), and a few transcriptome sequencing studies with the absence of whole-genome data (Liu C. et al., 2022; Liu et al., 2023; Wang et al., 2015; Wang et al., 2022; Zhu et al., 2021). Liu et al. have identified the genes and metabolic pathways in response to drought stress through RNA-sequencing by taking leaf tissues (Liu C. et al., 2022). But the drought response mechanism of each tissue in N. tangutorum with the prolongation of drought stress lacks comprehensive and systematic transcriptome data.
In light of the deficiencies in current research on drought resistance mechanisms of N. tangutorum, our objective is to address three key issues: How do the tissues of N. tangutorum respond to drought stress? What is the molecular mechanism underlying its drought resistance? Which key genes are involved in N. tangutorum’s response to drought? To find the answers, we conducted physiological analyses and transcriptome comparison in three tissues of N. tangutorum under drought stress stimulated by polyethylene glycol (PEG) 6,000 at 0 h, 6 h, and 24 h, using an integration of full-length and Illumina RNA sequencing data. The dynamics gene expression in each tissue, proposed potential molecular mechanisms and key genes underlying drought tolerance of N. tangutorum were investigated. The findings will enrich genetic resources underlying research on drought-tolerance and facilitate exploration of the novel genes for breeding plants with enhanced drought resistance.
2 Materials and methods
2.1 Plant materials and drought treatment
The N. tangutorum seeds were acquired from Nuomuhong farm, Haixi autonomous Prefecture, Qinghai Province in the northwest region of China (96°11′E, 36°33′N). They underwent a 24-h soaking period in sterile water, followed by surface sterilization with ethanol solution (75%, v/v) for 1 min and sterile water for five times. The clean seeds were sown in plastic containers (10 cm × 10 cm × 10 cm) filled with heat-sterilized river sand, containing 12 seeds per container. These containers were placed inside the greenhouse trays at Gansu Agricultural University (103°70′E, 36°09′N) with the temperature of 28°C ± 2°C/26°C ± 2°C (day/night), the photoperiod lasted for 16 h while darkness prevailed for 8 h, the relative humidity was maintained 60% ± 10%. After germination, the seedlings received weekly irrigation using half-strength Murashige and Skoog (MS) medium (Hope, Qingdao, China). The nutrient solution was prepared by dissolving 39.45 g of medium in 1 L of water and carefully pouring into the tray, ensuring complete saturation of the sand in the container. Poor seedlings were removed after 4 weeks of germination.
At the age of 8 weeks, the underperforming seedlings were selectively removed again to ensure the preservation of robust and uniformly sized seedlings. PEG 6,000 solution (20%, m/m) was applied to induce drought stress, in the same ways as irrigating with half-strength MS solution. Different tissues from N. tangutorum (roots, stems and leaves) were harvested at 0, 6, and 24 h after treatments (named as R0h, R6h, R24h; S0h, S6h, S24h; L0h, L6h, and L24h) (Supplementary Figure S1–S3). For each time point, three biological replicates were performed for each of the three tissues (3 tissues × 3 time points × 3 biological replications = 27 samples), with each replicate consisting of 12 individual seedlings. Root samples were collected from all seedlings’ root tissues, stem samples were collected from the middle section of the seedlings, approximately 3 cm in length, and leaf samples were taken from the leaves attached to stem samples (Supplementary Figure S4). All the samples immediately stored in liquid nitrogen for 4 h after collection, followed by freezing at −80°C in a refrigerator to facilitate RNA extraction and relevant experiment.
2.2 Preparation cDNA library of Illumina RNA-seq, PacBio SMRT-seq and sequencing
The EASYspin Plant microRNA Kit RN40 (Aidlab, Beijing, China) was utilized to extract total RNA from 27 samples. The quality of RNA samples was assessed by a NanoDrop 2000 instrument (Thermo Fisher Scientific, Waltham, MA, United States), while their integrity was evaluated by Agient 2,100 and LabChip GX instruments (PerkinElmer, Waltham, MA, United States). High-quality RNA samples were utilized for cDNA library construction employing the VAHTS Universal V6 RNA-seq Library Prep Kit for Illumina. Subsequently, sequencing of the libraries was performed on Illumina NovaSeq 6,000 platform (Illumina, San Diego, CA, United States) in 150-bp paired-end mode at Biomarker Technologies Corporation (Beijing, China).
To prepare the full-length cDNA library for PacBio SMRT-seq, a pooled sample comprising an equivalent quantity of total RNA from 27 samples was prepared. The SMRTbell library was performed utilizing the SMRTbell Template Prep Kit (Pacific Biosciences, Menlo Park, CA, United States), and the sequencing was established on Sequel II system (Pacific Biosciences, Menlo Park, CA, United States) at Biomarker Technologies Corporation (Beijing, China).
2.3 Sequences analysis
The RNA-seq raw data was filtered by fastp (v0.23.2) (Chen et al., 2018) to remove the adapter sequences, reads with over 10% N content, and low-quality reads, the resulting clean data was assembled into unigenes using trinity (v2.5.1) software (Grabherr et al., 2011).
The Iso-Seq3 pipeline (https://github.com/PacificBiosciences/IsoSeq) was utilized to analyze the PacBio raw subreads. Circular consensus (CCS) subreads were obtained from the subreads bam files using CCS (v6.2.0) with a minimum quality threshold set at 0.9 and full passes ≥3 for a zero-mode waveguide (ZMW). By examining the presence of the correct 5′primer, 3′primer, and polyA tail within the CCS sequence, we divided CCSs into full-length non-chimeric reads (FLNC) and non-full-length non-chimeric (nFL) reads. And then Lima (v2.1.0) and isoseq3 (v3.4.0) refine were employed to eliminate poly(A) tails and cDNA primers. High-quality consensus sequences for FLNC transcripts were derived using the ICE clustering algorithm. Classification based on post-correction accuracy exceeding 99% was performed for these high-quality FL consensus sequences. Cd-hit (identity >0.99) was used for removing redundancy of high-quality FL transcripts sequences, and then non-redundant FLNC transcripts were obtained.
Sequence alignment was performed using STAR (v2.5.0b) to align clean reads of RNA-seq with the non-redundant FLNC transcripts and obtain positional information on the transcript, using the non-redundant FLNC transcripts as a reference (Dobin et al., 2013). Kallisto software (v0.46.1) was utilized to compare the reads of Illumina RNA-seq with full-length transcripts and perform direct counting for transcript expression quantification (Bray et al., 2016). FPKM (Fragments Per Kilobase of transcript per Million mapped reads) was employed as a measure to estimate the level of transcript expression.
2.4 Identification of simple sequence repeats (SSR), prediction of lncRNA and open reading frame (ORF)
SSR were identified in transcript sequences exceeding 500 bp using MISA software (v1.0) (http://pgrc.ipk-gatersleben.de/misa/). The screening encompassed mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, representing six types of SSRs, and the corresponding minimum repeat parameters were 10 for mononucleotide, 6 for dinucleotides, and 5 for the other types.
The identification of potential lncRNA candidates was accomplished by integrating four computational methods, namely Coding-non-Coding Index (CNCI), Coding Potential Calculator (CPC), Coding Potential Assessment Tool (CPAT) and Protein family (Pfam) database. LncRNA candidates were selected from the transcripts that exceeded 200 nt in length and contained more than two exons, which were further refined using CPC (Kong et al., 2007), CNCI (Luo et al., 2014), CPAT (Wang et al., 2013) and Pfam. The finally lncRNA results were obtained by using a Venn diagram to compare the predicted lncRNAs from the four methods.
TransDecoder (v5.0.0) (https://github.com/TransDecoder/TransDecoder/releases) were used for ORF prediction (Haas et al., 2013).
2.5 Analysis of differentially expressed genes
The DESeq2 (v1.6.3) tool was utilized for identification the differentially expressed genes (DEGs) among sample groups (Anders and Huber, 2010). The criterion was |log2 (fold change) | >1 and FDR (false discovery rate) < 0.01, based on the FPKM values.
2.6 Weighted gene co-expression network analysis (WGCNA)
DEGs with FPKM values less than 1 were filtered out, resulting in 3,165 DEGs used for WGCNA analysis using the WGCNA (v1.47) package in R (Langfelder and Horvath, 2008). The weighted network was constructed with unsigned topological overlap matrix. The values of power, minimum number of genes for modules, and minimum height for merging modules are 5, 30, and 0.3045 respectively. Cytoscape (v3.10.2) was used to visualize the network results.
2.7 Quantitative real-time PCR (qRT-PCR) analysis
The TUREscript 1st Stand cDNA Synthesis Kit (Aidlab, Beijing, China) was used for performing reverse transcription. The qRT-PCR was conducted using 2 × SYBR® Green Master Mix (DF Biotech., Chengdu, China) on a qTOWER2.2 Quantitative Real-Time PCR Thermal Cyclers (Analytik Jena, Germany). The 18SrRNA gene from N. tangutorum was employed as a reference for normalizing the data. The relative expression of each transcript was determined using the 2−ΔΔCT method (Rao et al., 2013), and FPKM values were used for comparison. Each sample underwent three biological and three technical replicates. The Beacon designer software (v8.14) was used to design specific primers.
2.8 Measurement of physiological indexes
All physiological indexes were assessed utilizing commercially accessible kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The contents of H2O2, soluble sugar (SS), Malondialdehyde (MDA), proline (Pro) and total amino acid content (TAA) were measure by Hydrogen Peroxide assay kit (A064-1-1) (Zhou et al., 2020), Plant soluble sugar content test kit (A145-1-1) (Wang J. et al., 2021), Malondialdehyde (MDA) assay kit (A003-1-2) (Menhas et al., 2022), proline assay kit (A107-1-1) (Menhas et al., 2022) and Total Amino Acid assay kit (A026-1-1) (Sun et al., 2010), respectively. The activities of CAT, SOD and POD (Menhas et al., 2022) were measured by Catalase (CAT) assay kit (A007-1-1), Total Superoxide Dismutase (SOD) assay kit (A001-1-2) and Peroxidase (POD) assay kit (A084-3-1), respectively. All operational procedures were carried out according to the manufacturer’s instructions. Physiological measurements were obtained by averaging data from three biological samples (n = 3).
2.9 Data analysis
Excel 2021 was used for data collation and analysis. SPSS (v26.0) software was used to perform one-way variance analysis (ANOVA) on physiological indexes. The Duncan test was employed to analyze the means values of the data for statistical significance (p-values <0.05). The Venn diagrams were built using the free online tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html). The heatmaps were constructed using BMKCloud (www.biocloud.net) based on the FPKM values of DEGs.
3 Results
3.1 Overview of SMRT and RNA-seq sequencing data
A total of 23.92G of SMRT sequencing data were generated. 238,525 CCS reads and 188,559 (79.05%) full length non-chimeric (FLNC) reads were produced. Followed that, a total of 75,987 consensus sequences were generated and 99.99% (75,979) of them being recognized as high-quality sequences. Subsequently eliminating redundant reads led to the acquisition of 44,829 non-redundant FLNC transcripts (Supplementary Figure S5).
The Illumina RNA-seq generated a total of 184.60 Gb clean data (27 samples) with each sample containing more than 6.09 Gb. The Q30 percentages exceeded 93.91% and the GC contents exhibited a range of 44.78%–45.71% (Supplementary Table S1). A total of 47,808 unigenes were collected.
3.2 Identification of SSRs, prediction of lncRNA and ORF
In the study, 21,101 SSRs were identified. Mononucleotides accounts for the largest proportion at 62.67%, followed by dinucleotides (22.31%). Pentanucleotides exhibited the lowest abundance with a ratio of 0.31% (Figure 1A). And totally 11,615 lncRNAs were identified in N. tangutorum (Figure 1B). The prediction of ORF and their corresponding amino acid sequences in all non-redundant transcripts sequence was performed. A total of 18,343 complete ORFs were obtained, and the protein encoded by these ORFs had a length distribution ranging from 49aa to 2,427aa (Figure 1C).
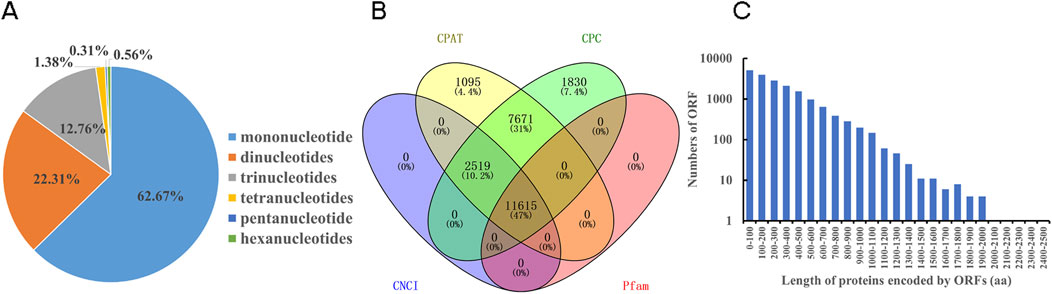
Figure 1. Identification of SSR, prediction of lncRNAs and ORFs. (A) Type distribution of SSRs. (B) Venn diagram of the predicted lncRNAs. (C) The length distribution of protein encoded by ORFs prediction (The vertical axis was scaled logarithmically).
3.3 Functional annotation of transcripts
44,829 transcripts were mapped into nine protein and nucleic acid databases by DIAMOND software. Out of the total transcripts, 33,703 (75.2%) obtained annotation information. Among them, 33,124 (98.3%) were annotated in NR (NCBI Non-Redundant protein sequences), 28,252 (83.8%) in GO (Gene Ontology), 23,081 (68.5%) in KEGG (Kyoto Encyclopedia of Genes and Genomes), 24,187 (71.8%) in Swissprot, 33,135 (98.3%) in TrEMBL (Translation of EMBL), 22,955 (68.1%) in Pfam (Protein family), 19,486 (57.8%) in KOG (euKaryotic Orthologous Groups), 9,795 (29.1%) in COG (Clusters of Orthologous Groups), and 28,849 (85.6%) in eggNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups) (Figure 2A). The identification of homologous species was achieved by aligning transcripts against the NR database. Among them, Pistacia vera (7,237, 21.85%) exhibited the highest sequence hits with N. tangutorum, followed by Acer yangbiense (4,252, 12.84%), Citrus sinensis (2,910, 8.79%), C. clementina (2,801, 8.46%), C. unshiu (1,046, 3.16%), Theobroma cacao (593, 1.79%) (Figure 2B).
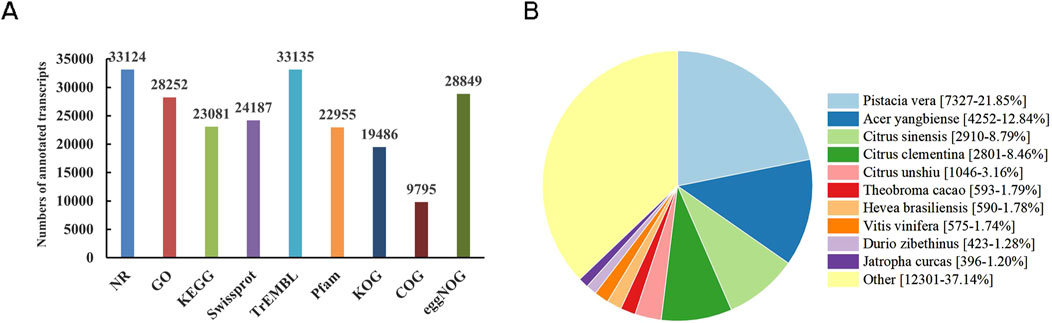
Figure 2. Functional annotation of non-redundant transcripts in N. tangutorum. (A) Functional annotation of transcripts in nine databases. (B) Analysis of the homologous species distribution.
3.4 Identification and KEGG classification of differentially expressed genes (DEGs)
The comparisons were conducted between the control group at 0 h and different time points of drought stress (6 h, 24 h) in specific tissues. A total of 682 and 731 DEGs were detected in the comparisons between L0h vs. L6h and L0h vs. L24h, 448 and 1,947 DEGs were found in R0h vs. R6h and R0h vs. R24h, 79 and 171 DEGs were identified in S0h vs. S6h and S0h vs. S24h, respectively (Figure 3A). And more than 90% of the DEGs were annotated (Supplementary Table S2). The leaves and roots exhibited the highest number of DEGs at 6 h and 24 h time points in six comparison groups, respectively. The Venn diagram analysis of annotated DEGs showed a limited overlap of DEGs were identified across different tissues following 6 h and 24 h drought treatment (Figures 3B, C). KEGG classification of exclusively expressed DEGs in each tissue at 6 h and 24 h indicated that the predominant clusters were all linked to “metabolic pathways” and “biosynthesis of secondary metabolites” in leaves, roots and stems (Supplementary Figures S6–S11). This implies a highly coordinated response to drought stress at the whole-plant level in N. tangutorum.
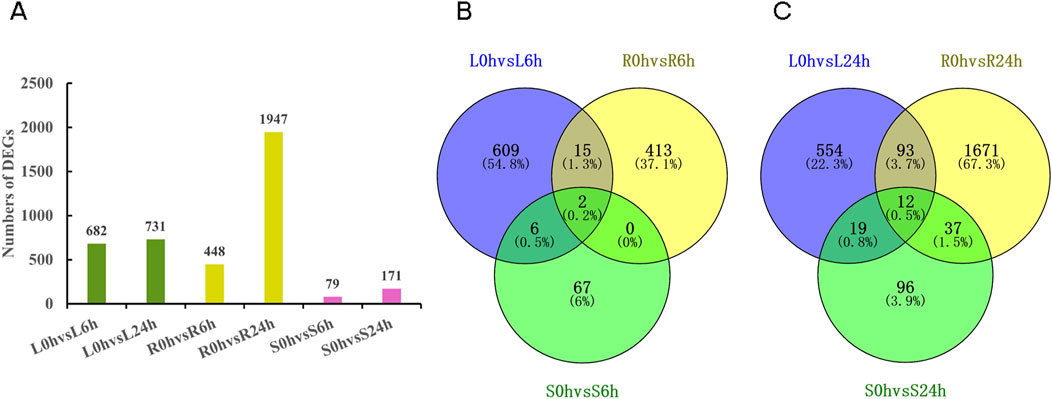
Figure 3. DEGs in N. tangutorum under drought stress. (A) The number of DEGs in different tissues in 0 h vs. 6 h and 0 h vs. 24 h comparison group (green: DEGs in leaves; yellow: DEGs in roots; fuchsia: DEGs in stems). (B) The Venn diagram of annotated DEGs in different tissues in 0 h vs. 6 h comparison groups. (C) The Venn diagram of annotated DEGs in different tissues in 0 h vs. 24 h comparison groups.
3.5 KEGG analysis of DEGs
The DEGs under drought stress were further studied using KEGG pathway enrichment analyses to explore the intricate biological behaviors. A total of 127 pathways exhibited enrichment among the DEGs. The top 20 notable pathways in the leaves, roots and stems were identified. In leaves, the pathways that showed the most significance during the stress process include “MAPK signaling pathway-plant” and “plant hormone signal transduction”. Additionally, at 6 h and 24 h, there is a significant enrichment of the “plant-pathogen interaction” and “starch and sucrose metabolism” pathways respectively (Figures 4A, B). In roots, there was an enrichment of DEGs in pathways related to “plant hormone signal transduction” and “MAPK signaling pathway-plant.” Additionally, “phenylpropanoid biosynthesis,” “starch and sucrose metabolism” and “biosynthesis of amino acids” pathway also showed active representation (Figures 4C, D). In stems, fewer DEGs were found after drought stress. The DEGs were enriched in “flavonoid biosynthesis” in 0 h vs. 6 h group (Figure 4E), while DEGs were dispersedly enriched in “plant hormone signal transduction” and “alpha-linolenic acid metabolism” pathway after 24 h drought stress (Figure 4F).
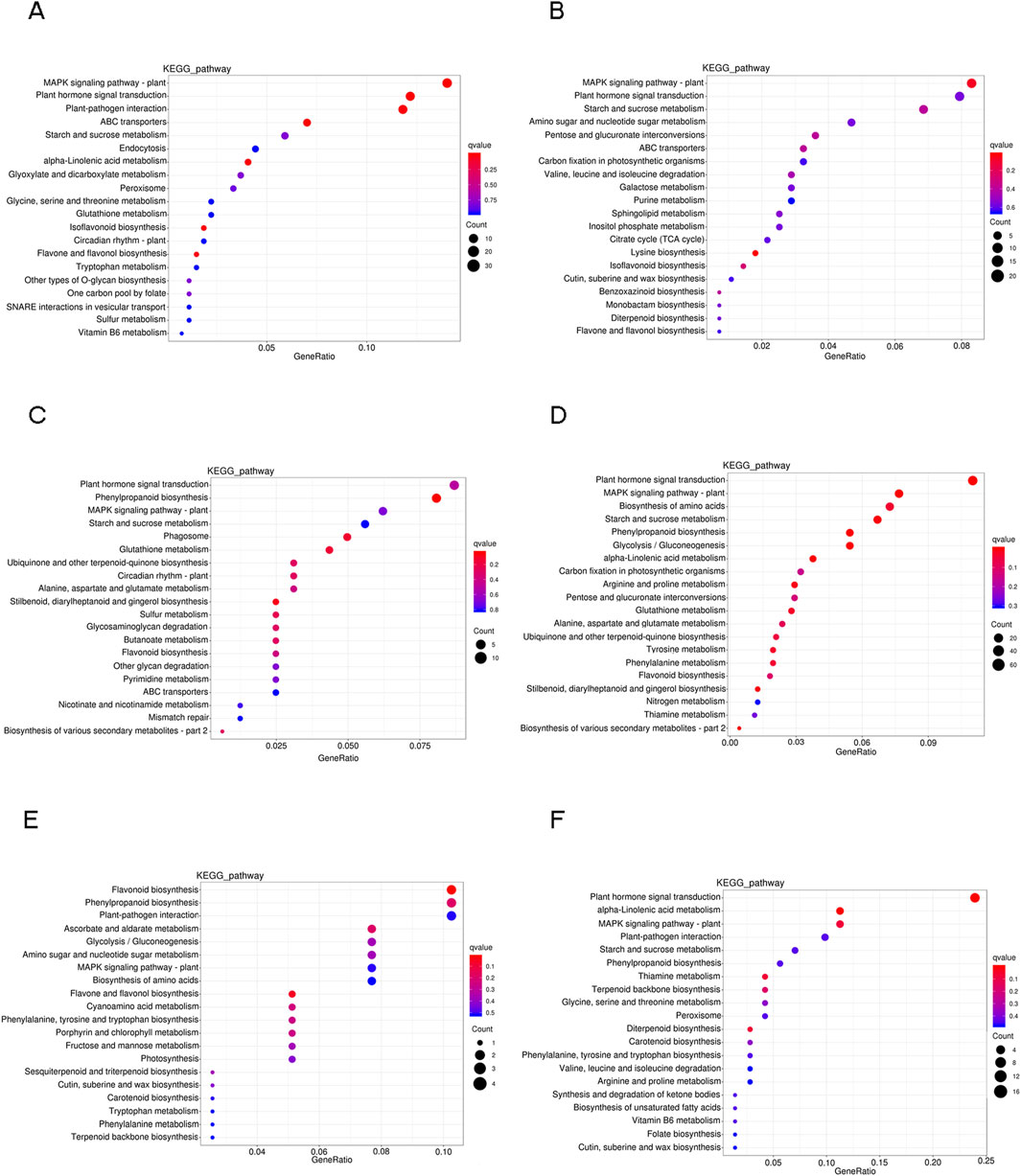
Figure 4. Scatterplot of KEGG pathway enrichment analysis for DEGs in six comparison groups. Only the top 20 pathways were shown. (A) L0h vs. L6h. (B) L0h vs. L24h. (C) R0h vs. R6h. (D) R0h vs. R24h. (E) S0h vs. S6h. (F) S0h vs. S24h.
3.6 DEGs associated with plant hormone signal transduction and mitogen-activated protein kinase (MAPK) cascade
The pathway of “plant hormone signal transduction” exhibits a notable enrichment in the whole plant. Hormones that undergo changes in gene expression include IAA, CTK, GA, ABA, ET, BR, JA, and SA (Figure 5). After 6 h drought stress, there were 33 DEGs associated with hormone signaling in leaves, 14 in roots, and 1 in stems. After a period of 24 h under drought conditions, the number changed to 22 DEGs in leaves, 79 in roots and 17 in stems (Figure 5; Supplementary Table S3). All eight hormones mentioned above were related to DEGs found in leaves and roots while only JA was mainly connected with DEGs found in stems.
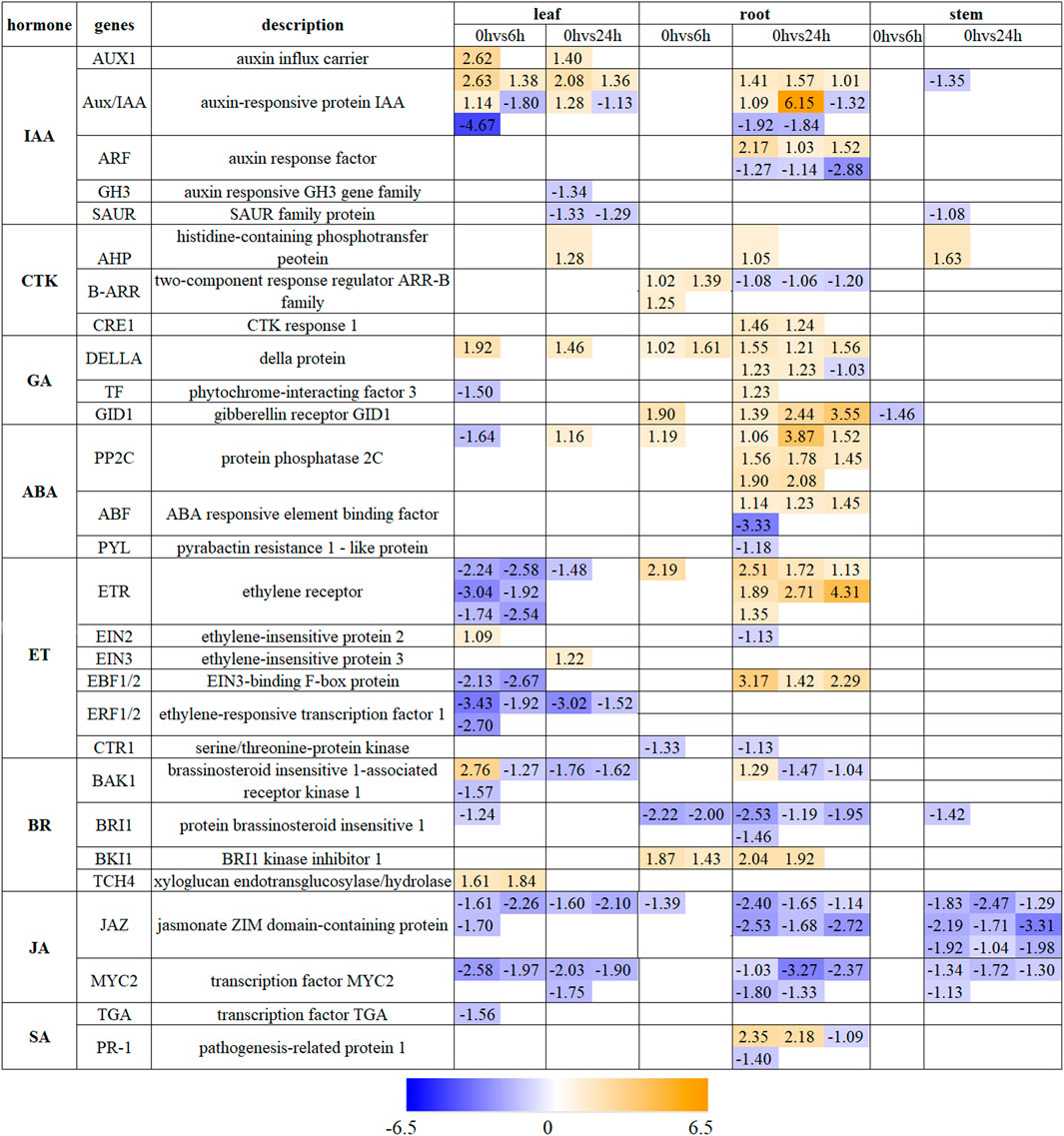
Figure 5. The expression profile of DEGs involved in “Plant hormone signal transduction” in leaves, roots and stems. The number represents the expression changes of DEGs defined as Log2FC from upregulation (orange) to downregulation (blue).
In the present investigation, two components (MAPK and MAPKKK) of the MAPK cascade were found to be DEGs in N. tangutorum (Figure 6; Supplementary Table S4). The differential expression of all MAPK genes and the majority of MAPKKK genes in N. tangutorum is observed in both leaves and roots, with only one MAPKKK gene showing differential expression in stems.
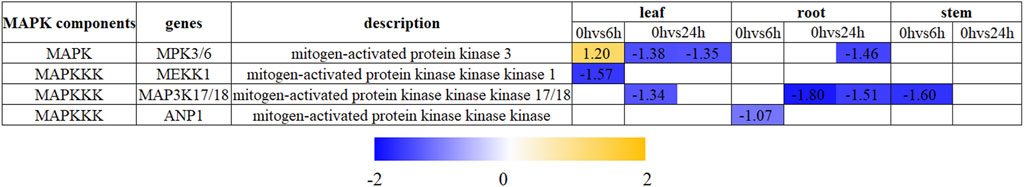
Figure 6. The expression profile of DEGs involved in “mitogen-activated protein kinase (MAPK) cascade” in leaves, roots and stems. The number represents the expression changes of DEGs defined as Log2FC from upregulation (orange) to downregulation (blue).
3.7 DEGs associated with the elimination of reactive oxygen species (ROS), starch and sucrose metabolism, phenylpropanoid biosynthesis, amino acid biosynthesis
The current study identified a total of 47 DEGs associated with ROS scavenging. These DEGs included 7 CAT genes, 4 SOD genes, 17 POD genes and 19 glutathione S-transferase (GST) genes (Supplementary Table S5). The expression alteration of both CAT and SOD genes were detected in all three tissues, while POD and GST genes mainly changed in roots. After 24 h of drought stress, 8 POD genes and 14 GST genes were upregulated in roots.
During the drought stress process at 6 h and 24 h time points, a total of 33 genes were involved in the “starch and sucrose metabolism” pathway in leaves (Figure 7A; Supplementary Table S6). 16 genes encoding sucrose synthase (SuS) and 2 genes encoding alpha-amylase (AMY) exhibited varying expression patterns. These findings imply the “starch and sucrose metabolism” may play a crucial role in N. tangutorum’s ability to cope with drought-induced stress.

Figure 7. Heatmap of DEGs involved in “starch and sucrose metabolism” pathway and “phenylpropanoid biosynthesis” pathway. (A) Heatmap of DEGs in “starch and sucrose metabolism” pathway in leaves. (B) Heatmap of DEGs in “phenylpropanoid biosynthesis” pathway in roots.
The transcriptome analysis revealed that 43 DEGs exhibited enrichment in the “phenylpropanoid biosynthesis” pathway in roots. It was particular noteworthy that the majority of the DEGs were upregulated after being exposed to drought, such as 3 phenylalanine ammonia-lyase (PAL) genes, 3 4-coumarate-CoA ligase (4CL) genes, 5 cinnamyl-alcohol dehydrogenase (CAD) and most of POD genes (Figure 7B; Supplementary Table S7). Moreover, these DEGs predominantly reside within the lignin biosynthesis pathway, which constitutes a pivotal branch of the phenylpropanoid biosynthesis (Dong and Lin, 2021).
The analysis revealed that the “biosynthesis of amino acid” pathway in R0h vs. R24h group exhibited enrichment with 52 DEGs. After 24h of drought stress, 44 out of 52 genes were found to be upregulated, including 7 gene encoding delta-1-pyrroline-5-carboxylate synthase (P5CS). In the early stage of drought at 6h, only 3 genes showed changes in expression, with one P5CS being upregulated (Supplementary Table S8).
3.8 Differentially expressed transcription factors (TFs)
The present study identified a total of 1,055 transcripts that were predicted to possess TF activity (Supplementary Table S9). The expression of 207 TFs from 35 different families was found to undergo alteration under drought stress conditions (Supplementary Table S10). The 207 TFs mainly enriched in AP2/ERF-ERF (32, 15.5%), WRKY (21, 10.1%), bHLH (16, 7.7%), NAC (15, 7.2%) and MYB (13, 6.3%) families. The number of differentially expressed TFs identified in pairwise comparisons were L0h vs. L6h (46), L0h vs. L24h (33), R0h vs. R6h (31), R0h vs. R24h (101), S0h vs. S6h (1), S0h vs. S24h (9), respectively (Figure 8A). Additionally, it is noteworthy that leaves, roots, and stems exhibited specific expression of 53, 97, and 9 TFs, respectively (Figure 8B). Most TFs showed differential expression only in one or two tissues. Notably, the expression levels of the 4 specific TFs, NB_transcript_24560 (C2H2), NB_transcript_63018 (MYB), NB_transcript_7810 (Tify), NB_transcript_38218 (Tify), changed across all three tissues (Figure 8C). Heat maps depicting tissue-specific TFs expression pattern revealed differences within or across tissues for TFs belonging to the same gene family (Figures 8D–F).
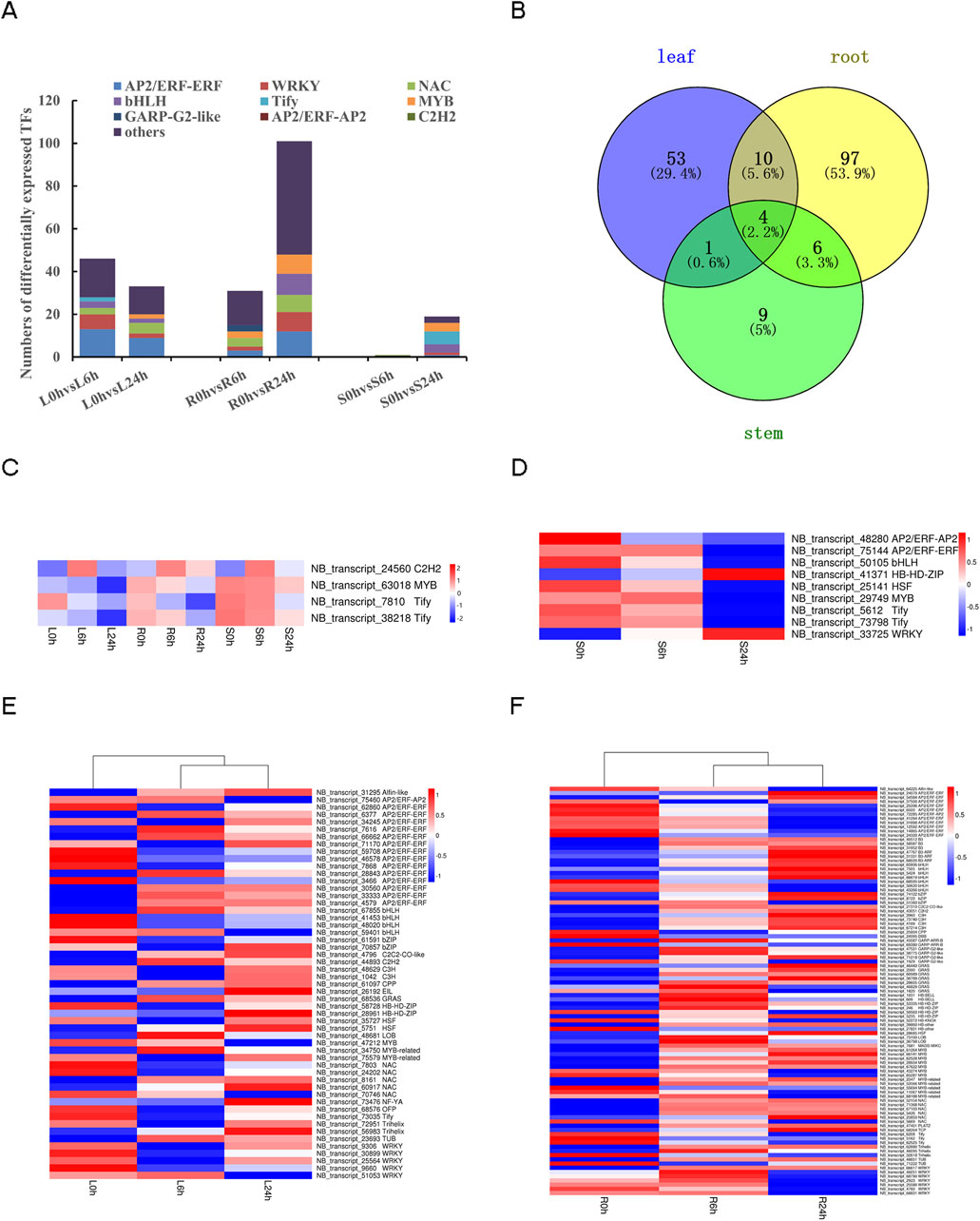
Figure 8. Analysis of differently expressed TFs under drought stress. (A) The TFs type composition in all six pairwise comparisons of 0 h, 6 h, 24 h in different tissues. (B) The Venn diagram of different expressed TFs in different tissues. (C) Heat map of tissue common TFs. (D) Heat map of tissue-specific TFs in stems. (E) Heat map of tissue-specific TFs in leaves. (F) Heat map of tissue-specific TFs in roots.
3.9 Physiological indexes of N. tangutorum under drought stress
We conducted measurement on eight physiological parameters to examine the impact of drought stress induced by PEG-6000 on N. tangutorum at the physiological level (Figure 9). After 6 h of drought stress, a slight increase in leaves proline content was observed, accompanied by minor changes in stems and roots. However, after 24 h drought stress, the proline content exhibited a significant rise of 78.4%, 45.6%, and 32.0% for leaves, stems, and roots respectively compared to the initial measurement at 0 h (Figure 9A). The content of MDA remained relatively stable after 6 h of drought stress, but increased significantly after 24 h. Compared with the content at 0h, it increased by 39.5%, 33.0%, and 24.1% in leaves, stems, and roots respectively (Figure 9B). Regarding ROS-related indexes, the H2O2 content followed different patterns of change in three tissues: it gradually increased in leaves, increased at 6 h and decreased at 24 h in stems, while roots showed an opposite trend compared to stems (Figure 9C). The activities of SOD, CAT and POD exhibited diverse trends after PEG-stimulated drought stress among tissues at different time point (Figures 9D–F). Additionally, POD activity in roots was 2.56, 2.04 times of that after 6 h and 24 h drought stress in leaves, and 2.41, 2.01 times of that after 6 h and 24 h drought stress in stems (Figure 9F). The soluble sugar (SS)content exhibited a decline at 6 h followed by an upward trend at 24 h after drought stress across all tissues (Figure 9G). The total amino acid (TAA) content remained unchanged during the entire duration of the treatment period for leaves and stems (including 6 h treatment in root), but significantly increased by 89.1% compared to initial level in roots after 24 drought treatment (Figure 9H).
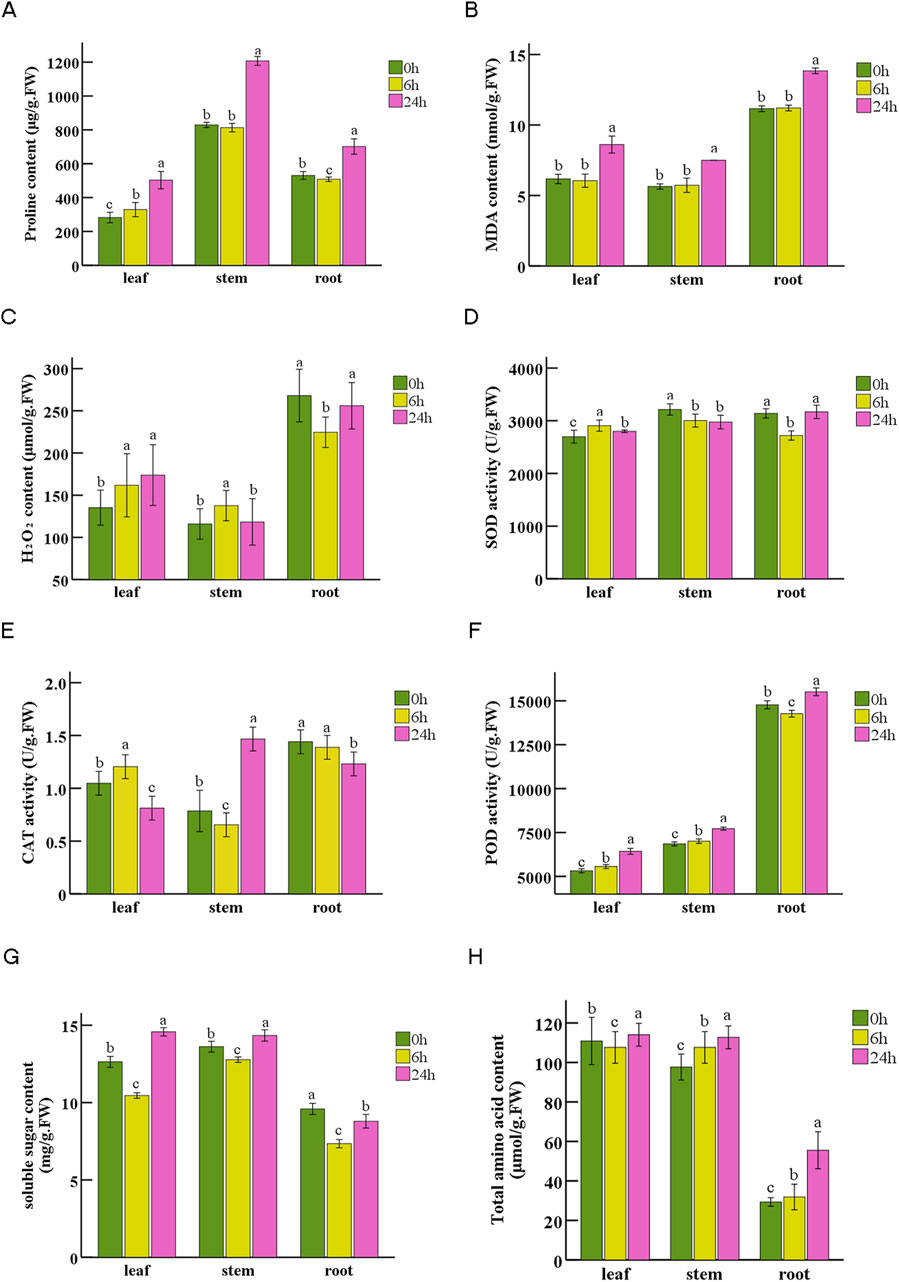
Figure 9. Physiological indexes of N. tangutorum in leaves, stems and roots at 0 h, 6 h, 24 h under drought stress. (A) Proline content. (B) Malondialdehyde (MDA) content. (C) H2O2 content. (D) Superoxide dismutase (SOD) activity. (E) Catalase (CAT) activity. (F) Peroxidase (POD) activity. (G) Soluble sugar content. (H) Total amino acid content. The data are shown as averages ±SD, n = 3. Different letters indicate the statistically significant differences at p < 0.05.
3.10 Weighted gene co-expression network analysis (WGCNA)
WGCNA was conducted to screen the modules and key genes associated with the drought tolerance of N. tangutorum. A total of 3,165 DEGs with FPKM>1 were screened for WGCNA analysis. Eight physiological indexes, including POD, H2O2, CAT, SOD, MDA, Pro, TAA, and SS were assessed as traits to evaluate the relationship among trait and modules (Figure 10A). Among the 10 identified modules, the number of genes ranged from a minimum of 37 (lightcyan) to a maximum of 1,062 (cyan) (Figure 10B; Supplementary Table S11). The cyan module displayed positive correlation with MDA, POD and H2O2 (correlation coefficients: 0.77, 0.7 and 0.51). The genes in cyan module were primarily enriched in “carbon metabolism,” “plant hormone signal transduction,” “biosynthesis of amino acid” and “glycolysis/gluconeogenesis” (Figure 10C). We selected the top 100 hub genes from the cyan module to construct a gene co-expression network. According to the annotation results of the 100 hub genes, there were 9 of the hub genes remain unannotated in neither of the nine databases in Section 3.3 (Supplementary Table S12), suggesting a substantial scope for further exploration in novel gene research within N. tangutorum underlying drought stress. Three genes encoding transcription factors (MYB, WRKY, AP2/ERF), one 1-aminocyclopropane-1-carboxylate oxidase 1 (ACO) gene, two glutathione S-transferase (GST) genes, three glyceraldehyde-3-phosphate dehydrogenases (GAPDH) genes were involved in the hub (Figure 10D).
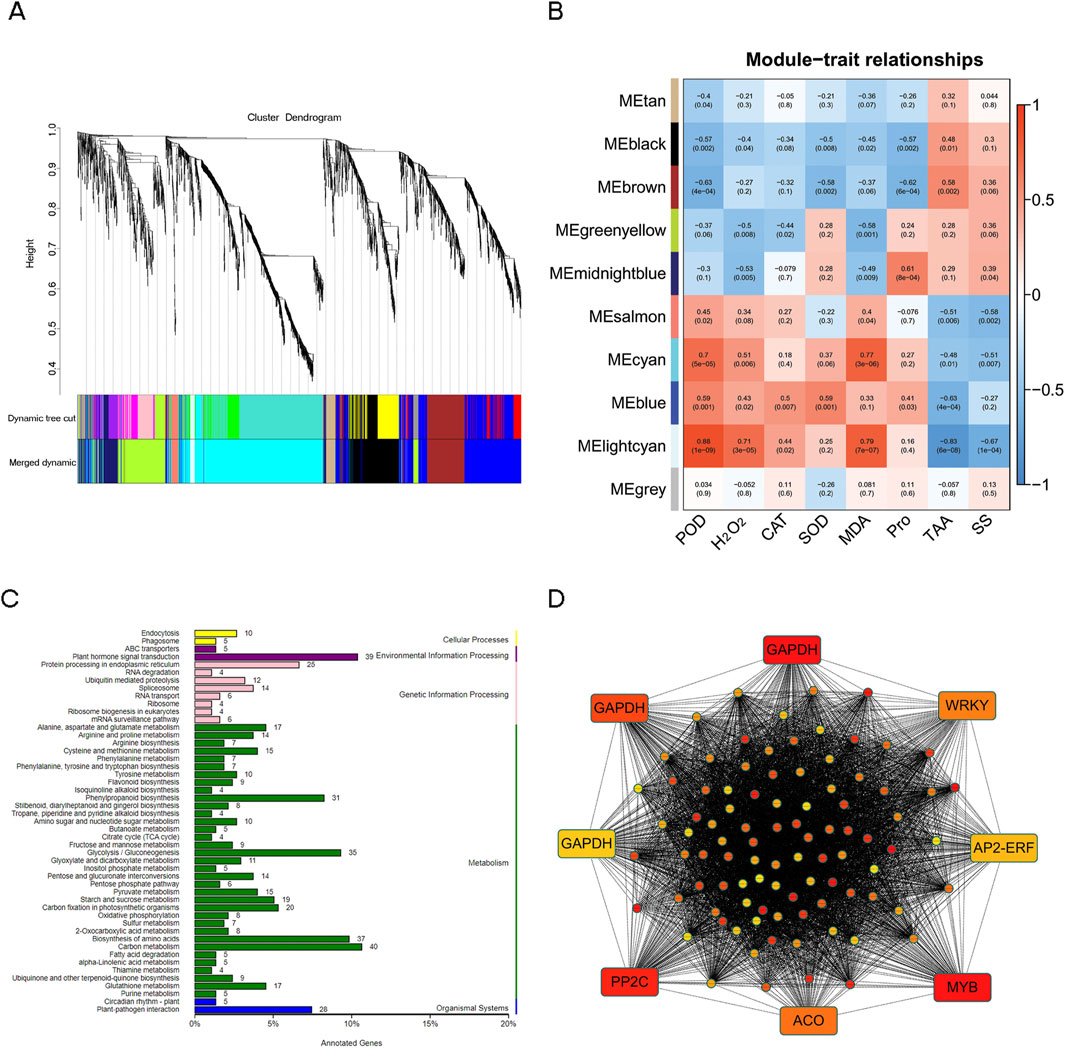
Figure 10. Weighted gene co-expression network analysis. (A) Clustering dendrogram of genes with original module (Dynamic Tree Cut) and merged module (Merged Dynamic). (B) Module-trait relationships between modules and physiological indexes. The numbers represent the correlation coefficient, and the numbers in parentheses mean p-value. (C) The KEGG pathway classification of the genes in cyan modules. (D) Gene network of the cyan module based on the top 100 hub genes.
3.11 Quantification and verification of gene expression level
The RNA-seq results were confirmed by performing qRT-PCR experiments. A total of twelve DEGs were selected randomly in three tissues for the validation. The results showed that the relative expression results obtained through qRT-PCR were consistent with the FPKM values provided by Illumina sequencing (Figure 11). The primer sequences of the twelve DEGs are listed in Supplementary Table S13.
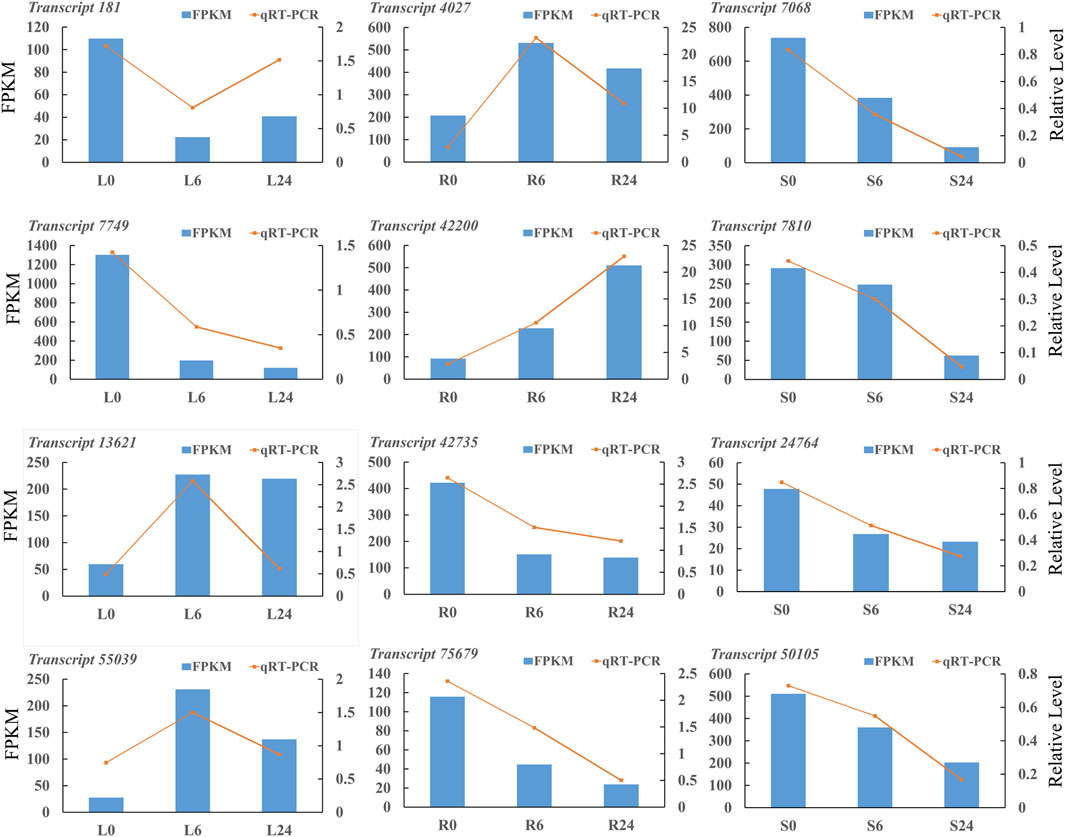
Figure 11. The qRT-PCR analyses of DEGs in N. tangutorum. The left vertical axis resents RNA-seq FPKM, the right vertical axis represents qRT-PCR relative expression, and the horizontal axis represents different samples (transcript 181: hypothetical protein CISIN_1g000438mg; transcript 7,749: hypothetical protein G4B88_011110; transcript 13,621: HIPP 24 heavy metal-associated isoprenylated plant protein 24; transcript 55,039: auxin-induced protein 22D-like; transcript 4,027: berberine bridge enzyme-like 13; transcript 42,200: glutathione S-transferase L3-like isoform X1; transcript 42,735: xyloglucan endotransglycosylase 3; transcript 75,679: nucleotide pyrophosphatase; transcript 7,068: protein TIFY 9 isoform X2; transcript 7,810: tify domain-containing protein; transcript 24,764: Auxin responsive SAUR protein; transcript 50,105: transcription factor MYC2-like).
4 Discussion
N. tangutorum is a plant with excellent survival ability in harsh desert environment and has a significant impact on maintaining ecological balance, as its dynamic gene expression regulation network and response mechanism in whole plant level under drought stress remain unexplored, unraveling the drought resistance mechanism of N. tangutorum is significant for resistance breeding. The interplay between tissue-common and tissue-specific pathways and genes is crucial for combating drought stress in N. tangutorum. We combined second-generation sequencing and third-generation sequencing technologies to comprehensively profile global gene expression across different tissues and time periods of N. tangutorum subjected to drought stress, aiming to uncover the genetic background underlying its drought resistance mechanism.
4.1 Signaling transduction under drought stress in N. tangutorum
Plant growth and development heavily rely on the presence of plant hormones, which also play a crucial role in improving the ability of plants to withstand drought conditions, especially for ABA, ET, BR, and JA (Verma et al., 2016). These hormones play the role of chemical messengers that facilitate the signal transduction process during periods of water scarcity (Ma et al., 2024; Ullah et al., 2018).
ABA is widely recognized as the primary stress hormone crucial for coping with various stress (Ullah et al., 2018). In this study, the core genes, such as PYL, PP2C, and ABF genes were observed in the modulation of ABA signaling. There are more ABA-related DEGs in the roots than in the leaves, and none were found in the stems. This may be due to the fact that ABA is mainly produced in the roots and subsequently transported to the leaves via stems to facilitate physiological regulation like closure stomata (Ullah et al., 2018). Additionally, the expression of PP2C genes increased during the drought period except one in leaves, similar results were found in Kentucky Bluegrass and Phormium tenax (Bai et al., 2017; Chen et al., 2019). Then ABF acted as an important transcription factor downstream of SnRK2 (sucrose non-fermenting 1-related protein kinase 2) triggering its participation in stomatal regulation (Purayil et al., 2020). By overexpression of PYL4 in wheat, the possibility of improving drought tolerance lies in reducing transpiration rates while simultaneously increasing photosynthetic activity (Mega et al., 2019). ET in plants exhibits a positive response to abiotic stress. In the current study, The ETR, EBF1/2, and EIN2 genes exhibited contrasting trends between leaf and root tissues. This indicates that the ET signal transduction mediated by drought stress show significant disparities in leaves and roots. Previous studies have revealed that the ET production is disturbed in leaves and flower buds, but has almost no effect on root under drought stress in coffee plants (Lima et al., 2021). The findings suggest that the production and signaling of ET in response to drought exhibit tissue-specific patterns. BRs play a crucial role in regulating plant development and their ability to cope with environmental stress. Previous research has indicated a negative effect of BRI1 on drought tolerance in Brachypodium distachyon and tomato (Feng et al., 2015; Nie et al., 2019), which was consistent in our result that all expression changes of BRI1 were downregulation in all tissues under drought stress. BKI1 acts as a suppressor, impeding the activation of BRI1, which in line with our observation of an upregulation in BKI1 gene expression and a downregulation in BRI1 in roots. The pivotal regulators in the JA signaling pathway are exemplified by the protein JAZ and TF MYC2, which occupy a central position. In our study, the expressions of both JAZ and MYC2 members were found to be downregulated. Notably, unlike other hormones, alterations in their expression were also observed in stem tissue. Previous studies have indicated that JAZ proteins function not only acts as repressor protein in JA signaling, but also serve as signaling hubs in hormone crosstalk (Wager and Browse, 2012).
The MAPK cascade is a crucial pathway for transmitting signals in plants, which enables the conversion of external stimuli like drought, cold and heat into internal responses. It consists of three crucial kinases: MAPKs (MPKs), MAPK kinases (MAPKKs, MAP2Ks or MEKs) and MAPK kinase kinases (MAPKKKs, MAP3Ks or MEKKs) (Zhang et al., 2021). In our study, the involvement of MPK3/6, MEKK1, MAP3K17/18, and ANP1 was observed in the transmission of signal under drought-induce conditions. The stress signaling hubs in other plants are primarily represented by the Arabidopsis MAP Kinases MPK6 and MPK3, which play crucial role in stomatal development (Hann et al., 2024; Pitzschke et al., 2009). Additionally, the abiotic stress-induced production of ROS leads to the activation of MPK3/6 (Jalmi and Sinha, 2015). The overexpression of TaMPK3 resulted in decreased drought tolerance (Liu Y. et al., 2022). On the contrary, the transformation of the MPK3 gene from cotton into Arabidopsis resulted in an improvement in abiotic stress resistance in transgenic plants (Sadau et al., 2021). The MPK3/6 showed upregulation in leaves after 6 h drought stress, while exhibited downregulation following 24 h drought stress in leaves and roots. In addition, the expression trend of MAP3K17/18 showed opposite patterns among different genotypes in broomcorn millet (Yuan et al., 2022). These results indicate that the components of MAPK cascades in different species have varying roles under drought stress, thus further research is needed to investigate the role of MAPK members in N. tangutorum.
4.2 Enzymes and DEGs related to ROS scavenging in N. tangutorum
Drought could cause higher production of ROS, resulting in the destruction of essential macromolecules, disturbance of the cellular redox equilibrium, the breakdown of chlorophyll and oxidative damage to membranes (Kumar et al., 2021). The MDA content increased 24 h after the induction of drought stress by PEG (Figure 9B), indicating that drought stress caused peroxidation of membrane lipid. Plants have evolved a protective mechanism that involves the enzymes SOD, POD, glutathione S-transferase (GST) and CAT to eliminate excess ROS (He et al., 2017). SOD is believed to catalyze the O2− into H2O2, while CAT and POD decompose H2O2 (Tian et al., 2012). In addition, GST is also a crucial enzymes member in ROS defense system (Yan et al., 2008). These antioxidant enzymes work synergistically to detoxify ROS (Sarker and Oba, 2018). In this study, the H2O2 content change varied in different tissues with prolonged drought stress, while the pattern of antioxidative enzymes showed inconsistency among the three tissues under different drought treatments time points. Correspondingly, the regulation of SOD, CAT, GST, and POD genes in N. tangutorum showed variations in expression levels when subjected to drought stress. The collaboration between antioxidant enzymes and associated DEGs ensures the equilibrium between ROS generation and detoxification, thereby safeguarding plants against oxidative stress caused by ROS.
4.3 The roles of drought-responsive pathways and metabolites in N. tangutorum
Drought stress not only induces the build-up of sugars and adjusts energy demands in plants but also facilitates the conversion of storage sugars (such as starch) into soluble forms to decrease cell water potential (Kaur et al., 2021). It has been established that the regulation of plant carbon metabolism is associated with the activities of sucrose synthase and alpha-amylase under drought stress (Min et al., 2020). Sucrose synthase (SuS) metabolizes sucrose into hexose phosphates, which are precursor for starch biosynthesis. Alpha-amylase (AMY) is a key enzyme related to the decomposition of starch and belong to the family 13 of glycosyl hydrolase (Dong and Beckles, 2019; Janecek, 1997). It was noticed that the expression of SuS and AMY genes exhibited differential regulation in leaves under drought stress. Additionally, the soluble sugar content increased at 6 h and decreased at 24 h in whole plant, suggesting that the starch and sugar conversion was dynamic and complex in N. tangutorum due to its dual role as an energy source and osmotic regulator for drought resistance, the ratio of cellular sugars and starch can be changed by stress-induced regulation of starch metabolism (Dong and Beckles, 2019). These finding implies that the drought resistance in N. tangutorum is linked with carbon metabolism.
The lignin pathways represent a prominent branch of the phenylpropanoid metabolism, and lignin play a role as mechanical support to cell wall and contributes to plant responses to drought stress by facilitating cell wall thickening, which reduces water permeability and maintains the plant cell turgor. The CAD and 4CL genes are all key enzyme genes encoding lignin synthesis (Dong and Lin, 2021). Upregulation of CAD gene in oriental melon can induce the accumulation of lignin and respond actively to drought stress (Liu et al., 2020; Silva et al., 2018). The Gh4CL7 gene of cotton plays a crucial role in the biosynthesis of lignin and confers enhanced drought resistance (Sun et al., 2020). During the process of lignin formation, peroxidases function as catalysts that polymerize monolignols into lignin (Dong and Lin, 2021). In current study, the upregulation of all 4CL and CAD genes and significantly higher activity of POD in roots indicate that the crucial role of lignin in drought resistance in N. tangutorum.
Amino acids serve as osmoprotectants in reaction to abiotic environmental factors (Batista-Silva, et al., 2019). In our study, the expression of 7 P5CS genes was upregulated in root tissues after 24 h of drought, while the expression of 2 genes encoding proline dehydrogenase (PRODH), a pivotal enzyme involved in proline degradation (Kamanga et al., 2018), were significantly downregulated in all three tissues after 24 h drought stress. These results contribute to an increase in proline content, which in consistent with the physiological tests showing increased proline content in leaves, stems and roots of N. tangutorum following 24 h of drought treatment. Furthermore, these findings highlighted the active response of PRODH to drought stress after 24 h. In addition to P5CS, the upregulation of a majority of genes in “biosynthesis of amino acids” pathway implies that amino acids may play additional roles in enhancing drought resistance of N. tangutorum. During the process of resisting drought in sugarcane, amino acid metabolism fulfilled dual function by signaling and providing carbon skeletons for energetic maintenance (Contiliani et al., 2022; Diniz et al., 2020).
4.4 Transcription factors play a vital role under drought stress in N. tangutorum
TFs act as the activation switch for cascades of signal transduction by recognizing the particular cis-elements located in promoter region of their target genes (Franco-Zorrilla et al., 2014). As regulatory genes, TFs have a significant impact on the regulation of numerous genes associated with the response to stress (Joshi et al., 2016; Singh and Laxmi, 2015). In our study, we observed significant alterations in the expression of AP2/ERF, WRKY, NAC, bHLH, and MYB family members following exposure to drought stress. The family of AP2/ERF is exclusive to plants that covers a large number of members in common plant species (e.g., 145 in Arabidopsis, 170 in rice, 200 in poplar and 291 in Chinese cabbage), playing a part in the ethylene-responsive transcription (Joshi et al., 2016). AP2/ERF-ERF as a subfamily belonging to the AP2/ERF superfamily, and the TFs from the ERF subfamily are considered tightly associated with responding to abiotic stresses and involved in hormone signaling pathway (Li et al., 2017; Mizoi et al., 2012). This finding aligns with our research results, indicating that the “plant hormone signal transduction” pathway exhibits a remarkably high degree of activity and there are numerous AP2/ERF-ERF TFs, which indicated that AP2/ERF-ERF subfamily significantly contributes to enhancing the ability of N. tangutorum to resist drought. The WRKY TFs take an active part as prominent TFs, similar to AP2/ERF-ERF, the two TFs constitute a quarter of the different expressed TFs, highlighting their important role as viable options for improving resistance to drought in N. tangutorum. WRKY transcription factors exhibited tissue-specific expression patterns. In leaf tissues, 4 out of 5 WRKYs showed downregulation followed by upregulation after 6 h and 24 h of drought stress, while in root tissues, differential regulation occurred for 6 out of 7 WRKYs after 6 h and downregulation was observed after 24 h. These results may suggest that WRKY members were involved in various biological processes in different tissues to respond to drought in N. tangutorum. MYB TFs contribute to the improvement of plant resilience against drought by controlling the activity of genes associated with metabolite production and stomatal response (Wang X. et al., 2021). And MYB TFs were implicated in the drought response of N. tangutorum across three distinct tissues.
It has been shown that the collaboration among TFs in drought response is crucial for the regulation of downstream gene expression (Singh and Laxmi, 2015). In the current study, more than 200 TFs altered their expression under drought stress. These master regulatory genes, TF families, hold great potential for mitigating drought stress, and overexpression these TFs through genetic engineering approaches can be employed to develop the stress-tolerant crops (Joshi et al., 2016; Shao et al., 2015). Additionally, N. tangutorum may serve as a valuable gene bank for breeding purposes.
4.5 Analysis of hub genes in response to drought in N. tangutorum
WGCNA analysis is a powerful tool for identifying hub genes by exploring the correlation between the gene module with similar expression patterns and physiological indexes (Zhu et al., 2024). In our study, the cyan module revealed PP2C, ACO, GST, and GAPDH genes as well as AP2/ERF, WRKY, MYB TFs to be identified as hub genes. PP2Cs plays a negative regulatory role in ABA signaling pathway (Mega et al., 2019), while ACO encodes the key enzyme in ET biosynthesis (Tang and Woodson, 1996), both were involved in the hormone regulation. The identification of two GST genes as hub genes further emphasizes the pivotal role in ROS scavenging for the drought resistance of N. tangutorum. GAPDH acts as an enzyme that facilitates the oxidation of triose phosphate in the process of glycolysis (Hildebrandt et al., 2015). It has been found that GAPDH also serves as a hub gene in enhancing drought tolerance by participating in photosynthetic adaptation of rice (Chintakovid et al., 2017). Despite being commonly used as a reference gene in transcriptomic and proteomic analyses, the upregulated expression of GAPDH genes during drought stress in N. tangutorum suggests the potential active response to drought stress through glycolysis process. The AP2/ERF, WRKY, and MYB TFs are closely associated with the regulation of downstream drought-responsive genes. The WGCNA results indicate that ABA and ET regulation, ROS scavenging, glycolysis are fundamental strategies employed by N. tangutorum to cope with drought.
4.6 Schematic model of the response to drought stress in N. tangutorum
We proposed a hypothetical model of drought responses in N. tangutorum based on the previous pathway and DEGs analysis (Figure 12). When plant suffer from drought stress, plant cell membranes are equipped with the ability to sense stress signals (Mahmood et al., 2020). In our study, we found that the top two ways of drought signaling in roots and leaves were based on hormones and MAPK components after 6 h and 24 h of drought stress. The same response was observed in stems after 24 h of drought stress. The DEGs related to hormone signaling involved eight types of plant hormones, IAA, GA, CTK, ABA, ET, BR, SA, and JA in leaves and roots at both investigated time point, which indicates that hormones are crucial in enabling N. tangutorum to endure stress caused by drought. Entire cascades of stress signal were transmitted in the nucleus, and TFs were activated to regulate a cluster of target genes. The AP2/ERF-ERF, WRKY, NAC, bHLH, MYB TFs are the main force in stress management in roots and leaves, while Tify TF play a principal role in stems after 24 h drought stress. These TFs interact with promoter regions of genes that exhibit sensitivity to stress, which involved in minimization of oxidative damage, metabolism adjustment (sugar, lignin and amino acid) and plant-pathogen interaction, upregulation or downregulation of these genes enhanced combinatorial tolerance against drought stress. In the process, leaves and roots share more common signal transduction pathway and TF, while stems have respective pathway with fewer DEGs, which may due to stem take an active part in integrating message from the roots and aerial part of N. tangutorum for its vascular system (Takahashi et al., 2020). In general, different tissues of N. tangutorum respond synergistically to drought with tissue-specific function, contributing to harmonization of plant tissues and overall stress tolerance on whole-plant level.
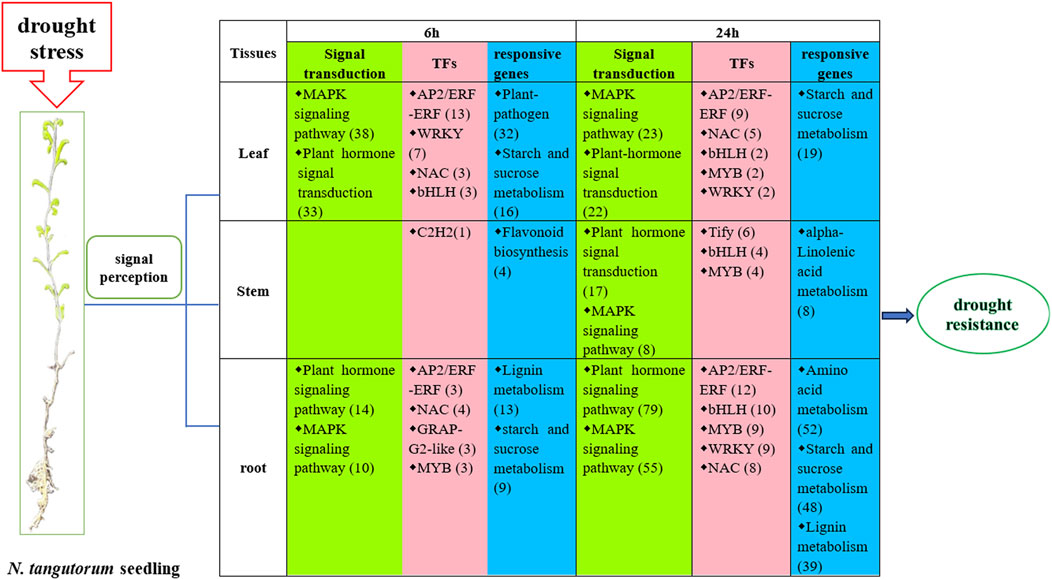
Figure 12. A schematic model of the transcriptional regulatory process in response to 6 h and 24 h drought stress in N. tangutorum at the whole-plant level (The numbers in bracket mean the number of related DEGs).
5 Conclusion
In this research, we investigated the response how N. tangutorum reacts to drought by conducting bioinformatics analysis and measuring physiological indexes. We examined three parts of the whole plant (leaves, roots and stems) and integrated Illumina RNA-seq and PacBio SMRT-seq to obtain comprehensive transcripts of N. tangutorum. The analysis of key pathways and DEGs revealed a synergistic modulation in response to the adverse effects of drought stress, N. tangutorum relied on hormones and the MAPK cascade for transmitting and responding to stress signals at the whole-plant level. Both leaf tissue and root tissue enhanced drought tolerance through carbon metabolism, while root tissue also employed lignin synthesis and amino acid metabolism, enabling N. tangutorum to effectively cope with unfavorable conditions. Furthermore, TFs analysis showed that AP2/ERF-ERF, WRKY, bHLH, NAC, MYB actively participated in responding to the stress induced by drought. The physiological indexes demonstrated the drought-induced physiological response of N. tangutorum. The WGCNA analysis revealed a candidate hub gene network related to drought stress. The results provide a basis for further exploration into the regulatory mechanisms involved in N. tangutorum’s adaptation to drought stress.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra/PRJNA1094917.
Author contributions
MW: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Writing–original draft. BW: Conceptualization, Investigation, Methodology, Writing–review and editing. CL: Investigation, Software, Validation, Writing–review and editing. XL: Data curation, Validation, Writing–review and editing. CH: Formal Analysis, Software, Writing–review and editing. YL: Conceptualization, Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by Regular Science and Technology Assistance Projects of the Ministry of Science and Technology to Developing Countries (KY202002011), Forestry and Grassland Science and Technology Innovation Project of Gansu Province (GLC2019-418-8), Gansu Provincial Department of Education 2023 Graduate Innovation Star Project (2023CXZX-640), Gansu Provincial Science and Technology Program (24JRRA677).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1474259/full#supplementary-material
References
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi:10.1186/gb-2010-11-10-r106
Bai, Z. Y., Wang, T., Wu, Y. H., Wang, K., Liang, Q. Y., Pan, Y. Z., et al. (2017). Whole-transcriptome sequence analysis of differentially expressed genes in Phormium tenax under drought stress. Sci. Rep. 7, 41700. doi:10.1038/srep41700
Bartels, D., and Sunkar, R. (2005). Drought and salt tolerance in plants. Crit. Rev. Plant Sci. 24, 23–58. doi:10.1080/07352680590910410
Batista-Silva, W., Heinemann, B., Rugen, N., Nunes-Nesi, A., Araújo, W. L., Braun, H. P., et al. (2019). The role of amino acid metabolism during abiotic stress release. Plant Cell. Environ. 42, 1630–1644. doi:10.1111/pce.13518
Bray, N. L., Pimentel, H., Melsted, P., and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi:10.1038/nbt.3519
Chen, L., Zhang, B., Xia, L. J., Yue, D. D., Han, B., Sun, W. A., et al. (2023). The GhMAP3K62-GhMKK16-GhMPK32 kinase cascade regulates drought tolerance by activating GhEDT1-mediated ABA accumulation in cotton. J. Adv. Res. 51, 13–25. doi:10.1016/j.jare.2022.11.002
Chen, S., Xu, X., Ma, Z. Y., Liu, J. X., and Zhang, B. (2021a). Organ-specific transcriptome analysis identifies candidate genes involved in the stem specialization of bermudagrass (Cynodon dactylon L.). Front. Genet. 12, 678673. doi:10.3389/fgene.2021.678673
Chen, S., Zhou, H., Zhang, G., Dong, Q., Wang, Z., Wang, H., et al. (2021b). Characterization, antioxidant, and neuroprotective effects of anthocyanins from Nitraria tangutorum Bobr. fruit. Food Chem. 353, 129435. doi:10.1016/j.foodchem.2021.129435
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890. doi:10.1093/bioinformatics/bty560
Chen, Y., Chen, Y., Shi, Z., Jin, Y., Sun, H., Xie, F., et al. (2019). Biosynthesis and signal transduction of ABA, JA, and BRs in response to drought stress of Kentucky Bluegrass. Int. J. Mol. Sci. 20, 1289. doi:10.3390/ijms20061289
Chintakovid, N., Maipoka, M., Phaonakrop, N., Mickelbart, M. V., Roytrakul, S., and Chadchawan, S. (2017). Proteomic analysis of drought-responsive proteins in rice reveals photosynthesis-related adaptations to drought stress. Acta Physiol. Plant. 39, 240. doi:10.1007/s11738-017-2532-4
Contiliani, D. F., de Oliveira Nebo, J. F. C., Ribeiro, R. V., Andrade, L. M., Peixoto Junior, R. F., Lembke, C. G., et al. (2022). Leaf transcriptome profiling of contrasting sugarcane genotypes for drought tolerance under field conditions. Sci. Rep. 12, 9153. doi:10.1038/s41598-022-13158-5
Didi, D. A., Su, S. P., Sam, F. E., Tiika, R. J., and Zhang, X. (2022). Effect of plant growth regulators on osmotic regulatory substances and antioxidant enzyme activity of Nitraria tangutorum. Plants-Basel 11, 2559. doi:10.3390/plants11192559
Diniz, A. L., da Silva, D. I. R., Lembke, C. G., Costa, M., ten-Caten, F., Li, F., et al. (2020). Amino acid and carbohydrate metabolism are coordinated to maintain energetic balance during drought in sugarcane. Int. J. Mol. Sci. 21, 9124. doi:10.3390/ijms21239124
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi:10.1093/bioinformatics/bts635
Dong, N. Q., and Lin, H. X. (2021). Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 63, 180–209. doi:10.1111/jipb.13054
Dong, S., and Beckles, D. M. (2019). Dynamic changes in the starch-sugar interconversion within plant source and sink tissues promote a better abiotic stress response. J. Plant Physiol. 234-235, 80–93. doi:10.1016/j.jplph.2019.01.007
Feng, Y., Yin, Y., and Fei, S. (2015). Down-regulation of BdBRI1, a putative brassinosteroid receptor gene produces a dwarf phenotype with enhanced drought tolerance in Brachypodium distachyon. Plant Sci. 234, 163–173. doi:10.1016/j.plantsci.2015.02.015
Franco-Zorrilla, J. M., López-Vidriero, I., Carrasco, J. L., Godoy, M., Vera, P., and Solano, R. (2014). DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. U. S. A. 111, 2367–2372. doi:10.1073/pnas.1316278111
Gao, Z., Zhang, J., Zhang, J., Zhang, W., Zheng, L., Borjigin, T., et al. (2022). Nitric oxide alleviates salt-induced stress damage by regulating the ascorbate-glutathione cycle and Na+/K+ homeostasis in Nitraria tangutorum Bobr. Plant Physiol. Bioch 173, 46–58. doi:10.1016/j.plaphy.2022.01.017
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi:10.1038/nbt.1883
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi:10.1038/nprot.2013.084
Hann, C. T., Ramage, S. F., Negi, H., Bequette, C. J., Vasquez, P. A., and Stratmann, J. W. (2024). Dephosphorylation of the MAP kinases MPK6 and MPK3 fine-tunes responses to wounding and herbivory in Arabidopsis. Plant Sci. 339, 111962. doi:10.1016/j.plantsci.2023.111962
He, F., Sheng, M., and Tang, M. (2017). Effects of Rhizophagus irregularis on photosynthesis and antioxidative enzymatic system in Robinia pseudoacacia L. Under drought stress. Front. Plant Sci. 8, 183. doi:10.3389/fpls.2017.00183
Hildebrandt, T., Knuesting, J., Berndt, C., Morgan, B., and Scheibe, R. (2015). Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 396, 523–537. doi:10.1515/hsz-2014-0295
Hirayama, T., and Shinozaki, K. (2010). Research on plant abiotic stress responses in the post-genome era: past, present and future. Plant J. 61, 1041–1052. doi:10.1111/j.1365-313X.2010.04124.x
Hu, N., Zheng, J., Li, W., and Suo, Y. (2014). Isolation, stability, and antioxidant activity of anthocyanins from Lycium ruthenicum murray and nitraria tangutorum Bobr of Qinghai-Tibetan plateau. Sep. Sci. Technol. 49, 2897–2906. doi:10.1080/01496395.2014.943770
Jalmi, S. K., and Sinha, A. K. (2015). ROS mediated MAPK signaling in abiotic and biotic stress-striking similarities and differences. Front. Plant Sci. 6, 769. doi:10.3389/fpls.2015.00769
Janecek, S. (1997). alpha-Amylase family: molecular biology and evolution. Prog. Biophys. Mol. Biol. 67, 67–97. doi:10.1016/s0079-6107(97)00015-1
Jiang, S., Chen, C., Dong, Q., Shao, Y., Zhao, X., Tao, Y., et al. (2021). Alkaloids and phenolics identification in fruit of Nitraria tangutorum Bobr. by UPLC-Q-TOF-MS/MS and their α-glucosidase inhibitory effects in vivo and in vitro. Food Chem. 364, 130412. doi:10.1016/j.foodchem.2021.130412
Joshi, R., Wani, S. H., Singh, B., Bohra, A., Dar, Z. A., Lone, A. A., et al. (2016). Transcription factors and plants response to drought stress: current understanding and future directions. Front. Plant Sci. 7, 1029. doi:10.3389/fpls.2016.01029
Kamanga, R. M., Mbega, E., and Ndakidemi, P. (2018). Drought tolerance mechanisms in plants: physiological responses associated with water deficit stress in Solanum lycopersicum. Adv. Crop Sci. Tech. 06 (06), 1000362. doi:10.4172/2329-8863.1000362
Kaur, H., Manna, M., Thakur, T., Gautam, V., and Salvi, P. (2021). Imperative role of sugar signaling and transport during drought stress responses in plants. Physiol. Plant 171, 833–848. doi:10.1111/ppl.13364
Kong, L., Zhang, Y., Ye, Z.-Q., Liu, X.-Q., Zhao, S.-Q., Wei, L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349. doi:10.1093/nar/gkm391
Kumar, A., Kumar, V., Dubey, A. K., Ansari, M. A., Narayan, S., Meenakshi, H., et al. (2021). Chickpea glutaredoxin (CaGrx) gene mitigates drought and salinity stress by modulating the physiological performance and antioxidant defense mechanisms. Physiol. Mol. Biol. Plants 27, 923–944. doi:10.1007/s12298-021-00999-z
Lamaoui, M., Jemo, M., Datla, R., and Bekkaoui, F. (2018). Heat and drought stresses in crops and approaches for their mitigation. Front. Chem. 6, 26. doi:10.3389/fchem.2018.00026
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinform 9, 559. doi:10.1186/1471-2105-9-559
Li, H., Wang, Y., Wu, M., Li, L. H., Li, C., Han, Z. P., et al. (2017). Genome-wide identification of AP2/ERF transcription factors in cauliflower and expression profiling of the ERF family under salt and drought stresses. Front. Plant Sci. 8, 946. doi:10.3389/fpls.2017.00946
Li, M., Li, H., Sun, A., Wang, L., Ren, C., Liu, J., et al. (2022a). Transcriptome analysis reveals key drought-stress-responsive genes in soybean. Front. Genet. 13, 1060529. doi:10.3389/fgene.2022.1060529
Li, W., Fu, Y., Lv, W., Zhao, S., Feng, H., Shao, L., et al. (2021b). Characterization of the early gene expression profile in Populus ussuriensis under cold stress using PacBio SMRT sequencing integrated with RNA-seq reads. Tree Physiol. 42, 646–663. doi:10.1093/treephys/tpab130
Li, W. L., Lee, J. B., Yu, S., Wang, F. D., Lv, W. Q., Zhang, X., et al. (2021a). Characterization and analysis of the transcriptome response to drought in Larix kaempferi using PacBio full-length cDNA sequencing integrated with de novo RNA-seq reads. Planta 253, 28. doi:10.1007/s00425-020-03555-3
Li, Z., Bai, D., Zhong, Y., Lin, M., Sun, L., Qi, X., et al. (2022b). Full-length transcriptome and RNA-seq analyses reveal the mechanisms underlying waterlogging tolerance in kiwifruit (Actinidia valvata). Int. J. Mol. Sci. 23, 3237. doi:10.3390/ijms23063237
Lima, A. A., Santos, I. S., Torres, M. E. L., Cardon, C. H., Caldeira, C. F., Lima, R. R., et al. (2021). Drought and re-watering modify ethylene production and sensitivity, and are associated with coffee anthesis. Environ. Exp. Bot. 181, 104289. doi:10.1016/j.envexpbot.2020.104289
Liu, C., Duan, N., Chen, X., Li, H., Zhao, X., Duo, P., et al. (2022a). Metabolic pathways involved in the drought stress response of Nitraria tangutorum as revealed by transcriptome analysis. Forests 13, 509. doi:10.3390/f13040509
Liu, C., Duan, N., Chen, X., Li, X., Zhao, N., Cao, W., et al. (2023). Transcriptome profiling and chlorophyll metabolic pathway analysis reveal the response of Nitraria tangutorum to increased nitrogen. Plants-Basel 12, 895. doi:10.3390/plants12040895
Liu, W., Jiang, Y., Wang, C., Zhao, L., Jin, Y., Xing, Q., et al. (2020). Lignin synthesized by CmCAD2 and CmCAD3 in oriental melon (Cucumis melo L.) seedlings contributes to drought tolerance. Plant Mol. Biol. 103, 689–704. doi:10.1007/s11103-020-01018-7
Liu, Y., Yu, T. F., Li, Y. T., Zheng, L., Lu, Z. W., Zhou, Y. B., et al. (2022b). Mitogen-activated protein kinase TaMPK3 suppresses ABA response by destabilising TaPYL4 receptor in wheat. New Phytol. 236, 114–131. doi:10.1111/nph.18326
Luo, H., Bu, D., Sun, L., Chen, R., and Zhao, Y. (2014). De novo approach to classify protein-coding and noncoding transcripts based on sequence composition. Methods Mol. Biol. 1182, 203–207. doi:10.1007/978-1-4939-1062-5_18
Ma, P., Guo, G., Xu, X., Luo, T., Sun, Y., Tang, X., et al. (2024). Transcriptome analysis reveals key genes involved in the response of Pyrus betuleafolia to drought and high-temperature stress. Plants 13, 309. doi:10.3390/plants13020309
Mahmood, T., Khalid, S., Abdullah, M., Ahmed, Z., Shah, M. K. N., Ghafoor, A., et al. (2020). Insights into drought stress signaling in plants and the molecular genetic basis of cotton drought tolerance. Cells 9, 105. doi:10.3390/cells9010105
Mega, R., Abe, F., Kim, J.-S., Tsuboi, Y., Tanaka, K., Kobayashi, H., et al. (2019). Tuning water-use efficiency and drought tolerance in wheat using abscisic acid receptors. Nat. Plants 5, 153–159. doi:10.1038/s41477-019-0361-8
Mei, L., Li, Z., Yan, Y., Wen, Z., Wen, X., Yang, Z., et al. (2020). Identification and functional study of oleoresin terpenoid biosynthesis-related genes in masson pine (Pinus massoniana L.) based on transcriptome analysis. Tree Genet. Genomes. 16, 53. doi:10.1007/s11295-020-01448-w
Menhas, S., Yang, X., Hayat, K., Niazi, N. K., Hayat, S., Amna, H., et al. (2022). Targeting Cd coping mechanisms for stress tolerance in Brassica napus under spiked-substrate system: from physiology to remediation perspective. Int. J. Phytoremediation 24, 622–636. doi:10.1080/15226514.2021.1960479
Min, X. Y., Lin, X. S., Ndayambaza, B., Wang, Y. R., and Liu, W. X. (2020). Coordinated mechanisms of leaves and roots in response to drought stress underlying full-length transcriptome profiling in Vicia sativa L. Bmc Plant Biol. 20, 165. doi:10.1186/s12870-020-02358-8
Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 1819, 86–96. doi:10.1016/j.bbagrm.2011.08.004
Nie, S. M., Huang, S. H., Wang, S. F., Mao, Y. J., Liu, J. W., Ma, R. L., et al. (2019). Enhanced brassinosteroid signaling intensity via SlBRI1 overexpression negatively regulates drought resistance in a manner opposite of that via exogenous BR application in tomato. Plant Physiol. Bioch 138, 36–47. doi:10.1016/j.plaphy.2019.02.014
Osmolovskaya, N., Shumilina, J., Kim, A., Didio, A., Grishina, T., Bilova, T., et al. (2018). Methodology of drought stress research: experimental setup and physiological characterization. Int. J. Mol. Sci. 19, 4089. doi:10.3390/ijms19124089
Pitzschke, A., Schikora, A., and Hirt, H. (2009). MAPK cascade signalling networks in plant defence. Curr. Opin. Plant Biol. 12, 421–426. doi:10.1016/j.pbi.2009.06.008
Purayil, F. T., Rajashekar, B., Kurup, S. S., Cheruth, A. J., Subramaniam, S., Tawfik, N. H., et al. (2020). Transcriptome profiling of Haloxylon persicum (bunge ex boiss and buhse) an endangered plant species under PEG-induced drought stress. Genes. 11, 640. doi:10.3390/genes11060640
Rao, X., Huang, X., Zhou, Z., and Lin, X. (2013). An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 3, 71–85.
Sadau, S. B., Ahmad, A., Tajo, S. M., Ibrahim, S., Kazeem, B. B., Wei, H. L., et al. (2021). Overexpression of GhMPK3 from cotton enhances cold, drought, and salt stress in Arabidopsis. Agron. (Basel). 11, 1049. doi:10.3390/agronomy11061049
Sarker, U., and Oba, S. (2018). Catalase, superoxide dismutase and ascorbate-glutathione cycle enzymes confer drought tolerance of Amaranthus tricolor. Sci. Rep. 8, 16496. doi:10.1038/s41598-018-34944-0
Shao, H. B., Wang, H. Y., and Tang, X. L. (2015). NAC transcription factors in plant multiple abiotic stress responses: progress and prospects. Front. Plant Sci. 6, 902. doi:10.3389/fpls.2015.00902
Silva, F. L. B., Vieira, L. G. E., Ribas, A. F., Moro, A. L., Neris, D. M., and Pacheco, A. C. (2018). Proline accumulation induces the production of total phenolics in transgenic tobacco plants under water deficit without increasing the G6PDH activity. Theor. Exp. Plant Phys. 30, 251–260. doi:10.1007/s40626-018-0119-0
Singh, B., Bohra, A., Mishra, S., Joshi, R., and Pandey, S. (2015). Embracing new-generation ‘omics’ tools to improve drought tolerance in cereal and food-legume crops. Biol. Plant. 59, 413–428. doi:10.1007/s10535-015-0515-0
Singh, D., and Laxmi, A. (2015). Transcriptional regulation of drought response: a tortuous network of transcriptional factors. Front. Plant Sci. 6, 895. doi:10.3389/fpls.2015.00895
Sun, S. C., Xiong, X. P., Zhang, X. L., Feng, H. J., Zhu, Q. H., Sun, J., et al. (2020). Characterization of the Gh4CL gene family reveals a role of Gh4CL7 in drought tolerance. Bmc Plant Biol. 20, 125. doi:10.1186/s12870-020-2329-2
Sun, Y., Cao, H., Yin, J., Kang, L. E., and Ge, F. (2010). Elevated CO2 changes the interactions between nematode and tomato genotypes differing in the JA pathway. Plant Cell. Environ. 33, 729–739. doi:10.1111/j.1365-3040.2009.02098.x
Takahashi, F., Kuromori, T., Sato, H., and Shinozaki, K. (2018). Regulatory gene networks in drought stress responses and resistance in plants. Adv. Exp. Med. Biol. 1081, 189–214. doi:10.1007/978-981-13-1244-1_11
Takahashi, F., Kuromori, T., Urano, K., Yamaguchi-Shinozaki, K., and Shinozaki, K. (2020). Drought stress responses and resistance in plants: from cellular responses to long-distance intercellular communication. Front. Plant Sci. 11, 556972. doi:10.3389/fpls.2020.556972
Tang, X., and Woodson, W. R. (1996). Temporal and spatial expression of 1-aminocyclopropane-1-carboxylate oxidase mRNA following pollination of immature and mature petunia flowers. Plant physiol. 112, 503–511. doi:10.1104/pp.112.2.503
Tian, Z., Wang, F., Zhang, W., Liu, C., and Zhao, X. (2012). Antioxidant mechanism and lipid peroxidation patterns in leaves and petals of marigold in response to drought stress. Hortic. Environ. Biotechnol. 53, 183–192. doi:10.1007/s13580-012-0069-4
Ullah, A., Manghwar, H., Shaban, M., Khan, A. H., Akbar, A., Ali, U., et al. (2018). Phytohormones enhanced drought tolerance in plants: a coping strategy. Environ. Sci. Pollut. R. 25, 33103–33118. doi:10.1007/s11356-018-3364-5
Ullah, A., Sun, H., Yang, X., and Zhang, X. (2017). Drought coping strategies in cotton: increased crop per drop. Plant Biotechnol. J. 15, 271–284. doi:10.1111/pbi.12688
Verma, V., Ravindran, P., and Kumar, P. P. (2016). Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 16, 86. doi:10.1186/s12870-016-0771-y
Waadt, R. (2020). Phytohormone signaling mechanisms and genetic methods for their modulation and detection. Curr. Opin. Plant Biol. 57, 31–40. doi:10.1016/j.pbi.2020.05.011
Wager, A., and Browse, J. (2012). Social network: JAZ protein interactions expand our knowledge of jasmonate signaling. Front. Plant Sci. 3, 41. doi:10.3389/fpls.2012.00041
Wang, J., Dang, Z., Zhang, H., Zheng, L., Borjigin, T., and Wang, Y. (2015). Gene transcript profiles in the desert plant Nitraria tangutorum during fruit development and ripening. Mol. Genet. Genomics 291, 383–398. doi:10.1007/s00438-015-1116-5
Wang, J., Liang, C., Yang, S., Song, J., Li, X., Dai, X., et al. (2021a). iTRAQ-based quantitative proteomic analysis of heat stress-induced mechanisms in pepper seedlings. PeerJ 9, e11509. doi:10.7717/peerj.11509
Wang, L., Du, M., Wang, B., Duan, H., Zhang, B., Wang, D., et al. (2022). Transcriptome analysis of halophyte Nitraria tangutorum reveals multiple mechanisms to enhance salt resistance. Sci. Rep. 12, 14031. doi:10.1038/s41598-022-17839-z
Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J.-P., and Li, W. (2013). CPAT: coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41, e74. doi:10.1093/nar/gkt006
Wang, X., Niu, Y., and Zheng, Y. (2021b). Multiple functions of MYB transcription factors in abiotic stress responses. Int. J. Mol. Sci. 22, 6125. doi:10.3390/ijms22116125
Yan, F., Yang, W.-k., Li, X.-y., Lin, T.-t., Lun, Y.-n., Lin, F., et al. (2008). A trifunctional enzyme with glutathione S-transferase, glutathione peroxidase and superoxide dismutase activity. BBA-Gen Subj. 1780, 869–872. doi:10.1016/j.bbagen.2008.03.003
Yang, F., and Lv, G. (2022a). Combined analysis of transcriptome and metabolome reveals the molecular mechanism and candidate genes of Haloxylon drought tolerance. Front. Plant Sci. 13, 1020367. doi:10.3389/fpls.2022.1020367
Yang, F., and Lv, G. H. (2022b). Characterization of the gene expression profile response to drought stress in Haloxylon using PacBio single-molecule real-time and Illumina sequencing. Front. Plant Sci. 13, 981029. doi:10.3389/fpls.2022.981029
Yang, Y., Shi, R., Wei, X., Fan, Q., and An, L. (2010a). Effect of salinity on antioxidant enzymes in calli of the halophyte Nitraria tangutorum Bobr. Plant Cell. Tiss. Org. 102, 387–395. doi:10.1007/s11240-010-9745-1
Yang, Y., Wei, X., Shi, R., Fan, Q., and An, L. (2010b). Salinity-induced physiological modification in the callus from halophyte nitraria tangutorum Bobr. J. Plant Growth Regul. 29, 465–476. doi:10.1007/s00344-010-9158-8
Yuan, Y. H., Liu, L., Gao, Y. B., Yang, Q. H., Dong, K. J., Liu, T. P., et al. (2022). Comparative analysis of drought-responsive physiological and transcriptome in broomcorn millet (Panicum miliaceum L.) genotypes with contrasting drought tolerance. Ind. Crop Prod. 177, 114498. doi:10.1016/j.indcrop.2021.114498
Zhang, A., Ji, Y., Sun, M., Lin, C., Zhou, P., Ren, J., et al. (2021). Research on the drought tolerance mechanism of Pennisetum glaucum (L.) in the root during the seedling stage. Bmc Genomics 22, 568. doi:10.1186/s12864-021-07888-5
Zhao, Q., He, L., Wang, B., Liu, Q.-L., Pan, Y.-Z., Zhang, F., et al. (2018). Transcriptome comparative analysis of salt stress responsiveness in Chrysanthemum (Dendranthema grandiflorum) roots by Illumina- and single-molecule real-time-based RNA sequencing. Dna Cell. Biol. 37, 1016–1030. doi:10.1089/dna.2018.4352
Zheng, L. L., Gao, Z., Wang, J., Zhang, H. R., and Wang, Y. C. (2014). Molecular cloning and functional characterization of a novel CBL-interacting protein kinase NtCIPK2 in the halophyte Nitraria tangutorum. Genet. Mol. Res. 13, 4716–4728. doi:10.4238/2014.July.2.1
Zhou, W., Wang, Y., Yang, F., Dong, Q., Wang, H., and Hu, N. (2019). Rapid determination of amino acids of nitraria tangutorum Bobr. From the qinghai-tibet plateau using HPLC-FLD-MS/MS and a highly selective and sensitive pre-column derivatization method. Molecules 24, 1665. doi:10.3390/molecules24091665
Zhou, Z., Wang, J., Zhang, S., Yu, Q., and Lan, H. (2020). Investigation of the nature of CgCDPK and CgbHLH001 interaction and the function of bHLH transcription factor in stress tolerance in Chenopodium glaucum. Front. Plant Sci. 11, 603298. doi:10.3389/fpls.2020.603298
Zhu, L., Lu, L., Yang, L., Hao, Z., Chen, J., and Cheng, T. (2021). The full-length transcriptome sequencing and identification of Na+/H+ antiporter genes in halophyte Nitraria tangutorum bobrov. Genes. 12, 836. doi:10.3390/genes12060836
Keywords: Nitraria tangutorum Bobr., drought stress, transcriptome analysis, plant hormone signal transduction, whole plant
Citation: Wei M, Wang B, Li C, Li X, He C and Li Y (2024) Integrated PacBio SMRT and Illumina sequencing uncovers transcriptional and physiological responses to drought stress in whole-plant Nitraria tangutorum. Front. Genet. 15:1474259. doi: 10.3389/fgene.2024.1474259
Received: 01 August 2024; Accepted: 12 September 2024;
Published: 01 October 2024.
Edited by:
Zhiqiang Wang, Chengdu Agriculture College, ChinaReviewed by:
Chenggong Liu, Chinese Academy of Forestry, ChinaTingting Chen, Nanjing Forestry University, China
Copyright © 2024 Wei, Wang, Li, Li, He and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Li, bGl5aUBnc2F1LmVkdS5jbg==
†Present address: Bo Wang, School of Bioengineering and Biotechnology, Tianshui Normal University, Tianshui, China