 Kira Mascho1
Kira Mascho1 Svetlana A. Yatsenko2
Svetlana A. Yatsenko2 Cecilia W. Lo2Xinxiu Xu2Jennifer Johnson3Lindsey R. Helvaty4Stephanie Burns Wechsler5
Cecilia W. Lo2Xinxiu Xu2Jennifer Johnson3Lindsey R. Helvaty4Stephanie Burns Wechsler5 Chaya N. Murali6
Chaya N. Murali6 Seema R. Lalani6
Seema R. Lalani6 Vidu Garg7Jennelle C. Hodge4Kim L. McBride7,8
Vidu Garg7Jennelle C. Hodge4Kim L. McBride7,8 Stephanie M. Ware9
Stephanie M. Ware9 Jiuann-Huey Ivy Lin2,3,10*
Jiuann-Huey Ivy Lin2,3,10*- 1Division of Pediatric Critical Care, Rainbow Babies and Children’s Hospital, Cleveland, OH, United States
- 2University of Pittsburgh, Pittsburgh, PA, United States
- 3UPMC Children’s Hospital of Pittsburgh, Pittsburgh, PA, United States
- 4Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN, United States
- 5Division of Medical Genetics, Emory University School of Medicine, Atlanta, GA, United States
- 6Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, United States
- 7Center for Cardiovascular Research and Heart Center, Nationwide Children’s Hospital, Columbus, OH, United States
- 8Department of Medical Genetics, University of Calgary, Calgary, AB, Canada
- 9Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, United States
- 10Department of Critical Care Medicine, University of Pittsburgh, Pittsburgh, PA, United States
Introduction: 5p deletion syndrome, also called Cri-du-chat syndrome 5p is a rare genetic syndrome with reports up to 36% of patients are associated with congenital heart defects. We investigated the association between left outflow tract obstruction and Cri-du-chat syndrome.
Methods: A retrospective review of the abnormal microarray cases with congenital heart defects in Children’s Hospital of Pittsburgh and the Cytogenomics of Cardiovascular Malformations Consortium.
Results: A retrospective review at nine pediatric centers identified 4 patients with 5p deletions and left outflow tract obstruction (LVOTO). Three of these patients had additional copy number variants. We present data suggesting an association of LVOTO with 5p deletion with high mortality in the presence of additional copy number variants.
Conclusion: A rare combination of 5p deletion and left ventricular outflow obstruction was observed in the registry of copy number variants and congenital heart defects.
Introduction
5p deletion syndrome or 5p minus syndrome, also called Cri-du-chat syndrome, is a rare genetic syndrome that was first described with the distinctive, high-pitched, cat-like cry in 1963 by Lejeunne et al. (Lejeune et al., 1963). In Frech, Cri-du-chat translates to “cry of the cat.” The most recognizable phenotypes are the characteristic shrill cry like the mewing of a cat, distinctive facial features, growth and developmental delay (Nguyen et al., 2015). However, there is a wide spectrum of features in the individuals with 5p deletion syndrome that may be attributed to the differences in their genotypes in whether terminal or interstitial 5p deletions occur at different breakpoints or the deleted genes in the 5p region (Nguyen et al., 2015). Cri-du-chat syndrome is a contiguous gene syndrome (Gersh et al., 1995). Studies by Overhauser et al. (1994) determined the deletion of 5p15.2 was correlated with facial dysmorphism and developmental delays and the deletion of 5p 15.3 was related to the characteristic cat-like cry (Gersh et al., 1995). Additional analysis by Zhang et al., in 2005 further localized the region of the cat-like cry to 5p 15.31, facial dysmorphism to 5p15.2 to 5p15.31, and speech delay to 5p15.32 to 5p 15.33 (5). Congenital heart defects (CHDs) are reported in approximately 15%–36% of 5p deletion syndrome, typically representing simple heart defects such as ventricular septal defect (VSD), atrial septal defect (ASD), patent ductus arteriosus (PDA), tetralogy of Fallot (TOF) or aortic stenosis (AS) (Hills et al., 2006; Mainardi et al., 2006; Nevado et al., 2021). Congenital heart defects are one of the most common causes of 5p deletion death (Nguyen et al., 2015). Left-sided lesions including left ventricular outflow tract obstruction (LVOTO) malformations have rarely been reported in 5p deletion syndrome. Nevado et al. (2021) observed up to 25% of additional copy number variants (CNVs) were noted in 5p deletion syndrome cohorts. Here we report two detailed case presentations of 5p deletion and LVOTO anomalies. Two more patients with 5p deletion in the presence of other CNVs were identified through a large multi-institution collaboration.
Methods
A retrospective review was performed on abnormal chromosomal microarray (CMA) cases (abnormal CNVs were detected either by SNP microarray or array CGH) with CHDs from UPMC Children’s Hospital of Pittsburgh (CHP) and the Cytogenomics of Cardiovascular Malformations (CCVM) Consortium (Landis et al., 2023). The expression of related genes in the human heart tissues was obtained from the GTEx portal (https://gtexportal.org/home/singleCellOverviewPage). Enrichment of transcription factor targets was rendered using Metascape for genes within 5p deletions identified in a separate cohort of four complex CHD patients with LVOTO.
Case description
Patient 1 (009–0116)
A male was born at 36–5/7-week gestational age with prenatal concerns for aortic valve stenosis, aortic arch abnormality, and intrauterine growth restriction (IUGR). At birth, his weight was 2.078 kg (1.24 percentile), length was 44 cm (1.23 percentile), and head circumference was 31 cm (0.12 percentile). He demonstrated microcephaly, prominent supraorbital ridge, hypertelorism, down-slanting palpebral fissures, prominent nasal root, and retrognathia. Right single palmar crease, tapered digits, and fifth digit clinodactyly were noted in his upper extremities. Bilateral metatarsus adductus and bilateral sandal gap between his first and second toes were noted in his lower extremities. An echocardiogram showed a dysplastic aortic valve with aortic stenosis (Figures 1B, C) and arch hypoplasia. The patient underwent aortic valvuloplasty at 2 days of life which was complicated by a posterior left ventricle (LV) pseudoaneurysm (Figure 1D). After the procedure there continued to be a significant gradient across the aortic valve and aortic arch with the concern of arch obstruction, therefore, he underwent arch reconstruction and aortic valvuloplasty at 10 days of life. He was noted to have a bicuspid aortic valve oriented in the anterior and posterior axis. There was a fusion of the commissures anteriorly. The aortic valve looked dysplastic and thickened. In addition, there was a raphe at the level of the junction between the right and left coronary cusps. His postoperative course was complicated by left vocal cord hypomobility and feeding difficulties. Subsequently, he was diagnosed with obstructive jaundice and was treated with biliary drainage (Figures 1E, F) when he was 2 months old. The drain was removed about a month later with no further issues. An echocardiogram at 5 months of age demonstrated the evolution of LV pseudoaneurysm. Given his congenital heart anomalies and multiple dysmorphic features, chromosomal microarray testing was ordered, which revealed a large deletion at 5p13.33-p13.2, specifically arr [GRCh37] 5p15.33p13.2 (22149_36232545) x1 encompassing 36.3 Mb and at least 320 genes (Figure 1A), including the critical region associated with Cri-du-chat syndrome which was identified in most studies as the region between 5p15.3-5p15.2 (Gersh et al., 1995; Overhauser et al., 1994; Church et al., 1995; Wu et al., 2005) and a 5.5 Mb deletion of 5p15.33-p15.32 (arr 5p15.33p15.32 (22178–5539182) x1 in a 3-generation family with atypical Cri-du-chat syndrome (Elmakky et al., 2014). This deletion also contains a 5p15 terminal (Cri-du-chat syndrome) region with sufficient evidence for haploinsufficiency (Zhang et al., 2005; Mainardi et al., 2006). Although he had multiple complications during his initial neonatal hospitalization and delayed milestones, he has been gaining weight and thriving on his growth curve (Figures 1G, H). He is 5 years old and is the only documented survivor in this case series, with the largest chromosomal deletion in the 5p region and no concurrent reported other CNVs. Based on ACMG guidelines this deletion is classified as pathogenetic CNV.
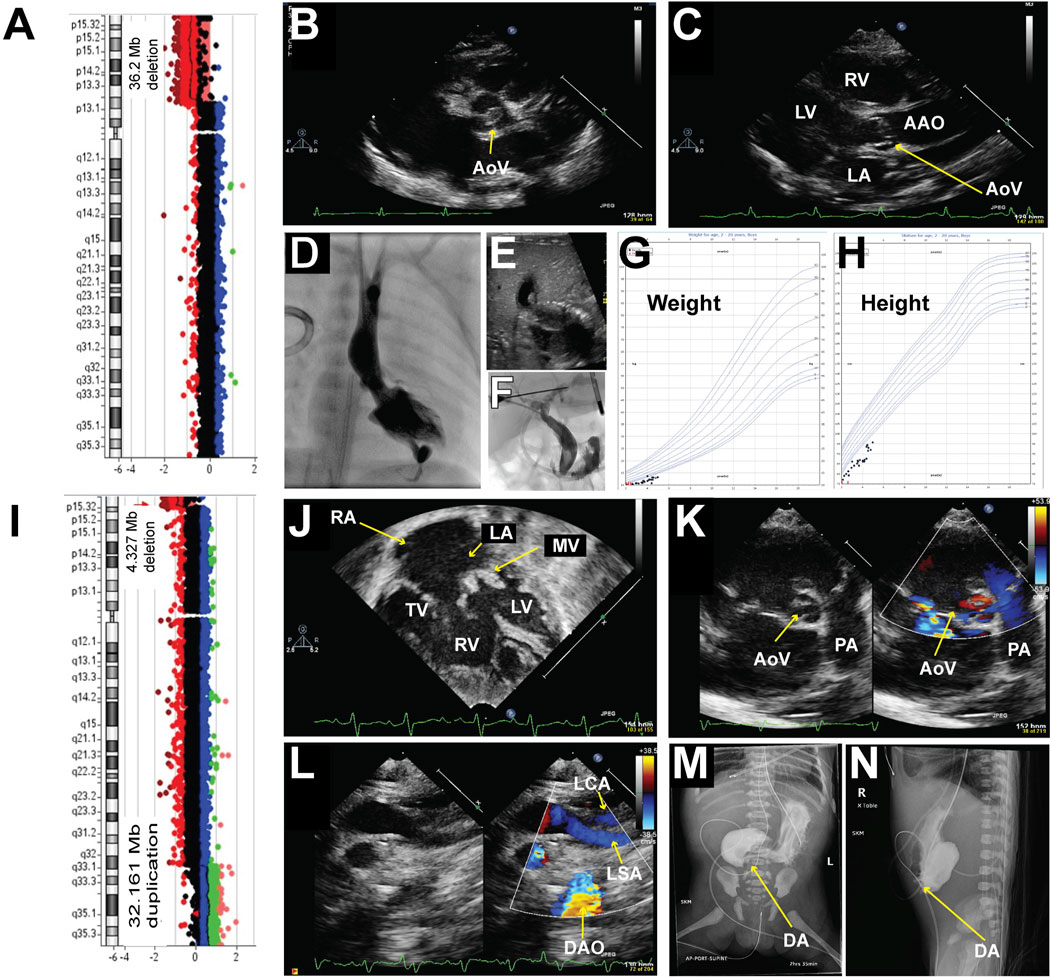
Figure 1. Chromosomal abnormalities and imaging from patients 1 and 2. Patient #1 (A–H) A Chromosomal microarray detected a 35.2 Mb deletion at 5p, specifically arr [GRCh37] 5p15.33p13.2 (22149_36232545) x1. (B–C): Representative echocardiogram imaging demonstrating the patient’s dysplastic aortic valve and post-stenotic dilatation of the ascending aorta. (D) Left ventriculogram demonstrating an apex-forming left ventricle with a hypoplastic aortic valve. The aortic valve annulus measured 4.46 mm in the anterior-posterior projection with mild post-stenotic dilation of the ascending aorta. A posterior pseudoaneurysm was noted with a small track extending apically through the myocardium. (E) Representative abdominal ultrasound image demonstrated dilation of the left intrahepatic biliary system as well as the intrahepatic common bile duct with gall sludge within the intrahepatic common bile duct. (F) An injection of contrast from a micro-puncture needle into a left hepatic duct filled a mildly dilated segmental biliary branch and communication with the dilated common hepatic and biliary ducts. (G) Growth chart of weight. (H) Growth chart of height. Patient #2 (I–N) (I) Chromosomal microarray detected a 4.327 Mb deletion at 5p, specifically arr [GRCh37] 5p15.33 (22149_4349495) x1, and a 32.161 Mb duplication at 5q, specifically arr [GRCh37] 5q32q35.3 (148535314_180696806) x3. (J–L): Representative echocardiogram imaging demonstrating a hypoplastic left ventricle, dysplastic and thickened mitral valve, large secundum atrial septal defect (ASD), large ventricular septal defect (VSD), and a bicuspid aortic valve with thickened leaflets. Color flow Doppler in image (L) showed a lack of continuity between the left subclavian artery and the descending aorta consistent with interrupted aortic arch type (A). (M–N): Upper GI with small bowel follow-through demonstrating opacification of the stomach and blind-ending proximal duodenum is consistent with duodenal atresia. AAO = Ascending Aorta, DAO = Descending Aorta, LA = Left Atrium, LV = Left Ventricle, LSA = Left Subclavian Artery, MV = Mitral Valve, PA = Pulmonary Artery, RA = Right Atrium, RV = Right Ventricle, TV = Tricuspid Valve.
Patient 2 (009–0119)
A 37–4/7-week gestational age female was prenatally diagnosed with Cri-du-chat syndrome, had a 4.327 Mb deletion at 5p, specifically arr [GRCh37] 5p15.33 (22149_4349495) x1, and a 32.161 Mb duplication on 5q, specifically arr [GRCh37] 5q32q35.3 (148535314_180696806) x3 (Figure 1I). This deletion also contains 5p15 terminal (Cri- du-chat syndrome) region with sufficient evidence for haploinsufficiency (Zhang et al., 2005; Mainardi et al., 2006). The duplication contains 5q35 recurrent (Sotos syndrome) region (includes NSD1) with sufficient evidence for triplosensitivity, which is associated with the Sotos phenotype (Rosenfeld et al., 2013). Prenatal ultrasound at around 20 weeks of gestational age demonstrated duodenal atresia and a decrease in LV function. She was born to a 26-year-old woman (gravida 7, para 1) with a history of six spontaneous abortions during prior pregnancies and a pericentric inversion- 46,XX,inv (Zhang et al., 2005) (p15.3q32). At birth, her weight was 1.665 kg (<0.01 percentile), length was 42.5 cm (<0.01 percentile), and head circumference was 30 cm (0.01 percentile). She was microcephalic, and her birth height and weight were below average. She was born without spontaneous respiratory effort, necessitating an emergent intubation in the delivery room. Her initial heart rate at birth was less than 60 bpm without spontaneous movement. Her Apgar scores were 1 at 1 min, 3 at 5 min, and 3 at 10 min. Multiple facial dysmorphisms, including hypertelorism, low-set ears, and micrognathia, were noted after delivery. An echocardiogram was obtained due to severe metabolic acidosis and a murmur which demonstrated a hypoplastic LV, severe mitral valve dysplasia, a large VSD, a large ASD secundum (Figure 1J), a dysplastic aortic valve (Figure 1K) with an interrupted aortic arch type A (Figure 1L) and a large PDA. The upper gastrointestinal series confirmed the diagnosis of duodenal atresia (Figures 1M, N). She required multiple inotropic agents with a maximum Wernovsky inotrope score of 20 after birth. In conjunction with her comorbidities and ongoing critical illness, surgical palliation was not offered to her. She passed away on the day of life 6. Based on ACMG guidelines, deletion and duplication are classified as pathogenetic CNV(14). Also, this result suggests a parental balance inversion origin, genetic counseling is recommended. We found that the patient’s mom carries a history of six spontaneous abortions during prior pregnancies and a pericentric inversion- 46, XX, inv (Zhang et al., 2005) (p15.3q32).
Additional patients
We identified five more patients with 5p deletion (Figure 2; Table 1; Supplementary Table S1) and LVOTO malformation from the CCVM Consortium (Hinton et al., 2015). This multi-site, cross-disciplinary collaboration has created a large database registry of patients with CHD who have had non-normal clinical CMA results. A total of 1,363 patients from nine pediatric centers across the United States were included in the study (Landis et al., 2023). There are 229 individuals with LVOTO in the CCVM Consortium. We excluded three patients who had small 5p deletion that is not consistent with the typical 5p deletion syndrome, with the size of deletion of 5p varying from 0.01 to 0.27 MB, which is less than the reported deletion size from 5 to 40 MB in Cri-du-chat syndrome and no genes in the deleted region that relate to heart development as well as not consistent with the typical 5p deletion syndrome (Simmons et al., 1995; Cerruti Mainardi, 2006) and these small deletions were not consistent with pathological CNV according to ACMG guidelines (Rosenfeld et al., 2013). These 3 patients also have additional CNVs that are known to cause CHDs (Supplementary Table S1). The major birth defects of these additional 2 patients we included are summarized in Table 1. In total, there are four patients with a deletion in regions associated with Cri-du-chat syndrome (5p15.2-5p15.3, patients 1–4) (Zhang et al., 2005; Mainardi et al., 2006) in this report, of whom three patients had additional CNVs. Patient 2 (009–0119) had a 5q duplication in the region containing HAND1 and Nkx2-5 (Supplementary Table S2). Patient 3 (001–0165) had a 7.1 Mb deletion arr [hg19] 5p15.33p15.31966648_7175604) x1, and an 8p duplication in the region containing MYOM2. Interstitial duplication of 8p23 was noted to be associated with CHDs but 8p trisomy has not been reported to be associated with LVOTO (Gug et al., 2020). Patient 3 has a complex CHD with a double outlet right ventricle (DORV) and both left and right ventricular outflow tract obstruction. Patient 4 (004–0090) had an 18.9 Mb deletion arr [GRCh37] 5p15.33p14.3 (22149_18927458) x1 and arr [GRCh37]5p14.3p11 (19049019_46115173) x3, the 18.0 Mb duplication and a complex rearrangement of chromosome 5. Of these 4 patients, 2 died before 12 months of age and one did not have updated clinical information.
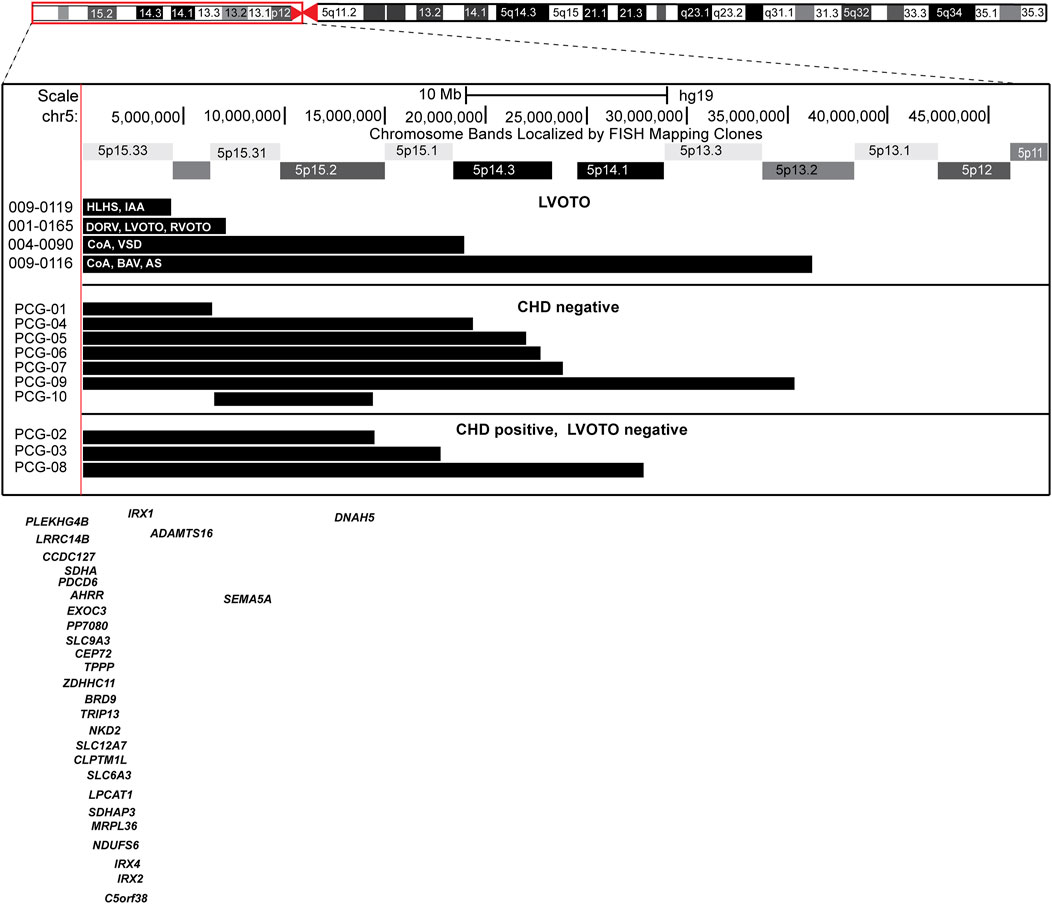
Figure 2. Ideogram of chromosome 5 with the expansion of the 5p region and location of the individual 5p deletions in each patient.
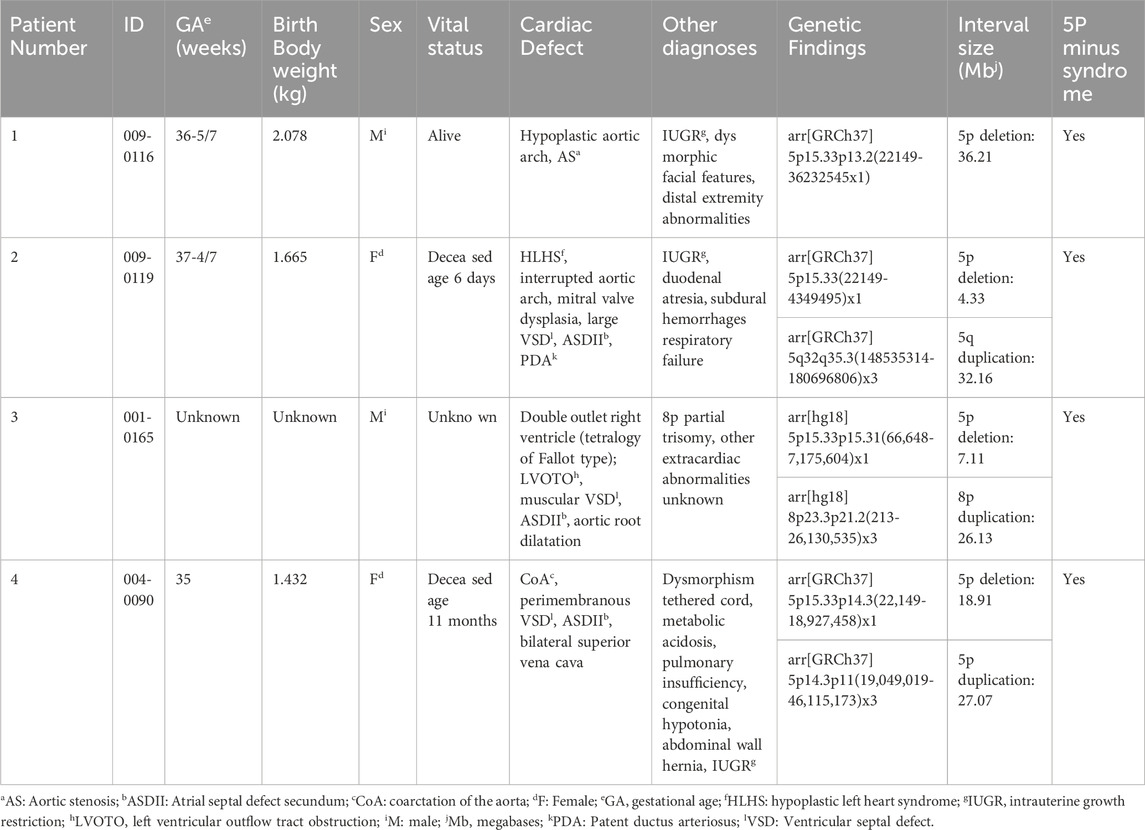
Table 1. Patients characteristics.
In summary, the 4 patients’ 5p deletions are classified as pathogenetic CNV based on ACMG guidelines (Riggs et al., 2020) with an incidence of 1.3% in this cohort (Landis et al., 2023). We reported the association of LVOTO and 5p deletion and observed an association of LVOTO and 5p deletion with high mortality in the presence of additional copy number variants. This also indicates an expansion of the cardiac abnormalities’ spectrum in the 5p deletion patients.
No common critical region for LVOTO defects in 5p deletion patients
To identify the specific region associated with LVOTO in patients with 5p deletion, we compared 10 patients with 5p deletion at CHP to the four with LVOTO. Of these 10, seven were without CHD and 3 had non-LVOTO CHD (Figure 2). There is not a common region associated with 5p deletion and LVOTO in this case series. This could be due to incomplete penetrance or the etiology of LVOTO may be concurrent copy abnormalities other than 5p deletion.
Discussion
Hills et al. reviewed a database of 98,000 congenital heart disease patients and identified twenty-one with Cri-du-chat syndrome (Hills et al., 2006). When characterized by the most hemodynamically significant lesion, twenty-one patients either had a VSD, PDA, Tetralogy of Fallot, or right ventricular outflow tract obstruction (Hills et al., 2006). The patients described in our case series all had septal defects, but uniquely all had more hemodynamically significant congenital anomalies causing LVOTO. 5p deletion syndrome was reported to be associated with CHDs in 18%–36% of patients including AS (7). When characterized by the most hemodynamically significant lesion, these patients either had a VSD, PDA, or TOF (7). The patients described in our case series had LVOTO malformation, three patients (patients 2–4) have additional CNVs. This could be due to incomplete penetrance. In addition, two of four patients pass away before 12 months of age with complex rearrangements at 5p/5q, complex critical congenital heart defects and intrauterine growth retardation, and low birth weight. Birth weight less than 1.5 kg with a critical congenital heart defect was known to be less likely to survive hospital discharge (Kim et al., 2021). Early observations of 5p deletion syndrome reported a 9.7% (32 of 341 individuals) mortality in childhood in 1978 with 90% of deaths within the first year (Niebuhr, 1978). The mortality rate decreased to 6.4% in the report in 2006 with 64% of deaths in the first year of life (Mainardi et al., 2006). Mainardi et al. also observed a higher mortality rate with unbalanced translocations that include a 5p deletion than the individuals with terminal deletions (18.5% vs. 4.8%) (Mainardi et al., 2006).
The etiology of CHD is multifactorial, and both epidemiologic studies and patient cohorts with chromosomal microarray testing have reported approximately 11%–18% of patients with CHD also have an identifiable syndromic genetic diagnosis (Helm et al., 2021), while 15%–21% of subjects with isolated CHD had LVOTO (Helm et al., 2021; Hoang et al., 2018). Genes known to be involved in cardiac expression are located on 5p (Supplementary Tables S3 and S4), including DNAH5 (5p15.33, in patients 2, 4), NDUFS6 (5p15.33, in patients 1–4), IRX4 (5p15.33, in patient 1–4), and ADAMTS16 (5p15.32, in patient 2–4) (Pervolaraki et al., 2018). ADAMTS16 p.H357Q variant is reported to be an inheritable human bicuspid aortic valve-related gene variant resulting from the fibronectin/ focal adhesion kinase (FAK) signal-mediated over-proliferation with extracellular matrix remodeling interruption (Lin et al., 2024). Patients 2 and 4 had a deletion in the DNAH5 gene, a disease-causing variant related to laterality defects resulting from immotile cilia that lack dynein arms (Nöthe-Menchen et al., 2019). Patients 1–4 had a deletion in the region containing both SDHA and NDUFS6, complexes related to mitochondrial oxidative phosphorylation, which are highly expressed in the left ventricle (Pervolaraki et al., 2018). Intrinsic mitochondrial defects contribute to the different prognoses of single ventricle CHD, especially for HLHS (Xu et al., 2022). Patients 1–4 also had a deletion in the region containing IRX4, a homeobox gene with an expression in the ventricular myocardium (Bruneau et al., 2000). We observed the enrichment of five transcription factor targets (Subramanian et al., 2005) that are associated with heart development or defects (Supplementary Table S5) from the analysis of the affected genes within the 5p deletions in our patients. e.g., DLX6 (Distal-Less Homeobox 6) directly regulates Basic helix-loop-helix transcription (bHLH) factor HAND2, which plays a crucial role for the development of the cardiac outflow tract (Holler et al., 2010). FOXJ2, a member of the Fork Head transcription factors family. Previous reports showed that there is a right/left heart difference in expression for FOXJ2 (Philip-Couderc et al., 2008). FOXJ2 also regulates Connexin-43 and E-Cadherin which may be associated with hypertrophic heart (Martin-de-Lara and Sanchez-Aparicio, 2008). These clues may explain how 5p deletion contributes to LVOTO.
The cases in this report suggest that the previous thought that 5p minus patients only have simple cardiac defects like ASD, VSD, and PDA, is not the case. There is a case report of a child with 5p minus and 20q duplication with Ebstein anomaly; the authors make a similar argument that this rare, complex congenital heart phenotype such as Ebstein has not ever been reported with 5p minus, though the 20q duplication may be part of the phenotype, too (Olivella et al., 2020). However, 5p deletion in the presence of other CNVs that are related to cardiac development may be associated with more complex hemodynamic significant cardiac defects. The correlation with LVOTO will be useful for clinical prognosis prediction on 5p deletion patients, as congenital heart defects are one of the most common causes of 5p deletion death (Nguyen et al., 2015). Significant hemodynamic complex congenital heart defects such as LVOTO increase the risk of mortality and morbidity. Further research is needed to elucidate an association between these genes and specific CHDs. In addition, further study of gene enhancers that may regulate gene expression from a distance or epigenetic regulation may provide additional insight into the critical regions for left heart formation and function on the short arm of chromosome 5.
Conclusion
We present data suggesting an association of LVOTO and 5p deletion with high mortality in the presence of additional CNVs. Discovering new genotype-phenotype correlations for rare CHDs, including expanding the associations with known syndromes, is an important ongoing process requiring large patient cohorts with deep phenotyping such as that collected by the CCVM Consortium.
Learning objectives
1. An association is suggested between LVOTO and 5p deletion, with high mortality occurring in patients with 5p deletion and other CNVs.
2. Expanding the phenotypic spectrum of known syndromes to include rare CHD requires multi-institution collaboration and expert-detailed heart phenotype evaluation.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by University of Pittsburgh/ Indiana University Institutional review Boards. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was obtained from the participants or the participant’s legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
KM: Writing–original draft, Writing–review and editing. SY: Data curation, Methodology, Validation, Writing–review and editing. CL: Conceptualization, Supervision, Writing–review and editing. XX: Data curation, Formal Analysis, Methodology, Writing–review and editing. JJ: Methodology, Visualization, Writing–review and editing. LH: Project administration, Writing–review and editing. SB: Data curation, Writing–review and editing. CM: Data curation, Validation, Writing–review and editing. SL: Data curation, Validation, Writing–review and editing. VG: Data curation, Validation, Writing–review and editing. JH: Data curation, Validation, Writing–review and editing. KLM: Data curation, Validation, Writing–review and editing. SW: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Resources, Supervision, Validation, Writing–review and editing. J-HI: Conceptualization, Data curation, Formal Analysis, Methodology, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Transformational Project Award (19TPA34850054) from American Heart Association (SMW).
Acknowledgments
The authors thank Angelo Arrigo, Olivia Phillips, Peizhao Zhang, and Aaron Tien for their assistance in preparing this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1451746/full#supplementary-material
Abbreviations
ASD, atrial septal defect; BAV, bicuspid aortic valve; CCVM, Cytogenomics of Cardiovascular Malformations Consortium; CHD, congenital heart defect; CMA, chromosomal microarray; CNV, copy number variants; CoA, coarctation of the aorta; HLHS, hypoplastic left heart syndrome; LVOTO, left outflow tract obstruction; VSD, ventricular septal defect.
References
Bruneau, B. G., Bao, Z.-Z., Tanaka, M., Schott, J.-J., Izumo, S., Cepko, C. L., et al. (2000). Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by nkx2-5 and dHand. Dev. Biol. 217 (2), 266–277. doi:10.1006/dbio.1999.9548
Cerruti Mainardi, P. (2006). Cri du Chat syndrome. Orphanet J. Rare Dis. 1, 33. doi:10.1186/1750-1172-1-33
Church, D. M., Bengtsson, U., Nielsen, K. V., Wasmuth, J. J., and Niebuhr, E. (1995). Molecular definition of deletions of different segments of distal 5p that result in distinct phenotypic features. Am. J. Hum. Genet. 56 (5), 1162–1172.
Elmakky, A., Carli, D., Lugli, L., Torelli, P., Guidi, B., Falcinelli, C., et al. (2014). A three-generation family with terminal microdeletion involving 5p15.33-32 due to a whole-arm 5;15 chromosomal translocation with a steady phenotype of atypical cri du chat syndrome. Eur. J. Med. Genet. 57 (4), 145–150. doi:10.1016/j.ejmg.2014.02.005
Gersh, M., Goodart, S. A., Pasztor, L. M., Harris, D. J., Weiss, L., and Overhauser, J. (1995). Evidence for a distinct region causing a cat-like cry in patients with 5p deletions. Am. J. Hum. Genet. 56 (6), 1404–1410.
Gug, C., Stoicanescu, D., Mozos, I., Nussbaum, L., Cevei, M., Stambouli, D., et al. (2020). De novo 8p21.3--> p23.3 duplication with t(4;8)(q35;p21.3) translocation associated with mental retardation, autism spectrum disorder, and congenital heart defects: case report with literature review. Front. Pediatr. 8, 375. doi:10.3389/fped.2020.00375
Helm, B. M., Landis, B. J., and Ware, S. M. (2021). Genetic evaluation of inpatient neonatal and infantile congenital heart defects: new findings and review of the literature. Genes (Basel) 12 (8), 1244. doi:10.3390/genes12081244
Hills, C., Moller, J. H., Finkelstein, M., Lohr, J., and Schimmenti, L. (2006). Cri du chat syndrome and congenital heart disease: a review of previously reported cases and presentation of an additional 21 cases from the Pediatric Cardiac Care Consortium. Pediatrics 117 (5), e924–e927. doi:10.1542/peds.2005-1012
Hinton, R. B., McBride, K. L., Bleyl, S. B., Bowles, N. E., Border, W. L., Garg, V., et al. (2015). Rationale for the Cytogenomics of cardiovascular malformations Consortium: a phenotype intensive registry based approach. J. Cardiovasc Dev. Dis. 2 (2), 76–92. doi:10.3390/jcdd2020076
Hoang, T. T. G. E., Roberts, A. E., Chung, W. K., Kline, J. K., Deanfield, J. E., Giardini, A., et al. (2018). The congenital heart disease genetic network study: cohort description. PLoS One 13 (1), e0191319. doi:10.1371/journal.pone.0191319
Holler, K. L., Hendershot, T. J., Troy, S. E., Vincentz, J. W., Firulli, A. B., and Howard, M. J. (2010). Targeted deletion of Hand2 in cardiac neural crest-derived cells influences cardiac gene expression and outflow tract development. Dev. Biol. 341 (1), 291–304. doi:10.1016/j.ydbio.2010.02.001
Kim, M., Okunowo, O., Ades, A. M., Fuller, S., Rintoul, N. E., and Naim, M. Y. (2021). Single-center comparison of outcomes following cardiac surgery in low birth weight and standard birth weight neonates. J. Pediatr. 238, 161–167.e1. doi:10.1016/j.jpeds.2021.06.059
Landis, B. J., Helvaty, L. R., Geddes, G. C., Lin, J. I., Yatsenko, S. A., Lo, C. W., et al. (2023). A multicenter analysis of abnormal chromosomal microarray findings in congenital heart disease. J. Am. Heart Assoc. 12, e029340. doi:10.1161/JAHA.123.029340
Lejeune, J., Lafourcade, J., Berger, R., Vialatte, J., Boeswillwald, M., Seringe, P., et al. (1963). 3 CASES OF PARTIAL DELETION OF THE SHORT ARM OF A 5 CHROMOSOME. C R. Hebd. Seances Acad. Sci. 257, 3098–3102.
Lin, Y., Yang, Q., Lin, X., Liu, X., Qian, Y., Xu, D., et al. (2024). Extracellular matrix disorganization caused by ADAMTS16 deficiency leads to bicuspid aortic valve with raphe formation. Circulation 149 (8), 605–626. doi:10.1161/CIRCULATIONAHA.123.065458
Mainardi, P. C., Pastore, G., Castronovo, C., Godi, M., Guala, A., Tamiazzo, S., et al. (2006). The natural history of Cri du Chat Syndrome. A report from the Italian Register. Eur. J. Med. Genet. 49 (5), 363–383. doi:10.1016/j.ejmg.2005.12.004
Martin-de-Lara, F., and Sanchez-Aparicio, P. (2008). Biological effects of FoxJ2 over-expression. Transgenic Res. 17 (6), 1131–1141. doi:10.1007/s11248-008-9214-3
Nevado, J., Bel-Fenellós, C., Sandoval-Talamantes, A. K., Hernández, A., Biencinto-López, C., Martínez-Fernández, M. L., et al. (2021). Deep phenotyping and genetic characterization of a cohort of 70 individuals with 5p minus syndrome. Front. Genet. 12, 645595. doi:10.3389/fgene.2021.645595
Nguyen, J. M., Qualmann, K. J., Okashah, R., Reilly, A., Alexeyev, M. F., and Campbell, D. J. (2015). 5p deletions: current knowledge and future directions. Am. J. Med. Genet. Part C Seminars Med. Genet. 169 (3), 224–238. doi:10.1002/ajmg.c.31444
Niebuhr, E. (1978). The Cri du Chat syndrome: epidemiology, cytogenetics, and clinical features. Hum. Genet. 44 (3), 227–275. doi:10.1007/BF00394291
Nöthe-Menchen, T., Wallmeier, J., Pennekamp, P., Höben, I. M., Olbrich, H., Loges, N. T., et al. (2019). Randomization of left-right asymmetry and congenital heart defects. Circulation Genomic Precis. Med. 12 (11), e002686. doi:10.1161/circgen.119.002686
Olivella, A., Manotas, H., Payan-Gomez, C., and Pineros, J. G. (2020). Ebstein anomaly associated with cri du chat (cat's cry) syndrome and 20q duplication. BMJ Case Rep. 13 (6), e233766. doi:10.1136/bcr-2019-233766
Overhauser, J., Huang, X., Gersh, M., Wilson, W., McMahon, J., Bengtsson, U., et al. (1994). Molecular and phenotypic mapping of the short arm of chromosome 5: sublocalization of the critical region for the cri-du-chat syndrome. Hum. Mol. Genet. 3 (2), 247–252. doi:10.1093/hmg/3.2.247
Pervolaraki, E., Dachtler, J., Anderson, R. A., and Holden, A. V. (2018). The developmental transcriptome of the human heart. Sci. Rep. 8 (1), 15362. doi:10.1038/s41598-018-33837-6
Philip-Couderc, P., Tavares, N. I., Roatti, A., Lerch, R., Montessuit, C., and Baertschi, A. J. (2008). Forkhead transcription factors coordinate expression of myocardial KATP channel subunits and energy metabolism. Circ. Res. 102 (2), e20–e35. doi:10.1161/CIRCRESAHA.107.166744
Riggs, E. R., Andersen, E. F., Cherry, A. M., Kantarci, S., Kearney, H., Patel, A., et al. (2020). Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22 (2), 245–257. doi:10.1038/s41436-019-0686-8
Rosenfeld, J. A., Kim, K. H., Angle, B., Troxell, R., Gorski, J. L., Westemeyer, M., et al. (2013). Further evidence of contrasting phenotypes caused by reciprocal deletions and duplications: duplication of NSD1 causes growth retardation and microcephaly. Mol. Syndromol. 3 (6), 247–254. doi:10.1159/000345578
Simmons, A. D., Goodart, S. A., Gallardo, T. D., Overhauser, J., and Lovett, M. (1995). Five novel genes from the cri-du-chat critical region isolated by direct selection. Hum. Mol. Genet. 4 (2), 295–302. doi:10.1093/hmg/4.2.295
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102 (43), 15545–15550. doi:10.1073/pnas.0506580102
Wu, Q., Niebuhr, E., Yang, H., and Hansen, L. (2005). Determination of the ‘critical region’ for cat-like cry of Cri-du-chat syndrome and analysis of candidate genes by quantitative PCR. Eur. J. Hum. Genet. 13 (4), 475–485. doi:10.1038/sj.ejhg.5201345
Xu, X., Jin, K., Bais, A. S., Zhu, W., Yagi, H., Feinstein, T. N., et al. (2022). Uncompensated mitochondrial oxidative stress underlies heart failure in an iPSC-derived model of congenital heart disease. Cell Stem Cell. 29 (5), 840–855.e7. doi:10.1016/j.stem.2022.03.003
Keywords: 5p deletion, congenital heart defect, genetic disorder, left ventricular outflow tract obstruction, copy number variant
Citation: Mascho K, Yatsenko SA, Lo CW, Xu X, Johnson J, Helvaty LR, Burns Wechsler S, Murali CN, Lalani SR, Garg V, Hodge JC, McBride KL, Ware SM and Lin J-HI (2024) Case Report: An association of left ventricular outflow tract obstruction with 5p deletions. Front. Genet. 15:1451746. doi: 10.3389/fgene.2024.1451746
Received: 19 June 2024; Accepted: 25 September 2024;
Published: 18 October 2024.
Edited by:
Elaine T. Lim, University of Massachusetts Medical School, United StatesReviewed by:
Julian Nevado, Fundación para la Investigación Biomédica del Hospital Universitario La Paz (FIBHULP), SpainKhanh Tran, University of Massachusetts Medical School, United States
Copyright © 2024 Mascho, Yatsenko, Lo, Xu, Johnson, Helvaty, Burns Wechsler, Murali, Lalani, Garg, Hodge, McBride, Ware and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiuann-Huey Ivy Lin, aml1YW5uaHVleS5saW41QHVwbWMuZWR1