 Mengting Zhu1†
Mengting Zhu1† Pengfei Li1,2†
Pengfei Li1,2† Weiwei Wu2*Wenxin Zheng2*Juncheng Huang2Hanikzi Tulafu2Changchun Lin1,2Weikun Tao2Qi Aladaer1
Weiwei Wu2*Wenxin Zheng2*Juncheng Huang2Hanikzi Tulafu2Changchun Lin1,2Weikun Tao2Qi Aladaer1- 1College of Animal Science, Xinjiang Agricultural University, Urumqi, China
- 2Institute of Animal Science, Xinjiang Academy of Animal Sciences, Urumqi, China
Xinjiang is a major province of sheep breeding in China, which plays an important role in meeting people’s needs for meat products, increasing farmers’ income and sustainable development of animal husbandry. However, the genetic differentiation relationship between breeds was not clear, and most sheep had low fecundity, which seriously restricted the efficient development of sheep industry. Therefore, this study used the whole genome resequencing to detect the genetic variation of Dexin mutton and fine-wool sheep, explored the selected regions and important genes of the litter size traits, analyzed the genetic mechanism of reproductive traits, and provided new insights for the high fecundity breeding of sheep. A total of 5,236.338 G genome data and 35,884,037 SNPs were obtained. Furthermore, we identified 39 selection signals spanning candidate genes, 99 genes were significantly associated related to growth, reproduction and immunity, among which, BRIP1, BMPR1B, BMP4, NGF, etc. genes, and MAKP signaling pathway, Fanconi anemia pathway and Thyroid hormone signaling pathway and other signaling pathways were significantly correlated with litter size trait. Among them, we identified NGF, TrKA and BRIP1 genes was the important genes for sheep litter size traits and the mutation frequencies of 9 SNPs in BRIP1 gene were significantly different in domestic sheep in the world. The research provided new insights for the breeding of self-cultivated meat fine-wool sheep.
Background
Sheep have domesticated about 11,000 years ago in the Fertile Crescent and provided necessary basis for human economy and culture (Alberto et al., 2018). China is a large agricultural country, animal husbandry was one of the main industries and played an important role in national economic and social development. Mutton is favored by consumers because of its unique flavor, high protein and low cholesterol. In recent years, the shortage of mutton-based sheep products had led to a straight upward trend mutton prices in China. The contradiction between the rapid rised in Chinese mutton demand and the low production capacity of domestic mutton had become increasingly prominent. Improving the production performance of ewes and mutton production had become a major industrial demand at this stage. Reproductive traits were the most important production traits in important economic traits (reproduction, meat and fur) of sheep (Wang et al., 2019; Zhang et al., 2022; Zhong et al., 2024). Whereas, Multiple lambing (high fecundity) was an important basis for efficient breeding and sustainable development of sheep production industry. With the upgrading of sequencing technology, the cost of sequencing had also been reduced. In the study of major economic animals, the whole genome resequencing analysis strategy had been widely used. More and more researchers had carried out whole genome sequencing research in order to identify a large number of candidate genes related to important economic traits of candidate groups at the genomic level. Prof. Li’s team deep resequencing of 248 sheep including the wild ancestor (O. orientalis), landraces, and improved breeds individuals from all over the world at an average sequencing depth of ∼25.7 ×, providing insights into the demographic history of sheep and a valuable genomic resource for future genetic studies and improved genome-assisted breeding of sheep and other domestic animals (Li et al., 2020). Liu and his team revealed the genetic basis of year-round estrus, drought tolerance, hypoxia resistance, and cold tolerance traits of Xinjiang sheep breeds resistance, and cold tolerance traits of Xinjiang sheep breeds (Zhang et al., 2023). Furthermore, Li et al. (2022) revealed that PAPSS2 was used to high-altitude adaptability in Tibetan goats, following interspecific introgression and natural selection. Similarly, Zhu et al. (2023) provides insights into the microevolution and identifies genes associated with reproduction traits via whole genome resequencing.
A fine breed was an important foundation to ensure the healthy development of sheep industry and the key to enhance the core competitiveness of sheep industry. The project team members had gone through 16 years (2008–2023) in the early stage, with German Merino sheep as the male parent and Chinese Merino sheep as the female parent. Through three stages of progressive hybridization, cross fixation and breeding improvement, after four generations of continuous breeding, a new breed of fine-wool sheep with uniform body shape, stable genetic performance and suitable for both meat and wool in the northern pastoral area and the farming-pastoral ecotone was bred. The main characteristics are fast growth and development, good meat performance, strong adaptability, resistance to roughage, suitable for feeding in the majority of agricultural and pastoral areas, and high breeding efficiency. At present, the population size of Dexin mutton and fine-wool sheep has reached 84,000, of which the number of core breeding groups and breeding groups has reached 15,000, and the number of families has reached more than 8. In terms of reproductive traits, Dexin mutton and fine-wool sheep reached sexual maturity at 9 months of age, and ewes reached sexual maturity at 8 months of age. The age of ewes was 15 months, and the age of rams was 18 months. The estrous cycle of ewes was 17–21 days, the duration of estrus was 36–48 h, and the gestation period was about 150 days. The lambing rate of multiparous ewes was 130%, and the survival rate of lambs was more than 95%.
Based on this, this study used whole genome resequencing technology to study the selected regions and candidate genes of the litter size trait of Dexin mutton and fine-wool sheep, and identified the specific selection signals. These genes contribute to the reproductive characteristics of the species and deepen our understanding of the unique genomic structure of the species, so as to further improve and sustainable use.
Materials and methods
Ethics approval and consent to participate
All animal treatments were according to the recommendation of the Regula-tions for the Administration of Affairs Concerning Experimental Animals of China, and approved by the Animal Care Committee of Xinjiang Agricultura University responsible for overseeing the ethical use of animals in research within the university. All methods are reported in accordance with ARRIVE guidelines for the reporting of animal experiments (2024004).
Sample collection
Whole-blood samples (5 mL) were collected from 51 Dexin mutton and fine-wool sheep (single-lamb group: 31 and double-lamb group: 20), randomly selected from Keping County, Aksu, southern Xinjiang. DNA was extracted from blood using the TIANamp Blood DNA Kit (TIANGEN, China) according to the manufacturer’s instructions. To investigate the genetic basis of Dexin mutton and fine-wool sheep. The whole-genome sequencing (WGS) 10 × was performed using the Illumina HiSeq2500 Sequencer (Illumina Inc.) by Tianjin Compass Biotechnology Co., Ltd. (Tianjin, China). To obtain high-quality data, reads containing adapters, reads with ≥10% unidentified nucleotides (N) were removed, and low-quality reads (>50% of the read bases with a Phred quality score (i.e., Q-score) ≤5) were also removed.
Additionally, one hundred genomic DNA samples were obtained from healthy Dexin mutton and fine-wool sheep by intravenous blood collection in Tianxin Breeding Farm, Aksu, southern Xinjiang, China (4 years old and weighed 45 ± 3.44 kg). Another one hundred and sixteen genomic DNA samples were obtained from healthy Dexin mutton and fine-wool sheep by intravenous blood collection in Wenquan Breeding Farm, Aksu, southern Xinjiang, China (4 years old and weighed 48 ± 2.94 kg). These sample DNA were used for SNPs validation and application.
Mapping and variant calling
To obtain high-quality data, the effective sequencing data was compared to the sheep reference genome sequence (ARS-UI Ramb v2.0) by using BWA (V0.7.12) with default parameters (mem -t 4 -k 32 -M) (Li and Durbin, 2009). SAMtools version 1.9 was employed to convert SAM files to the BAM format and subsequently sort the resulting BAM files by contigs (Li et al., 2009). The SNPs were detected by SAMtools (v1.2) with the following parameters “mpileup -m 2 -F 0.002 -d 1,000” (Li et al., 2009). Moreover, to exclude SNP callings errors, variant sites with QD < 2.0, MQ < 20, FS > 60.0 were discarded, then variants were annotated by using ANNOVAR v.21-Jun-2013. The VCF file was obtained by Haplotyper and GVCFtyper, and the data were filtered by PLINK software (v.1.9). After variant calling and obtaining the variant call set, we performed genomic diversity analysis to explore the patterns of genetic variation within populations. The loci with MAF less than 0.05, the loci with markers less than 1e-5 deviating from Hardy-Weinberg equilibrium (--maf 0.05 -hwe 1e-6), and the loci with deletion rate more than 10% were excluded (--geno 0.10). Principal component analysis (PCA) of the 51 samples was conducted by EIGENSOFT (Price et al., 2006). Based on neighbor-joining (NJ) method, phylogenetic tree was constructed by PHYLIP (v.3.6) (Felsenstein, 1989), and displayed by Newick Utilities.
Genome-wide selective sweep regions
In this study, Nucleotide diversity (π) and fixation index (FST) were calculated using the vcftools v.0.1.14, with a sliding window approach (100-kb windows with 50-kb step length) (Zhang et al., 2021). The parameters for the VCFtools program were as follows: “--fst-window -size 100,000 --fst-window-step 50,000”. The selected region was to apply the top significant 1% for FST and π ratio. The intersection selective regions considered as under strong selective sweep and subsequently examined for potential candidate genes.
Functional enrichment analysis
Gene Ontology (GO) and KEGG pathways were retrieved to determine clusters of functionally related genes (Harris et al., 2004; Jiao et al., 2012). KEGG pathway enrichment analysis was performed using KOBAS 2.0 (http://kobas.cbi.pku.edu.cn/), and a corrected P-value 0.05 was set as the threshold. Then the genome database (http://animal.omics.pro/code/index.php/SheepVar) was used to retrieve the mutation frequency distribution of SNPs in the world's domestic sheep breeds, and analyzed the differences in mutation frequency.
PCR for BRIP1 gene
Based on the results of whole genome resequencing in this study and combined with previous literature, a correlation was found between the BRIP1 gene and breast cancer and ovarian cancer. Additionally, it was observed that the expression level of BRIP1 in cervical cancer tissues is significantly lower than that in normal cervical tissues. Furthermore, upregulation of BRIP1 gene expression has been reported to increase the risk of breast cancer and have a significant impact on the reproductive function of female mammals. Therefore, it is speculated that the BRIP1 gene may be a potential candidate gene affecting litter size in sheep. In this study, we selected the BRIP1 gene as a candidate gene affecting litter size, analyzed its polymorphism in the population of Dexin mutton and fine wool sheep, and investigated its association with litter size. BRIP1 (Gene ID: 101107879) were designed with reference to the sheep. The PCR was performed in a 25 μL reaction volume containing 2 μL of DNA (50 ng), 1 μL of each primer, 12.5 μL of PCR Mix and 8.5 μL of water. The cycling protocol was 2 min at 95°C, followed by 36 cycles of 94°C for 30 s, TA C annealing for 30 s and 72°C for 45 s (TA for all markers are presented in Table 1).

Table 1. PCR primer sequences for BRIP1.
The primers used in this study were designed with reference to the sheep TrKA (Gene ID: 101109763) and NGF (Gene Bank ID:101104540) mRNA sequences published in GenBank. Table 1 shows the primer sequence information. The PCR was performed in a 25 μL reaction volume containing 2 μL of DNA (50 ng), 1 μL of each primer, 12.5 μL of PCR Mix and 8.5 μL of water. The cycling protocol was 2 min at 95°C, followed by 36 cycles of 94°C for 30 s, TA°C annealing for 30 s and 72°C for 45 s (TA for all markers are presented in Table 2). Additionally, the gene polymorphism was detected by PCR-RFLP technique. The TrKA gene was digested by restriction enzyme Sac II, and the NGF gene was digested by restriction enzyme SfiI. In a 15 μL digestion reaction system, including 7 μL of PCR product, 1 μL of restriction enzyme Sac II or Sfi I, 1 μL of 10 × Buffer and 5 μL of ddH2O. Then water bath at 37°C for 5 h.
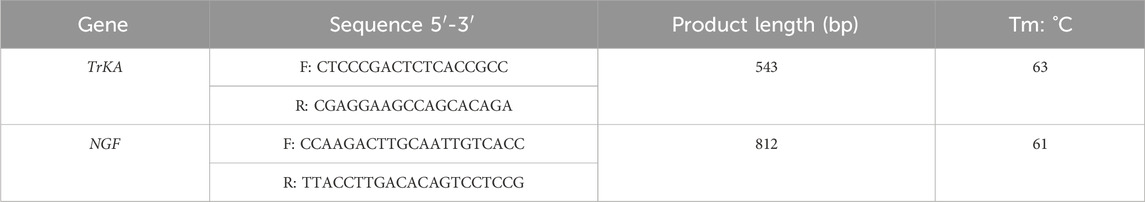
Table 2. PCR primer sequences for TrKA and NGF.
Statistical analysis
Genotypic and allelic frequencies and the Hardy-Weinberg equilibriums were estimated using the SPSS 20.0 (IBM Corp., Armonk, NY, United States). The logistic regression model was used to analyse the association of polymorphisms with litter size in different sheep breeding farm: Yij = μ+Fi + Gj+Sk + eijk, in which Y was the phenotypic value of the litter size, μ was the overall population mean, Fi was the farm effect, Gi was genotype effect, Sk was the fixed effect of season (k = 1, 2, 3, 4), and eijk was the random residual. Additionally, the logistic regression model was used to analyse the association of polymorphisms with ovulation rate in different sheep populations: Yij = μ+Gi + eij, in which Y was the phenotypic value of the ovulation rate, μ was the overall population mean, Gi was genotype effect, and eij was the random residual. Analysis was performed using the logistic regression model procedure of SAS (Ver 9.2; SAS Institute Inc., Cary, NC, United States). The data are expressed as the mean ± standard error (SE). Differences between means were accepted as statistically significant at P < 0.05 and as a trend at 0.05 < P < 0.10.
Results
Reads and mapping
High-depth whole-genome resequencing of the 51 samples of Dexin mutton and fine-wool sheep (Figure 1; Supplementary Data S1) generated a total of 5,236.338G Gb, the ratio of the total sequenced bases to the genome size, that is, the minimum sequencing depth, was 4.32×; the maximum sequencing depth was 11.55×,the average sequencing depth was 7.45×, and an average genome coverage of 98.20% (Q20 ≥ 98.17% and Q30 ≥ 96.71%), The Mapping rate after alignment with the reference genome reached more than 99%. The GC base contents ranged from 42.10% to 45.34%, with an average of 43.24%. (Supplementary Data S1).

Figure 1. Female (A) and male (B) Dexin mutton and fine-wool sheep.
Population genetic structure
SNPs had a different distribution on chromosomes (Figure 2A). We conducted a principal component analysis. PCA analysis provided corroborating evidence for the two groups. These two groups could not be distinguished, was gathered together (Figure 2B). For another, the NJ tree revealed that the two populations were clustered into one group, which was consistent with the PCA results (Figure 2C).
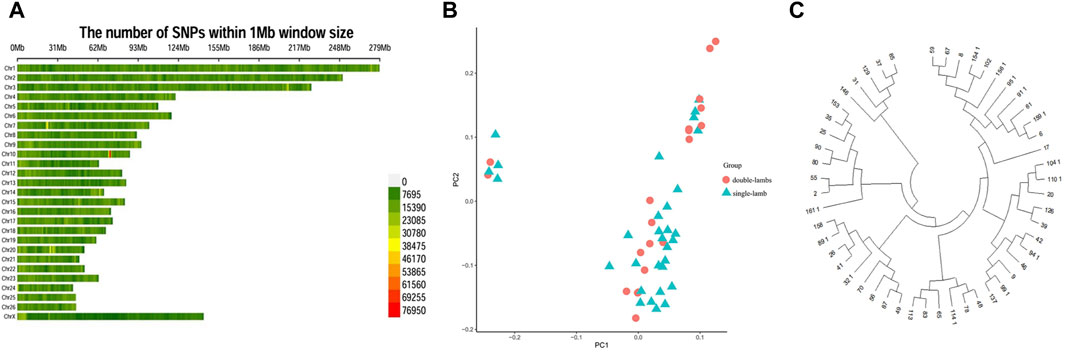
Figure 2. Population genetic structure. (A) Distribution of SNPs on chromosomes. (B) PCA analysis. (C) NJ tree.
Selective imprints of litter size trait of dexin mutton and fine-wool sheep
In order to screen candidate genes for litter size trait, the experimental population was divided into single-lamb group and double-lamb group according to the litter size record. Two methods of FST and Pi-ratio were used to screen candidate genes for reproductive traits and growth traits.
The top 1% region of FST (>0.027) and Pi-ratio (>1.48), and annotating the first 1% region by ANNOVAR (v.21-Jun-2013), 663 genes were retained in FST after removing duplicate genes (Figure 3A; Supplementary Data S2), and 612 genes were retained in Pi-ratio (Figure 3B; Supplementary Data S3). The intersection of FST and Pi-ratio annotated genes included 99 genes (Figures 3C, D).

Figure 3. (A) The distribution of FST on autosomes of single lamb and double lamb of Dexin mutton and fine-wool sheep. (B) The distribution of Pi-ratio on autosomes of single lamb and litter size of Dexin mutton and fine-wool sheep. (C) Selection region and selection signal. (D) The intersection results of FST and Pi-ratio methods.
Through the above two selection methods, we screened the corresponding signal regions in 26 autosomes of single and double lamb populations (Table 3). Consistent signal regions were found on chromosomes 11 and 12. For example, the position was in Chr 11: 10,900,000-10,950,000 region includes BRIP1 and INTS2 genes; the position was in Chr 12: 79,960,000–80,010,000 region includes ELF3, COMMD8, LOC121816089, IP09, SHISA4, LOC121816077, LMOD1 genes. Additionally, this study also screened the “maker gene” BMPR1B which was significantly related to sheep reproduction.
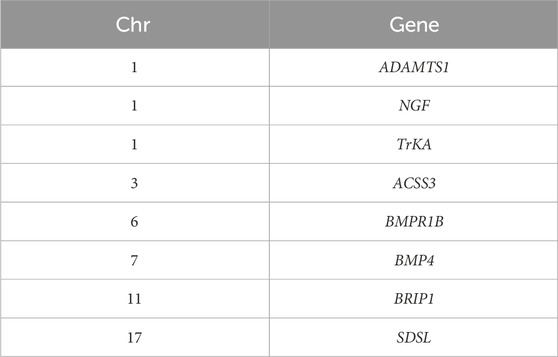
Table 3. Candidate genes related to reproductive performance.
Functional enrichment analysis
GO enrichment and KEGG pathway analysis of the selected candidate genes were performed using DAVID and KOBAS. A total of 99 genes were annotated (P < 0.05) and enriched in important economic traits such as growth traits, reproductive traits, immune adaptability, etc. Notably, candidate genes were mainly enriched in ATP binding, DNA binding, G-protein coupled receptor binding, nucleus and positive regulation of gene expression, etc. Through the KEGG pathway analysis, candidate genes BRIP1, NGF, ACSS3 BMP4, BMPR1B and other genes were enriched in Fanconi anemia pathway, MAKP signaling pathway, Thyroid hormone signaling pathway and other signaling pathways. Some genes are also enriched in cancer-related signaling pathways such as Pathway in cancer (Figure 4).
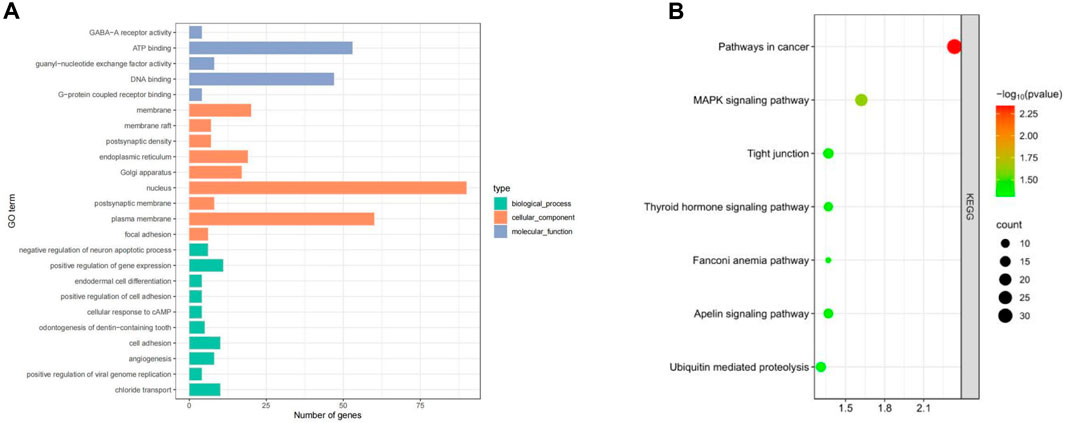
Figure 4. Enrichment results of candidate genes in Dexin mutton and fine-wool sheep. (A) GO Enrichment. (B) KEGG pathways.
Candidate genes and mutation sites
There were obvious signal differences and haplotype differences in the chromosome 11 region between single-lamb and double-lamb populations (Figure 5). The SnpEff software annotation showed that there were 9 missense mutation SNPs in the BRIP1 gene, which were Chr 11:10,972,067 c.1136C > T p.Thr379Ile, Chr 11:11,098,249 c.2900G > A p.Arg967Gln, Chr 11:11,098,267 c.2918G > A p.Ser973Asn,Chr 11:11,098,431 c.3082G > A p.Asp1028Asn, Chr 11:11,098,497 c.3148A > G p.Asn1050Asp, Chr 11:11,098,533 c.3184A > G p.Ile1062Val, Chr 11:11,098,537 c.3188T > C p.Val1063Ala, Chr 11:11,098,543 c.3194A > T p.Glu1065Val, Chr 11:11,098,683 c.3334G > A p.Gly1112Arg.
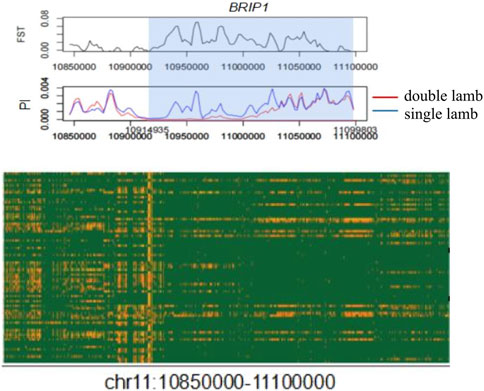
Figure 5. Local signal map and corresponding haplotype heat map of chromosome 11 region.
The mutation frequency distribution of 9 missense mutations in the BRIP1 gene in the world domestic sheep breeds is shown in Figure 6 by sheep genome database (http://animal.omics.pro/code/index.php/SheepVar), and the mutation frequency in the world domestic sheep is quite different.
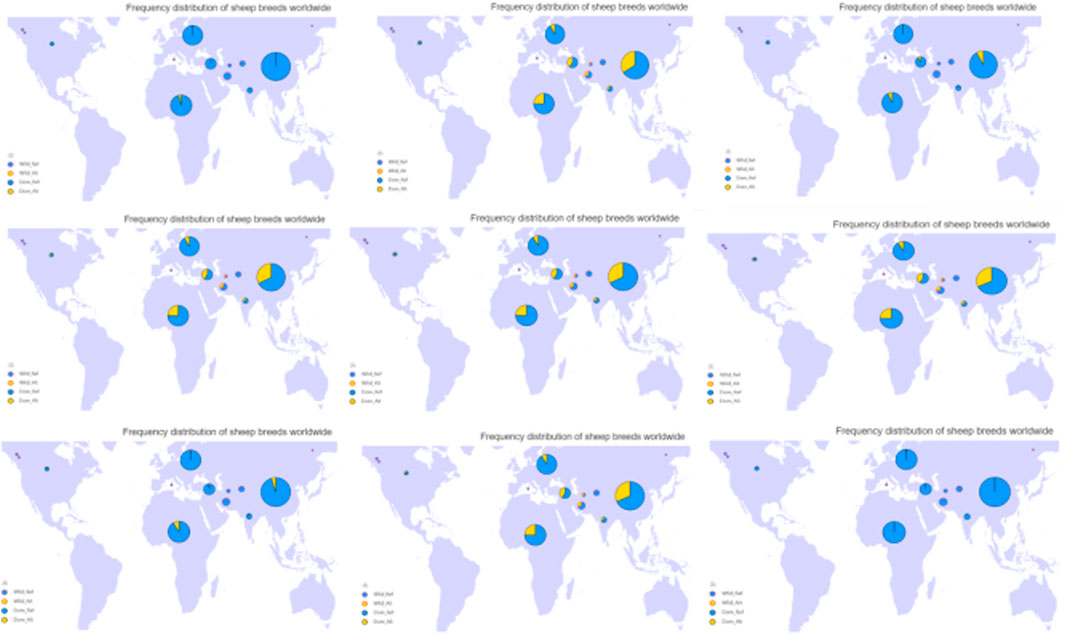
Figure 6. The frequency distribution map of in domestic sheep in the world.
Identification of litter size traits genes
The PCR products of BRIP1 (Figures 7A, B) gene was detected by PCR. The results showed that the size of the PCR products was consistent with the expected results. Then, the SNPs of BRIP1 gene was detected by Sanger sequencing. 9 SNPs mutations were identified, and the results was shown in Table 5. Further, it was found that the g:11098497 A > G and g:11098533 A > G loci deviated from Hardy-weinberg equilibrium among the 9 SNPs mutation loci of BRIP1 gene exon (P < 0.05) (Table 4). The sequencing results of different genotypes were shown in Figure 8.
Figure 7. (A,B) PCR amplificationfor BRIP1 gene.
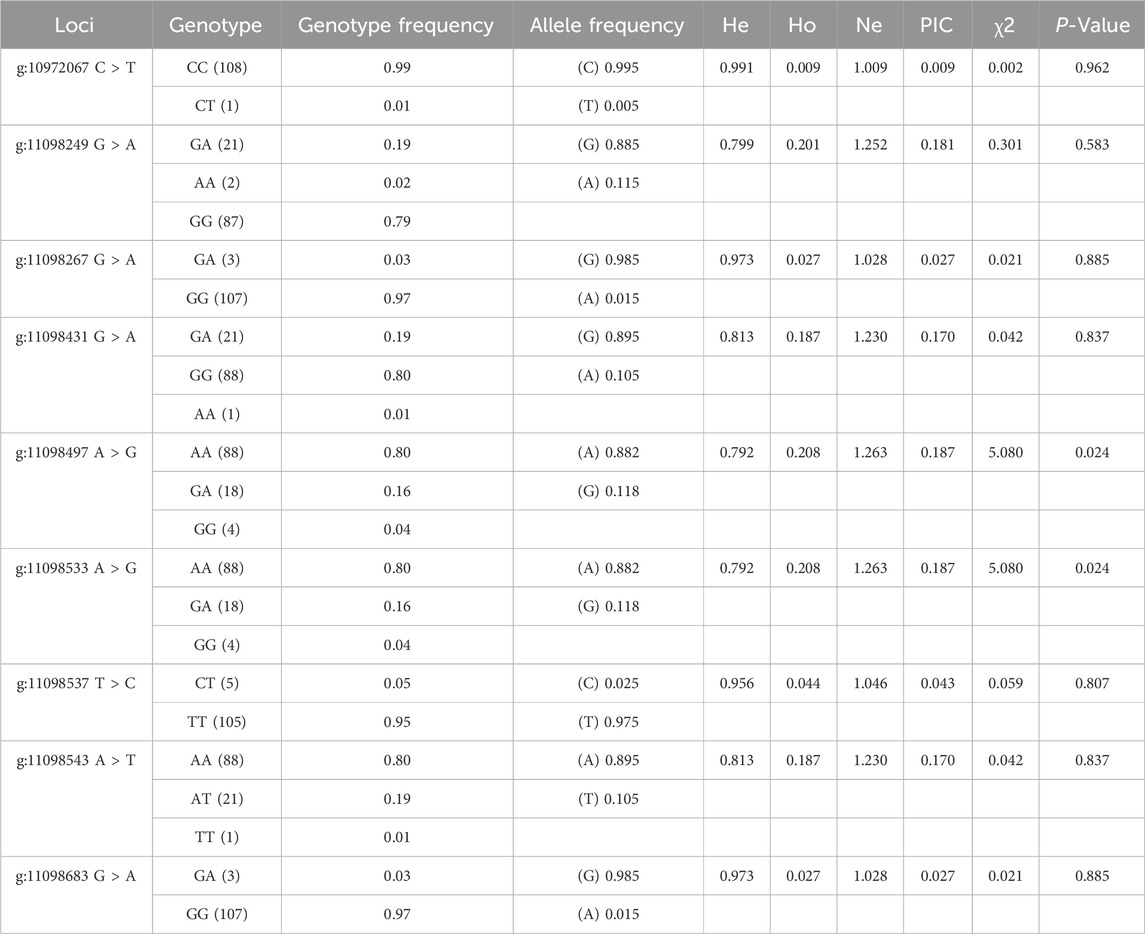
Table 4. Population genetic analysis of the BPIP1 gene.
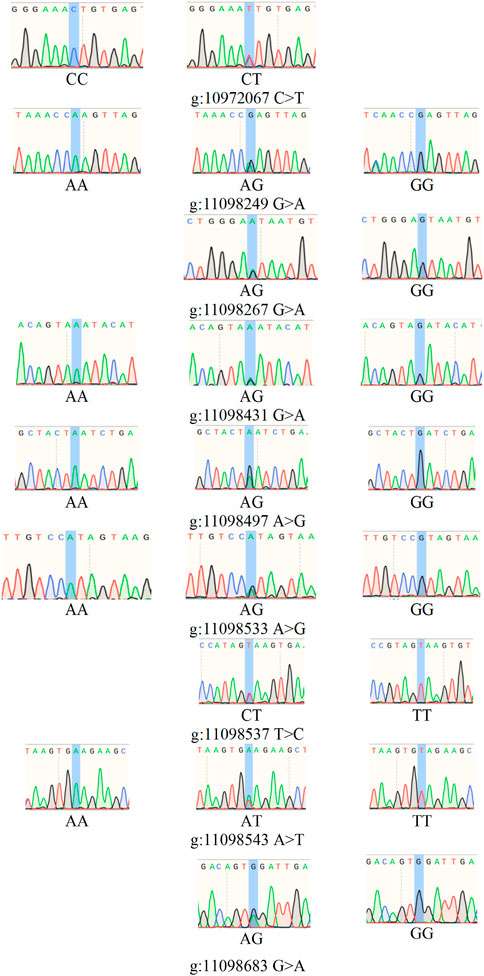
Figure 8. SNPs of BRIP1 gene.
In this study, the association between the litter size and different genotypes of BRIP1 gene g.11098497 A > G and g.11098533 A > G mutation sites of 255 Dexin mutton and fine-wool sheep was analyzed. The results are shown in Table 6. There was a strong linkage effect at the three mutation sites of BRIP1 gene, and the litter size with GG and AG genotypes was significantly higher than that in AA genotypes (P < 0.05) (Table 5).
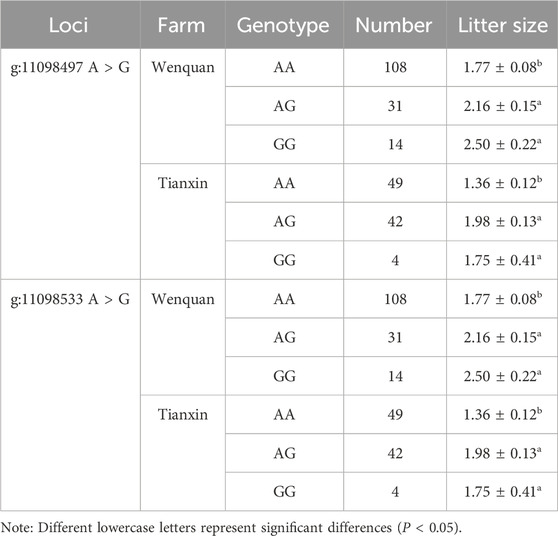
Table 5. Association analysis between genotypes of BRIP1 of litter size.
The PCR products of TrKA (Figure 9A) and NGF (Figure 9B) genes were detected by PCR. The results showed that the size of the PCR products was consistent with the expected results. Using the PCR-RFLP method, the PCR product of TrKA digested by Sac II produced two bands of 432 bp and 111 bp, which were identified as CC genotype. A 543 bp band was identified as TT genotype. When the locus was in the CT heterozygous type, three bands of 543 bp, 432 bp, and 111 bp were produced, but the 111bp band was not shown because the 1% agarose gel had no obvious separation effect on the 111 bp band (Figure 9C). For NGF gene, the PCR product of NGF digested by Sfi I produced two bands of 205 bp and 607 bp, respectively, and the genotype was identified as GG type. A 812 bp band was identified as AA genotype. Three bands of 812 bp, 205 bp, and 607 bp were produced, and the genotype was identified as GA type (Figure 9D). Finally, the bands of the digested product were consistent with the expected size. The results of PCR sequencing peak map were also consistent (Figures 9E, F).
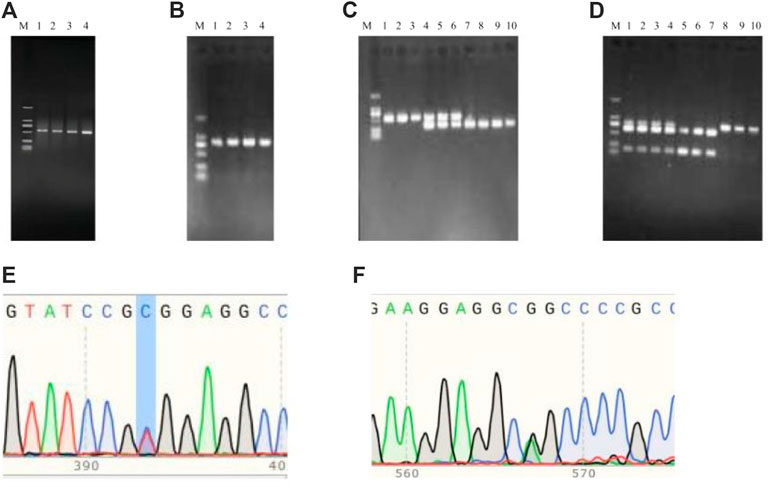
Figure 9. PCR amplification and PCR product sequencing peak diagrams and PCF-RFLP results of TrKA (A,C,E) and NGF (B,D.F). M: MarkerD2K (The size of each band from bottom to top was 100, 250, 500, 750, 1,000, 2000, respectively).
Early breeding molecular markers for litter size traits
Hardy-Weinberg equilibrium test of TrKA gene showed that the locus was moderately polymorphic in both fields. The frequencies of CC, CT and TT genotypes in Tianxin breeding farm were 0.17, 0.33 and 0.51, respectively. The frequencies of CC, CT and TT genotypes in Wenquan breeding farm were 0.1, 0.45 and 0.45, respectively. TT was the dominant genotype. Chi-square test showed that the site was deviated from Hardy-Weinberg disequilibrium (P< 0.05) in Tianxin breeding farm, suggesting that the site had strong selection potential in Tianxin (Table 6). Hardy-Weinberg equilibrium test of NGF gene showed that the locus was in moderate polymorphism in both farms. The frequencies of GG, GA and AA genotypes in Tianxin breeding farm were 0.11, 0.19 and 0.71, respectively, and GG was the dominant genotype. The frequencies of GG, GA and AA genotypes in Wenquan breeding farm were 0.07, 0.24 and 0.68, respectively, and AA was the dominant genotype. Chi-square test showed that both Tianxin and Wenquan were deviated from Hardy-Weinberg disequilibrium (P < 0.01), suggesting that the locus had strong selection potential in this population (Table 7).
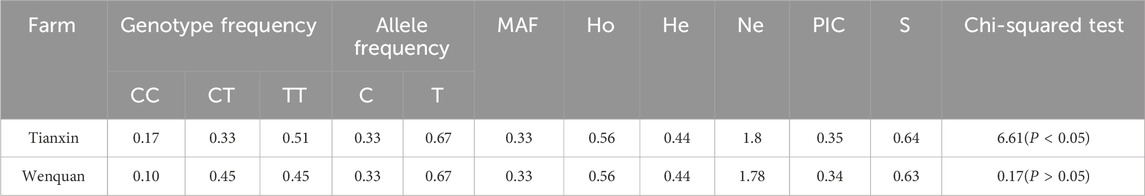
Table 6. Population genetic analysis of TrKA gene in different breeding farm.
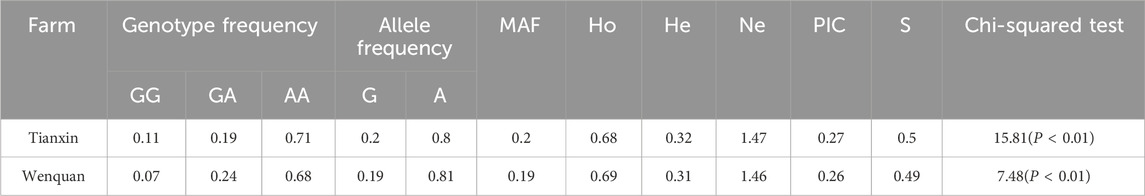
Table 7. Population genetic analysis of NGF gene in different breeding farm.
Correlation analysis showed that the TT genotype of TrKA was the dominant genotype, which could increase the litter size. The GG genotype of NGF gene was the dominant genotype, which also increased the litter size (Table 8). The results showed that TrKA gene and NGF gene had an effect on the litter size of Dexin mutton and fine-wool sheep.

Table 8. Association analysis between genotypes of TrKA and NGF of litter size.
Discussion
Sheep was one of the earliest domesticated animals. It was not only providing important means of living such as meat, milk, skin and hair for human beings, but also was an important part of the spread of human civilization (Li et al., 2020). China had rich sheep variety resources and had good development potential. It occupied an important position in animal husbandry and played an important role in promoting the increase of farmers and herdsmen’s income. Since the late 1980s, China had vigorously developed animal husbandry and had become the country with the largest number of sheep breeding, slaughter and mutton production. However, on the whole, there was still a big gap between the mutton sheep industry and the developed countries. One of the main reasons was that the low fecundity of most sheep was one of the main factors limiting the development of sheep industry. While Litter size was an important economic trait affected by many factors such as heredity, and its heritability is low. It was impossible to achieve the effect of multi-gene polymerization by using traditional techniques to mine candidate genes affecting sheep litter size. Importantly, for some breeding programs of mutton sheep, litter size was one of the main selection objectives. Therefore, it was of great significance to reveal the genetic basis of reproductive traits by using whole genome resequencing technology to mine the selected regions and candidate genes of sheep litter size traits, and to clarify the germplasm characteristics.
Dexin mutton and fine-wool sheep is a new breed cultivated by our breeding team after 16 years in Xinjiang, China. German merino sheep was used as the female parent and Chinese merino sheep was used as the male parent. The breeding process was progressive hybridization, cross fixation and breeding improvement. The molecular breeding was combined to form a high prolificacy and fast growth and development mutton performance. In the study, we obtained a total of 5,236.338 Gb, with an average depth of 7.45× per individual and an average genome coverage of 98.20% (Q20 ≥ 98.17% and Q30 ≥ 96.71%), and the Mapping rate after alignment with the reference genome reached more than 99%. Additionally, the single lamb and double lambs were were clustered into one group. The PCA results were consistent with the NJ tree results. The important genes of litter size traits were identified by FST and Pi-ratio. A total of 99 genes were screened that may affect important economic traits such as growth, reproduction and immune adaptability of sheep. This was also consistent with the breeding process of Dexin mutton and fine-wool sheep.
The reproductive performance of sheep was an important factor affecting its breeding efficiency, and the litter size was an important trait to measure the reproductive traits of sheep. Improving the litter size of sheep was an important means to promote the vigorous development of sheep industry in China. Therefore, it is of great significance to identify the important genes of the litter size traits of the self-cultivated meat fine-wool sheep in Xinjiang. Interestingly, in the process of identifying the trait genes of litter size traits, we found that in addition to the maker gene “BMPR1B” which was significantly related to the litter size trait, ACSS3, SDSL BMP4, ADAMTS1, NGF and BRIP1 were also found. These candiate genes mainly enriched in MAKP signaling pathway, Thyroid hormone signaling pathway and some genes were also enriched in cancer-related signaling pathways such as Pathway in cancer. MAPK signaling pathway and thyroid hormone signaling pathway play an important role in sheep reproduction (Su et al., 2023; Wang et al., 2023; Zhu et al., 2023). The MAPK signaling pathway was involved in regulating the physiological and biochemical processed of the sheep reproductive system, while the thyroid hormone signaling pathway affected the reproductive development and sex hormone regulation of sheep. The MAPK signaling pathway belonged to the classical conserved three-level enzymatic cascade reaction pathway, and its reaction mainly depended on the phosphorylation of protein kinases at different levels. MAPKKKs phosphorylate and activate specific MAPKKs, and MAPKKs then phosphorylate and activate downstream MAPKs, and then act on transcription factors to regulate specific gene expression (Wang et al., 2023). The previous study of the project team members found that miR-200c mediated MAPK8 to promote the development of sheep ovarian granulosa cells through MAPK signaling pathway and regulate the reproductive performance of sheep (Yang et al., 2016; Yang et al., 2018; Zhai et al., 2018; Zhu et al., 2022). Similarly, Hai (2023) demonstrated that leptin could inhibit the oxidation process of sheep endometrial epithelial cells through the MAPK pathway, thus playing a positive regulatory role in sheep embryo implantation. Lv et al. (2023) pointed that UNC5C、BMPR1B and GLIS1 may affect the reproductive traits of sheep through MAPK signaling pathway. Yang and his team employed high-throughput sequencing approach to investigate genome-wide CNVs between Tibetan sheep and White Suffolk, found that thyroid hormone signaling pathway and CTNNB1 gene that would be responsible for differential biological characteristics of breeds, especially reproductive traits (Yang et al., 2023).
Interestingly, BRIP1 gene also called FANCJ. BRIP1 gene-related reports are related to breast cancer and ovarian cancer. BRIP1 was lowly expressed in cervical cancer tissues compared with normal cervical tissues and was closely related to poor prognosis. BRIP1 gene was closely related to the function of breast cancer susceptibility BRCA1 (breast cancer 1, early onset), also known as BACH1 or FANCJ, located on 17q22-q24, about 18Kb, composed of 20 exons and 19 introns. BRIP1 gene single nucleotide polymorphism could affect the susceptibility of cervical cancer, and BRIP1 gene mutation was closely related to the occurrence and development of cervical cancer (Voutsadakis, 2020). Rafnar et al. (2011) confirmed that BRIP1 gene was one of the representative tumor suppressor genes in ovarian cancer, suggesting that this mutation in BRIP1 gene may also increase the risk of other cancers. Similarity, it had also been reported that the expression of BRIP1 was increased, which was easy to cause breast cancer and had a significant effect on the reproductive function of female mammals (Zou et al., 2016; Gupta et al., 2017). Moreover, this study also found that the frequency distribution of 9 SNPs of BRIP1 gene was quite different in the world sheep. Additionally, g:11098497 A > G and g:11098533 A > G showed a strong linkage effect, the number of each genotype was the same, and the results of the association analysis between genotype and litter size were the same.
While, the NGF and BMP4 genes were also found to be significantly correlated with reproduction traits. Nerve growth factor (NGF) played an important physiological role in the regulation of hypoxic-ischemic adaptation of central nervous system tissues or cells mainly by binding its high-affinity receptor TrkA. Abnormal expression of NGF and its receptors could cause diabetes, depression, Alzheimer’s disease and other diseases (Li et al., 2005; Wiener et al., 2015). In addition, the NGF gene also played an important role in some non-nervous systems such as reproduction and immunity. NGF was transported to the nucleus and binds to it by binding to cell surface receptors. So as to initiate cell activity, caused a series of biological effects, and played its function. Studies had shown that the synthesis of NGF in the ovary increases, which could increase the activity of the ovary and affect the reproductive performance of the female (Lansdown and Rees, 2012). NGF gene had a certain amount of expression in the animal reproductive system such as ovary, oviduct and uterus. It played an important physiological role in follicular growth and development, ovulation, ovarian hormone synthesis, gamete transport, sperm capacitation, fertilization and early embryos (Li et al., 2005; MTakeuchi and Harada, 2011). Also, a studies had found that NGF could promote the proliferation of granulosa cells and increase the pregnancy rate by inhibiting the downregulation of CDKN1A gene mediated by ESR2 (Wang et al., 2015; Zheng et al., 2019). Naicy et al. found that the relative expression of NGF gene in the ovaries of prolific goats was significantly higher than that of singleton goats (Naicy et al., 2016). Herein, we showed that the TT genotype of TrKA was the dominant genotype, which could increased the litter size. The GG genotype of NGF gene was the dominant genotype, which also increased the litter size. The results showed that TrKA gene and NGF gene had an effect on the litter size of Dexin mutton and fine-wool sheep. Also, in the study, NGF (g: 92654175G > A) and TrKA (g: 106814725C > T) was transformed and applied, and it was found that the two SNPs in Wenquan Breeding Farm, Aksu, southern Xinjiang, China and Tianxin Breeding Farm, Aksu, southern Xinjiang, China could significantly increase litter size. The economic value of the current transformation was ¥50,000. Further transformation and application were in progress. Studies had also confirmed that the expression of NGF and the expression of BMP4 had a synergistic effect, which promoted the ovulation of the female, thereby increased the number of lambs and improved the reproductive efficiency (Guang et al., 2010). Bone morphogenetic proteins (BMPs) are members of the transforming growth factor-β (TGF-β) superfamily and play an important role in the development process. BMP4 was an important member of the BMPs family and a key gene in the TGF-β signaling pathway. It was widely expressed in organisms, and its biological functions involved almost all systems from embryo to adult, especially in reproductive activity (Hu et al., 2004). The function of the BMP family was mainly dependent on the classic BMP/Smad signaling pathway (Da Cunha et al., 2017). Xu et al. (2010) proved that BMP4 could regulate ovulation rate via the BMP/Smad pathway and closely related molecules. Faure et al. found that the relative expression of BMP4 in the sheep prolific group was significantly higher than that in the single group (Faure et al., 2005). With the deepening of research, the polymorphism of BMP4 gene had been proved to be associated with human diseases (Carstens and Michael, 2015), production performance of livestock and poultry (Fang et al., 2009). Fang et al. found that the polymorphism of BMP4 gene was closely related to the growth and reproductive traits of goats (Fang et al., 2009). Similarly, Pan et al.found a missense mutation g.63454744T > G in the coding region of sheep BMP4 gene by whole genome resequencing technology, which had a significant effect on the reproductive performance of ewes, which was consistent with the results of this study that BMP4 gene may be an important gene affecting twinning traits (Zhangyuan et al., 2018). It was speculated that BRIP1, NGF, BMP4 and other genes may regulate the growth, litter size traits of Dexin mutton and fine-wool.
As a new strain bred by German merino sheep was used as the female parent and Chinese merino sheep was used as the male parent, we used genome-wide resequencing to explore the selected regions for growth, reproduction, and immune adaptation, and identified the selection signals and important genes for litter size traits. The candidate genes were mainly enriched in MAPK signaling pathway, Fanconi anemia pathway, Thyroid hormone signaling pathway and other signaling pathways. It provides a new insights for molecular breeding of new breeds of Dexin mutton and fine-wool sheep, and also provides target genes and functional sites for the breeding of other new sheep breeds.
Conclusion
We identified several major candidate genes and markers under selection in Dexin mutton and fine-wool sheep by whole genome re-sequencing. It played essential roles in reproduction, growth and immunity as well as other economic traits. We also explored a large number of genetic variants and genetic genetic basis underlying different phenotypes. Additionally, We identified NGF, TrKA and BRIP1 genes was the important genes for sheep litter size traits. Our results contribute to understand the genetic basis of Dexin mutton and fine-wool sheep and provide valuable information for future breeding and improvement of new breeds. This study provided a comprehensive insight to the litter size traits of Dexin mutton and fine-wool sheep, which will underpin more-accurate identification of high prolific gene variants in the near future and facilitate novel breeding strategies.
Data availability statement
The data presented in the study are deposited in the NCBI, accession number PRJNA1084106.
Ethics statement
The animal studies were approved by All animal treatments were according to the recommendation of the Regula-tions for the Administration of Affairs Concerning Experimental Animals of China, and approved by the Animal Care Committee of Xinjiang Agricultura University responsible for overseeing the ethical use of animals in research within the university. All methods are reported in accordance with ARRIVE guidelines for the reporting of animal experiments (2024004). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
MZ: Formal Analysis, Methodology, Software, Writing–original draft, Writing–review and editing. PL: Formal Analysis, Methodology, Writing–original draft. WW: Conceptualization, Funding acquisition, Resources, Visualization, Writing–original draft, Writing–review and editing. WZ: Conceptualization, Resources, Writing–original draft, Writing–review and editing. JH: Resources, Writing–original draft, Writing–review and editing. HT: Resources, Software, Writing–review and editing. CL: Data curation, Formal Analysis, Software, Writing–original draft. WT: Resources, Writing–original draft. QA: Validation, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. his research was funded by Regional collaborative innovation project of autonomous region (Science and Technology Aid Xinjiang Program) (2022E02015) and the special fund project of the central government to guide local scientific and technological development (ZYYD2022B08). National Key Research and Development Program of China (2021YFD1300905), And Special Fund Project for Central Leading Local Science and Technology Development (ZYYD2022C14).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1457634/full#supplementary-material
References
Alberto, F. J., Boyer, F., Orozco-terWengel, P., Streeter, I., Servin, B., de Villemereuil, P., et al. (2018). Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 9, 813. doi:10.1038/s41467-018-03206-y
Carstens, , and Michael, H. (2015). Single nucleotide polymorphism of BMP4 Gene: a risk factor of non-syndromic cleft lip with or without cleft palate. Indian J. Plastic Surg. 48, 164–165. doi:10.4103/0970-0358.163054
Da Cunha, E. D. V., De Souza, G. B., Passos, J. R. d.S., Silva, A. W. B., Dau, A. M., Saraiva, M. V. A., et al. (2017). Effects of bone morphogenetic protein 4 (BMP4) on in vitro development and survival of bovine preantral follicles enclosed in fragments ovarian tissue. Zygote 1. doi:10.1017/S0967199417000089
Fang, X., Xu, H., Zhang, C., Chen, H., Hu, X., Gao, X., et al. (2009). Polymorph. BMP4 gene its Assoc. growth traits goats. Mol. Biol. Rep. 36 (6), 1339–1344. doi:10.1007/s11033-008-9317-1
Faure, M. O., Nicol, L., Fabre, S., Fontaine, J., Mohoric, N., McNeilly, A., et al. (2005). BMP-4 inhibits follicle-stimulating hormone secretion in Ewe pituitary. BMP-4 inhibits follicle-stimulating hormone Secret. ewe Pituit. 186, 109–121. doi:10.1677/joe.1.05988
Felsenstein, J. (1989). PHYLIP-phylogeny inference package (version 3.2). Cladistics-the Int. J. Willi Hennig Soc. 5, 164–166. doi:10.1086/416571
Guang, R., Hanlin, Z., Guanyu, H., Dongjin, W., and Junming, Z. (2010). Cloning and sequence analysis of the rDNA-ITS of trichuris sp. in black goat in hainan. Acta Agric. Boreali-Occidentalis Sin. 19, 27–30. doi:10.3724/SP.J.1142.2010.40491
Gupta, I., Ouhtit, A., Al-Ajmi, A., Rizvi, S. G. A., Tamimi, Y., Al-Riyami, M., et al. (2017). BRIP1 overexpression is correlated with clinical features and survival outcome of luminal breast cancer subtypes. Endocr. Connect. 7, 65–77. EC-17-0173. doi:10.1530/EC-17-0173
Hai, S. (2023). Role of leptin in the regulation of endometrial epithelial cell function and MAPK signaling pathway in shee. Xinjiang: Shihezi University.
Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., et al. (2004). The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261. doi:10.1093/nar/gkh036
Hu, J., Chen, Y. X., Wang, D., Qi, X., Li, T. G., Hao, J., et al. (2004). Developmental expression and function of Bmp4 in spermatogenesis and in maintaining epididymal integrity. Dev. Biol. 276, 158–171. doi:10.1016/j.ydbio.2004.08.034
Jiao, X., Sherman, B. T., Huang da, W., Stephens, R., Baseler, M. W., Lane, H. C., et al. (2012). DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 28, 1805–1806. doi:10.1093/bioinformatics/bts251
Lansdown, A., and Rees, D. A. (2012). The sympathetic nervous system in polycystic ovary syndrome: a novel therapeutic target? Clin. Endocrinol. 77, 791–801. doi:10.1111/cen.12003
Li, C., Wu, Y., Chen, B., Cai, Y., Guo, J., Leonard, A. S., et al. (2022). Markhor-derived introgression of a genomic region encompassing PAPSS2 confers high-altitude adaptability in Tibetan goats. Mol. Biol. Evol. 39, msac253. doi:10.1093/molbev/msac253
Li, C., Watanabe, G., Jin, W. Z., Furuta, , Suzuki, A. K., Kawaguchi, T., et al. (2005). Expression of nerve growth factor (NGF), and its receptors TrkA and p75 in the reproductive organs of the adult male rats. Zool. Sci. 22 (8), 933–937. doi:10.2108/zsj.22.933
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Li, X., Yang, J., Shen, M., Xie, X. L., Liu, G. J., Xu, Y. X., et al. (2020). Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 11, 2815. doi:10.1038/s41467-020-16485-1
Lv, X., Chen, W., Wang, S., Cao, X., Yuan, Z., Getachew, T., et al. (2023). Whole-genome resequencing of Dorper and Hu sheep to reveal selection signatures associated with important traits. Anim. Biotechnol. 34, 3016–3026. doi:10.1080/10495398.2022.2127409
Mtakeuchi, K. M., and Harada, M. (2011). Differentiation of benign and malignant uterine corpus tumors by using proton MR spectroscopy at 3 T:preliminary study. Int. J. Med. Radiology 21, 850–856. doi:10.1007/s00330-010-1974-5
Naicy, T., Venkatachalapathy, R. T., Aravindakshan, T. V., Radhika, G., Raghavan, K. C., Mini, M., et al. (2016). Nerve Growth Factor gene ovarian expression, polymorphism identification, and association with litter size in goats. Theriogenology 86, 2172–2178.e3. doi:10.1016/j.theriogenology.2016.07.011
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi:10.1038/ng1847
Rafnar, T., Gudbjartsson, D. F., Sulem, P., Jonasdottir, A., Sigurdsson, A., Jonasdottir, A., et al. (2011). Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 43, 1104–1107. doi:10.1038/ng.955
Sharma, S., Sharma, P. M., Mistry, D. S., Chang, R. J., Olefsky, J. M., Mellon, P. L., et al. (2011). PPARG regulates gonadotropin-releasing hormone signaling in LbetaT2 cells in vitro and pituitary gonadotroph function in vivo in mice. Biol. Reproduction 84, 466–475. doi:10.1095/biolreprod.110.088005
Su, Y., Gao, X., Wang, Y., Li, X., Zhang, W., and Zhao, J. (2023). Astragalus polysaccharide promotes sheep satellite cell differentiation by regulating miR-133a through the MAPK/ERK signaling pathway. Int. J. Biol. Macromol. Struct. Funct. Interact. 239, 124351. doi:10.1016/j.ijbiomac.2023.124351
Voutsadakis, I. A. (2020). Landscape of BRIP1 molecular lesions in gastrointestinal cancers from published genomic studies. World J. Gastroenterology 26, 1197–1207. doi:10.3748/wjg.v26.i11.1197
Wang, P., Jia, X., Lu, B., Huang, H., Liu, J., Liu, X., et al. (2023). Erianin suppresses constitutive activation of MAPK signaling pathway by inhibition of CRAF and MEK1/2. Signal Transduct. Target Ther. 8, 96–1888. doi:10.1038/s41392-023-01329-3
Wang, W., Zhang, X., Zhou, X., Zhang, Y., La, Y., Zhang, Y., et al. (2019). Deep genome resequencing reveals artificial and natural selection for visual deterioration, plateau adaptability and high prolificacy in Chinese domestic sheep. Front. Genet. 10, 300. doi:10.3389/fgene.2019.00300
Wang, Y., Liu, W., Du, J., Yu, Y., Liang, N., Liang, M., et al. (2015). NGF promotes mouse granulosa cell proliferation by inhibiting ESR2 mediated down-regulation of CDKN1A. Mol. and Cell. Endocrinol. 406, 68–77. doi:10.1016/j.mce.2015.02.024
Wiener, C. D., de Mello Ferreira, S., Pedrotti Moreira, F., Bittencourt, G., de Oliveira, J. F., Lopez Molina, M., et al. (2015). Serum levels of nerve growth factor (NGF) in patients with major depression disorder and suicide risk. J. Affect. Disord. 184, 245–248. doi:10.1016/j.jad.2015.05.067
Xu, Y., Li, E., Han, Y., Chen, L., and Xie, Z. (2010). Differential expression of mRNAs encoding BMP/Smad pathway molecules in antral follicles of high- and low-fecundity Hu sheep. Animal Reproduction Sci. 120, 47–55. doi:10.1016/j.anireprosci.2010.02.009
Yang, H., Lin, S., Lei, X., Yuan, C., Tian, Z., Yu, Y., et al. (2016). Identification and profiling of microRNAs from ovary of estrous Kazakh sheep induced by nutritional status in the anestrous season. Anim. Reprod. Sci. 175, 18–26. doi:10.1016/j.anireprosci.2016.10.004
Yang, H., Liu, X., Hu, G., Xie, Y., Lin, S., Zhao, Z., et al. (2018). Identification and analysis of microRNAs-mRNAs pairs associated with nutritional status in seasonal sheep. Biochem. Biophys. Res. Commun. 499, 321–327. doi:10.1016/j.bbrc.2018.03.155
Yang, P., Wang, G., Jiang, S., Chen, M., Zeng, J., Pang, Q., et al. (2023). Comparative analysis of genome-wide copy number variations between Tibetan sheep and White Suffolk sheep. Anim. Biotechnol. 34, 986–993. doi:10.1080/10495398.2021.2007937
Zhai, M., Xie, Y., Liang, H., Lei, X., and Zhao, Z. (2018). Comparative profiling of differentially expressed microRNAs in estrous ovaries of Kazakh sheep in different seasons. Gene 664, 181–191. doi:10.1016/j.gene.2018.04.025
Zhang, C. L., Zhang, J., Tuersuntuoheti, M., Zhou, W., Han, Z., Li, X., et al. (2023). Landscape genomics reveals adaptive divergence of indigenous sheep in different ecological environments of Xinjiang, China. Sci. Total Environ. 15, 166698. doi:10.1016/j.scitotenv.2023.166698
Zhang, D. Y., Zhang, X. X., Li, F. D., Yuan, L. F., Li, X. L., Zhang, Y. K., et al. (2022). Whole-genome resequencing reveals molecular imprints of anthropogenic and natural selection in wild and domesticated sheep. Zool. Res. 43 (5), 695–705. doi:10.24272/j.issn.2095-8137.2022.124
Zhang, Y., Xue, X., Liu, Y., Abied, A., Zhao, Q., Zhao, S., et al. (2021). Genome-wide comparative analyses reveal selection signatures underlying adaptation and production in Tibetan and Poll Dorset sheep. Sci. Rep. 11, 2466. doi:10.1038/s41598-021-81932-y
Zhangyuan, P., Shengdi, L., Qiuyue, L., Zhen, W., Zhengkui, Z., Ran, D., et al. (2018). Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. Gigascience 4. doi:10.1093/gigascience/giy019
Zheng, Q., Fu, X., Jiang, J., Zhang, N., Chen, H., Wang, W., et al. (2019). Umbilical cord mesenchymal stem cell transplantation prevents chemotherapy-induced ovarian failure via the NGF/TrkA pathway in rats. BioMed Res. Int. 2019, 6539294–6539299. doi:10.1155/2019/6539294
Zhong, T., Hou, D., Zhao, Q., Zhan, S., Wang, L., Li, L., et al. (2024). Comparative whole-genome resequencing to uncover selection signatures linked to litter size in Hu Sheep and five other breeds. BMC Genomics 25 (1), 480. doi:10.1186/s12864-024-10396-x
Zhu, M., Yang, Y., Yang, H., Zhao, Z., Zhang, H., Blair, H. T., et al. (2023). Whole-genome resequencing of the native sheep provides insights into the microevolution and identifies genes associated with reproduction traits. BMC Genomics 24, 392. doi:10.1186/s12864-023-09479-y
Zhu, M., Zhang, H., Yang, H., Zhao, Z., Blair, H. T., Liang, H., et al. (2022). Targeting GNAQ in hypothalamic nerve cells to regulate seasonal estrus in sheep. Theriogenology 181, 79–88. doi:10.1016/j.theriogenology.2022.01.005
Keywords: Dexin mutton and fine-wool sheep, litter size trait, selection signature, candidate genes, whole-genome resequencing
Citation: Zhu M, Li P, Wu W, Zheng W, Huang J, Tulafu H, Lin C, Tao W and Aladaer Q (2024) The genetic characterization of germplasm and identification of the litter size trait associated candidate genes in Dexin mutton and fine-wool sheep. Front. Genet. 15:1457634. doi: 10.3389/fgene.2024.1457634
Received: 01 July 2024; Accepted: 31 July 2024;
Published: 14 August 2024.
Edited by:
Ran Di, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Hua Yang, Xinjiang Academy of Agricultural and Reclamation Sciences (XAARS), ChinaWeimin Wang, Lanzhou University, China
Zhao Zongsheng, Shihezi University, China
Copyright © 2024 Zhu, Li, Wu, Zheng, Huang, Tulafu, Lin, Tao and Aladaer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiwei Wu, d3V3ZWl3ZWlncEBmb3htYWlsLmNvbQ==; Wenxin Zheng, end4MjAyMEAxMjYuY29t
†These authors have contributed equally to this work