 Jing Chen
Jing Chen Shuo Yang
Shuo Yang He Wang1,2
He Wang1,2 Hongjing Wang
Hongjing Wang Yuanyuan Xiao
Yuanyuan Xiao Shanling Liu
Shanling Liu- 1Department of Medical Genetics/Prenatal Diagnostic Center, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, Chengdu, China
- 3Department of Obstetrics and Gynecology, West China Second University Hospital, Sichuan University, Chengdu, China
Background: The Ankyrin Repeat Domain Containing Protein 17 (ANKRD17, OMIM:615929) gene is a protein-coding gene associated with diseases such as Chopra-Amiel-Gordon Syndrome and Non-Specific Syndromic Intellectual Disability. The protein encoded by ANKRD17 gene belongs to the ankyrin repeat-containing protein family, which is one of the most widely existing protein domains that exclusively mediate protein-protein interactions. To date, the research and reports on the ANKRD17 gene are limited.
Case presentation: Trio whole exome sequencing (Trio-WES) was conducted on the proband and his unaffected parents to elucidate the genetic etiology in the proband, who was clinically diagnosed with developmental delay and other phenotypes. Subsequently, Sanger sequencing was employed for validation of the identified candidate variant. Furthermore, RNA analysis was utilized to ascertain the impact of the variant on splicing. The WES revealed a novel heterozygous ANKRD17 splicing variant (c.7248 + 1G>A) in the proband, but not detected in his unaffected parents. And the presence of the splicing variant of the ANKRD17 gene was valided by the Sanger sequencing subsequently. And the RNA analysis confirmed that the novel variant was predicted to result in loss of donor splice site, and the analysis at mRNA level confirmed that it leads to exon 32 skipping (r.7100_7278del179) and causes premature termination of translation to the protein (p.D2357fs), therefore is classified as pathogenic.
Conclusion: Our study reported a novel splicing variant in ANKRD17 gene, which may be associated with partial eating, frequent urination, and tic syndrome. This finding expands both the phenotypic and genotypic spectrum of ANKRD17 gene. Although there is currently no curative therapy available for ANKRD17 gene variants, a definitive diagnosis of its genetic etiology is significant for genetic counseling and family planning purposes. Furthermore, this is the first reported case of the ANKRD17 gene in China.
Background
The ANKRD17 gene, a downstream effector of cyclin E/CDK2, plays a pivotal role in cell cycle and DNA progression (Deng et al., 2009). The ANKRD17 gene engages with molecules that facilitate immune responses towards bacteria and viruses. It also has been proposed that knockdown and overexpression of ANKRD17 is functionally implicated in NOD2 and NOD1 mediated responses to bacteria in diverse human cell lines. Furthermore, the ankyrin repeat-containing protein, encoded by the ANKRD17 gene, is one of the most widely existing protein domains that exclusively mediate protein-protein interactions. Additionally, this gene’s encoded protein participates in both DNA replication and various functions (Wang et al., 2012; Menning and Kufer, 2013; Li et al., 2006). The clinical phenotype associated with this gene is characterized by Chopra-Amiel-Gordon syndrome (MIM:619504). The major phenotypic characteristic of 34 individuals from 32 families with Chopra-Amiel-Gordon syndrome were developmental delay (variable), intellectual disability (variable), language develop delay, feeding difficulties, non-specific MRI and EEG abnormalities, seizures, a predisposition to recurrent infections, and others. Furthermore, a significant number of individuals exhibit similar facial dysmorphism (Chopra et al., 2021).
The infrequency of reports concerning the ANKRD17 gene is notable. To date, only 30 damage mutation (DM) variants have been documented in the Human Gene Mutation Database (HGMD, http://www.hgmd.org), including 14 missense variants, two splice site variants, eight small deletions, two small insertions, two in-frame insertion-deletions, and one microdeletion (spanning 1.16 Mb). Except for cases in which parental samples could not be obtained, the other variants were de novo. In this study, we firstly report a novel splicing variant (c.7248 + 1 G>A) that leads to the skipping of exon 32 in the ANKRD17 gene. Our findings not only expand the genotypic and phenotypic spectrum of ANKRD17 gene but also provide valuable information for genetic counseling. Furthermore, this is the first case report of the ANKRD17 gene in China.
Case presentation
Clinical data
The proband is a 5-year-old male child. At the age of 2 years, the proband exhibited frequent blinking and involuntary laryngeal vocalization without apparent triggers. Additionally, the proband primarily demonstrated attention deficit and hyperactivity disorder (ADHD), mental and language retardation and feeding difficulty. No abnormalities were observed in the proband during the fetus or neonatal period. Detailed description of clinical manifestations was presented in Table 1 and Figure 1. This study was granted ethical approval by the Medical Ethics Committee of West China Second University Hospital, Sichuan University (Chengdu, China).
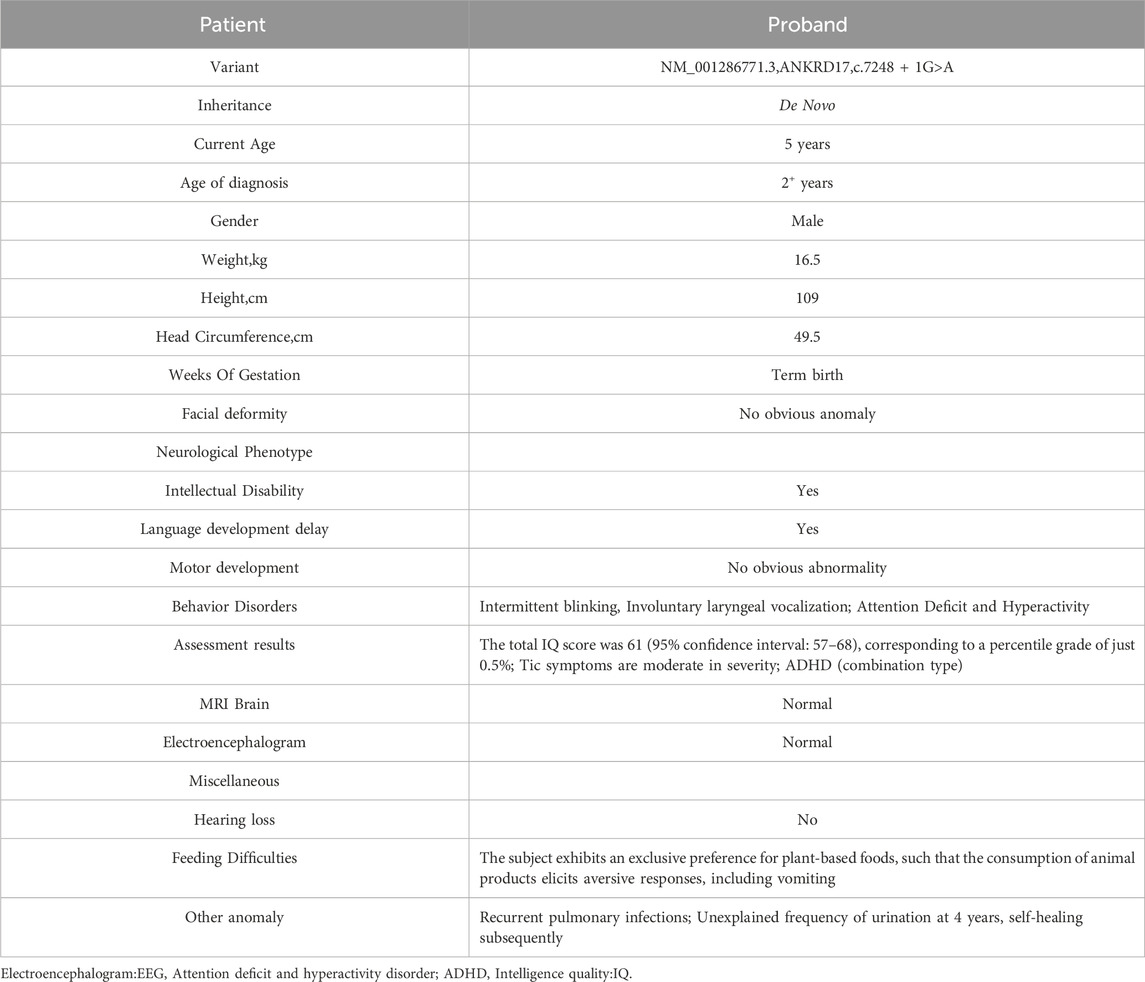
Table 1. The Clinical features of the proband.
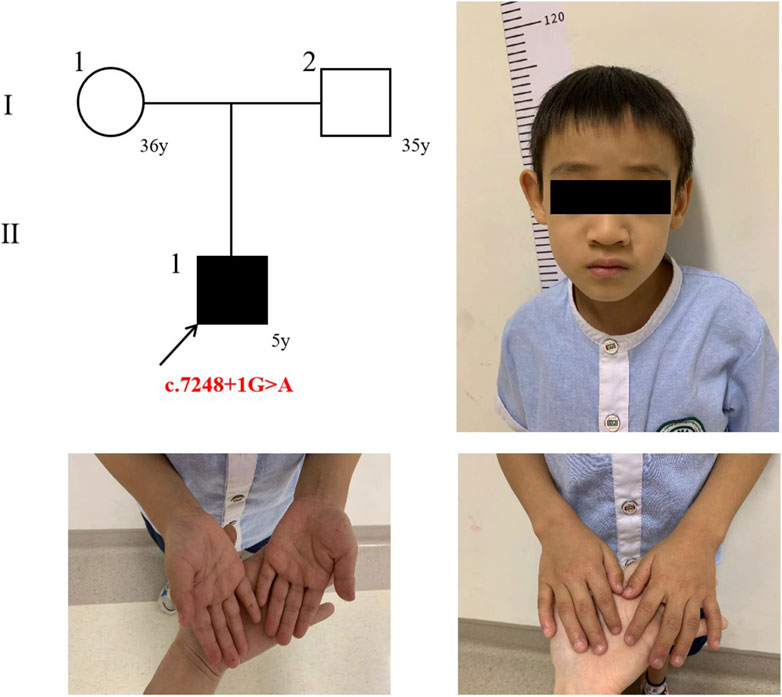
Figure 1. The trio’s pedigree and the proband’s photograph.
Whole exome sequencing and analysis
The gDNA was extracted from the proband and parents’ (II1, I1 and I2) peripheral blood leukocytes using QIAamp DNA Blood Mini Kit (QIAGEN), following the manufacturer’s instruction. WES was performed using gDNA from II1, I1 and I2. Exome capture sequencing was performed using NanoWES Human Exome V1 (Berry genomics) according to the manufacturer’s protocol. The exon-enriched libraries were sequenced through Illunima NovaSeq6000 platform. Then, all reads were aligned to the reference human genome GRCh38/hg38 with Burrows-Wheeler Aligner (BWA). Then, singlenucleotide variants (SNVs) and small insertions or deletions (InDels) were identified by GATK Unified Genotyper (broadinstitute.org/). The ENLIVEN variants annotation interpretation system (Berry genomics) were used for functional annotation. High frequency variants (frequency over 5%) from public databases including GnomAD (http://gnomad.broadinstitute.org/) and 1000 Genomes Project (1000G) (http://browser.1000genomes.org) were excluded. Pathogenicity of SNVs was evaluated based on related scientific literature and disease databases, including ClinVar (http://www.ncbi.nlm.nih.gov/clinvar), PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), OMIM (http://www.omim.org), HGMD (http://www.hgmd.org), etc. And the candidate pathogenic variants related to proband’s phenotypes were evaluated according to American Society for Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015).Variations were classified into five categories according to the ACMG guidelines: pathogenic, likely pathogenic, uncertain significance, likely benign and benign. After ensuring data quality control for WES in this study, variation analysis was conducted according to the established process. Please refer to Supplemental Figure 1 for a detailed analysis flow chart.
Validation by Sanger sequencing and transcript analysis
The suspected splicing variant of the ANKRD17 gene was validated by Sanger sequencing, and sequencing was performed using PCR primers designed with Primer Premier 5. Te sequences of the gDNA primers used was ANKRD17-F:5′-GGATAGGTGCTACTGGAGGAA-3′ and ANKRD17-R:5′-TGGAGCGAGATAGTACAGGAAT-3′. PCR products were sequenced using an ABI 3500 Genetic Analyser (Termo Fisher Scientifc) for ANKRD17 c.7248 + 1G>A.
To figure out if the variant affects splicing, RNA was extracted from fresh peripheral blood of the trio (I1, I2, and II1) using the RNApure Blood Kit (CWBIO, CW0582S), according to the manufacturer’s instructions. Complementary DNA (cDNA) was then amplified using a primer set designed to amplify ANKRD17 from exon 31 to exon 33; F:5′-GTAGGACATAGTGGCATCTG-3′, R:5′-ATAGGTGCTACTGGAGGAAT-3′. The PCR product were electrophoresed in 2% agarose gel, and bands extracted from agarose gel bands and subjected to Sanger sequencing.
Results
Following WES, we identified a novel heterozygous variant (NM_001286771.3, c.7248 + 1G>A) in the ANKRD17 gene for the proband (II1), which was not present in his parents (I1, I2). The reference transcript of ANKRD17 gene in the HGMD database is NM_001286771.3. The average sequencing depth of WES on the ANKRD17 gene was 87×. The presence of the variant was further validated through Sanger sequencing (Figure 2). The proband, who was clinically diagnosed with DD and other clinical phenotypes, had the variant, while it was not detected in either the proband’s mother or father, as expected. Currently, this variant has not been reported in population-control databases, such as ExAC Browser, 1000 Genomes Project, gnomAD, or in disease databases, such as HGMD or ClinVar database.
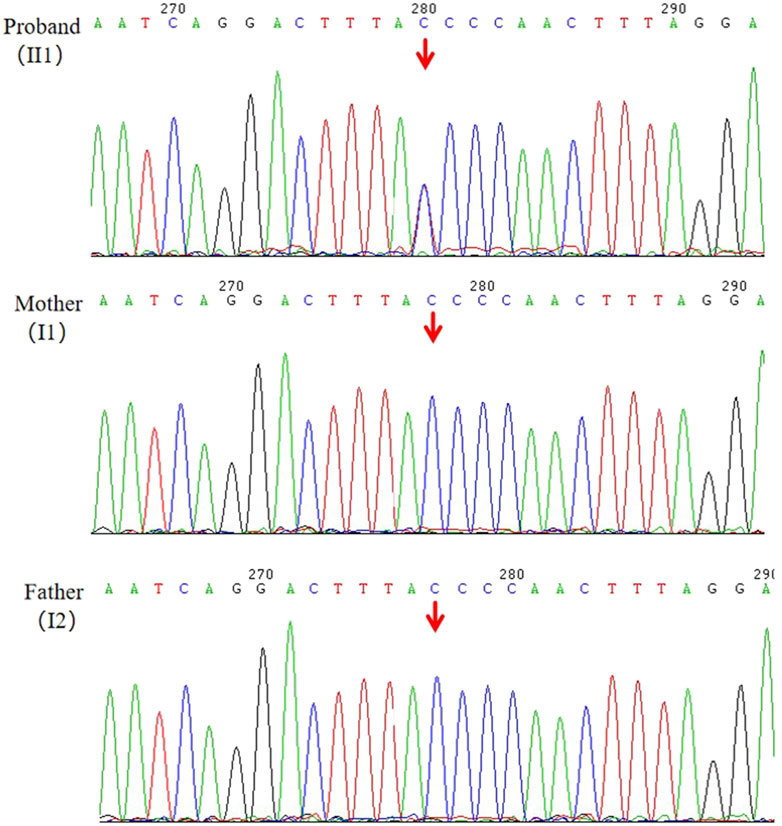
Figure 2. Validation of the candidate variant (ANKRD17,c.7248 + 1G>A) of gDNA (II1, I1, I2) by Sanger sequencing.
The three splicing-predict software (SPIDEX: 1.977, dbscSNV ADA _SCOR: 0.999991058, dbscSNV_RF_SCORE: 0.918 and spliceAI = T|ANKRD17|0.00|0.00|0.03|0.99|7|39|7|1) predict that this variant may influence splice. Thus, cDNA from the proband and his parents was acquired and the target PCR products were sequenced. Two bands (marked as Band one and two in Figure 3A) were observed after electrophoresis for the proband, while only one band (Band 1) was observed in his parents. The size of the target band is 281 bp, which is consistent with band one in both the proband and his parents. However, only the proband exhibits a second band with an approximate size of 102 bp. Direct sequencing of these cDNA PCR products with both forward and reverse primers (Figure 3B). For the splicing variant (c.7248 + 1G>A), a 179 bp deletion of ANKRD17 mRNA was detected (Figure 3D). Instead of the classic transcription product shared by the family, the distinct splicing products presented only in the affected proband would indicate dysfunction of the ANKRD17 gene, leading to the development of the disease.
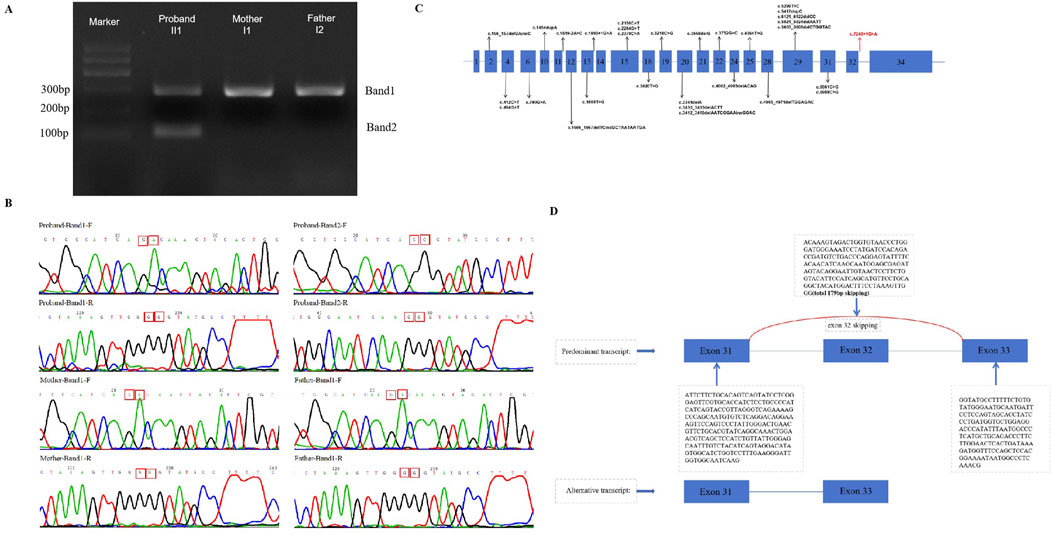
Figure 3. (A) Gel electrophoresis of the targeted cDNA fragments was performed (II1,I1,I2); (B) Sanger sequencing results of cDNA PCR products (Band one and 2) were obtained using forward and reverse primers, respectively; (C) A schematic of the ANKRD17 gene (NM_001286771.3)is shown, indicating the reported variants and locations of all disease-causing mutations (DM) in the HGMD database (HGMD® Professional 2023.3). The novel splicing variants found in this study are represented by red arrows; (D) The electropherograms of the cDNA fragment sequencing reveal exon 32 skipping of the ANKRD17 gene.
According to the ACMG (Richards et al., 2015) criteria, the novel splicing variant c.7248 + 1G>A within the ANKRD17 gene is classified as “pathogenic” (PVS1, PS2_M, PM2) based on the above results. According to the PLINK software (Purcell et al., 2007), the genetic relationship between the proband and his parents was consistent, and the variation was not found in the peripheral blood of the parents, indicating that the variation was de novo in the proband, and the phenotype of the ANKRD17 gene was consistent with that of the proband, but did not have specificity, so PS2 was downgraded to PS2_M. In addition, WES analysis revealed no other likely pathogen or pathogenic SNVs or CNVs associated with the proband’s clinical phenotype.
Discussion and conclusion
Chopra-Amiel-Gordon syndrome, also known as ANKRD17-Related Neurodevelopmental Syndrome, is caused by the heterozygous loss-of-function (LOF) pathogenic variants in the ANKRD17 gene. The ANKRD17 gene is located on chromosome 4q13 and inherited in an autosomal dominant (AD) pathogenic pattern. The protein encoded by the ANKRD17 gene is a member of the ankyrin repeat-containing protein family, possessing two distinct arrays of ankyrin repeats in its amino-terminal region: one with 15 ankyrin repeats and the other with 10 ankyrin repeats. Ankyrin repeats serve as protein-protein interaction modules and are associated with various biological processes. The ANKRD17 gene has been demonstrated to contribute to DNA replication during the S-phase of the cell cycle in human cells (Deng et al., 2009; National Center for Biotechnology Information, 2023; Ghosh et al., 2012). Additionally, the ANKRD17 gene is also likely to exert a significant influence on meiosis and the response to mismatch repair (Ray et al., 2012). The Ankyrin repeat protein ANKRD17 interacts with Retinoic acid-inducible gene-I (RIG-I), melanoma differentiation-associated gene 5 (MDA5), and virus-induced signaling adaptor (VISA) to enhance RLR-mediated immune signaling. Overexpression or knockout of ANKRD17 expression both impairs RLR signaling (Wang et al., 2012). The proband in our study exhibited a history of recurrent lung infections and frequent urination, potentially indicating immune impairment. However, further mechanistic investigations are required to substantiate this association in future studies.
Chopra-Amiel-Gordon syndrome is an ultra-rare disorder, characterized by developmental delay (DD), intellectual disability (ID), speech delay, behavioral disorders (spectrum disorder and ADHD), facial dysmorphism (including a triangular face, high anterior hairline, and low-set ears, etc.), and various other features (Chopra et al., 2021; Silverstein et al., 2022). The observed phenotype of the proband in our study is essentially consistent with this finding. Development and cognitive abilities are variable, with disabilities ranging from borderline to severe. The presence of normal cognition has also been documented in patients. However, almost all individuals with ANKRD17 gene variants exhibit speech delay, including those with intellectual abilities within the normal range. The mutation spectrum of ANKRD17-associated neurodevelopmental syndrome, coupled with the gene’s intolerance to loss of function in the general population (gnomAD pLI score = 1), strongly suggests haploinsufficiency as the underlying disease mechanism (Sveden et al., 1993).
Only one case report suggests a potential association between the variant of the ANKRD17 gene (c.6988A>G) and neonatal aneurysm rupture, which leads to subarachnoid hemorrhage (Silverstein et al., 2022). Qiu-Xia Yu, et al. reported that the de novo missense variant of the ANKRD17 gene (c.1525C>T) may be associated with prenatal isolated clubfoot (Yu et al., 2022). What’s more, the ANKRD17 gene is also related to cell migration ability and plays a role in tumor metastasis, such as oral cancer and ovarian cancer,etc (Liu et al., 2014; D'Souza et al., 2017). The majority of the reported ANKRD17 gene variants are de novo, and there have been no documented cases of incomplete penetrance, which is consistent with the findings presented in our article. However, since the first reported case with this splicing variation in the ANKRD17 gene, further investigations into the underlying mechanisms are warranted in future studies.
In our study, we have confirmed that the c.7248 + 1G>A variant results in the loss of a donor splice site, leading to exon 32 skipping (r.7100_7278del179), premature termination of translation. The transcript (NM_001286771.3) of the ANKRD17 gene consists of 34 exons, which encode a total of 2,490 amino acids. Notably, exon 32 skipping results in approximately a 2% loss of the amino acid sequences. Therefore, it can be classified as a pathogenic variant according to established ACMG (Richards et al., 2015) classification criteria. Furthermore, there is currently limited research reports on the ANKRD17 gene. The HGMD database currently includes only two splicing variants (c.1619-2A>C and c.1890 + 1G>A) and our novel variant contributes to the overall expansion of reported ANKRD17 gene variants (Figure 3C). Our finding expands the genotypic spectrum of ANKRD17 gene. Furthermore, further elucidation of the specific pathogenic mechanism of this variant requires additional in vitro and in vivo experiments.
Our study suggests that the variants of the ANKRD17 gene may be associated with partial eating, frequent urination, and tic syndrome, in addition to established phenotypes such as developmental delay. Our findings expands the phenotypic and genotypic spectrum of ANKRD17 gene. Currently, there is no curative therapy available for ANKRD17-Related Neurodevelopmental Syndrome, however, a definitive diagnosis of its genetic etiology is significant for genetic counseling and family planning. What’s more, this is the first case report of the ANKRD17 gene being reported in China.
Data availability statement
The variation data reported in this paper have been deposited in the Genome Variation MapGVM in National Genomies Data Center, Beijing Institute of Genomics. Chinese Academy of Sciences and China National Center for Bioinformation, under accession number GVM000811 (https://ngdc.cncb.ac.cn/gvm/).
Ethics statement
The studies involving humans were approved by The Medical Ethics Committee of West China Second University Hospital, Sichuan University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
JC: Writing–original draft, Writing–review and editing. SY: Writing–original draft. HeW: Writing–review and editing. HoW: Writing–review and editing. YX: Writing–review and editing. SL: Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Key Research and Development Program of China (2022YFC2703300 and 2022YFC2703302).
Acknowledgments
We appreciate the invaluable contribution of patients and their families to this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1422469/full#supplementary-material
References
Chopra, M., McEntagart, M., Clayton-Smith, J., Platzer, K., Shukla, A., Girisha, K. M., et al. (2021). Heterozygous ANKRD17 loss-of-function variants cause a syndrome with intellectual disability, speech delay, and dysmorphism. Am. J. Hum. Genet. 108 (6), 1138–1150. doi:10.1016/j.ajhg.2021.04.007
Deng, M., Li, F., Ballif, B. A., Li, S., Chen, X., Guo, L., et al. (2009). Identification and functional analysis of a novel cyclin e/cdk2 substrate ankrd17. J. Biol. Chem. 284 (12), 7875–7888. doi:10.1074/jbc.M807827200
D’Souza, W., Pradhan, S., and Saranath, D. (2017). Multiple single nucleotide polymorphism analysis and association of specific genotypes in FHIT, SAMD4A, and ANKRD17 in Indian patients with oral cancer. Head. Neck 39 (8), 1586–1595. doi:10.1002/hed.24798
Ghosh, G., Wang, V. Y., Huang, D. B., and Fusco, A. (2012). NF-κB regulation: lessons from structures. Immunol. Rev. 246 (1), 36–58. doi:10.1111/j.1600-065X.2012.01097.x
Li, J., Mahajan, A., and Tsai, M. D. (2006). Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45 (51), 15168–15178. doi:10.1021/bi062188q
Liu, M., Zhang, X., Hu, C. F., Xu, Q., Zhu, H. X., and Xu, N. Z. (2014). MicroRNA-mRNA functional pairs for cisplatin resistance in ovarian cancer cells. Chin. J. Cancer 33 (6), 285–294. doi:10.5732/cjc.013.10136
Menning, M., and Kufer, T. A. (2013). A role for the Ankyrin repeat containing protein Ankrd17 in Nod1-and Nod2-mediated inflammatory responses. FEBS Lett. 587 (14), 2137–2142. doi:10.1016/j.febslet.2013.05.037
National Center for Biotechnology Information (2023). U.S. National Library of Medicine, ANKRD17 ankyrin repeat domain 17 [Homo sapiens (human)]. Available at: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=26057 (Accessed November 13, 2023).
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Ray, D., Hogarth, C. A., Evans, E. B., An, W., Griswold, M. D., and Ye, P. (2012). Experimental validation of Ankrd17 and Anapc10, two novel meiotic genes predicted by computational models in mice. Biol. Reprod. 86 (4), 102. doi:10.1095/biolreprod.111.095216
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Silverstein, R., Kuwabara, M., and Appavu, B. (2022). Neonatal aneurysm rupture in a child with a de novo variant to ANKRD17. Child. Neurol. Open 9, 2329048X221134600. doi:10.1177/2329048X221134600
Sveden, A., Gordon, C. T., Amiel, J., and Chopra, M. (1993–2023). “ANKRD17-Related neurodevelopmental syndrome,” in GeneReviews®. Editors M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Beanet al. (Seattle (WA): University of Washington, Seattle).
Wang, Y., Tong, X., Li, G., Li, J., Deng, M., and Ye, X. (2012). Ankrd17 positively regulates RIG-I-like receptor (RLR)-mediated immune signaling. Eur. J. Immunol. 42 (5), 1304–1315. doi:10.1002/eji.201142125
Keywords: whole exome sequencing, Chopra-Amiel-Gordon syndrome, ANKRD17, mRNA analysis, splicing abnormality
Citation: Chen J, Yang S, Wang H, Wang H, Xiao Y and Liu S (2024) Case report: Whole exome sequencing reveals a novel splicing variant of ANKRD17 gene in a Chinese male juvenile with developmental delay and transient tic disorder. Front. Genet. 15:1422469. doi: 10.3389/fgene.2024.1422469
Received: 24 April 2024; Accepted: 28 August 2024;
Published: 09 September 2024.
Edited by:
Dora Janeth Fonseca, Rosario University, ColombiaReviewed by:
Yanru Huang, Xiamen University, ChinaAruna Pal, West Bengal University of Animal and Fishery Sciences, India
Copyright © 2024 Chen, Yang, Wang, Wang, Xiao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanyuan Xiao, eXl4aWFvMjAxMEAxNjMuY29t; Shanling Liu, c3Vubnk2MzBAMTI2LmNvbQ==
†These authors have contributed equally to this work