 Jiang Wu
Jiang Wu Yang Gao1
Yang Gao1 Jinyao Wang
Jinyao Wang Aihua Guo
Aihua Guo Sen Li
Sen Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 26 July 2024
Sec. Genomics of Plants and the Phytoecosystem
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1433548
This article is part of the Research TopicApplied Research on Plant Organelle Genomes and Key GenesView all 3 articles
Members of the genus Hemerocallis have significant value as ornamental, edible, and medicinal plants, particularly in China, where they have been utilized for thousands of years as both a vegetable and Traditional Chinese Medicine. Hemerocallis species exhibit strict control over flowering time, with individuals flowering either diurnally or nocturnally. However, our understanding of the evolutionary history of this genus, especially concerning important horticultural traits, remains limited. In this study, sequencing and assembly efforts were conducted on 73 samples within the Hemerocallis genus. All accessions were classified into two distinct groups based on their diurnal (daylilies) or nocturnal (nightlilies) flowering habits. Comparative analysis of the chloroplast genomes from these two groups identified fifteen variant hotspot regions, including fourteen SNPs and one deletion, which hold promise for the development of molecular markers for interspecific identification. Phylogenetic trees, generated through both maximum-likelihood and Bayesian inference methods using 76 shared protein-coding sequences, revealed that diurnal flowering evolved prior to nocturnal flowering. The divergence between the two groups is estimated to have occurred approximately 0.82 MYA (95% CI: 0.35–1.45 MYA). The ancestral state of Hemerocallis is hypothesized to have featured diurnal flowering with orange yellow petals. This study marks the first reconstruction of the evolutionary history and ancestral state of the genus Hemerocallis. The findings contribute significantly to our understanding of the adaptation and speciation history within the genus.
The species of Hemerocallis (Asphodelaceae: Hemerocallidoideae) (Group, 2016) are mainly distributed in East Asia, with China as the center of origin. These species exhibit extraordinary morphological diversity, particularly in the flowers of hybrid cultivars, and have therefore been embraced by ornamental plant breeders around the world (Stout, 1934). The pre-blossom flower buds of Hemerocallis citrina Barni have been utilized as a fresh and dried vegetable for thousands of years in China (Qing et al., 2021). In addition, the genus is considered to possess exceptional medical value in Traditional Chinese Medicine (Wang et al., 2018; Ma et al., 2023; Zhao et al., 2024).
All members of genus Hemerocallis exhibit two distinct features. The first is strict control over flower opening time, with flowers opening either diurnally or nocturnally (Kang and Chung, 2000; Hasegawa et al., 2006; Ren et al., 2021). The second is that the blossom duration for individual flowers never exceeds 24 h (Rodriguez-Enriquez and Grant-Downton, 2013; Ren et al., 2021; Yang et al., 2021). These peculiarities have earned the daytime flowering members the moniker “daylily,” and likewise, the nighttime flowering members the moniker “nightlily” (Li et al., 2021). Among the 11 wild Hemerocallis species native to China, eight bloom diurnally (Hemerocallis fulva, H. esculenta, H. nana, H. dumortieri, H. middendorfii, H. multiflora, H. plicata, and H. forrestii) and three bloom nocturnally (H. lilioasphodelus, H. citrina, and H. minor) (Hirota et al., 2021). In general, diurnal daylilies bloom between 4:30 and 7:30 UTC, while nocturnal nightlilies bloom between 16:30 and 20:30 UTC (Hasegawa et al., 2006).
Because both groups bloom during completely different intervals, they are preferentially pollinated by different insects (Hasegawa et al., 2006; Yasumoto and Yahara, 2006). Despite this, the two groups are not reproductively isolated, and fertile F1 generations can be produced following pollination (Stout and Chandler, 1933; Kawano, 1961; Stout, 1986; Hasegawa et al., 2006). Research into the evolutionary history of genus Hemerocallis suggests that the differentiation of flowering time-associated traits may be an internal mechanism for maintaining genetic diversity. Variation in flowering time is directly related to environmental adaptation, and therefore understanding the molecular mechanism underlying variation in flowering time is crucial for understanding Hemerocallis adaptation, reproductive isolation, and domestication (Stinchcombe et al., 2004; Kollmann and Bañuelos, 2004; Kawakami et al., 2011; Dijk and Hautekèete, 2014; Gaudinier and Blackman, 2019). However, the whole evolutionary history of genus Hemerocallis is still poorly resolved, particularly those events pertaining to the development of important traits.
Chloroplasts, key centers of photosynthesis and biosynthesis within plant cells, are semi-autonomous organelles possessing autonomous genetic systems (Sugiura, 1995). Current evidence suggests that chloroplasts originated from endosymbiotic cyanobacteria (Zhang et al., 2023). In most angiosperms, the chloroplast genome has a low recombination rate and is passed to the next-generation as a complete haplotype. Compared to the nuclear and mitochondrial genomes, the chloroplast genome exhibits a moderate nucleotide replacement rate. Due to these characteristics, the chloroplast genome is well-suited for phylogenetic reconstruction and evolutionary route tracing, particularly among closely-related species or members of the same population. Over the past decade, the chloroplast genome has become an important resource for phylogenetics and divergence time estimation (Sugiura, 1995).
A phylogenetic tree that accurately represents the relationships among members of the genus Hemerocallis is crucial for comprehending the origin and evolutionary history of important traits. In this study, we utilized the chloroplast genome to construct phylogenetic relationships within the genus Hemerocallis, aiming to elucidate the relationships among branches. Subsequently, we examined the variations and hotspot mutation regions among groups. Additionally, we identified differentiation events and reconstructed the ancestral traits of Hemerocallis, highlighting the importance of adaptation associated with flowering time during the evolutionary history of this genus. The findings of this study will enhance our understanding of the evolutionary trajectory of Hemerocallis and holds significance for the genetic enhancement and breeding of germplasm resources.
All materials used in this study were collected from plants cultivated at the experimental orchard located at Shanxi Agriculture University. The majority of the samples were sourced from China, with some originating from other countries and regions. Due to the inherent instability and inconsistency of horticultural traits in newly-transplanted plants, all re-sequenced accessions were selected from plants that had been transplanted for more than 3 years and exhibited stable and consistent traits. After years of phenotypic evaluation and statistical analysis, a total of 73 accessions were selected (Supplementary Table S1). Genomic DNA was extracted using the plant genomic DNA kit (TaKaRa, Kusatsu, Japan). DNA quality was assessed using a Nanodrop 2000 (Thermo Scientific, Waltham, MA, United States) and by 1% (w/v) agarose gel electrophoresis. Libraries were constructed and sequenced in 150 bp paired-end reads using the Illumina HiSeq 2000 platform at the Genedio Company (Guangzhou, China).
For each sample, approximately 1G of reads were extracted from the clean data to assemble the chloroplast genome. We employed GetOrganelle 1.7.5 (Jin et al., 2018) for de novo assembly with recommended parameters, using our previously assembled chloroplast genome as reference (NC_064967.1). Novowrap (Wu et al., 2021) was utilized to adjust the direction of the inverted repeat (IR) region and the starting site of the newly assembled genome. To validate circularization of assembled sequences, Bandage v0.9.0 (Wick et al., 2015) was employed. In cases of incompletely assembled genomes, SAMtools v1.20 (Danecek et al., 2021) was used to obtain the bam file at the breakpoint to further verify the read depth. Subsequently, the chloroplast genomes were submitted to CPGAVS2 (Shi et al., 2019) for annotation, combined with manual corrections by Geneious Prime v2021. Annotation of tRNAs was conducted using tRNAscan-SE v2.0 (Chan et al., 2021). The complete chloroplast genome was visualized using OGDRAW v1.3.1 (Lohse et al., 2007). All chloroplast genomes were deposited in GenBank (Supplementary Table S1).
The phylogenetic tree was constructed using the whole chloroplast genome. To avoid impacts resulting from poor alignments caused by gaps, the intergenic sequences between trnT-UGU and trnL-UAA were removed from all sample. Then samples with identical genome sequences were filtered, retaining only one representative for further analysis. Subsequently, the sequences were aligned using Mafft v7.487 (Katoh et al., 2002). The dataset were inspected manually and automatically trimmed using trimAl v2.0 (Capella-Gutierrez et al., 2009). jModeltest v2.1.10 (Darriba et al., 2012) was employed to identify the best nucleotide evolution model, selected based on the Akaike Information Criterion (AIC). IQ-TREE2 (Minh et al., 2020) was utilized to build the ML tree, specifying the molecular substitution models GTR + I + G, with the bootstrap set to 1,000 cycles. The final result was visualized using FigTree (http://beast.bio.ed.ac.uk/FigTree).
The IRscope (Amiryousefi et al., 2018) online tool was employed to analyze and visualize the contraction and expansion of the IR regions and the SSC (short single copy) region. Variant hotspot regions were identified by aligning all samples using mVISTA v2.0 (Frazer et al., 2004) with the LAGAN (Brudno et al., 2003) alignment program, using S129 as the reference sequence. Nucleotide polymorphism was computed by DnaSP v6.12.03 (Rozas et al., 2017), with window and step sizes set to 500bp and 50bp, respectively. The results were visualized using R. To further analyze the distribution of variants between the two clades, Geneious was utilized to find the location of all variants and calculate the variant counts among different samples.
In order to avoid interference resulting from interspecific hybridization, we developed a new dataset consisting of nine representative samples belonging to six Hemerocallis species. Four Asparagales species served as outgroups. Phylogenetic relationships were evaluated based on whole-genome sequences. With the inclusion of outgroups, after aligning the sequences in MAFFT, the Phylosuite v1.2.3 (Zhang et al., 2020) module “treesuite” was utilized for sequence substitution saturation analysis to assess the suitability of the data for phylogenetic reconstruction. Both the maximum-likelihood (ML) and bayesian inference (BI) methods were used, and analysis of the ML tree was the same as that of the unrooted tree. Bayesian phylogenetic analysis was executed using MrBayes v3.1.2 (Ronquist et al., 2012), applying the best fit model identified according to the AIC as determined by jModeltest2. The Markov Chain Monte Carlo (MCMC) analysis was run for 1,000,000 generations, with trees sampled every 1,000 generations, “lset nst” set to “6,” “rates” set to “invgamma,” and two MCMC chains run independently. Each chain began with a random tree with default prior values and was sampled every 100 generations, with the top 25% discarded as burn-in. The operation was deemed stable and then terminated automatically when the aggregate diagnostic value reached ≤ 0.01 (stopval = 0.01). Tracer (Lee et al., 2021) was used to evaluate convergence. Both ML and BI trees were visualized using Figtree.
MCMCtree was employed in PAML v4.7 (Yang, 2007) to estimate the speciation time among members of Hemerocallis. The topological structure obtained as described above served as the input tree file. Given the absence of a fossil record for the study species, the molecular clock was calibrated twice based on information from the TimeTree database (http://www.timetree.org/) (Kumar and Hedges, 2017). Initially, the tree was calibrated by fixing the crown origin at 71.9 MYA. Subsequently, the occurrence of Asphodelaceae was constrained to the time range of 52.2–67.4 MYA.
In order to reconstruct the ancestral status of Hemerocallis, we retained the tree derived from BI analysis to infer the flowering time and petal color of the common ancestor. The maximum parsimony (MP) method was conducted in Mesquite v3.81 (Chung-hau et al., 2006) to reconstruct the evolutionary history of the two phenotypic traits. The character states were treated as unordered and coded as follows: 0, yellow; 1, golden; 2, orange yellow; 3, reddish orange; 0, daylily; 1, nightlily. The data for each morphological trait were obtained from both published literature (Li et al., 2021) and the field observations conducted by our research group. The data matrix can be found in Supplementary Table S8.
Of the 73 samples assembled, 26 resulted in complete chloroplast genomes while 47 remained uncyclized. We used Bandage to visualize the imcomplete genome and found that all samples exhibited discontinuities in the large single copy (LSC) region. Further examination of the annotation files revealed that gaps in the chloroplast genome were consistently appearing in the trnT-UGU and trnL-UAA intergenic region. The complete chloroplast genomes ranged in length from 156052 to 156102 base pairs (bp) (Figure 1). The LSC regions varied from 18501 to 18531bp, the SSC regions ranged from 84804 to 84843bp, and IR measured 26369bp in length (Supplementary Table S3).
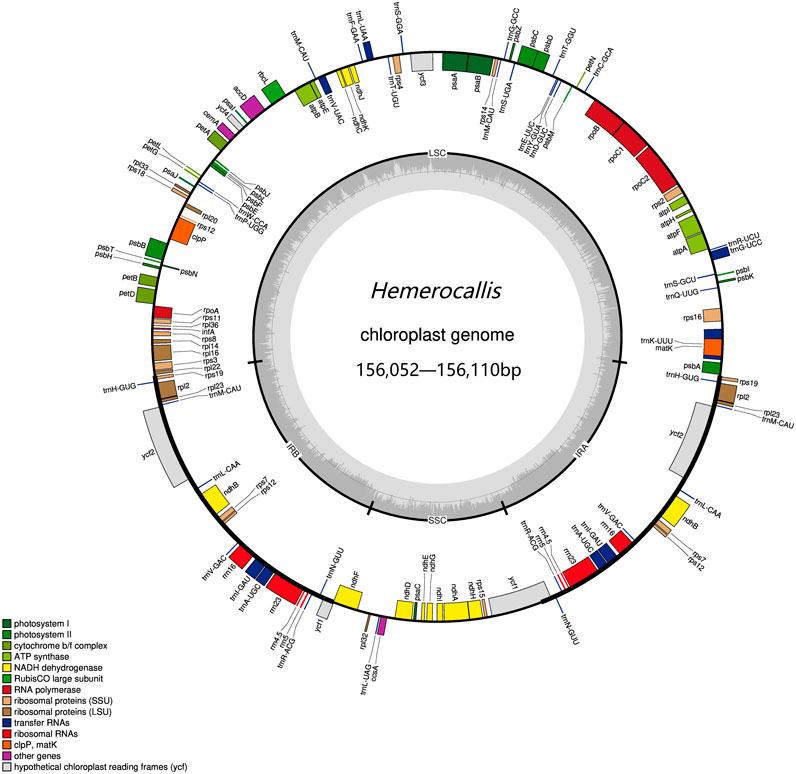
Figure 1. The Hemerocallis chloroplast genome. On the outer circle, the inner genes were transcribed counterclockwise and the outer genes were transcribed clockwise. On the inner circle, the dark gray bars indicated the GC content.
The Hemerocallis chloroplast genome contains 110 genes, including 78 protein-coding, 4 rRNA, and 28 tRNA genes. Among these, 19 genes were duplicated in the inverted repeat regions, encompassing eight tRNA, four rRNA, and seven protein-coding genes. Nine genes (atpF, rpoC1, petB, petD, rps16, trnG-UCC, trnK-UUU, trnL-UAA, and trnV-UAC) contain a single intron, while two genes (clpP and ycf3) contain two introns (Supplementary Table S2). No gene rearrangements were observed in any of the chloroplast genomes (complete and incomplete), and the number of genes remained relatively conserved across samples. The overall guanine-cytosine (GC) content ranged from 37.21% to 38.45%, with the LSC region containing 35.08% GC, the SSC region containing 31.96% GC, and the IR region containing 42.87% GC (Supplementary Table S3). Overall, these results are consistent with previous reports (Jia et al., 2024).
After removing duplicate genome sequences (Supplementary Table S4), a total of 30 samples remained. An unrooted ML trees and BI tree were constructed using the whole chloroplast genome as input data, and the main branch bootstrap values were all greater than 90 (Figure 2). Considering the trait of flowering time, the phylogenetic tree can be broadly divided into two groups, with most samples within each group exhibiting consistent flowering characteristics. The diurnal flowering group included H. fulva (S26), H. middendorffii (S41), H. multiflora (S45), and H. fulva var. aurantiaca (S40). Meanwhile, the nocturnal flowering group included H. lilioasphodelus (S8), H. minor (S7), H. citrina var. altissima (S46) and H. citrina (S6, S128, etc.). Notably, the diurnal flowering group also included a nocturnal flowering samples (S182), while the nocturnal flowering group included three diurnal flowering sample (S140, S40, and S198). Among these four samples, all of them were cultivars.
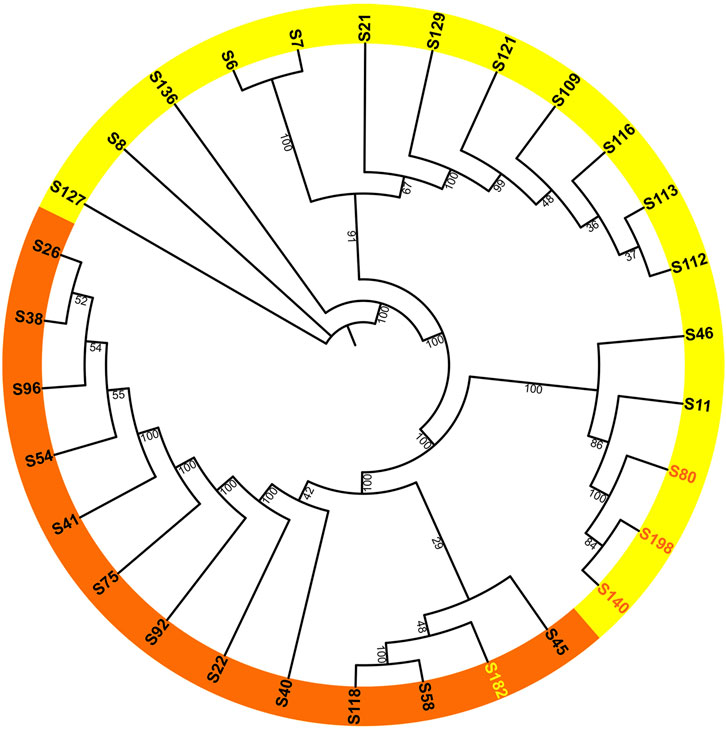
Figure 2. Maximum-likelihood phylogenetic reconstruction of 30 Hemerocallis accessions based on chloroplast genomes. The daylilies are highlighted in orange, while the nightlilies are highlighted in yellow. Bootstrap values are derived from 1,000 bootstrap replicates.
The results from IRscope (Supplementary Figure S1) analysis indicated that there were no significant differences in the lengths of the IRs and SSC regions between daylilies and nightlilies. The length difference of the ycf1 gene located at the IRa and SSC boundary region was minimal, while the lengths of the ycf1 and ndhF genes located at the IRb and SSC boundary region were identical between the two groups. Examination of the results from mVISTA revealed that non-coding regions of Hemerocallis chloroplast genomes exhibited higher polymorphism than coding regions, consistent with observations in most plants (Figure 3). According to calculations from DnaSP, the IR region being the most conservative (Pi value: 0.000361), the SSC region exhibiting the highest polymorphism (Pi value: 0.002577), and the LSC region having a medium level of polymorphism (Pi value: 0.001582). Additionally, the figure displayed the top ten nucleotide polymorphism sites on the genome, which served as hotspot regions of variation. Among them, seven peaks were located in intergenic regions (trnS-GGA_trnG-UCC, psaA_ycf3, psbE_petL, ndhF_rpl32, rpl32_trnL-UAG, psaC_ndhE, rps15_ycf1), while the remaining three peaks were located within gene sequences (ycf3, rpl32, rps15) (Figure 4; Supplementary Table S5). Based on the annotation file of the reference genome, fifteen variants specific to the Daylily group were identified, including fourteen SNP variants and one deletion (Supplementary Table S6). These variant sites could be further developed as molecular markers for distinguishing.
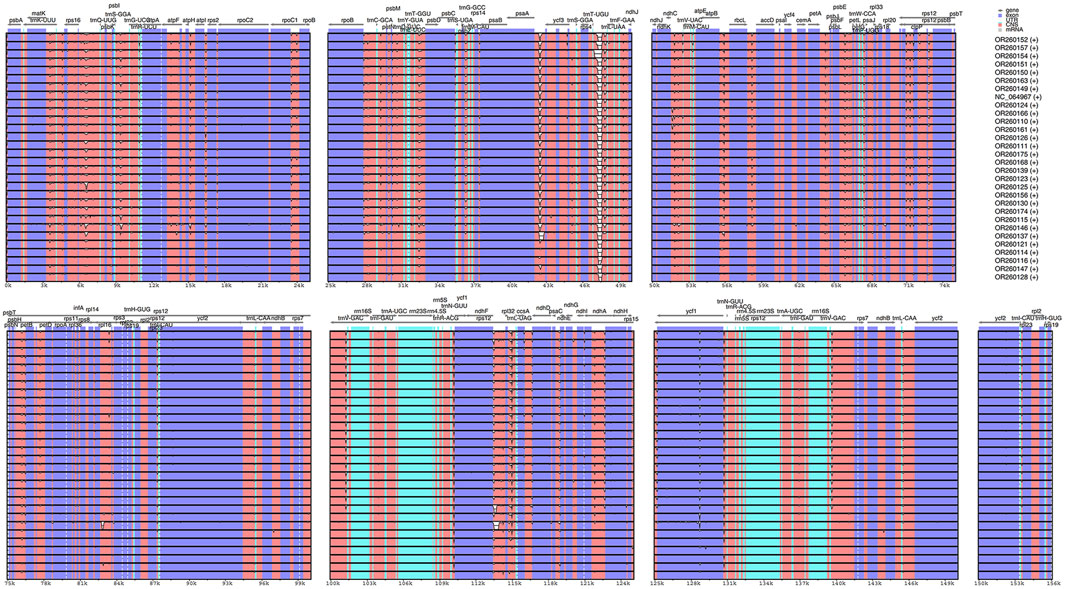
Figure 3. Sequence alignment of the plastome sequences of 30 Hemerocallis samples with OR260163 (S129) as reference. The gray arrows indicate the direction of genes. Blue and white correspond to coding regions and non-coding regions, respectively.
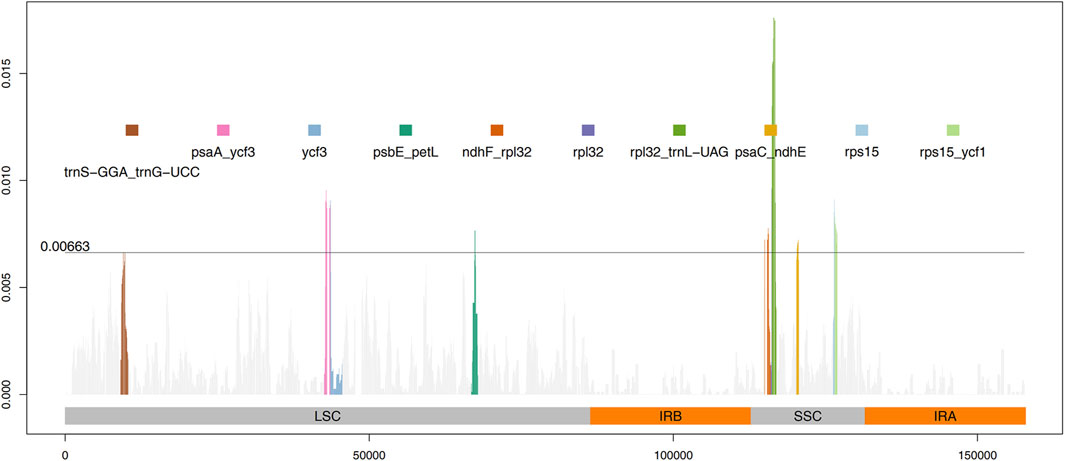
Figure 4. Sliding window calculation of nucleotide diversity (Pi) in the Hemerocallis chloroplast genomes with window width 500 bp and step size 50 bp. The X-axis represents the position of the midpoint of a window, and the Y-axis represents the nucleotide diversity of each window. The threshold line indicates the 10th peak of Pi value.
Both ML and BI trees were constructed using 76 shared protein-coding sequences (Supplementary Figure S3). Overall, the phylogenetic topologies obtained using the two methods were essentially identical, with high support at most nodes. Hemerocallis and Xanthorrhoea, two genera in subfamily Hemerocallidoideae, first clustered together and then clustered with members of family Asphodelaceae. The genus of Hemerocallis contained two sub-groups: daylily and nightlily. The daylily group compromised H. fulva (S27) and its variety (S40), H. multiflora (S45), and H. middendorfii (S41), which all exhibited daytime flowering. On the other hand, the nightlily group consisted of H. lilioasphodelus (S8), H. minor (S7) and two cultivars of H. citrina (S6, S128), all of which bloom at night. Meanwhile, the daylily was positioned closer to the root. These results suggest that nightlilies were derived from daylilies.
In order to reconstruct the divergence history of the two Hemerocallis groups, we performed a time divergence analysis based on a phylogenetic tree with multiple calibrations (Figure 5). The MCMCtree results revealed that the common ancestor of Hemerocallis occurred during the late Paleocene, approximately 56.13MYA (95% CI: 51.31–63.54 MYA). Differentiation occurred gradually during the second stage of the Miocene, approximately 19.2 MYA, which was somewhat earlier than previously reported (Hirota et al., 2021). The ancestor split into the daylily and nightlily at approximately 0.82 MYA (95% CI: 0.35–1.45MYA). Notably, the daylilies emerged earlier than the nightlilies. The divergence of multiple species within Hemerocallis mainly occurred within the time range approximately 1.92–0.03MYA. This result is fairly consistent with previously report (Hirota et al., 2021).
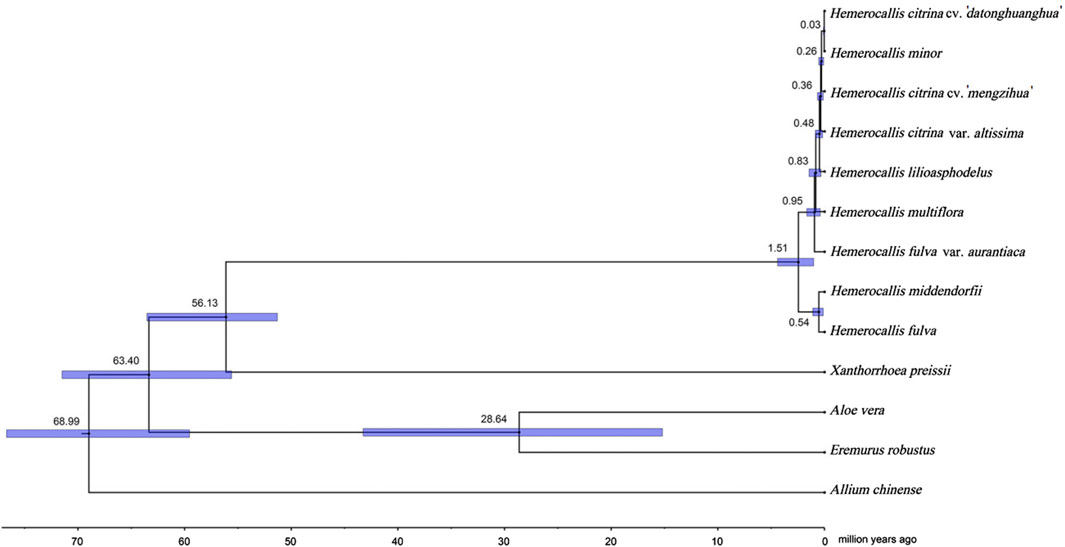
Figure 5. Hemerocallis divergence time estimation based on Bayesian inference tree. Divergence time analysis was conducted using MCMCtree. All estimates are shown with their 95% confidential intervals (95% CI) highlighted in blue.
To further investigate population differentiation within Hemerocallis, we explored evolutionary trends associated with the flowering time and petal color traits (Figure 6). The ancestral state was reconstructed using the Cp genome-based BI tree (Supplementary Figure S3). The ancestor of Hemerocallis most likely had orange yellow petals and flowered diurnally, suggesting that nocturnal flowering and yellow evolved later.
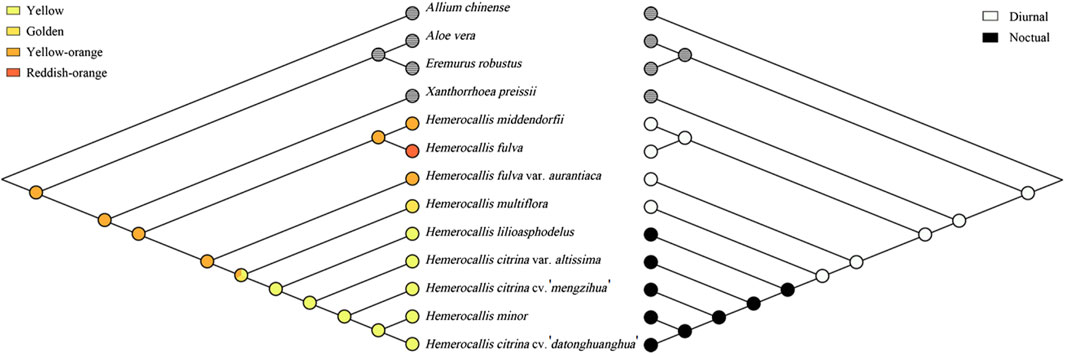
Figure 6. Ancestral states of petal color (left) and flowering time (right). Trees were reconstructed in Mesquite using the maximum parsimony method. Each trait was coded as follows: 0, yellow; 1, golden; 2, orange yellow; 3, reddish orange; 0, daylily; 1, nightlily. Traits for outgroups were left blank.
The Hemerocallis chloroplast genome was relatively conserved, characterized by a typical quadripartite structure consisting of LSC region, one SSC region, and two IRs. In all incompletely assembled genomes, breakpoints occurred at the same position. This outcome was primarily attributed to the GC bias of certain second-generation sequencing platforms (Browne et al., 2020). According to statistical analysis, the GC content in the LSC region of Hemerocallis chloroplast genomes was notably low (17.27%–17.44%) (Supplementary Table S3), resulting in low coverage of reads in this region and leading to gaps. Similar occurrences have also been noted during the assembly of chloroplast genomes in other species (Xue et al., 2012; Wang et al., 2013; Chen et al., 2023b).
Modern Hemerocallis cultivars have originated from hybridization among a limited number of species, which subsequently served as the parental lines for many ornamental cultivars (Tomkins et al., 2001). Due to the maternal inheritance of chloroplast genomes in Hemerocallis, and flowering time is controlled by genes of the nuclear genome (Chaves-Silva et al., 2018; Sunil and Shetty, 2022), classification based on flowering time traits may not align perfectly with molecular sequences. For instance, in our study, some cultivars (e.g., S198, S140, S40, and S182) were all misclassified when considering only chloroplast data, suggesting the need to combine nuclear genome data for accurate classification.
Intensive breeding has significantly reduced genetic diversity (Rodriguez-Enriquez and Grant-Downton, 2013). In this study, the Pi value of the whole chloroplast genome for all samples was only 0.00128, further confirming the narrowing of genetic diversity due to artificial selection pressure. Despite the high variability in floral traits among cultivars, they shared the same chloroplast genome (Supplementary Figure S4), a trend particularly evident in ornamental varieties. However, the phylogenetic structure generally aligns with flowering time traits, further supporting the rationality of subgrouping Hemerocallis into daylilies and nightlilies after removing hybrid species interference (Supplementary Figure S3).
Comparative analysis revealed no significant differences in the IR/SSC boundary positions between daylilies and nightlilies, suggesting minimal evolutionary shifts in these regions. Non-coding regions exhibited higher polymorphism compared to coding regions, consistent with most plants (Chen et al., 2023a; Li et al., 2024). Additionally, fifteen variations showed significant differences between daylilies and nightlilies. According to the reference genome annotation file, four SNPs were located within genes (ycf1, ycf2, ycf4) (Supplementary Table S6), indicating that these genes might have undergone strong selection pressure over the course of evolution. These variations might have a critical influence on the flowering time in Hemerocallis. These variations could serve as potential molecular markers for the identification of Hemerocallis species, aiding the breeding of new Hemerocallis cultivars with desired traits, and assisting in conservation efforts by identifying genetic diversity hotspots.
Our study included most wild species within the Hemerocallis, providing a relatively accurate reflection of the phylogenetic relationship between daylilies and nightlilies. ML and BI trees shared the same topology, indicating that nightlilies evolved from daylilies. These findings are consistent with previous results (Hirota et al., 2021). Continuous variation in external morphology among different species complicates classification, as seen with H. lilioasphodelus, H. minor, and H. citrina. They share highly similar external morphologies and karyotypes, leading some to suggest treating H. minor and H. citrina as two subspecies of H. lilioasphodelus (Zhi-Ting et al., 1996; Zhi-Ting and Singchi, 1997). Our topology suggests that H. lilioasphodelus appeared first within the nightlily group. However, the evolutionary relationship between H. minor and H. citrina cv. ‘datonghuanghua’ was closer, with both appearing around 0.23MYA, later than H. citrina cv. ‘mengzihua’. H. citrina cv. ‘mengzihua’ is a well-known landrace of H. citrina in Hunan Province, China, while H. citrina cv. ‘datonghuanghua’ is another landrace in Shanxi Province, China, both with a long cultivation history. According to literature records, wild H. citrina is mainly distributed south of the Qinling Mountains, while wild H. minor is more prevalent in northern and northeastern China (Wu, Raven, and Hong, 2000). Therefore, we speculate that H. citrina cv. ‘datonghuanghua’ might originally belong to H. minor, although this conjecture required further verification.
The divergence time analysis indicated a rapid species expansion during the Pleistocene, a period characterized by significant climatic fluctuations. These environmental changes likely exerted selective pressures that drove trait divergence within the genus. For example, the shift from diurnal to nocturnal flowering may have been a strategy to avoid competition for pollinators or to exploit different pollinator species active at night. This adaptive flexibility would have been advantageous in the face of varying Pleistocene climates, promoting speciation and diversification within the genus.
Reconstructing the evolutionary history of Hemerocallis is crucial for understanding key traits. Our study utilized outgroups to root the phylogenetic tree and inferred that the ancestral state featured orange-yellow petals and diurnal flowering. This ancestral trait set provides significant insights into the adaptive strategies and ecological niches occupied by early Hemerocallis species.
However, our current understanding is based on a limited number of species, which constrains the robustness of our conclusions. The limited sampling could overlook key variations and evolutionary events that are critical to fully understanding the evolutionary trajectory of Hemerocallis. For instance, incorporating more wild species, especially those from underrepresented regions or habitats, could reveal additional adaptive traits and evolutionary pathways. Furthermore, integrating data from nuclear genomes alongside chloroplast genomes could provide a more comprehensive view of the evolutionary dynamics. Nuclear genomes, which are subject to different selective pressures and inheritance patterns compared to chloroplast genomes, can offer insights into gene flow, hybridization events, and the role of polyploidy in the evolution of Hemerocallis. Future studies should also consider the ecological interactions between Hemerocallis species and their pollinators, herbivores, and symbiotic partners. Understanding these interactions can shed light on the selective pressures that have shaped the evolution of key traits.
In this study, we selected a total of 74 samples to construct the phylogenetic relationships, further confirming the division of Hemerocallis into daylilies and nightlilies. Comparative analysis of the chloroplast genomes of the two groups revealed a relatively conserved chloroplast structure and boundary regions of the two IRs. Fifteen variant sites were identified, which showed group-specific characteristics and could serve as molecular markers for identifying daylilies and nightlilies in future studies. The phylogenetic relationships with outgroups not only reaffirmed the earlier appearance of the daylily group compared to the nightlily group but also indicated that Hemerocallis underwent evolutionary radiation in its long history. The ancestral state was inferred to feature diurnal flowering and orange yellow petals. To our knowledge, this is the first prediction of the ancestral state of Hemerocallis, and the identification of molecular markers for the two groups can also provide a basis for molecular breeding.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
JaW: Writing–original draft, Writing–review and editing. YG: Data curation, Software, Writing–original draft. JnW: Data curation, Writing–original draft. AG: Validation, Writing–original draft. NQ: Resources, Writing–review and editing. GX: Funding acquisition, Supervision, Writing–original draft. SL: Project administration, Supervision, Writing–original draft.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was co-supported by Research and Demonstration of Key Technologies for Conservation, Utilization, and Industrial Upgrading of Hemerocallis Germplasm (202102140601009) and Research and Application Demonstration of Key Technologies in the Daylily Industry (2021YFD1600301).
We are grateful to Dr. Zhiqiang Wu who works in Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences for his help in revising this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1433548/full#supplementary-material
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi:10.1093/bioinformatics/bty220
Browne, P. D., Nielsen, T. K., Kot, W., Aggerholm, A., Gilbert, M. T. P., Puetz, L., et al. (2020). GC bias affects genomic and metagenomic reconstructions underrepresenting GC-poor organisms. GigaScience 9, giaa008. doi:10.1093/gigascience/giaa008
Brudno, M., Do, C. B., Cooper, G. M., Kim, M. F., Davydov, E., Green, E. D., et al. (2003). LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13, 721–731. doi:10.1101/gr.926603
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi:10.1093/bioinformatics/btp348
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi:10.1093/nar/gkab688
Chaves-Silva, S., Santos, A. L. D., Chalfun-Júnior, A., Zhao, J., Peres, L. E. P., and Benedito, V. A. (2018). Understanding the genetic regulation of anthocyanin biosynthesis in plants - tools for breeding purple varieties of fruits and vegetables. Phytochemistry 153, 11–27. doi:10.1016/j.phytochem.2018.05.013
Chen, J., Wang, F., Zhao, Z., Li, M., Liu, Z., and Peng, D. (2023a). Complete chloroplast genomes and comparative analyses of three Paraphalaenopsis (Aeridinae, Orchidaceae) species. Int. J. Mol. Sci. 24, 11167. doi:10.3390/ijms241311167
Chen, J., Zang, Y., Shang, S., Yang, Z., Liang, S., Xue, S., et al. (2023b). Chloroplast genomic comparison provides insights into the evolution of seagrasses. BMC Plant Biol. 23, 104. doi:10.1186/s12870-023-04119-9
Chung-hau, L., Sigal, B., Arne Ø, M. A., Ambuj, S., and Oakley, T. H. (2006). CoMET: a Mesquite package for comparing models of continuous character evolution on phylogenies. Evol. Bioinforma. Online 2, 183–186. doi:10.1177/117693430600200021
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. doi:10.1093/gigascience/giab008
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi:10.1038/nmeth.2109
Dijk, H. V., and Hautekèete, N. (2014). Evidence of genetic change in the flowering phenology of sea beets along a latitudinal cline within two decades. J. Evol. Biol. 27, 1572–1581. doi:10.1111/jeb.12410
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi:10.1093/nar/gkh458
Gaudinier, A., and Blackman, B. K. (2019). Evolutionary processes from the perspective of flowering time diversity. New Phytol. 225, 1883–1898. doi:10.1111/nph.16205
Group, T. A. P. (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical J. Linn. Soc. 181, 1–20. doi:10.1111/boj.12385
Hasegawa, M., Yahara, T., Yasumoto, A., and Hotta, M. (2006). Bimodal distribution of flowering time in a natural hybrid population of daylily (Hemerocallis fulva) and nightlily (Hemerocallis citrina). J. Plant Res. 119, 63–68. doi:10.1007/s10265-005-0241-3
Hirota, S. K., Yasumoto, A. A., Nitta, K., Tagane, M., Yahara, T., Suyama, Y., et al. (2021). Evolutionary history of Hemerocallis in Japan inferred from chloroplast and nuclear phylogenies and levels of interspecific gene flow. Mol. Phylogenetics Evol. 164, 107264. doi:10.1016/j.ympev.2021.107264
Jia, M., Wang, J., Cao, D., Jiang, C., Li, W., Tembrock, L. R., et al. (2024). The pan-plastome of Hemerocallis citrina reveals new insights into the genetic diversity and cultivation history of an economically important food plant. BMC Plant Biol. 24, 44. doi:10.1186/s12870-023-04668-z
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., Li, D. Z., Yi, T. S., et al. (2018). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 1, 241. doi:10.1186/s13059-020-02154-5
Kang, S. S., and Chung, M. G. (2000). High levels of allozyme variation within populations and low allozyme divergence within and among species of Hemerocallis (Liliaceae). Am. J. Bot. 87, 1634–1646. doi:10.2307/2656740
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi:10.1093/nar/gkf436
Kawakami, T., Morgan, T. J., Nippert, J. B., Ocheltree, T. W., Ungerer, M. C., Dhakal, P., et al. (2011). Natural selection drives clinal life history patterns in the perennial sunflower species, Helianthus maximiliani. Mol. Ecol. 20, 2318–2328. doi:10.1111/j.1365-294X.2011.05105.x
Kawano, S. (1961). On the natural hybrid population of Hemerocallis. Can. J. Bot. 39, 667–681. doi:10.1139/b61-054
Kollmann, J., and Bañuelos, M. J. (2004). Latitudinal trends in growth and phenology of the invasive alien plant Impatiens glandulifera (Balsaminaceae). Divers. Distribution 10, 377–385. doi:10.1111/j.1366-9516.2004.00126.x
Kumar, S., and Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Syst. Appl. Microbiol. 40, 256–259. doi:10.1016/j.syapm.2017.01.004
Lee, M. S., Shin, W. S., and Han, S. W. (2021). TRACER: extreme attention guided salient object tracing network. Syst. Biol. 70, syy032. doi:10.1093/sysbio/syy032
Li, L., Wu, Q., Zhai, J., Wu, K., Fang, L., Li, M., et al. (2024). Comparative chloroplast genomics of 24 species shed light on the genome evolution and phylogeny of subtribe Coelogyninae (Orchidaceae). BMC Plant Biol. 24, 31. doi:10.1186/s12870-023-04181-y
Li, S., Ji, F., Hou, F., Shi, Q., Xing, G., Chen, H., et al. (2021). Morphological, palynological and molecular assessment of Hemerocallis core collection. Sci. Hortic. 285, 110181. doi:10.1016/j.scienta.2021.110181
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi:10.1007/s00294-007-0161-y
Ma, T., Sun, Y., Lin, J., Wang, J., Zhang, X., Yan, T., et al. (2023). Chemical constituents and mechanisms from Hemerocallis citrina Baroni with anti-neuroinflammatory activity. J. Funct. Foods 102, 105427. doi:10.1016/j.jff.2023.105427
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi:10.1093/molbev/msaa015
Qing, Z., Liu, J., Yi, X., Liu, X., Zeng, J., Lao, J., et al. (2021). The chromosome-level Hemerocallis citrina Borani genome provides new insights into the rutin biosynthesis and the lack of colchicine. Hortic. Res. 8, 89. doi:10.1038/s41438-021-00539-6
Ren, Y., Gao, Y., and Zhang, Q. (2021). Morning and evening alarm of the circadian clock for flower opening times in Hemerocallis. Plant Sci. 311, 110992. doi:10.1016/j.plantsci.2021.110992
Rodriguez-Enriquez, M. J., and Grant-Downton, R. T. (2013). A new day dawning: Hemerocallis (daylily) as a future model organism. AoB Plants 5, pls055. doi:10.1093/aobpla/pls055
Ronquist, F., Teslenko, M., Mark, P. V. D., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi:10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi:10.1093/molbev/msx248
Shi, L., Chen, H., Jiang, M., Wang, L., Wu, X., Huang, L., et al. (2019). CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73. doi:10.1093/nar/gkz345
Stinchcombe, J. R., Weinig, C., Ungerer, M., Olsen, K. M., Mays, C., Halldorsdottir, S. S., et al. (2004). A latitudinal cline in flowering time in Arabidopsis thaliana modulated by the flowering time gene FRIGIDA. PNAS 101, 4712–4717. doi:10.1073/pnas.0306401101
Stout, A. B. (1986). The wild species and garden clones, both old and new, of the genus Hemerocallis. New York: American Hemerocallis Society.
Stout, A. B., and Chandler, C. (1933). Pollen-tube behavior in Hemerocallis with special reference to incompatibilities. Bull. Torrey Botanical Club 60, 397–416. doi:10.2307/2480493
Sunil, L., and Shetty, N. P. (2022). Biosynthesis and regulation of anthocyanin pathway genes. Appl. Microbiol. Biotechnol. 106, 1783–1798. doi:10.1007/s00253-022-11835-z
Tomkins, J. P., Wood, T. C., Barnes, L. S., Westman, A., and Wing, R. A. (2001). Evaluation of genetic variation in the daylily (Hemerocallis spp.) using AFLP markers. Theor. Appl. Genet. 102, 489–496. doi:10.1007/s001220051672
Wang, J., Hu, D., Hou, J., Li, S., Wang, W., Li, J., et al. (2018). Ethyl acetate fraction of Hemerocallis citrina baroni decreases tert-butyl hydroperoxide-induced oxidative stress damage in BRL-3A cells. Oxidative Med. Cell. Longev. 2018, 1526125–1526213. doi:10.1155/2018/1526125
Wang, S., Shi, C., and Gao, L. Z. (2013). Plastid genome sequence of a wild woody oil species, prinsepia utilis, provides insights into evolutionary and mutational patterns of rosaceae chloroplast genomes. PLOS ONE 8, e73946. doi:10.1371/journal.pone.0073946
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi:10.1093/bioinformatics/btv383
Wu, P., Xu, C., Chen, H., Yang, J., Zhang, X., and Zhou, S. (2021). NOVOWrap: an automated solution for plastid genome assembly and structure standardization. Mol. Ecol. Resour. 6, 2177–2186. doi:10.1111/1755-0998.13410
Wu, Z. Y., Raven, P. H., and Hong, D. Y. (2000). Flora of China, 24. Beijing: Science Press: St. Louis: Missouri Botanical Garden Press.
Xue, J. H., Wang, Y., and Zhou, S. L. (2012). Polymorphic chloroplast microsatellite loci in Nelumbo (Nelumbonaceae). Am. J. Bot. Primer Note and Protoc. Plant Sci. 99, e240–e244. doi:10.3732/ajb.1100547
Yang, Y., Qin, N., Huang, J., Guo, A., Kang, X., Li, S., et al. (2021). Dynamic changes of pectin epitopes and daylily tepals during flower opening and senescence of Hemerocallis citrina. Sci. Hortic. 288, 110367. doi:10.1016/j.scienta.2021.110367
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi:10.1093/molbev/msm088
Yasumoto, A. A., and Yahara, T. (2006). Post-pollination reproductive isolation between diurnally and nocturnally flowering daylilies, Hemerocallis fulva and Hemerocallis citrina. J. Plant Res. 119, 617–623. doi:10.1007/s10265-006-0028-1
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Wang, G. T., Li, W. X., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi:10.1111/1755-0998.13096
Zhang, Y., Tian, L., and Lu, C. (2023). Chloroplast gene expression: recent advances and perspectives. Plant Commun. 4, 100611. doi:10.1016/j.xplc.2023.100611
Zhao, R., Luo, J., and Xu, B. (2024). Insights into secondary metabolites and health promoting effects of edible flower Hemerocallis citrina Baroni. J. Funct. Foods 116, 106133. doi:10.1016/j.jff.2024.106133
Zhiting, X., and Singchi, C. (1997). Numerical cytotaxonomic studies of Hemerocallis (liliaceae) from China. J. Syst. Evol. 36, 215–218. doi:10.1111/j.1444-5113.1997.tb01123.x
Keywords: Hemerocallis, daylily, nightlily, chloroplast genome, ancestor state
Citation: Wu J, Gao Y, Wang J, Guo A, Qin N, Xing G and Li S (2024) Comparative analysis of chloroplast genome and evolutionary history of Hemerocallis. Front. Genet. 15:1433548. doi: 10.3389/fgene.2024.1433548
Received: 16 May 2024; Accepted: 07 June 2024;
Published: 26 July 2024.
Edited by:
Fan Zhang, Jiangsu Province and Chinese Academy of Sciences, ChinaReviewed by:
Linhe Sun, Jiangsu Province and Chinese Academy of Sciences, ChinaCopyright © 2024 Wu, Gao, Wang, Guo, Qin, Xing and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoming Xing, eGluZ2d1b21pbmdAMTYzLmNvbQ==; Sen Li, c2F1bGlzZW5AMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.