
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 18 November 2024
Sec. Genetics of Common and Rare Diseases
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1412442
 Salia Bamba1,2
Salia Bamba1,2 Lala Sidibé1,3
Lala Sidibé1,3 Seybou H. Diallo1,4
Seybou H. Diallo1,4 Lassana Cissé1
Lassana Cissé1 Kékouta Dembélé5
Kékouta Dembélé5 Abdoulaye Yalcouyé1
Abdoulaye Yalcouyé1 Weizhen Ji2
Weizhen Ji2 Mohamed Emile Dembélé1
Mohamed Emile Dembélé1 Salimata Diarra1,2
Salimata Diarra1,2 Alassane dit Baneye Maiga1
Alassane dit Baneye Maiga1 Oumou Traoré1
Oumou Traoré1 Salimata Diallo4
Salimata Diallo4 Samuel Ephrata Mefoung1
Samuel Ephrata Mefoung1 Amadou Touré3,6
Amadou Touré3,6 Adama Koné6
Adama Koné6 Lauren Jeffries2
Lauren Jeffries2 Cheick O. Guinto1,7
Cheick O. Guinto1,7 Emily K. Mis2
Emily K. Mis2 Kenneth H. Fischbeck8
Kenneth H. Fischbeck8 Mustafa K. Khokha2
Mustafa K. Khokha2 Saquib A. Lakhani2
Saquib A. Lakhani2 Guida Landouré1,7*
Guida Landouré1,7*Background and Objectives: Developmental and epileptic encephalopathies (DEEs) are a group of neurological disorders characterized by early-onset seizures that are often resistant to treatment, by electroencephalographic abnormalities, and by developmental delay or regression. Their genetic basis remains largely unelucidated, especially in sub-Saharan Africa (SSA). We investigated the genetic bases of DEE in three Malian families.
Methods: Patients underwent clinical evaluation, and DNA was obtained for whole exome sequencing (WES). Putative variants were screened in all available family members and in silico prediction analyses were performed to assess pathogenicity.
Results: Five patients from three unrelated families with DEEs had symptoms that started during the neonatal period with seizures and myoclonus that became refractory to antiepileptic medications. WES identified previously unreported variants in all three families: homozygous variants in GRIN1 and SYNJ1, and compound heterozygous variants in RARS2. These variants affected protein structure by in silico tools and were classified as variants of uncertain significance hot, pathogenic/likely pathogenic respectively according to ACMG criteria.
Discussion: We identified rare variants in three genes (GRIN1, SYNJ1, and RARS2) associated with early onset of DEEs in SSA, expanding their genetic and epidemiological spectrum. Larger cohort studies in SSA may unravel more variants with potential clinical implications and further our understanding of the disease mechanism.
Developmental and epileptic encephalopathies (DEEs) are a group of neurological disorders characterized by early-onset seizures that are often resistant to treatment, by electroencephalographic abnormalities, and by developmental delay or regression (Happ and Carvill, 2020). Previously thought to be caused by acquired factors, the discovery of monogenic mutations through next-generation sequencing has revealed a genetic basis for some DEE subtypes (McTague et al., 2016). While the genetic etiology of DEEs is increasingly recognized worldwide, only a few cases have been genetically diagnosed in sub-Saharan Africa (SSA) due to limited access to genetic facilities (Esterhuizen et al., 2018). In this study, we report novel variants causing DEEs in the Malian population.
This study was in compliance with the declaration of Helsinki and ethics approval was obtained from the Faculté de Médecine et d’Otondostomalogie, Université des Sciences, des Techniques et des Technologies de Bamako (N°2020/129/CE/FMOS/FAPH). Written informed consent/assent was obtained from all participants and/or legal guardians.
Patients were examined by neurologists, pediatricians, and medical geneticists. Blood chemistries, brain imaging, and electroencephalography (EEG) were performed in selected available patients to rule out acquired causes and refine phenotypic descriptions.
DNA was extracted from peripheral blood using the Puregene Blood DNA kit C (Qiagen, Germantown, MD) following the manufacturer’s instructions. WES was performed in trios where possible for each family. Variant calling, annotation, and prioritization as well as prediction for deleteriousness are detailed in Supplementary Material S1. Segregation of candidate variants in available family members was done by Sanger sequencing. Variants were classified according to American College of Medical Genetics (ACMG) criteria (Richards et al., 2015).
Protein sequences of relevant protein domains for GRIN1 (NP_000823.4), SYNJ1 (NP_001153774.1) and RARS2 (NP_001337434.1) were obtained from the National Center of Biotechnology Information (NCBI). Three-dimensional (3D) structures of mutant proteins were modelled on SWISS-MODEL server and newly predicted structures were refined on Galaxy Web server (https://galaxy.seoklab.org/). Pymol served for structure visualization and hydrogen bonds analysis.
Clinical and genetic findings are summarized in Table 1.
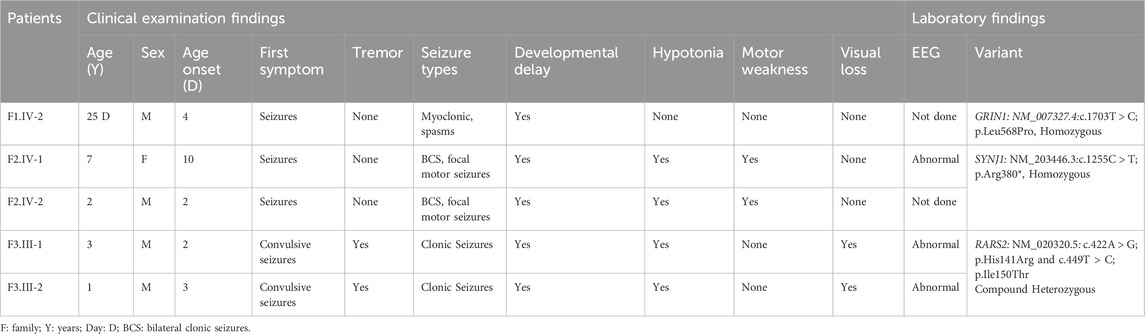
Table 1. Phenotypic and genetic findings in patients with DEE.
Family 1 (Figure 1A): A 25-days-old male from a consanguineous marriage and Soninké ethnicity, born after normal pregnancy and delivery, was referred for uncontrolled neonatal seizures and developmental delay. Symptoms started 4 days after birth with myoclonic seizures, spasms in flexion of the upper limbs, and tonic seizures involving the four limbs. Mother reported five miscarriages and premature death of another one-year-old child. WES identified a novel homozygous missense variant in GRIN1 (NM_007327.4: c.1703T > C; p. (Leu568Pro)) in the proband, categorized as “hot” variant of uncertain significance (VUS-Hot) by ACMG criteria (PM1,PP3, PM2). The Leu568 residue is conserved across a wide range of species (Figure 1B) and the variant is segregating with the disease in the family (Figure 1C). The variant was predicted to disrupt protein 3D conformations and folding, including the loss of four hydrogen bonds between the native Leu589 and Phe591 (Figure 1D). The patient was started on intravenous Sodium Valproate and Clonazepam with no effect and died shortly afterwards.
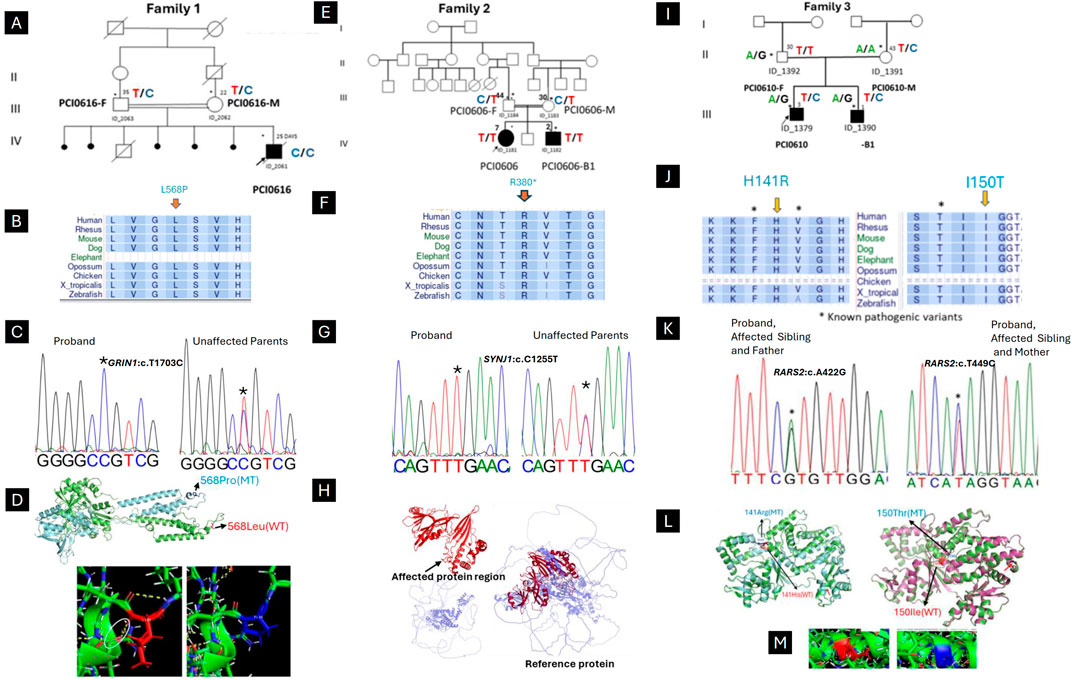
Figure 1. Clinical and genetic findings of DEE patients (Family 1,2,3). (A) Pedigree of the case with neonatal myoclonic epilepsy showing recessive inheritance pattern (arrow indicates the proband and asterisks the participants seen in clinic). (B) The horizontal rectangle at the bottom represents the amino acid alignment around the mutated residue and their corresponding conservation among selected species using the UCSC Genome Browser. (C) Chromatogram showing the c.1703T > C variant in GRIN1. (D) Superimposed structure of GRIN1 wild type (green) and mutant (light blue), showing the shift of 3D conformational between. Hydrogen bond analysis showing the loss of bonding interaction of wild type leucine (white arrow) with phenylalanine in the mutant. (E) Pedigree of the case with infantile epileptic encephalopathy showing recessive inheritance pattern (arrow indicates the proband and asterisks the participants seen in clinic). (F) The horizontal rectangle at the bottom represents the amino acid alignment around the mutated residue and their corresponding conservation among selected species using the UCSC Genome Browser. (G) Chromatogram showing the (C) 1138C > T variant in SYNJ1. (H) Structure of the SYNJ1 reference protein, with the affected region highlighted in red. The truncated protein (in red) and the remaining portion of the reference protein (in light blue) illustrate the consequences of a premature stop codon, leading to the rupture of the full-length structure and the loss of both the α-helix and β-sheet regions. (I) Pedigree of the case with Ponto-cerebellar hypoplasia with seizures and showing recessive inheritance pattern (arrow indicates the proband and asterisks the participants seen in clinic). (J) The horizontal rectangle at the bottom represents the amino acid alignment around the mutated residue and their corresponding conservation among selected species using the UCSC Genome Browser.). (K) Chromatogram showing the compound heterozygous in RARS2 with both affected children (c.422A > G inherited from Father and c.449T > C inherited from Mother). (L) Superimposed structure of wild type RARS2 (green) and mutant (p.His141Arg; light blue) showing the partial loss of helix in the mutant (red arrow). (M) Superimposed structure of wild type RARS2 (magenta) and mutant (p.Ile150Thr; green) showing the partial loss of helix in the mutant (red arrow). Hydrogen bond analysis showing that there is not significant difference in bonding interaction between Isoleucine-150 and Threonine-150.
Family 2 (Figure 1E): A 7-year-old female proband and her 2-year-old brother from healthy consanguineous parents of Songhai ethnicity, were referred for seizures and motor acquisition delay. Parents reported that around 1 week after birth both patients began having 4–10 bilateral clonic seizures daily. The proband showed delayed motor acquisition, hypotonia with axial weakness, generalized muscle atrophy in the four limbs, multidirectional nystagmus with bilateral Babinski sign, and scoliosis. EEG recorded when the proband was 7 years old, revealed right temporal spike waves and significantly slowed background activity, along with low voltage in the left hemisphere (Figure 2A). Her brother also had motor acquisition delay with generalized hypotonia with inability to sit, muscle atrophy in shoulders and lower limbs, and lumbar hyperlordosis. He had absent tendon reflexes in the four limbs, Babinski sign on the left and multidirectional nystagmus on vertical gaze. WES identified a novel homozygous nonsense variant in SYNJ1 (NM_203446.3: c.1138C > T; p. (Arg380*)) in both children, classified as likely pathogenic (ACMG criteria PVS1, PM2). The Arg380 residue is conserved across a wide range of species (Figure 1F) and the variant is segregating with the disease in the family (Figure 1G). The premature stop codon, leading to disrupture of the full-length structure of SYNJ1 reference protein and the loss helical structure in the Sac domain and β-sheet regions (Figure 1H). Despite treatment with multiple medications including Carbamazepine, Sodium Valproate, and Clonazepam, both patients continue to experience frequent seizures.
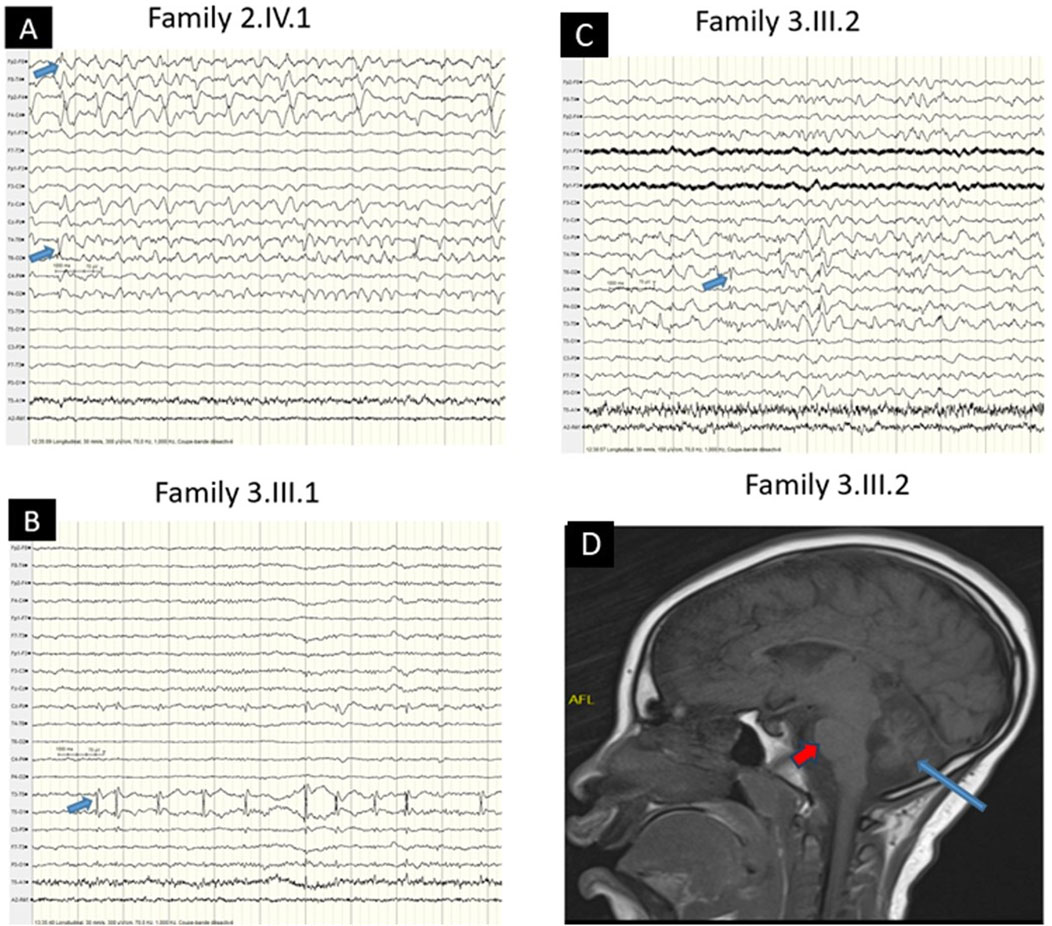
Figure 2. EEG and MRI Characteristics (A) EEG showing right temporal spike waves with left low-voltage background activity (blue arrow) in the older patient (Family 2, IV.1). (B) EEG showing left temporal spike waves (blue arrow) in the patient (Family 3, III.1). (C) EEG showing focal (right temporoparietal) spike waves (blue arrow) in the patient (Family 3, III.2). (D) Brain MRI showing cerebellar atrophy (blue arrow) sparing the pons (red arrow) (Family 3, III.2).
Family 3 (Figure 1I): Male siblings, one and 3 years old, from non-consanguineous healthy parents of Mandingo ethnicity were seen for seizures, psychomotor delay, and visual loss. Symptoms started with tonic clonic seizures within 1 month after birth, and both patients had psychomotor acquisition delay, visual loss, and clonic seizures. Tendon reflexes were brisk in the lower limbs and normal in the upper limbs. In addition, the older patient had a hyperactivity disorder. Sleeping EEG showed left temporal predominant spike waves in the older brother (Figure 2B) and focal right temporoparietal paroxysmal spike waves in the younger (Figure 2C). Brain magnetic resonance imaging of the older child showed cerebellar atrophy sparing the pons (Figure 2D). WES revealed novel compound heterozygous variants in RARS2 (NM_020320.5: c.422A > G; p. (His141Arg), and c.449T > C; p. (Ile150Thr)). The His141 and Ile150 residues are conserved across a wide range of species (Figure 1J). The p. (His141Arg) and p. (Ile150Thr) variants have been classified as likely pathogenic according to ACMG criteria, with p. (His141Arg) meeting the PP3, PM1, PP5, and PM2 criteria, and p. (Ile150Thr) by PP3, PP5, PM2, and PM3 criteria, respectively. The first variant is inherited from the father and second is inherited from the mother (Figure 1K). Molecular modeling revealed major predicted changes including the loss of helical structures (Figures 1L,M). The patients were being treated with multiple medications including Sodium Valproate, Clonazepam, and Piracetam. However, the treatment was unsuccessful, and the patients continue to experience frequent seizures.
The global prevalence of epilepsy is higher in SSA as compared to other regions of the world (Paul et al., 2012). Although this is partially attributable to factors such as infection and malnutrition, limited access to DNA sequencing undermines the ability to delineate genetic causes (Esterhuizen et al., 2023). Still, the rapid evolvement and decreasing costs of sequencing technologies has facilitated the identification epilepsy genes in SSA, including for DEEs (Esterhuizen et al., 2023). In a recent South African study, genetic variants were detected in 51 of 234 children with DEEs, with SCN1A being the most frequently implicated gene (Hebbar and Mefford, 2020). Besides this study, however, reports of molecularly diagnosed DEEs are scarce in SSA. Our work is the first study in Mali examining the genetics of DEEs. We identified unrelated families with DEEs caused by novel pathogenic variants in previously reported genes, GRIN1, SYNJ1 and RARS2.
The GRIN1 gene plays a pivotal role in the proper functioning of N-methyl-D-aspartate (NMDAR) receptor, essential for brain synaptic mechanisms. It is linked to a spectrum of neurological disorders, ranging from DEE to neurodevelopmental disorders, with or without seizures and hyperkinetic movements, showcasing its significant variability in phenotypic expression (Lemke et al., 2016a). An intriguing case from Morocco highlighted a novel mutation associated with intellectual challenges and autism-like features, underlining the gene’s broad impact (Blakes et al., 2022). Our study adds to this with a case from the Sub-Saharan Africa population, emphasizing a rare but classic developmental epileptic encephalopathy phenotype. The p. (Leu568Pro) variant meets the PP3 criterion is supported by the pathogenicity prediction score of 0.911 for human nonsynonymous SNVs (nsSNVs) using the MetaRNN model, which falls between 0.841 and 0.939, indicating that the variant is of moderate pathogenicity. Additionally, the variant satisfies the PM1 criterion, as many missense variants cluster in the transmembrane region close to the mutated Leu568 residue. Therefore, this variant is classified as VUS-Hot, suggesting a probable pathogenic role by the consistent genotype-phenotype correlation and the recessive inheritance pattern observed in the pedigree, which shows severe features associated with a biallelic variant, like the p. Gln556* case (Lemke et al., 2016b), and by the absence of other plausible point or copy number variants in genes associated with DEEs.
The SYNJ1 gene’s product is key in synaptic vesicle dynamics, with mutations known to lead to early-onset Parkinson’s disease or DEE53, based on the affected protein domain (Hardies et al., 2016). This paper reports on a DEE53 case, characterized by intractable epilepsy and developmental delay, linked to a critical domain mutation (Sac-domain). The p. Arg380* variant is classified as “likely pathogenic” because it satisfies ACMG criteria PVS1 and PM2 based on the fact that it is a null variant with potential loss-of-function mechanism and on its absence in variome databases including gnomAD.
RARS2 encodes for the mitochondrial arginine-tRNA synthetase, with specific mutations causing pontocerebellar hypoplasia type 6 (PCH6) (Lühl et al., 2016). Clinical spectrum of RARS2 mutations typically include neurological symptoms such as encephalopathy with intractable seizures and severe developmental delay, primarily affecting the brain. Other organ systems, such as the cardiac, ocular, renal, or hepatic systems, are not commonly involved in this disease (Edvardson et al., 2007). The phenotype varies between the presence or absence of pontocerebellar hypoplasia (PCH). This study describes patients without PCH. While these cases are generally considered to have a milder phenotype, with developmental milestones being relatively normal up to around 6 months, (Nishri et al., 2016) in this study, the two patients contrast with the typical clinical manifestations starting with early infantile psychomotor delay (Lühl et al., 2016). Additionally, the patients have had an unusually prolonged lifespan compared to other affected patients. The phenotypic differences could be due to potential genetic modifiers or be stochastic. According to the ACMG criteria, the first variant in RARS2 meets the PM1 criterion (moderate) because it is located in the Aminoacyl-tRNA Synthetase Domain of the RARS2 protein, specifically in a short sequence motif HIGH region which has seven reported missense or in-frame deletion variants including three pathogenic or likely pathogenic and four VUS. UniProt (2024).
In summary, we have identified rare genetic variants in GRIN1, SYNJ1, and RARS2 associated with early onset of DEEs in a SSA population from Mali. Our results expand the genetic and epidemiological spectrum of this disease. Larger cohort studies, particularly in other understudied populations, may unravel additional variants that could have implications for their populations and be important in furthering knowledge of the disease mechanism.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author/s.
The studies involving humans were approved by the Faculté de Médecine et d’Otondostomalogie, Université des Sciences, des Techniques et des Technologies de Bamako (N°2020/129/CE/FMOS/FAPH). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
SB: Formal Analysis, Methodology, Data curation, Software, Validation, Visualization, Writing–original draft. LN: Resources, Writing–review and editing, Investigation, Visualization. SD: Data curation, Resources, Writing–review and editing, Investigation. LC: Data curation, Formal Analysis, Investigation, Resources, Writing–review and editing. KD: Investigation, Writing–review and editing. AY: Data curation, Investigation, Methodology, Visualization, Writing–review and editing. WJ: Data curation, Formal Analysis, Writing–review and editing. MD: Investigation, Resources, Writing–review and editing. SD: Investigation, Resources, Writing–review and editing. AB: Investigation, Resources, Writing–review and editing. OT: Investigation, Resources, Writing–review and editing. SD: Investigation, Writing–review and editing. SM: Investigation, Writing–review and editing. AT: Investigation, Resources, Writing–review and editing. AK: Investigation, Writing–review and editing. LJ: Investigation, Methodology, Writing–review and editing. OG: Data curation, Investigation, Writing–review and editing. EM: Data curation, Investigation, Writing–review and editing. KF: Supervision, Writing–review and editing. MK: Data curation, Investigation, Supervision, Writing–review and editing. SL: Funding acquisition, Investigation, Supervision, Validation, Writing–review and editing. GL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grant numbers U01HG007044 under the H3Africa initiative and R01NS118522 funded by the National Institute of Neurological Disorders and Stroke (NINDS), the Fogarty International Center and the Centre Hospitalier Universitaire du Point “G”, Bamako, Mali.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1412442/full#supplementary-material
Blakes, A. J. M., English, J., Banka, S., and Basu, H. (2022). A homozygous GRIN1 null variant causes a more severe phenotype of early infantile epileptic encephalopathy. Am. J. Med. Genet. Part A 188 (2), 595–599. doi:10.1002/ajmg.a.62528
Edvardson, S., Shaag, A., Kolesnikova, O., Gomori, J. M., Tarassov, I., Einbinder, T., et al. (2007). Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 81 (4), 857–862. doi:10.1086/521227
Esterhuizen, A. I., Carvill, G. L., Ramesar, R. S., Kariuki, S. M., Newton, C. R., Poduri, A., et al. (2018). Clinical application of epilepsy genetics in Africa: is now the time? Front. neurology 9, 276. doi:10.3389/fneur.2018.00276
Esterhuizen, A. I., Tiffin, N., Riordan, G., Wessels, M., Burman, R. J., Aziz, M. C., et al. (2023). Precision medicine for developmental and epileptic encephalopathies in Africa—strategies for a resource-limited setting. Genet. Med. 25 (2), 100333. doi:10.1016/j.gim.2022.11.002
Happ, H. C., and Carvill, G. L. (2020). A 2020 view on the genetics of developmental and epileptic encephalopathies. Epilepsy Curr. 20 (2), 90–96. doi:10.1177/1535759720906118
Hardies, K., Cai, Y., Jardel, C., Jansen, A. C., Cao, M., May, P., et al. (2016). Loss of SYNJ1 dual phosphatase activity leads to early onset refractory seizures and progressive neurological decline. Brain 139 (9), 2420–2430. doi:10.1093/brain/aww180
Hebbar, M., and Mefford, H. C. (2020). Recent advances in epilepsy genomics and genetic testing. F1000Research 9, doi:10.12688/f1000research.21366.1
Lemke, J. R., Geider, K., Helbig, K. L., Heyne, H. O., Schütz, H., Hentschel, J., et al. (2016a). Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology 86 (23), 2171–2178. doi:10.1212/WNL.0000000000002740
Lemke, J. R., Geider, K., Helbig, K. L., Heyne, H. O., Schütz, H., Hentschel, J., et al. (2016b). Delineating the GRIN1 phenotypic spectrum: a distinct genetic NMDA receptor encephalopathy. Neurology 86 (23), 2171–2178. doi:10.1212/WNL.0000000000002740
Lühl, S., Bode, H., Schlötzer, W., Bartsakoulia, M., Horvath, R., Abicht, A., et al. (2016). Novel homozygous RARS2 mutation in two siblings without pontocerebellar hypoplasia - further expansion of the phenotypic spectrum. Orphanet J. rare Dis. 11 (1), 140. doi:10.1186/s13023-016-0525-9
McTague, A., Howell, K. B., Cross, J. H., Kurian, M. A., and Scheffer, I. E. (2016). The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurology 15 (3), 304–316. doi:10.1016/S1474-4422(15)00250-1
Nishri, D., Goldberg-Stern, H., Noyman, I., Blumkin, L., Kivity, S., Saitsu, H., et al. (2016). RARS2 mutations cause early onset epileptic encephalopathy without ponto-cerebellar hypoplasia. Eur. J. Paediatr. neurology EJPN official J. Eur. Paediatr. Neurology Soc. 20 (3), 412–417. doi:10.1016/j.ejpn.2016.02.012
Paul, A., Adeloye, D., George-Carey, R., Kolčić, I., Grant, L., and Chan, K. Y. (2012). An estimate of the prevalence of epilepsy in Sub–Saharan Africa: a systematic analysis. J. Glob. health 2, 020405. doi:10.7189/jogh.02.020405
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. official J. Am. Coll. Med. Genet. 17 (5), 405–424. doi:10.1038/gim.2015.30
UniProt (2024). Q5T160 - SYRM_HUMAN. Available at: https://www.uniprot.org/uniprotkb/Q5T160/variant-viewer
Keywords: developmental epileptic encephalopathies (DEEs), exome sequencing, novel variant, sub-Saharan, Mali
Citation: Bamba S, Sidibé L, Diallo SH, Cissé L, Dembélé K, Yalcouyé A, Ji W, Dembélé ME, Diarra S, Maiga AdB, Traoré O, Diallo S, Mefoung SE, Touré A, Koné A, Jeffries L, Guinto CO, Mis EK, Fischbeck KH, Khokha MK, Lakhani SA and Landouré G (2024) Case report: Novel variants cause developmental and epileptic encephalopathy in three unrelated families from Mali. Front. Genet. 15:1412442. doi: 10.3389/fgene.2024.1412442
Received: 05 April 2024; Accepted: 28 October 2024;
Published: 18 November 2024.
Edited by:
Edoardo Monfrini, University of Milan, ItalyReviewed by:
Tahir Khan, Stanley Manne Children’s Research Institute, United StatesCopyright © 2024 Bamba, Sidibé, Diallo, Cissé, Dembélé, Yalcouyé, Ji, Dembélé, Diarra, Maiga, Traoré, Diallo, Mefoung, Touré, Koné, Jeffries, Guinto, Mis, Fischbeck, Khokha, Lakhani and Landouré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guida Landouré, Z3VpZGFAaWNlcm1hbGkub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.