 Shuang Li
Shuang Li Li Liu1†
Li Liu1† Zulfiqar Ahmed
Zulfiqar Ahmed Fuwen Wang
Fuwen Wang Chuzhao Lei
Chuzhao Lei
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 25 July 2024
Sec. Livestock Genomics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1435793
Introduction: To enhance the beef cattle industry, Heilongjiang Province has developed a new Crossbred beef cattle variety through crossbreeding with exotic commercial breeds. This new variety exhibits relatively excellent meat quality, and efficient reproductive performance, catering to market demands.
Method: This study employed whole genome resequencing technology to analyze the genetic pedigree and diversity of 19 Heilongjiang Crossbred beef cattle, alongside 59 published genomes from East Asian, Eurasian, and European taurine cattle as controls. In addition, genes related to production traits were also searched by identifying Runs of Homozygosity (ROH) islands and important fragments from ancestors.
Results: A total of 14,427,729 biallelic SNPs were discovered, with the majority located in intergenic and intron regions and a small percentage in exon regions, impacting protein function. Population genetic analyses including Principal Component Analysis (PCA), Neighbor-Joining (NJ) tree, and ADMIXTURE identified Angus, Holstein, and Mishima as the main ancestors of Crossbred beef cattle. In genetic diversity analysis, nucleotide diversity, linkage disequilibrium, and inbreeding coefficient analysis reveal that the genetic diversity of Crossbred beef cattle is at a moderate level, and a higher inbreeding coefficient indicates the need for careful breeding management. In addition, some genes related to economic traits are identified through the identification of Runs of Homozygosity (ROH) islands and important fragments from ancestors.
Conclusion: This comprehensive genomic characterization supports the targeted improvement of economically important traits in Crossbred beef cattle, facilitating advanced breeding strategies.
Domesticated cattle, primarily categorized into Bos taurus taurus and Bos taurus indicus, serve as fundamental livestock for agriculture and transportation. These animals are pivotal to agricultural societies by fulfilling social demands for meat and milk production, food supply, and crop production by enhancing soil fertility through manure (Decker et al., 2014). The distribution of B. taurus taurus and B. taurus indicus is closely related to climatic conditions. Bos taurus taurus adapt to temperate and cold climates and are mainly distributed in the Northern Hemisphere (Buggiotti et al., 2021; Xia et al., 2023). B. taurus indicus have strong heat resistance and immunity, adapt to tropical and subtropical climates, and are mainly distributed around the equator and the Southern Hemisphere (Utsunomiya et al., 2019; Li et al., 2023). Recently, the research reports that domestic cattle worldwide can be categorized into five distinctly different populations by whole genome sequencing analysis: European taurine, Eurasian taurine, East Asian taurine, Chinese indicine, and Indian indicine (Chen et al., 2018). In addition, African taurine and African indicine are also very important domestic cattle populations (Pitt et al., 2019). At the same time, many studies have analyzed the genomic variation characteristics of local cattle in China through whole genome sequencing technology, such as the cold tolerance of Yanbian cattle (Shen et al., 2020), the faster growth rate and higher feed conversion rate of Jiaxian Red Cattle (Xia et al., 2021), the heat resistance and better immunity of Dianzhong cattle (Zhang et al., 2021), the heat resistance and higher meat production performance of Chaling cattle (Li et al., 2023), the higher immune performance and fine meat quality of Sanjiang cattle (Lyu et al., 2023). This provides a scientific basis for the scientific protection and genetic improvement of the genetic resources of local cattle breeds in China.
Heilongjiang Province is a major producer of dairy cattle and beef cattle (Jia et al., 2022). To promote the genetic improvement of beef cattle and accelerate the process of beef cattle breeding, based on the original varieties of cattle, pure breeds and commercial varieties such as Angus and Holstein were introduced from abroad, and new Heilongjiang Crossbred cattle varieties were formed through hybridization (Bu et al., 2021; Nie and Han, 2023). However, there are shortcomings in the development and utilization of cattle variety resources in Heilongjiang Province. Due to the excessive dependence on imported breeds and backward breeding technology, the province has no local characteristic breeds (Li et al., 2021). Therefore, it is urgent to combine advanced technologies such as genome and transcriptome with traditional breeding ideas to further promote the development of the beef cattle industry.
The Heilongjiang Crossbred cattle in this study is a breed developed to improve the beef production and quality in Heilongjiang Province. The original beef cattle with poor productivity in Heilongjiang Province were crossed with Angus to improve beef production, then crossed with Holstein to improve the reproductive ability, and finally crossed with other commercial cattle to improve beef quality. The carcass weight of Heilongjiang Crossbred cattle is more than 300 kg, the slaughter rate can reach about 55%, and the meat production is significantly improved compared with that before. At the same time, the calving age of Heilongjiang Crossbred cattle is before 2.5 years old, and the conception rate is about 80%, which has a high reproductive ability. The production performance of Heilongjiang Crossbred cattle meets the market requirements, but there is a lack of specific research on its genetic resources. To ascertain the different lineage origins and identify genes associated with economically significant traits in Heilongjiang Crossbred cattle, we conducted whole-genome resequencing on 19 individuals using the B. taurus reference genome assembly (ARS-UCD1.2). This process involved identifying SNPs and comparing these SNPs with those from various commercial breeds globally to assess the genetic diversity and clustering within Heilongjiang Crossbred cattle. Subsequently, we pinpointed Runs of Homozygosity (ROH) islands within the Heilongjiang Crossbred cattle genomes and identified genes implicated in meat quality and reproductive efficiency located within high-frequency ROH segments. Additionally, introgression analysis was performed to detect genes associated with valuable economic traits in the introgressed fragments of the Heilongjiang Crossbred cattle genomes. These findings provide a scientific foundation for the genetic enhancement of hybrid beef cattle, facilitating improvements in traits crucial for economic viability and productivity.
Ear tissue samples of Heilongjiang Crossbred cattle (n = 19) were collected by random sampling from Huanan County, Jiamusi City, Heilongjiang Province of China. All samples are from designated farm for beef cattle molecular breeding research and genetic resource mining project. The collected samples adhered to the breed characteristics of Heilongjiang Crossbred cattle, and according to the pedigree information of the farm, there was no kinship between individuals. The Genomic DNA of the ear tissue samples was extracted using a standard phenol/chloroform-based protocol. The DNA library was constructed for each sample (500 bp insert size). The Sequencing was done via Illumina NovaSeq 6000 with a 2 × 150 bp model at Novogene Bioinformatics Institute, Beijing, China, and 150 bp paired-end sequence data were generated (the BioProject accession number PRJNA1101862). Furthermore, we obtained sequence data from 59 cattle, including East Asian taurine cattle (15 Hanwoo and 8 Mishima cattle), Eurasian taurine cattle (6 Gelbvieh and 8 Simmental cattle), and European taurine cattle (14 Angus and 8 Holstein cattle) available publicly (Supplementary Table S2). In total, 78 whole genomes of cattle were used for the subsequent analysis.
The clean reads were mapped onto the B. taurus reference genome assembly ARS-UCD1.2 using BWA-MEM (0.7.13-r1126) with default parameters (Li and Durbin, 2009). After mapping, the SNPs were detected by using Samtools (Li et al., 2009), Picard tools (http://broadinstitute.github.io/picard), and Genome Analysis Toolkit (GATK, version 3.6-0-g89b7209). The raw SNPs were called using the “HaplotypeCaller,” “GenotypeGVCFs” and “SelectVariants” of GATK. After SNP calling, we used the “VariantFiltration” to discard sequencing and alignment artifacts from the SNPs with the parameters “QD < 2.0, FS > 60.0, MQ < 40.0, MQRankSum <−12.5, ReadPosRankSum <−8.0 and SOR > 3.0” and mean sequencing depth of variants (all individuals) “<1/3× and >3×.” By using the ANNOVAR (Wang et al., 2010), the SNPs were annotated based on the latest reference assembly (ARS-UCD1.2).
After pruning in PLINK with the parameter (--maf 0.01 --indep-pair-wise 50 5 0.2), a set of SNPs was generated for the following analyses. Genetic distance was constructed by PLINK (--distance-matrix) and an unrooted neighbor-joining (NJ) tree was constructed based on the matrix of pairwise genetic distances using MEGA v7.0 (Kumar et al., 2016) and iTOL v5 (Letunic and Bork, 2021). The principal component analysis (PCA) was performed using the smartPCA of the EIGENSOFT v5.0 package (Patterson et al., 2006). The Population structure analysis was assessed with genetic clusters K ranging from 2 to 6 using the ADMIXTURE v1.3 (Alexander and Lange, 2011) and plotted with ggplot2 package of R.
We used VCFtools to estimate the nucleotide diversity of each breed in window sizes of 50 kb with 50 kb increments (Danecek et al., 2011). The Linkage disequilibrium (LD) decay with physical distance between SNPs was calculated and visualized by using PopLDdecay software with default parameters (Zhang et al., 2019). We evaluated inbreeding coefficient by proportion of the genome covered by runs of homozygosity (FROH) calculated with PLINK, which was estimated based on the total length of ROH divided by the length of autosomes per individual (McQuillan et al., 2008). The plot as mentioned above was depicted using ggplot2 package of R (R Core Team, 2023). Finally, calculate the number of breed-specific SNPs using Python (3.8.16). The idea of the Python script is to compare the SNP dataset of the target breed with the SNP datasets of other breeds, filter out duplicate SNP of the target breed and other breeds, and obtain breed specific SNP.
The runs of homozygosity (ROHs) were identified using the --homozyg option implemented in the PLINK (Purcell et al., 2007), which slides a window of 100 SNPs (-homozyg-window-snp 100 --homozyg-snp 200 --homozyg-kb 100 --homozyg-gap 1000 --homozyg-window-threshold 0.05 --homozyg-window-het 1) across the genome estimating homozygosity. In this study, we defined ROH islands by using the “- homozygous group” option in PLINK to uniformly overlap homozygous regions with frequencies higher than 0.60.
Candidate genes were obtained by annotating the identified ROH islands with gene transfer format file (ARS-UCD1.2_genomic.gtf). To better understand the gene functions and signaling pathways of the identified candidate genes, functional enrichment analysis of GO terms and KEGG pathways was performed by KOBAS 3.0 (Xie et al., 2011). The GO and KEGG pathways were considered to be significantly enriched only when the Corrected P value [Benjamini and Hochberg (1995)] was lower than 0.05. In addition, this study downloaded the cattle QTLs data (QTLdb_cattleARS_UCD1.gff) from the Animal QTlLdb (https://www.animalgenome.org/cgi-bin/QTLdb/index), and used Bedtools (v2.31.0) to find the QTL loci of candidate genes of ROH island related to economic traits.
LOTER (Dias-Alves et al., 2018) was used to infer taurine ancestry along the genomes of Heilongjiang Crossbred cattle. We selected Angus, Holstein, and Mishima as reference panels based on the population structure. Then, the length and frequency of ancestral segments in each reference group were calculated by LOTER (Lyu et al., 2023).
To identify functional genes that are relevant to Heilongjiang Crossbred cattle in ancestral segments, we detected the selection signatures within Heilongjiang Crossbred cattle using the integrated haplotype score (iHS), which is calculated by selscan and then processed with 50 kb window and a step of 20 kb. We selected genes satisfying the following two conditions as candidate genes: 1) the top 1% in the ancestral segments; 2) iHS ≥ 2 and P value < 0.05. To better understand the gene functions and signaling pathways of the identified candidate genes, functional enrichment analysis of GO terms and KEGG pathways was performed by KOBAS 3.0 (Xie et al., 2011). The GO and KEGG pathways were considered to be significantly enriched only when the Corrected P value [Benjamini and Hochberg (1995)] was lower than 0.05.
The samples of 19 Heilongjiang Crossbred cattle were sequenced using whole-genome resequencing technology (Supplementary Table S1), along with 59 published whole genome sequencing data, including East Asian taurine cattle, Eurasian taurine cattle, and European taurine cattle as control (Supplementary Table S2). A total of about 665G of raw data were obtained from 19 Heilongjiang Crossbred cattle, generating 5.53 billion clean reads and aligning to the reference genome ARSUCD1.2_Btau5.0.1Y.fa with an average depth of 15.65×.
We annotated 14,427,729 biallelic SNPs that were discovered in 19 Heilongjiang Crossbred cattle. Functional annotation of the polymorphic sites revealed that 8,854,919 SNPs (61.37%) were located in the intergenic region, and 5,315,992 (36.85%) SNPs were located in the intron region. Exon accounted for 0.82% of the total SNPs, including 50,153 nonsynonymous and 63,048 synonymous SNPs (Figure 1).
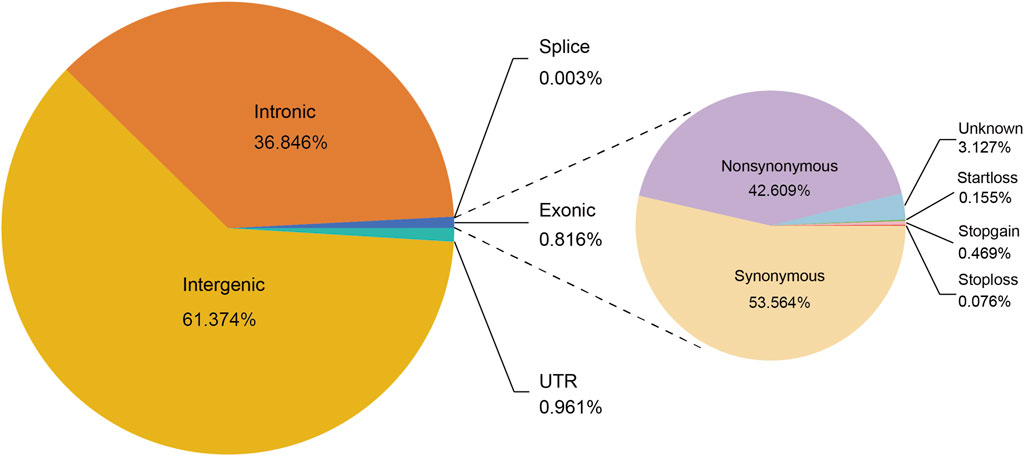
Figure 1. Functional classification of detected SNPs in 19 Heilongjiang Crossbred cattle genomes.
To investigate the genetic relationship between Heilongjiang Crossbred cattle and other taurine cattle breeds, we conducted principal component analysis (PCA), neighbor-joining (NJ) tree, and ADMIXTURE analysis based on genomic SNPs (Figure 2). PCA shows that East Asian taurine cattle, Eurasian taurine cattle, and European taurine cattle form separate clusters. The lineage of Heilongjiang Crossbred cattle is relatively complex and scattered in distribution (Figure 2A). The NJ tree provides similar results to the above conclusions of PCA (Figure 2B). Admixture analysis revealed that when K = 6, all breeds used as reference groups are independent, and it is found that Heilongjiang Crossbred cattle are mainly of Angus and Holstein ancestry, with a small amount of Mishima ancestry (Figure 2C).
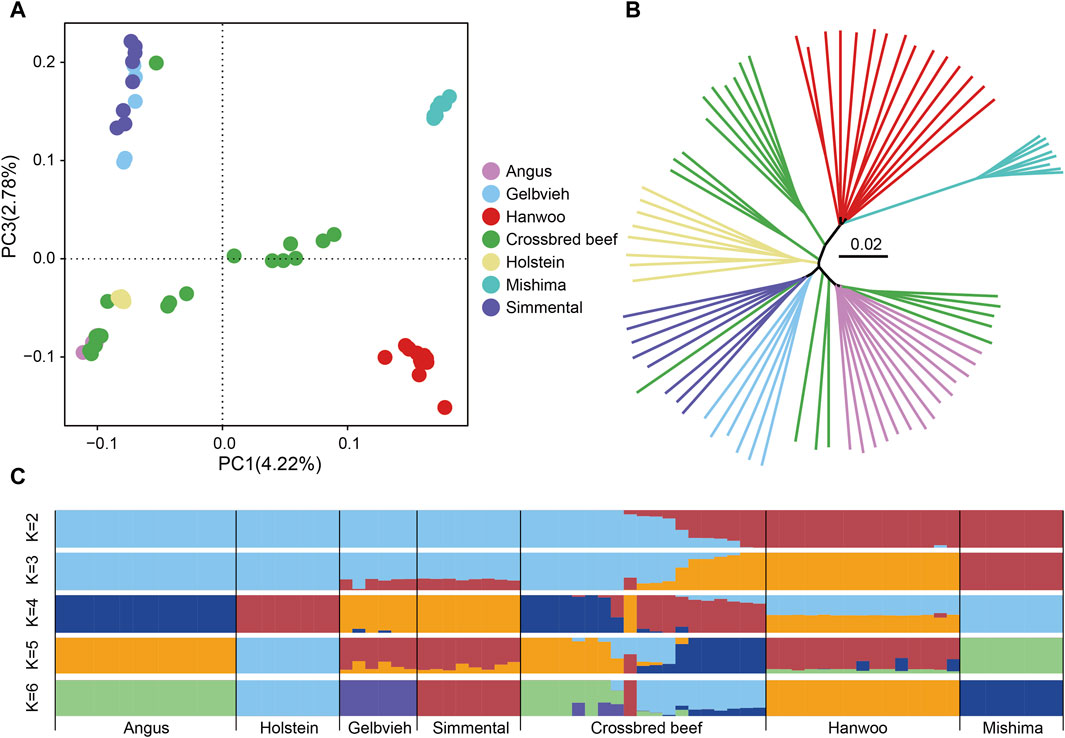
Figure 2. Population structure analysis of Heilongjiang Crossbred cattle. (A) The principal component analysis of cattle populations with PC1 against PC2. (B) The Neighbor-joining tree of relationships among populations. (C) Genetic structure of cattle populations using ADMIXTURE from K = 2 to K = 6.
To understand the genomic diversity of Heilongjiang Crossbred cattle, we conducted nucleotide diversity analysis, LD analysis, and inbreeding coefficient analysis on four populations of East Asian taurine cattle, Eurasian taurine cattle, European taurine cattle, and Heilongjiang Crossbred cattle. As shown in Figure 3A, the nucleotide diversity of the four populations is similar, and the nucleotide diversity of Heilongjiang Crossbred cattle is slightly higher. Similarly, in the LD decay analysis, the four populations also showed a consistent trend, with little difference in the rate (Figure 3B). The analysis of the inbreeding coefficient elucidates that some East Asian taurine cattle have relatively high values for inbreeding coefficients, and there is also a certain degree of inbreeding in Heilongjiang Crossbred cattle, which may be related to the use of inbreeding methods to fix traits in the breeding process (Figure 3C; Supplementary Table S3). In addition, we also annotated the SNPs of each breed individually. There is not much difference in the number of SNPs among taurine cattle, with East Asian taurine cattle having the highest number (15,874,716). Furthermore, we found that East Asian taurine cattle possessed the highest number of specific SNPs (3,479,775), European taurine cattle possessed the lowest number of specific SNPs (866,141), and Heilongjiang Crossbred cattle possessed 909.238 specific SNPs (Figure 3D). In addition to the analysis according to the populations, we also made statistics for each breed, and the results showed that the trend of genetic diversity between breeds and populations was the same (Supplementary Figure S1). In particular, among East Asian taurine cattle, the inbreeding coefficient of Hanwoo is lower, but the inbreeding coefficient of Mishima is higher.
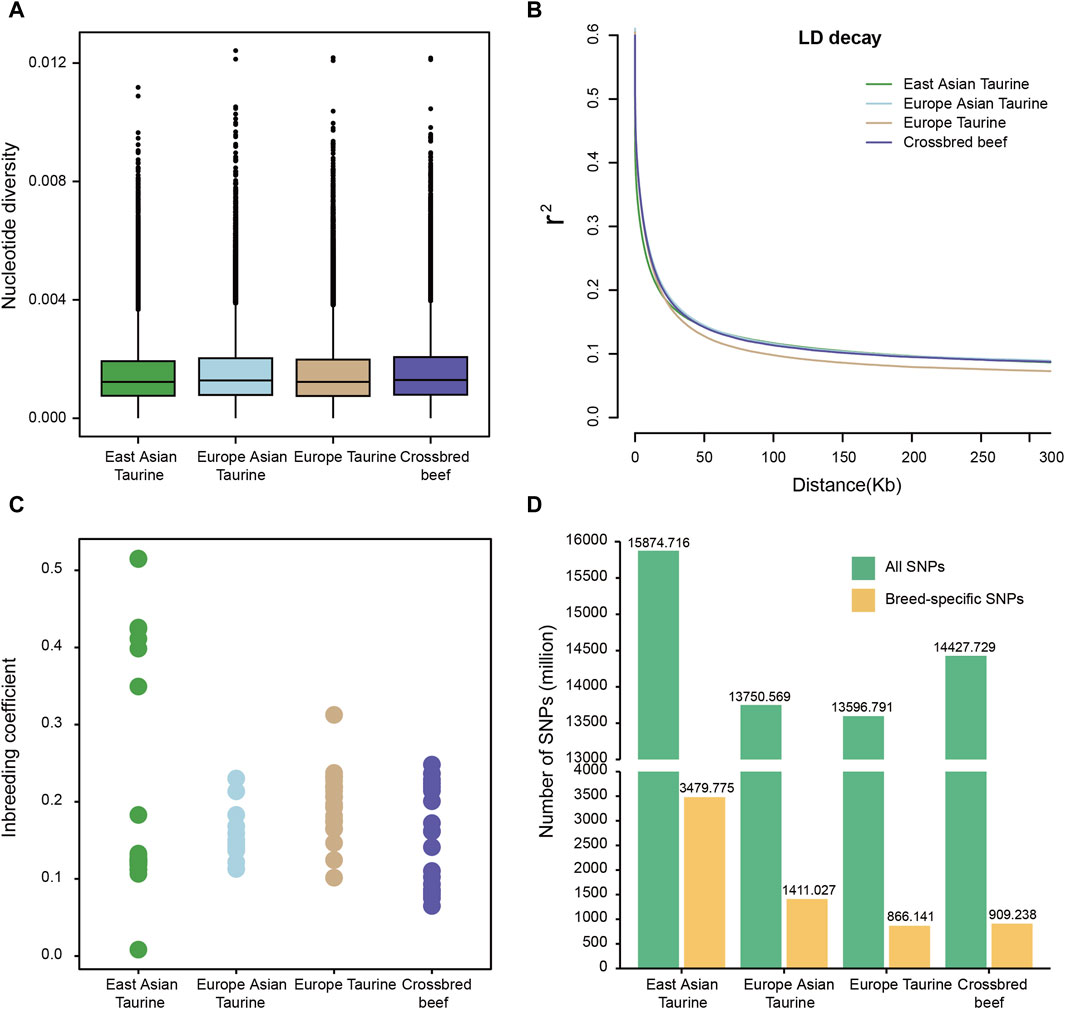
Figure 3. Genetic diversity among four populations. (A) Box plots of the nucleotide diversity for each population. The points which were on the outside of the whiskers showed outliers. (B) Decay of linkage disequilibrium on cattle autosomes estimated from each population. (C) Inbreeding coefficient for each population. (D) Number of SNPs for each population.
Runs of homozygosity (ROHs) is a continuous homozygous region in the DNA sequence of diploid organisms. We utilized whole genome sequencing data of 78 individuals for ROH analysis. Firstly, the ROH of each individual in each population was identified and shown in Figure 4A. Except for the large difference in the length and number of ROH in East Asian taurine cattle, there was little difference between individuals in other populations. Therefore, we analyzed each breed, and the results showed that the larger difference in East Asian taurine cattle was due to the larger number and length of ROH in Mishima (Supplementary Figure S2A). After that, we divided ROH into five categories: < 0.5 Mb, 0.5–1 Mb, 1–2 Mb, 2–4 Mb, and > 4 Mb. We found that the proportion of various ROHs in each population and breed were similar, and the length of most ROHs was less than 0.5 Mb (Figure 4B; Supplementary Figure S2). Figures 4C, D show the distribution of ROH on chromosomes in Heilongjiang Crossbred cattle. It can be seen that ROH is abundantly distributed on chromosome 1 (2,123 ROHs) and least distributed on chromosome 25 (459 ROHs). In addition, ROH length is also the longest on chromosome 1 (500,103 Mb), but the shortest on chromosome 28 (97.872 Mb) (Supplementary Table S4).
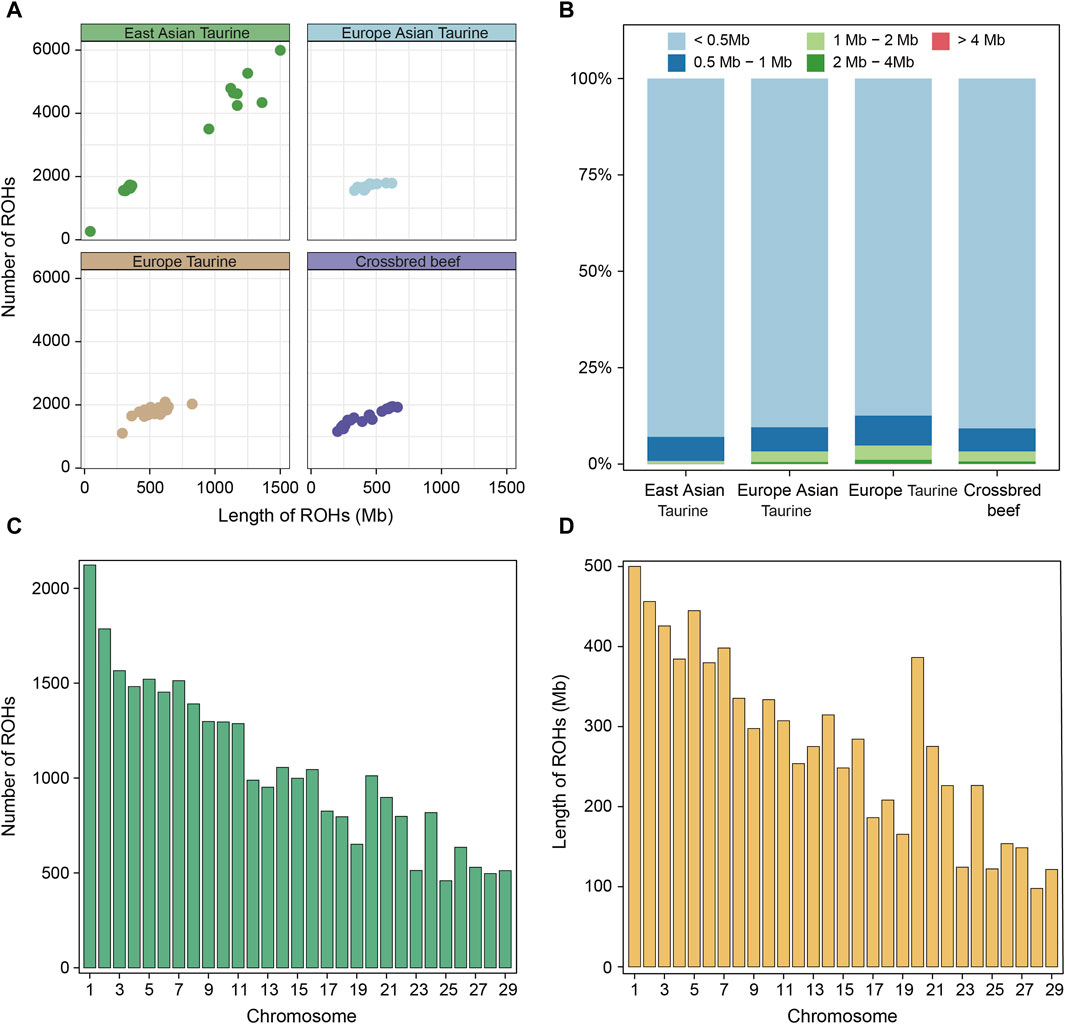
Figure 4. ROH analysis among four populations. (A) Total length and total number of ROHs per individual in each population. (B) The proportion of different categories of ROHs in each population. (C) The number of ROHs on each chromosome in Heilongjiang Crossbred cattle. (D) The length of ROHs on each chromosome in Heilongjiang Crossbred cattle.
ROH are suited to detect signatures of selection via ROH islands, we next calculated the frequency of ROH and identified ROH islands for each chromosome. The ROH frequencies are presented in Figure 5A. We regarded ROH with a frequency larger than 0.6 as ROH islands and searched for candidate genes overlapping with those ROH islands. In total, we identified 127 ROH islands. We found that the most frequent ROH island among all ROH islands is located in chromosome 17.
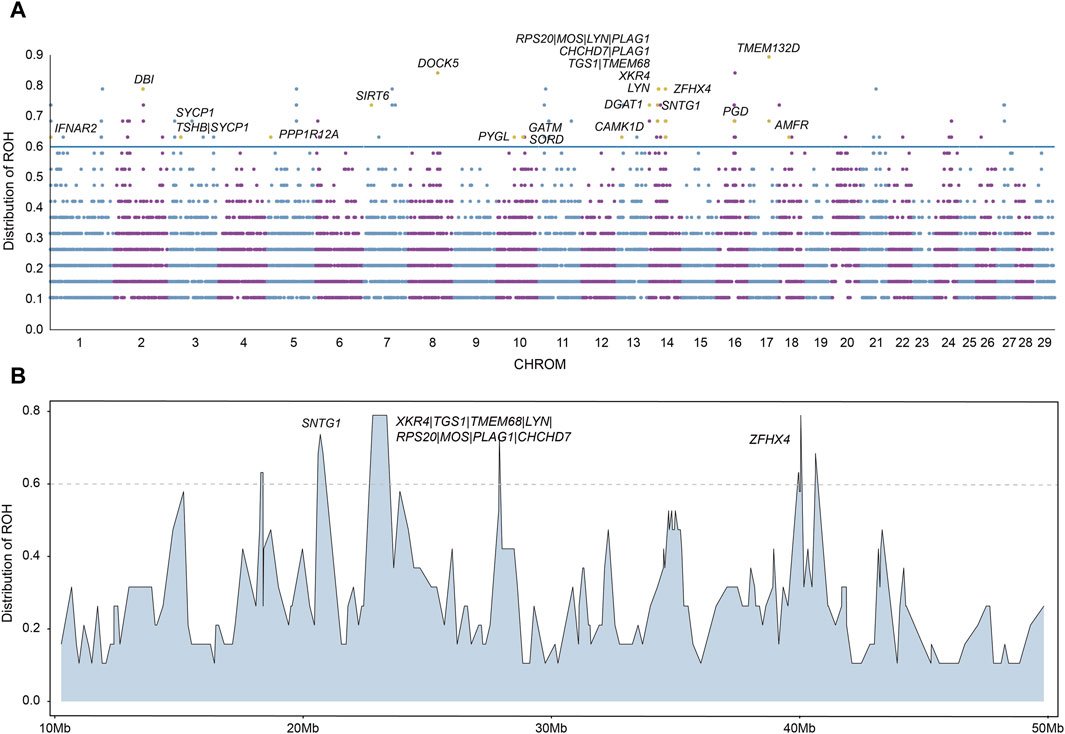
Figure 5. Candidate regions on ROH islands. (A) The distribution of ROH across autosomes. The X-axis represents the genomic coordinate, and the Y-axis displays the frequency of overlapping ROH among individuals. (B) Distribution of ROH on a long segment of chromosome 14.
We annotated the 127 ROH islands identified and obtained a total of 89 candidate genes (Supplementary Table S5). We performed functional enrichment analysis using KEGG pathways and Gene Ontology (GO) for candidate genes. The results represented significant enrichment of 1 KEGG pathway term and 11 GO terms (corrected P value < 0.05, Supplementary Tables S6, S7). Among these 89 candidate genes with important functions were found, including reproduction (AMFR, CAMK1D, GATM, IFNAR2, PGD, PYGL, SORD, SYCP1, TMEM132D), growth (MOS, PLAG1, TSHB, ZFHX4) and meat quality (CHCHD7, DBI, DGAT1, DOCK5, LYN, PPP1R12A, RPS20, SIRT6, SNTG1, TGS1, TMEM68, XKR4) (Table 1). In particular, in chromosome 14, a long segment annotated with multiple genes is found (Figure 5B). In order to verify that these genes are indeed related to economic traits, ROH islands containing candidate genes were compared with cattle QTLs data (QTLdb_cattleARS_UCD1.gff), and the results showed that these genes do have QTLs related to economic traits (Supplementary Table S8).
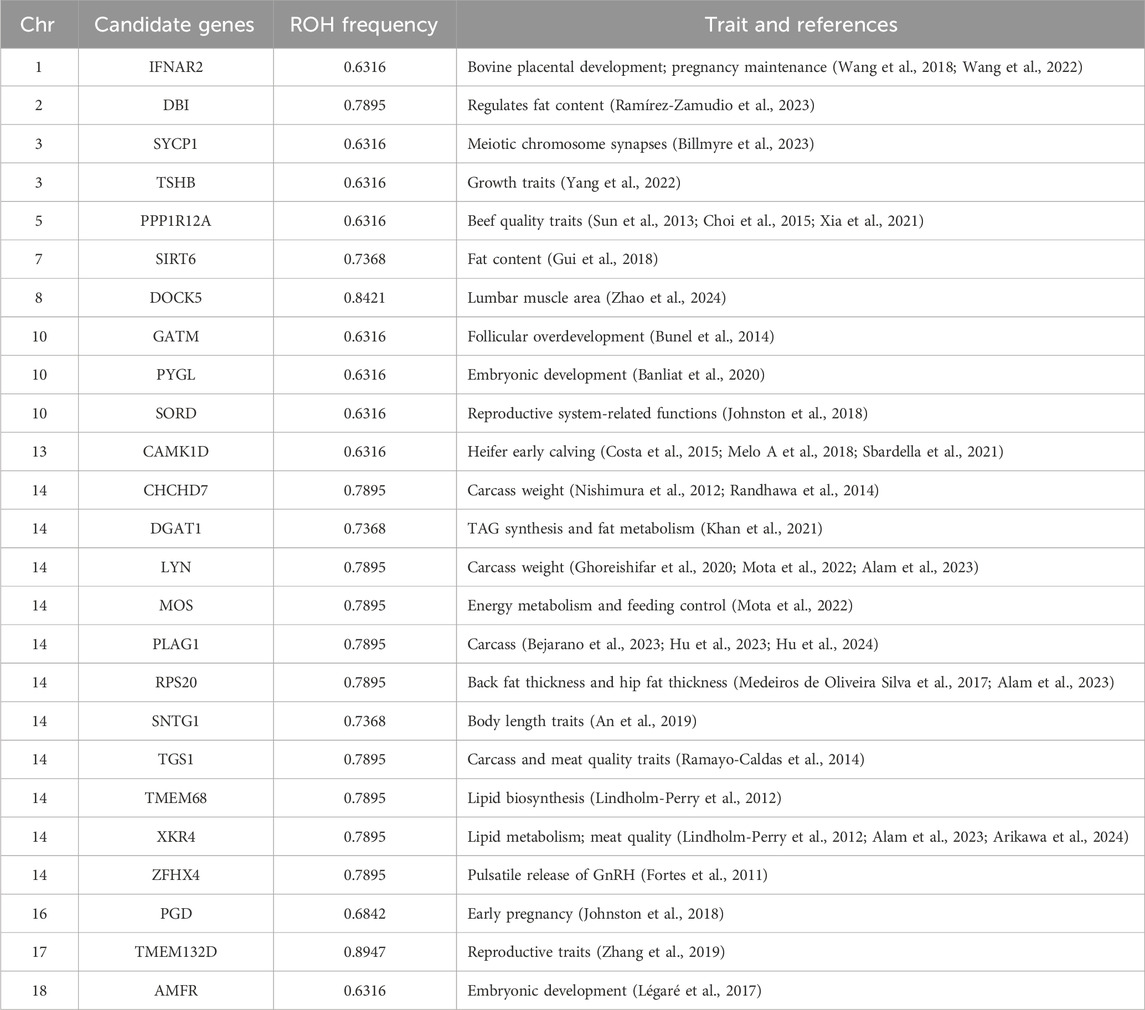
Table 1. Candidate genes on ROH islands.
We used LOTER to infer the taurine cattle ancestry in the Heilongjiang Crossbred cattle genome (Dias-Alves et al., 2018). According to the previous population structure analysis, Angus, Holstein, and Mishima were selected as the reference group. The results showed that Angus cattle had the highest infiltration frequency, followed by Holstein, and Mishima had the lowest infiltration frequency. To screen the ancestral segments that are important for Heilongjiang Crossbred cattle, we detected the selection signatures within Heilongjiang Crossbred cattle using an integrated haplotype score (iHS). Finally, the fragments with the top 1% introgression frequency overlapping with the selected fragments in the iHS method (iHS ≥ 2 and P value < 0.05) were considered high-frequency ancestral fragments. After that, these fragments were annotated, resulting in 165 (Angus), 90 (Holstein), and 103 (Mishima) genes, respectively (Supplementary Tables S9–S11). Among these genes, we found some genes related to the economic traits of Heilongjiang Crossbred cattle (Figure 6), including reproductive traits (BCAR3, CDH13, CDH18, KMT2C, MACROD2, PCSK5, PPP1R1C, SCAPER, ZPBP) and meat production traits (AGBL4, ARFGAP3, AUTS2, DPH6, DPP6, EEPD1, GRID2, LEKR1, LINGO2, MAP3K5, NEGR1, NKAIN2, PLCD1, PRKN, RASAL2, RHOBTB1, TRIM64, TRIM77, WWOX) (Table 2), reflecting the role of ancestral segments for Heilongjiang Crossbred cattle.
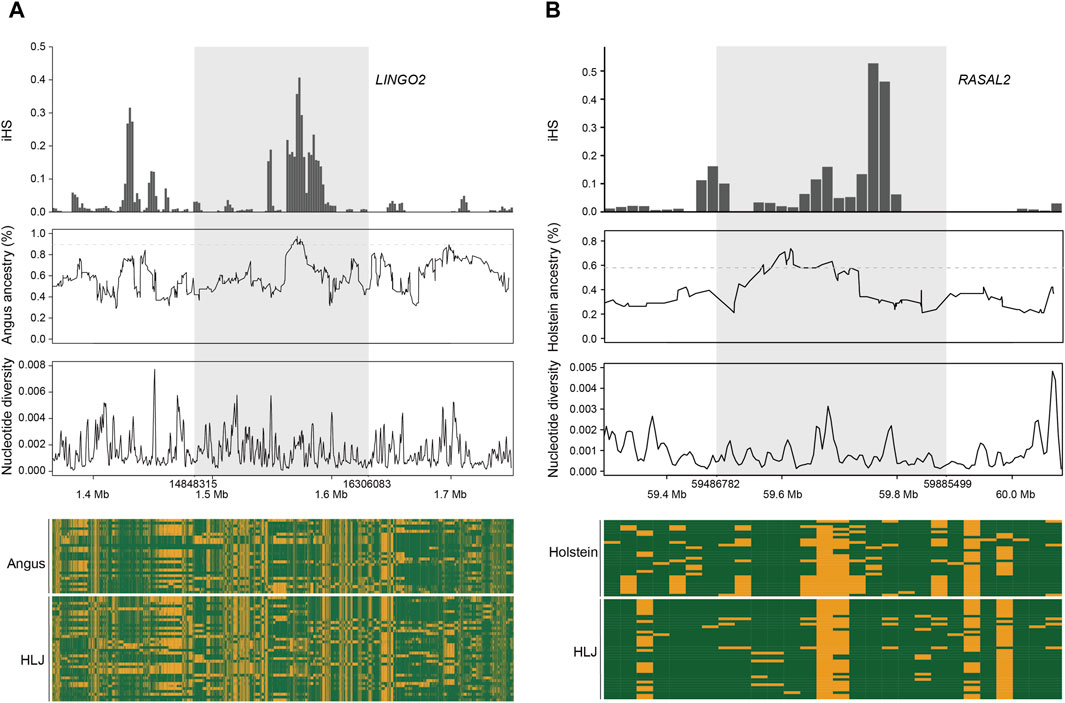
Figure 6. Example of candidate selective loci with taurine ancestry. (A) LINGO2 gene. (B) RASAL2 gene.
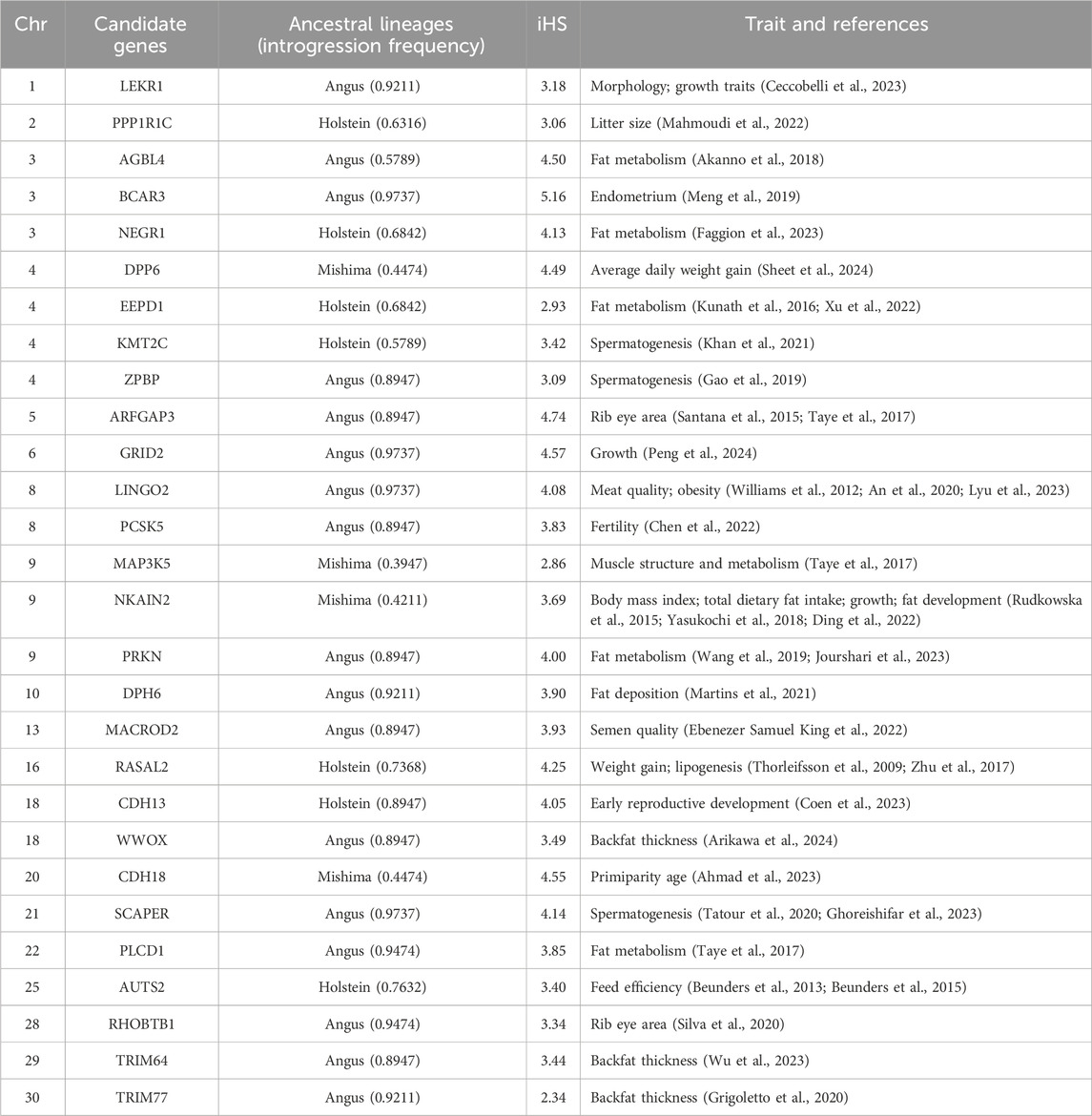
Table 2. Candidate genes with taurine ancestry.
As living standards have risen, there has been a corresponding increase in consumer demand for beef and dairy products in recent years. Heilongjiang Province, serving as a principal production hub for dairy and beef cattle, has hybridized native breeds with commercially purebred cattle to develop a new crossbred variety. This innovation aims to satisfy the escalating consumer demand for beef. However, at present, there is little understanding of the genetic resource of Heilongjiang Crossbred cattle, so it is of great significance to study the genetic diversity and population structure of Heilongjiang Crossbred cattle through whole genome sequencing data.
We resequenced the whole genome of 19 Heilongjiang Crossbred cattle and selected possible ancestors: East Asian taurine cattle, Eurasian taurine cattle, and European taurine cattle as reference populations for comparative analysis. We annotated the final generated SNP set, and the results showed that most SNPs were located in intergenic regions, and SNPs located in exons accounted for only 0.82% of the total SNPs, which was consistent with previous studies (Shen et al., 2020; Xia et al., 2021; Lyu et al., 2023). Among the SNPs located in exons, 42.61% were nonsynonymous SNPs and 53.56% were synonymous SNPs (Figure 1).
Next, we used the whole genome data of 78 individuals to analyze the genetic relationship between Heilongjiang Crossbred cattle and other breeds. We performed PCA, NJ tree, and ADMIXTURE based on genomic SNPs. The results of PCA and NJ tree are similar. East Asian taurine cattle, Eurasian taurine cattle, and European taurine cattle form independent clusters, which play a reference role. Heilongjiang Crossbred cattle are scattered among different breeds, representing the existence of hybridization of different lineages, which are more complex. In the ADMIXTURE, the CV value is the smallest when K = 2, and it is evident that Heilongjiang Crossbred cattle have lineage hybridization. But when K = 2, the specific bloodline composition of Heilongjiang hybrid cattle is not obvious. When K = 6, all breeds used as the reference population are independent. At this time, it can be seen that the Heilongjiang Crossbred cattle are mainly of Angus and Holstein ancestry, followed by the Mishima ancestry, and a very small number of Gelbvieh and Simmental ancestry. PCA, NJ tree and ADMIXTURE analysis all proved that Heilongjiang Crossbred cattle came from the hybridization of different lineages.
Then the SNP data set of 78 individuals was used to analyze the genetic diversity of Heilongjiang Crossbred cattle. Because the four populations belong to taurine, the genetic diversity is not different. Among them, the nucleotide diversity of Heilongjiang Crossbred cattle was slightly higher (Figure 3A), the LD decay rate was slightly faster (Figure 3B), and the genetic diversity was relatively high. According to the inbreeding coefficient analysis of each individual, it was found that the inbreeding degree of some individuals in East Asian taurine cattle was relatively high, and there was also a certain degree of inbreeding in Heilongjiang Crossbred cattle (Figure 3C), which may be related to the inbreeding method adopted in the breeding process of Heilongjiang Crossbred cattle, and the degree of inbreeding needs to be properly controlled in the subsequent breeding process. In addition, the total number of SNPs and the number of specific SNPs in each population were also counted. The results showed that the four populations had little difference, and the Heilongjiang Crossbred cattle belonged to the medium level (Figure 3D), which was consistent with the results of genetic diversity. At the same time, we counted the number of SNPs of each breed separately (Supplementary Figure S1), and the results showed that the number of SNPs of Heilongjiang Crossbred cattle was higher than that of each ancestral source, which may be due to the heterosis generated by hybridization.
In addition, we also conducted a systematic analysis of the ROHs of 78 individuals. The presence of long ROHs results from inbreeding, while shorter ROHs reflect the influence of ancient ancestors (Purfield et al., 2012). First, we compared the total length and total number of ROHs of each individual in each population (Figure 4A), and the results showed that except for the large individual differences in East Asian taurine cattle, the ROHs of the other three populations were similar in number and length. The large individual differences in East Asian taurine cattle are due to the large difference between Hanwoo and Mishima (Supplementary Figure S2), which may be due to the high degree of artificial selection of Mishima. After that, the ROHs of each population were divided into five categories according to < 0.5 Mb, 0.5–1 Mb, 1–2 Mb, 2–4 Mb, and > 4 Mb, and the proportion of different categories of ROHs in each population was shown (Figure 4B). The results showed that the length with the largest proportion in each population was <0.5 Mb, but the number of long-segment ROHs of East Asian taurine cattle was the least, and the number of long-segment ROHs of European taurine cattle was the most. The long segment ROHs of Heilongjiang Crossbred cattle belonged to the medium level in the four populations, indicating that there was a certain degree of hybridization in Heilongjiang Crossbred cattle, which was consistent with the results of the inbreeding coefficient. Finally, we counted the number and length of ROHs in each chromosome of Heilongjiang Crossbred cattle (Figures 4C, D). The number of ROHs on the chromosome is the largest (2, 123 ROHs) and the length is the longest (500, 103 Mb). The lowest number of ROHs was on chromosome 25 (459 ROHs), followed by chromosome 28 (497 ROHs) and chromosome 29 (512 ROHs). The shortest length of ROHs is chromosome 28 (97.872 Mb), followed by chromosome 29 (121.593 Mb) and chromosome 25 (122.323 Mb). The trend of the number and length distribution of ROHs on all chromosomes was consistent with previous studies (Zhao, 2021).
In addition to the statistics of ROHs of each population, we also identified ROH islands of hybrid beef cattle and detected some genes related to economic traits. We regarded ROHs with a frequency greater than 0.6 in Heilongjiang Crossbred cattle as ROH islands, and a total of 127 ROH islands were identified. These ROH islands were annotated and a total of 89 candidate genes were obtained (Supplementary Table S5). We performed functional enrichment analysis using KEGG pathways and Gene Ontology (GO) for candidate genes. The results represented significant enrichment of 1 KEGG pathway term and 11 GO terms (corrected P-value < 0.05, Supplementary Tables S6, S7). Among these 89 candidate genes, we found some genes with important functions (Figure 5A), including reproduction (AMFR, CAMK1D, GATM, IFNAR2, PGD, PYGL, SORD, SYCP1, TMEM132D), growth (MOS, PLAG1, TSHB, ZFHX4) and meat quality (CHCHD7, DBI, DGAT1, DOCK5, LYN, PPP1R12A, RPS20, SIRT6, SNTG1, TGS1, TMEM68, XKR4). Comparing these genes with cattle QTLs data, the results also proved that these genes were related to economic traits (Supplementary Table S8).
Heilongjiang Crossbred cattle are bred to meet the growing demand of people, and have good reproductive ability and meat production ability. Therefore, some genes related to reproductive ability have also been found in ROH Islands (AMFR, CAMK1D, GATM, IFNAR2, PGD, PYGL, SORD, SYCP1, TMEM132D). AMFR and PYGL play important roles in embryonic development (Légaré et al., 2017; Banliat et al., 2020). CAMK1D, GATM, IFNAR2, and PGD have effects on the reproductive function of cows and affect the reproductive function of cows from different aspects. CAMK1D was found to be associated with heifer early calving until 30 months and stability traits in Nelore cattle (Costa et al., 2015; Melo A et al., 2018; Sbardella et al., 2021). GATM is associated with bovine follicular overdevelopment, affecting follicular function and oocyte quality (Bunel et al., 2014). IFNAR2 plays an important role in bovine placental development and pregnancy maintenance (Wang et al., 2018; Wang et al., 2022). PGD is a potential marker of early pregnancy (Johnston et al., 2018). In addition, SORD exerts reproductive system-related functions and affects the reproductive ability of bulls (Johnston et al., 2018). SYCP1 is essential for meiotic chromosome synapses in vivo and plays an important role in sexual reproduction (Billmyre et al., 2023). TMEM132D is a candidate gene that constitutes the potential genetic structure of porcine reproductive traits (Zhang et al., 2019). It is also selected in Heilongjiang Crossbred cattle, and it is presumed to have the same effect on the reproductive traits of cattle. These genes play roles in reproductive traits from different aspects, which may be the reason why Heilongjiang Crossbred cattle have higher reproductive efficiency.
In addition to reproductive traits, the functional genes identified in ROH islands also have genes related to meat production ability, including growth (MOS, PLAG1, TSHB, ZFHX4) and meat quality (CHCHD7, DBI, DGAT1, DOCK5, LYN, PPP1R12A, RPS20, SIRT6, SNTG1, TGS1, TMEM68, XKR4). Four genes were identified in terms of growth traits. MOS has an impact on energy metabolism and feeding control, affecting the growth rate of beef cattle by affecting feed efficiency (Mota et al., 2022). IGF-2 is a cell growth and differentiation factor that plays an important role in muscle growth and differentiation in cattle (Huang et al., 2014; Júnior et al., 2016), PLAG1 regulates IGF-2 expression (Van Dyck et al., 2007; Akhtar et al., 2012; Utsunomiya et al., 2017). Therefore, PLAG1 plays an important role in carcass and has been selected in many studies on cattle (Bejarano et al., 2023; Hu et al., 2023; Hu et al., 2024). TSHB was identified to be associated with growth traits in chickens (Yang et al., 2022). It was also screened in this study, so we speculated that it would play a similar role in Cross-bred beef cattle. ZFHX4 regulates the onset of publication by affecting the pulsatile release of GnRH, thereby promoting beef cattle growth (Fortes et al., 2011). A total of 12 genes were identified in meat production traits, which can be divided into two aspects. The first is fat synthesis and fat metabolism. Intramuscular fat, which determines the tenderness and flavor of beef to a certain extent, is an important part of beef production (Hunt et al., 2014). DBI regulates fat content by upregulating the PPAR signaling pathway (Gross et al., 2017; Chen et al., 2022; Ramírez-Zamudio et al., 2023). Triacylglycerol (TAG) is the main component of intramuscular fat, and DGAT1 affects intramuscular fat content by participating in tag synthesis and fat metabolism (Khan et al., 2021). PPP1R12A is one of the genes highly expressed in porcine intramuscular adipose tissue and has been selected in other studies on beef quality traits (Sun et al., 2013; Choi et al., 2015; Xia et al., 2021). SIRT6 and TGS1 regulate fat content and meat quality traits by affecting transcript levels (Ramayo-Caldas et al., 2014; Gui et al., 2018). TMEM68 is expressed in rumen, abdominal stomach, intestine and adipose tissue of cattle and may affect lipid biosynthesis (Lindholm-Perry et al., 2012). The protein encoded by XKR4 plays an important biological role in cellular and lipid metabolism (Lindholm-Perry et al., 2012), and has been reported in several genes related to meat quality traits (Alam et al., 2023; Arikawa et al., 2024). These genes affect the intramuscular fat content of Heilongjiang Crossbred cattle from different pathways and play an important role in meat quality. The second part is the genes that affect other meat production indicators of beef cattle. CHCHD7 has been strongly selected in studies on bovine body size (Randhawa et al., 2014), and was considered one of the three major QTLs for carcass weight in Japanese black cattle (Nishimura et al., 2012). DOCK5 was identified to be associated with the lumbar muscle area (Zhao et al., 2024). LYN is related to the carcass weight of beef cattle in several studies (Ghoreishifar et al., 2020; Mota et al., 2022; Alam et al., 2023). RPS20 is the reason for the difference in back fat thickness and hip fat thickness in Nelore cattle (Medeiros de Oliveira Silva et al., 2017), and was selected in the study of carcass traits of Korean Hanwoo Cattle (Alam et al., 2023). Genome-wide association study reveals that SNTG1 is associated with increasing body length traits in Chinese Wagyu beef cattle (An et al., 2019). These genes affected the body size data and carcass weight of Heilongjiang Crossbred cattle and improved the meat production of Heilongjiang Crossbred cattle.
In particular, in chromosome 14, a long segment annotated with multiple genes is found (Figure 5B). This fragment contains a total of 10 genes, which affect Heilongjiang Crossbred cattle from different aspects so that Heilongjiang Crossbred cattle can produce more high-quality beef.
Through the analysis of population structure, we found that the Heilongjiang Crossbred cattle were mainly of Angus, Holstein, and Mishima pedigrees. To study the influence of ancestral lineage on Heilongjiang Crossbred cattle, we calculated the introgression proportion of the three ancestral lineages with LOTER. The results showed that Angus had the highest penetration frequency, followed by Holstein, and Mishima had the lowest penetration frequency, which was consistent with the results of the population structure analysis. To screen the ancestral segments that have important effects on Heilongjiang Crossbred cattle, we combined the iHS method to detect the selection signatures within Heilongjiang Crossbred cattle. Finally, the fragments with the top 1% introgression frequency overlapped with the selected fragments in the iHS method (iHS ≥ 2, P value < 0.05), which were considered to be high-frequency ancestral fragments with important roles. These fragments were then annotated, resulting in 165 (Angus), 90 (Holstein), and 103 (Mishima) genes, respectively (Supplementary Tables S9–S11). Among these genes, we found some genes related to economic traits of Heilongjiang Crossbred cattle (Figure 6), including reproductive traits (BCAR3, CDH13, CDH18, KMT2C, MACROD2, PCSK5, PPP1R1C, SCAPER, ZPBP) and meat production traits (AGBL4, ARFGAP3, AUTS2, DPH6, DPP6, EEPD1, GRID2, LEKR1, LINGO2, MAP3K5, NEGR1, NKAIN2, PLCD1, PRKN, RASAL2, RHOBTB1, TRIM64, TRIM77, WWOX). These genes reflect the role of high-frequency ancestral segments for Heilongjiang Crossbred cattle.
Among the genes related to reproductive traits, five genes are related to male reproduction, three genes are related to female reproduction, and the remaining one is related to embryonic development. CDH13 plays an important role in the early reproductive development of bulls (Coen et al., 2023). MACROD2 variation was associated with semen quality and was a biomarker of bull semen quality (Ebenezer Samuel King et al., 2022). SCAPER is highly expressed in testis and found to be involved in spermatogenesis in multiple species (Tatour et al., 2020; Ghoreishifar et al., 2023). KMT2C and ZPBP were identified as target genes of bull sexual maturation and associated with spermatogenesis (Gao et al., 2019; Khan et al., 2021). These five genes improve the fertility of bulls from three aspects: hormone regulation, semen quality, and spermatogenesis. BCAR3 is expressed in the endometrium (Meng et al., 2019), the resulting variation is related to the number of stillborn pigs and was selected in the study on Nellore cattle (Wang et al., 2022; Ahmad et al., 2023). CDH18 and PCSK5 are related to the age of first calving and the fertility of Holstein cattle, respectively (Chen et al., 2022; Ahmad et al., 2023). While PPP1R1C was reported to be associated with litter size (Mahmoudi et al., 2022).
Genes that affect carcass quality fat production and metabolism are crucial for optimizing the genetic selection process of beef cattle. We also found some genes related to meat quality traits in the introgressed genes. AGBL4, EEPD1, NEGR1, PLCD1 and PRKN are all related to fat metabolism and affect beef quality by affecting intramuscular fat content (Kunath et al., 2016; Taye et al., 2017; Akanno et al., 2018; Wang et al., 2019; Xu et al., 2022; Faggion et al., 2023; Jourshari et al., 2023). DPH6, LINGO2, NKAIN2, and RASAL2 are related to weight gain and fat generation and development, and affect meat quality by affecting fat deposition (Thorleifsson et al., 2009; Williams et al., 2012; Rudkowska et al., 2015; Zhu et al., 2017; Yasukochi et al., 2018; An et al., 2020; Martins et al., 2021; Ding et al., 2022; Lyu et al., 2023). MAP3K5 affects meat tenderness by participating in muscle structure and metabolism (Taye et al., 2017). These genes may be the reason for the delicate meat quality of Heilongjiang Crossbred cattle.
In addition, there are some genes related to meat production. ARFGAP3 and RHOBTB1 are related to the rib eye area (REA) of Nellore cattle and have been selected in other studies on beef cattle (Santana et al., 2015; Taye et al., 2017; Silva et al., 2020). AUTS2 is associated with digestive system diseases (Beunders et al., 2013; Beunders et al., 2015), affecting the feed efficiency of cattle and thus growth speed (Hu et al., 2023). DPP6 is a candidate gene for average daily weight gain (ADG), which is associated with the growth rate of cattle (Sheet et al., 2024). In the study of sheep, LEKR1 was found to be related to morphology and growth traits (Ceccobelli et al., 2023), and we speculated that it has a similar role in beef cattle. Meanwhile, GRID2 was also identified to be associated with growth (Peng et al., 2024). TRIM64, TRIM77, and WWOX were detected as candidate genes for backfat thickness (BFT) (Grigoletto et al., 2020; Wu et al., 2023; Arikawa et al., 2024). These genes help Heilongjiang Crossbred cattle grow rapidly to produce more beef.
Among the numerous genes related to economic traits, we selected two for display (Figure 6). The higher iHS frequency and lower nucleotide diversity represent that genes are selected and fixed in Heilongjiang Crossbred cattle. According to the introgression frequency of the ancestral fragment and the haplotype heatmap, it can be seen that the gene is an introgression fragment from the ancestor and the haplotype of Heilongjiang Crossbred cattle is the same as that of the ancestor. Therefore, these two genes are thought to be derived from the ancestral introgression and fixed in Heilongjiang Crossbred cattle. They play an important role in the economic traits of Heilongjiang Crossbred cattle: improving the reproductive efficiency of Heilongjiang Crossbred cattle and making Heilongjiang Crossbred cattle produce better quality beef.
In summary, our study utilized whole genome sequencing to explore the population structure of Heilongjiang Crossbred cattle, elucidate their genetic diversity, and identify genes related to economic traits through ROH islands and ancestral infiltration fragments. The results showed that Heilongjiang Crossbred cattle are mainly of Angus ancestry, followed by Holstein ancestry, and a small amount of Mishima ancestry with modest genetic diversity. In addition, we identified key candidate genes related to reproductive efficiency and beef quality through ROH islands and high-frequency ancestral infiltration fragments, providing new insights into the genetic basis of higher reproductive efficiency and stronger meat production ability in Heilongjiang Crossbred cattle. These findings not only enhance our understanding of the unique characteristics of Heilongjiang Crossbred cattle but also have significance in promoting crossbreeding improvement in beef cattle. However, the number of populations in this study is relatively small, and the number of populations can be expanded for more detailed research in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
This study was reviewed and approved by the Institutional Animal Care and Use Committee of Northwest A&F University (FAPWCNWAFU, Protocol number, NWAFAC 1008). The study was conducted in accordance with the local legislation and institutional requirements.
SL: Data curation, Formal Analysis, Visualization, Writing–original draft. LL: Data curation, Formal Analysis, Writing–original draft. ZA: Writing–review and editing. WF: Writing–review and editing. CL: Conceptualization, Funding acquisition, Methodology, Resources, Writing–review and editing. FS: Conceptualization, Funding acquisition, Methodology, Resources, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Key R&D Program of Heilongjiang Province, China (2022ZX02B09) and the China Agriculture Research System of MOF and MARA (CARS-37).
The authors would like to thank the High-Performance Computing (HPC) of Northwest A&F University (NWAFU), China for providing computing resources.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1435793/full#supplementary-material
Ahmad, S. F., Singh, A., Gangwar, M., Kumar, S., Dutt, T., and Kumar, A. (2023). Haplotype-based association study of production and reproduction traits in multigenerational Vrindavani population. Gene 867, 147365. doi:10.1016/j.gene.2023.147365
Akanno, E. C., Chen, L., Abo-Ismail, M. K., Crowley, J. J., Wang, Z., Li, C., et al. (2018). Genome-wide association scan for heterotic quantitative trait loci in multi-breed and crossbred beef cattle. Genet. Sel. Evol. GSE 50 (1), 48. doi:10.1186/s12711-018-0405-y
Akhtar, M., Holmgren, C., Göndör, A., Vesterlund, M., Kanduri, C., Larsson, C., et al. (2012). Cell type and context-specific function of PLAG1 for IGF2 P3 promoter activity. Int. J. Oncol. 41 (6), 1959–1966. doi:10.3892/ijo.2012.1641
Alam, M. Z., Haque, M. A., Iqbal, A., Lee, Y., Ha, J., Jin, S., et al. (2023). Genome-wide association study to identify QTL for carcass traits in Korean Hanwoo cattle. Animals open access J. MDPI 13 (17), 2737. doi:10.3390/ani13172737
Alexander, D. H., and Lange, K. (2011). Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinforma. 12, 246. doi:10.1186/1471-2105-12-246
An, B., Xia, J., Chang, T., Wang, X., Xu, L., Zhang, L., et al. (2019). Genome-wide association study reveals candidate genes associated with body measurement traits in Chinese Wagyu beef cattle. Anim. Genet. 50 (4), 386–390. doi:10.1111/age.12805
An, B., Xu, L., Xia, J., Wang, X., Miao, J., Chang, T., et al. (2020). Multiple association analysis of loci and candidate genes that regulate body size at three growth stages in Simmental beef cattle. BMC Genet. 21 (1), 32. doi:10.1186/s12863-020-0837-6
Arikawa, L. M., Mota, L. F. M., Schmidt, P. I., Frezarim, G. B., Fonseca, L. F. S., Magalhães, A. F. B., et al. (2024). Genome-wide scans identify biological and metabolic pathways regulating carcass and meat quality traits in beef cattle. Meat Sci. 209, 109402. doi:10.1016/j.meatsci.2023.109402
Banliat, C., Tsikis, G., Labas, V., Teixeira-Gomes, A., Com, E., Lavigne, R., et al. (2020). Identification of 56 proteins involved in embryo-maternal interactions in the bovine oviduct. Int. J. Mol. Sci. 21 (2), 466. doi:10.3390/ijms21020466
Bejarano, D. H., Martínez, R. A., and Rocha, J. F. (2023). Genome-wide association study for growth traits in Blanco Orejinegro and Romosinuano cattle. Trop. Anim. Health Prod. 55 (6), 358. doi:10.1007/s11250-023-03743-9
Beunders, G., de Munnik, S. A., Van der Aa, N., Ceulemans, B., Voorhoeve, E., Groffen, A. J., et al. (2015). Two male adults with pathogenic AUTS2 variants, including a two-base pair deletion, further delineate the AUTS2 syndrome. Eur. J. Hum. Genet. EJHG 23 (6), 803–807. doi:10.1038/ejhg.2014.173
Beunders, G., Voorhoeve, E., Golzio, C., Pardo, L. M., Rosenfeld, J. A., Talkowski, M. E., et al. (2013). Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 92 (2), 210–220. doi:10.1016/j.ajhg.2012.12.011
Billmyre, K. K., Kesler, E. A., Tsuchiya, D., Corbin, T. J., Weaver, K., Moran, A., et al. (2023). SYCP1 head-to-head assembly is required for chromosome synapsis in mouse meiosis. Sci. Adv. 9 (42), eadi1562. doi:10.1126/sciadv.adi1562
Bu, Y., Sun, F., Wang, C., Zhao, X., Liu, L., Xu, S., et al. (2021). Suggestions on the development of beef cattle genetic improvement in Heilongjiang Province. HEILONGJIANG J. ANIMAL REPRODUCTION 29 (05), 40–43. doi:10.19848/j.cnki.ISSN1005-2739.2021.08.0007
Buggiotti, L., Yurchenko, A. A., Yudin, N. S., Vander Jagt, C. J., Vorobieva, N. V., Kusliy, M. A., et al. (2021). Demographic history, adaptation, and NRAP convergent evolution at amino acid residue 100 in the world northernmost cattle from siberia. Mol. Biol. Evol. 38 (8), 3093–3110. doi:10.1093/molbev/msab078
Bunel, A., Nivet, A. L., Blondin, P., Vigneault, C., Richard, F. J., and Sirard, M. A. (2014). Cumulus cell gene expression associated with pre-ovulatory acquisition of developmental competence in bovine oocytes. Reproduction, Fertil. Dev. 26 (6), 855–865. doi:10.1071/RD13061
Ceccobelli, S., Landi, V., Senczuk, G., Mastrangelo, S., Sardina, M. T., Ben-Jemaa, S., et al. (2023). A comprehensive analysis of the genetic diversity and environmental adaptability in worldwide Merino and Merino-derived sheep breeds. Genet. Sel. Evol. GSE 55 (1), 24. doi:10.1186/s12711-023-00797-z
Chen, H., Peng, T., Shang, H., Shang, X., Zhao, X., Qu, M., et al. (2022). RNA-seq analysis reveals the potential molecular mechanisms of puerarin on intramuscular fat deposition in heat-stressed beef cattle. Front. Nutr. 9, 817557. doi:10.3389/fnut.2022.817557
Chen, N., Cai, Y., Chen, Q., Li, R., Wang, K., Huang, Y., et al. (2018). Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 9 (1), 2337. doi:10.1038/s41467-018-04737-0
Chen, S., Schenkel, F. S., Melo, A. L. P., Oliveira, H. R., Pedrosa, V. B., Araujo, A. C., et al. (2022). Identifying pleiotropic variants and candidate genes for fertility and reproduction traits in Holstein cattle via association studies based on imputed whole-genome sequence genotypes. BMC Genomics 23 (1), 331. doi:10.1186/s12864-022-08555-z
Choi, J., Choi, B., Lee, S., Lee, S., Kim, H., Yu, D., et al. (2015). Whole-genome resequencing analysis of Hanwoo and yanbian cattle to identify genome-wide SNPs and signatures of selection. Mol. Cells 38 (5), 466–473. doi:10.14348/molcells.2015.0019
Coen, S., Keogh, K., Lonergan, P., Fair, S., and Kenny, D. A. (2023). Early life nutrition affects the molecular ontogeny of testicular development in the young bull calf. Sci. Rep. 13 (1), 6748. doi:10.1038/s41598-022-23743-3
Costa, R. B., Camargo, G. M. F., Diaz, I. D. P. S., Irano, N., Dias, M. M., Carvalheiro, R., et al. (2015). Genome-wide association study of reproductive traits in Nellore heifers using Bayesian inference. Genet. Sel. Evol. GSE 47 (1), 67. doi:10.1186/s12711-015-0146-0
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinforma. Oxf. Engl. 27 (15), 2156–2158. doi:10.1093/bioinformatics/btr330
Decker, J. E., McKay, S. D., Rolf, M. M., Kim, J., Molina Alcalá, A., Sonstegard, T. S., et al. (2014). Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 10 (3), e1004254. doi:10.1371/journal.pgen.1004254
Dias-Alves, T., Mairal, J., and Blum, M. G. B. (2018). Loter: a software package to infer local ancestry for a wide range of species. Mol. Biol. Evol. 35 (9), 2318–2326. doi:10.1093/molbev/msy126
Ding, R., Zhuang, Z., Qiu, Y., Ruan, D., Wu, J., Ye, J., et al. (2022). Identify known and novel candidate genes associated with backfat thickness in Duroc pigs by large-scale genome-wide association analysis. J. Anim. Sci. 100 (2), skac012. doi:10.1093/jas/skac012
Ebenezer Samuel King, J. P., Kumaresan, A., Talluri, T. R., Sinha, M. K., Raval, K., Nag, P., et al. (2022). Genom-wide analysis identifies single nucleotide polymorphism variations and altered pathways associated with poor semen quality in breeding bulls. Reproduction Domest. animals = Zuchthygiene 57 (10), 1143–1155. doi:10.1111/rda.14185
Faggion, S., Boschi, E., Veroneze, R., Carnier, P., and Bonfatti, V. (2023). Genomic prediction and genome-wide association study for boar taint compounds. Animals open access J. MDPI 13 (15), 2450. doi:10.3390/ani13152450
Fortes, M. R. S., Reverter, A., Nagaraj, S. H., Zhang, Y., Jonsson, N. N., Barris, W., et al. (2011). A single nucleotide polymorphism-derived regulatory gene network underlying puberty in 2 tropical breeds of beef cattle. J. Anim. Sci. 89 (6), 1669–1683. doi:10.2527/jas.2010-3681
Gao, Y., Li, S., Lai, Z., Zhou, Z., Wu, F., Huang, Y., et al. (2019). Analysis of long non-coding RNA and mRNA expression profiling in immature and mature bovine (Bos taurus) testes. Front. Genet. 10, 646. doi:10.3389/fgene.2019.00646
Ghoreishifar, M., Vahedi, S. M., Salek Ardestani, S., Khansefid, M., and Pryce, J. E. (2023). Genome-wide assessment and mapping of inbreeding depression identifies candidate genes associated with semen traits in Holstein bulls. BMC Genomics 24 (1), 230. doi:10.1186/s12864-023-09298-1
Ghoreishifar, S. M., Eriksson, S., Johansson, A. M., Khansefid, M., Moghaddaszadeh-Ahrabi, S., Parna, N., et al. (2020). Signatures of selection reveal candidate genes involved in economic traits and cold acclimation in five Swedish cattle breeds. Genet. Sel. Evol. GSE 52 (1), 52. doi:10.1186/s12711-020-00571-5
Grigoletto, L., Ferraz, J. B. S., Oliveira, H. R., Eler, J. P., Bussiman, F. O., Abreu Silva, B. C., et al. (2020). Genetic architecture of carcass and meat quality traits in Montana Tropical® composite beef cattle. Front. Genet. 11, 123. doi:10.3389/fgene.2020.00123
Gross, B., Pawlak, M., Lefebvre, P., and Staels, B. (2017). PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 13 (1), 36–49. doi:10.1038/nrendo.2016.135
Gui, L., Raza, S. H. A., Garcia, M., Sun, Y., Ullah, I., and Han, Y. (2018). Genetic variants in the SIRT6 transcriptional regulatory region affect gene activity and carcass quality traits in indigenous Chinese beef cattle (Bos taurus). BMC Genomics 19 (1), 785. doi:10.1186/s12864-018-5149-0
Hu, M., Jiang, H., Lai, W., Shi, L., Yi, W., Sun, H., et al. (2023). Assessing genomic diversity and signatures of selection in Chinese red steppe cattle using high-density SNP array. Animals open access J. MDPI 13 (10), 1717. doi:10.3390/ani13101717
Hu, M., Shi, L., Yi, W., Li, F., and Yan, S. (2024). Identification of genomic diversity and selection signatures in Luxi cattle using whole-genome sequencing data. Anim. Biosci. 37 (3), 461–470. doi:10.5713/ab.23.0304
Hu, Z., Boschiero, C., Li, C., Connor, E. E., Baldwin, R. L. T., and Liu, G. E. (2023). Unraveling the genetic basis of feed efficiency in cattle through integrated DNA methylation and CattleGTEx analysis. Genes. 14 (12), 2121. doi:10.3390/genes14122121
Huang, Y., Zhan, Z., Li, X., Wu, S., Sun, Y., Xue, J., et al. (2014). SNP and haplotype analysis reveal IGF2 variants associated with growth traits in Chinese Qinchuan cattle. Mol. Biol. Rep. 41 (2), 591–598. doi:10.1007/s11033-013-2896-5
Hunt, M. R., Garmyn, A. J., O'Quinn, T. G., Corbin, C. H., Legako, J. F., Rathmann, R. J., et al. (2014). Consumer assessment of beef palatability from four beef muscles from USDA Choice and Select graded carcasses. Meat Sci. 98 (1), 1–8. doi:10.1016/j.meatsci.2014.04.004
Jia, Y., Wang, C., Jia, Y., Wei, C., and Yang, H. (2022). Analysis of Heilongjiang beef cattle industry development trend in 2022. J. Heilongjiang Bayi Agric. Reclam. Univ. 34 (05), 68–71. doi:10.3969/j.issn.1002-2090.2022.05.011
Johnston, D., Malo Estepa, I., Ebhardt, H. A., Crowe, M. A., and Diskin, M. G. (2018). Differences in the bovine milk whey proteome between early pregnancy and the estrous cycle. Theriogenology 114, 301–307. doi:10.1016/j.theriogenology.2018.04.008
Jourshari, M. G., Shadparvar, A. A., Ghavi Hossein-Zadeh, N., Rafeie, F., Banabazi, M. H., and Johansson, A. M. (2023). Genome-wide association study on abdomen depth, head width, hip width, and withers height in native cattle of Guilan (Bos indicus). PLoS One 18 (8), e0289612. doi:10.1371/journal.pone.0289612
Júnior, G. A. F., Costa, R. B., de Camargo, G. M. F., Carvalheiro, R., Rosa, G. J. M., Baldi, F., et al. (2016). Genome scan for postmortem carcass traits in Nellore cattle. J. Anim. Sci. 94 (10), 4087–4095. doi:10.2527/jas.2016-0632
Khan, I. M., Liu, H., Zhuang, J., Khan, N. M., Zhang, D., Chen, J., et al. (2021). Circular RNA expression and regulation profiling in testicular tissues of immature and mature wandong cattle (Bos taurus). Front. Genet. 12, 685541. doi:10.3389/fgene.2021.685541
Khan, M. Z., Ma, Y., Ma, J., Xiao, J., Liu, Y., Liu, S., et al. (2021). Association of DGAT1 with cattle, Buffalo, goat, and sheep milk and meat production traits. Front. Vet. Sci. 8, 712470. doi:10.3389/fvets.2021.712470
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi:10.1093/molbev/msw054
Kunath, A., Hesselbarth, N., Gericke, M., Kern, M., Dommel, S., Kovacs, P., et al. (2016). Repin1 deficiency improves insulin sensitivity and glucose metabolism in db/db mice by reducing adipose tissue mass and inflammation. Biochem. Biophys. Res. Commun. 478 (1), 398–402. doi:10.1016/j.bbrc.2016.07.038
Légaré, C., Akintayo, A., Blondin, P., Calvo, E., and Sullivan, R. (2017). Impact of male fertility status on the transcriptome of the bovine epididymis. Mol. Hum. Reprod. 23 (6), 355–369. doi:10.1093/molehr/gax019
Letunic, I., and Bork, P. (2021). Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic. acids. Res. 49 (W1), W293–W296. doi:10.1093/nar/gkab301
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinforma. Oxf. Engl. 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, S., Lei, H., Li, J., Sun, A., Ahmed, Z., Duan, H., et al. (2023). Analysis of genetic diversity and selection signals in Chaling cattle of southern China using whole-genome scan. Anim. Genet. 54 (3), 284–294. doi:10.1111/age.13305
Li, W., Han, Y., Song, X., Zhang, Z., Zhao, J., Ding, D., et al. (2021). Analysis of viral load in different specimen types and serum antibody levels of COVID-19 patients. Mod. Anim. Husb. Technol. 19 (06), 30–31. doi:10.1186/s12967-020-02693-2
Lindholm-Perry, A. K., Kuehn, L. A., Smith, T. P. L., Ferrell, C. L., Jenkins, T. G., Freetly, H. C., et al. (2012). A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle(1). Anim. Genet. 43 (2), 216–219. doi:10.1111/j.1365-2052.2011.02232.x
Lyu, Y., Guan, X., Xu, X., Wang, P., Li, Q., Panigrahi, M., et al. (2023). A whole genome scan reveals distinct features of selection in Zhaotong cattle of Yunnan province. Anim. Genet. 54 (6), 731–742. doi:10.1111/age.13363
Lyu, Y., Ren, Y., Qu, K., Quji, S., Zhuzha, B., Lei, C., et al. (2023). Local ancestry and selection in admixed Sanjiang cattle. Stress Biol. 3 (1), 30. doi:10.1007/s44154-023-00101-5
Mahmoudi, P., Rashidi, A., Nazari-Ghadikolaei, A., Rostamzadeh, J., Razmkabir, M., and Huson, H. J. (2022). Genome-wide association study reveals novel candidate genes for litter size in Markhoz goats. Front. Vet. Sci. 9, 1045589. doi:10.3389/fvets.2022.1045589
Martins, R., Brito, L. F., Machado, P. C., Pinto, L. F. B., Silva, M. R., Schenkel, F. S., et al. (2021). Genome-wide association study and pathway analysis for carcass fatness in Nellore cattle measured by ultrasound. Anim. Genet. 52 (5), 730–733. doi:10.1111/age.13129
McQuillan, R., Leutenegger, A., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., et al. (2008). Runs of homozygosity in European populations. Am. J. Hum. Genet. 83 (3), 359–372. doi:10.1016/j.ajhg.2008.08.007
Medeiros de Oliveira Silva, R., Bonvino Stafuzza, N., de Oliveira Fragomeni, B., Miguel Ferreira de Camargo, G., Matos Ceacero, T., Noely Dos Santos Gonçalves Cyrillo, J., et al. (2017). Genome-wide association study for carcass traits in an experimental Nelore cattle population. PLoS One 12 (1), e0169860. doi:10.1371/journal.pone.0169860
Melo A, T., Bresolin, T., Mota, L., Fortes, M., Albuquerque, L., and Carvalheiro, R. (2018). “Multi-trait meta-analysis for sexual precocity traits in Nellore cattle,” in Proceedings of the 11th world congress on genetics applied to livestock production (WCGALP 2018) (Auckland, New Zealand: WCGALP), 178.
Meng, X., Liu, J., Wang, H., Chen, P., and Wang, D. (2019). MicroRNA-126-5p downregulates BCAR3 expression to promote cell migration and invasion in endometriosis. Mol. Cell. Endocrinol. 494, 110486. doi:10.1016/j.mce.2019.110486
Mota, L. F. M., Santos, S. W. B., Júnior, G. A. F., Bresolin, T., Mercadante, M. E. Z., Silva, J. A. V., et al. (2022). Meta-analysis across Nellore cattle populations identifies common metabolic mechanisms that regulate feed efficiency-related traits. BMC Genomics 23 (1), 424. doi:10.1186/s12864-022-08671-w
Nie, D., and Han, D. (2023). Introduction and development suggestions of beef cattle industry in Heilongjiang province. Animal Husb. China (04), 21–22. doi:10.3969/j.issn.2095-2473.2023.04.010
Nishimura, S., Watanabe, T., Mizoshita, K., Tatsuda, K., Fujita, T., Watanabe, N., et al. (2012). Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 13, 40. doi:10.1186/1471-2156-13-40
Patterson, N., Price, A. L., and Reich, D. (2006). Population structure and eigenanalysis. PLoS Genet. 2 (12), e190. doi:10.1371/journal.pgen.0020190
Peng, W., Fu, C., Shu, S., Wang, G., Wang, H., Yue, B., et al. (2024). Whole-genome resequencing of major populations revealed domestication-related genes in yaks. BMC Genomics 25 (1), 69. doi:10.1186/s12864-024-09993-7
Pitt, D., Sevane, N., Nicolazzi, E. L., MacHugh, D. E., Park, S. D. E., Colli, L., et al. (2019). Domestication of cattle: two or three events? Evol. Appl. 12 (1), 123–136. doi:10.1111/eva.12674
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Purfield, D. C., Berry, D. P., McParland, S., and Bradley, D. G. (2012). Runs of homozygosity and population history in cattle. BMC Genet. 13, 70. doi:10.1186/1471-2156-13-70
Ramayo-Caldas, Y., Fortes, M. R. S., Hudson, N. J., Porto-Neto, L. R., Bolormaa, S., Barendse, W., et al. (2014). A marker-derived gene network reveals the regulatory role of PPARGC1a, HNF4G, and FOXP3 in intramuscular fat deposition of beef cattle. J. Anim. Sci. 92 (7), 2832–2845. doi:10.2527/jas.2013-7484
Ramírez-Zamudio, G. D., Ganga, M. J. G., Pereira, G. L., Nociti, R. P., Chiaratti, M. R., Cooke, R. F., et al. (2023). Effect of cow-calf supplementation on gene expression, processes, and pathways related to adipogenesis and lipogenesis in longissimus thoracis muscle of F1 Angus × Nellore cattle at weaning. Metabolites 13 (2), 160. doi:10.3390/metabo13020160
Randhawa, I. A. S., Khatkar, M. S., Thomson, P. C., and Raadsma, H. W. (2014). Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep. BMC Genet. 15, 34. doi:10.1186/1471-2156-15-34
R Core Team (2023). R: a language and environment for statistical computing. Vienna, Austria: Foundation for Statistical Computing.
Rudkowska, I., Pérusse, L., Bellis, C., Blangero, J., Després, J., Bouchard, C., et al. (2015). Interaction between common genetic variants and total fat intake on low-density lipoprotein peak particle diameter: a genome-wide association study. J. nutrigenetics nutrigenomics 8 (1), 44–53. doi:10.1159/000431151
Santana, M. H. A., Ventura, R. V., Utsunomiya, Y. T., Neves, H. H. R., Alexandre, P. A., Oliveira Junior, G. A., et al. (2015). A genomewide association mapping study using ultrasound-scanned information identifies potential genomic regions and candidate genes affecting carcass traits in Nellore cattle. J. animal Breed. Genet. = Zeitschrift fur Tierzuchtung und Zuchtungsbiologie 132 (6), 420–427. doi:10.1111/jbg.12167
Sbardella, A. P., Watanabe, R. N., da Costa, R. M., Bernardes, P. A., Braga, L. G., Baldi Rey, F. S., et al. (2021). Genome-wide association study provides insights into important genes for reproductive traits in Nelore cattle. Animals open access J. MDPI 11 (5), 1386. doi:10.3390/ani11051386
Sheet, S., Jang, S. S., Kim, J. H., Park, W., and Kim, D. (2024). A transcriptomic analysis of skeletal muscle tissues reveals promising candidate genes and pathways accountable for different daily weight gain in Hanwoo cattle. Sci. Rep. 14 (1), 315. doi:10.1038/s41598-023-51037-9
Shen, J., Hanif, Q., Cao, Y., Yu, Y., Lei, C., Zhang, G., et al. (2020). Whole genome scan and selection signatures for climate adaption in yanbian cattle. Front. Genet. 11, 94. doi:10.3389/fgene.2020.00094
Silva, D. B. S., Fonseca, L. F. S., Pinheiro, D. G., Magalhães, A. F. B., Muniz, M. M. M., Ferro, J. A., et al. (2020). Spliced genes in muscle from Nelore Cattle and their association with carcass and meat quality. Sci. Rep. 10 (1), 14701. doi:10.1038/s41598-020-71783-4
Sun, W. X., Wang, H. H., Jiang, B. C., Zhao, Y. Y., Xie, Z. R., Xiong, K., et al. (2013). Global comparison of gene expression between subcutaneous and intramuscular adipose tissue of mature Erhualian pig. Genet. Mol. Res. GMR 12 (4), 5085–5101. doi:10.4238/2013.October.29.3
Tatour, Y., Bar-Joseph, H., Shalgi, R., and Ben-Yosef, T. (2020). Male sterility and reduced female fertility in SCAPER-deficient mice. Hum. Mol. Genet. 29 (13), 2240–2249. doi:10.1093/hmg/ddaa113
Taye, M., Kim, J., Yoon, S. H., Lee, W., Hanotte, O., Dessie, T., et al. (2017). Whole genome scan reveals the genetic signature of African Ankole cattle breed and potential for higher quality beef. BMC Genet. 18 (1), 11. doi:10.1186/s12863-016-0467-1
Taye, M., Lee, W., Jeon, S., Yoon, J., Dessie, T., Hanotte, O., et al. (2017). Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mammalian genome official J. Int. Mammalian Genome Soc. 28 (11-12), 528–541. doi:10.1007/s00335-017-9715-6
Thorleifsson, G., Walters, G. B., Gudbjartsson, D. F., Steinthorsdottir, V., Sulem, P., Helgadottir, A., et al. (2009). Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 41 (1), 18–24. doi:10.1038/ng.274
Utsunomiya, Y. T., Milanesi, M., Fortes, M. R. S., Porto-Neto, L. R., Utsunomiya, A. T. H., Silva, M. V. G. B., et al. (2019). Genomic clues of the evolutionary history of Bos indicus cattle. Anim. Genet. 50 (6), 557–568. doi:10.1111/age.12836
Utsunomiya, Y. T., Milanesi, M., Utsunomiya, A. T. H., Torrecilha, R. B. P., Kim, E., Costa, M. S., et al. (2017). A PLAG1 mutation contributed to stature recovery in modern cattle. Sci. Rep. 7 (1), 17140. doi:10.1038/s41598-017-17127-1
Van Dyck, F., Braem, C. V., Chen, Z., Declercq, J., Deckers, R., Kim, B., et al. (2007). Loss of the PlagL2 transcription factor affects lacteal uptake of chylomicrons. Cell. Metab. 6 (5), 406–413. doi:10.1016/j.cmet.2007.09.010
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic. acids. Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, W., Liu, R., Liang, X., Zhao, Q., Qu, P., Yao, K., et al. (2018). Expression of IFNAR1 and IFNAR2 in cattle placenta during early pregnancy. Reproduction Domest. animals = Zuchthygiene 53 (2), 385–392. doi:10.1111/rda.13118
Wang, X., Chen, C., Wang, L., Su, Y., Li, B., Xiao, L., et al. (2022). Specific activation of embryonic IFNAR1 and endometrial IFNAR2 induced by embryonic IFNτ directs normal uterine fate for bovine early implantation. J. Reprod. Immunol. 153, 103677. doi:10.1016/j.jri.2022.103677
Wang, X., Wang, L., Shi, L., Zhang, P., Li, Y., Li, M., et al. (2022). GWAS of reproductive traits in large white pigs on chip and imputed whole-genome sequencing data. Int. J. Mol. Sci. 23 (21), 13338. doi:10.3390/ijms232113338
Wang, Z., Ma, H., Xu, L., Zhu, B., Liu, Y., Bordbar, F., et al. (2019). Genome-wide scan identifies selection signatures in Chinese Wagyu cattle using a high-density SNP array. Animals open access J. MDPI 9 (6), 296. doi:10.3390/ani9060296
Williams, M. J., Almén, M. S., Fredriksson, R., and Schiöth, H. B. (2012). What model organisms and interactomics can reveal about the genetics of human obesity. Cell. Mol. life Sci. CMLS 69 (22), 3819–3834. doi:10.1007/s00018-012-1022-5
Wu, J., Wu, T., Xie, X., Niu, Q., Zhao, Z., Zhu, B., et al. (2023). Genetic association analysis of copy number variations for meat quality in beef cattle. Foods Basel, Switz. 12 (21), 3986. doi:10.3390/foods12213986
Xia, X., Qu, K., Wang, Y., Sinding, M. S., Wang, F., Hanif, Q., et al. (2023). Global dispersal and adaptive evolution of domestic cattle: a genomic perspective. Stress Biol. 3 (1), 8. doi:10.1007/s44154-023-00085-2
Xia, X., Zhang, S., Zhang, H., Zhang, Z., Chen, N., Li, Z., et al. (2021). Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data. BMC Genomics 22 (1), 43. doi:10.1186/s12864-020-07340-0
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic. acids. Res. 39 (Web Server issue), W316–W322. doi:10.1093/nar/gkr483
Xu, Z., Wu, J., Zhou, J., Zhang, Y., Qiao, M., Sun, H., et al. (2022). Integration of ATAC-seq and RNA-seq analysis identifies key genes affecting intramuscular fat content in pigs. Front. Nutr. 9, 1016956. doi:10.3389/fnut.2022.1016956
Yang, C., Teng, J., Ning, C., Wang, W., Liu, S., Zhang, Q., et al. (2022). Effects of growth-related genes on body measurement traits in wenshang barred chickens. J. Poult. Sci. 59 (4), 323–327. doi:10.2141/jpsa.0210138
Yasukochi, Y., Sakuma, J., Takeuchi, I., Kato, K., Oguri, M., Fujimaki, T., et al. (2018). Identification of three genetic variants as novel susceptibility loci for body mass index in a Japanese population. Physiol. Genomics 50 (3), 179–189. doi:10.1152/physiolgenomics.00117.2017
Zhang, C., Dong, S., Xu, J., He, W., and Yang, T. (2019). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinforma. Oxf. Engl. 35 (10), 1786–1788. doi:10.1093/bioinformatics/bty875
Zhang, X., Qu, K., Jia, P., Zhang, J., Liu, J., Lei, C., et al. (2021). Assessing genomic diversity and productivity signatures in Dianzhong cattle by whole-genome scanning. Front. Genet. 12, 719215. doi:10.3389/fgene.2021.719215
Zhang, Z., Chen, Z., Ye, S., He, Y., Huang, S., Yuan, X., et al. (2019). Genome-wide association study for reproductive traits in a duroc pig population. Animals open access J. MDPI 9 (10), 732. doi:10.3390/ani9100732
Zhao, G. (2021). Association and Prediction of traits Based on genomie homozygous Segments in beef cattle: Chinese academy of agricultural. Sci. Thesis. doi:10.27630/d.cnki.gznky.2021.000654
Zhao, X., Qiu, Y., Meng, F., Zhuang, Z., Ruan, D., Wu, J., et al. (2024). Genome-wide association studies for loin muscle area, loin muscle depth and backfat thickness in DLY pigs. Anim. Genet. 55 (1), 134–139. doi:10.1111/age.13386
Keywords: Heilongjiang crossbred beef cattle, functional gene, genetic diversity, population genetic structure, whole genome
Citation: Li S, Liu L, Ahmed Z, Wang F, Lei C and Sun F (2024) Identification of Heilongjiang crossbred beef cattle pedigrees and reveals functional genes related to economic traits based on whole-genome SNP data. Front. Genet. 15:1435793. doi: 10.3389/fgene.2024.1435793
Received: 21 May 2024; Accepted: 08 July 2024;
Published: 25 July 2024.
Edited by:
Mario Barbato, University of Messina, ItalyReviewed by:
Arianna Bionda, University of Milan, ItalyCopyright © 2024 Li, Liu, Ahmed, Wang, Lei and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuzhao Lei, bGVpY2h1emhhbzExMThAMTI2LmNvbQ==; Fang Sun, aGxqeG1zZkAxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.