 Chan Liu1
Chan Liu1 Feixia Zheng
Feixia Zheng- 1Department of Physical Medicine and Rehabilitation Center, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
- 2Department of Pediatrics Neurology, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, China
Background: Minichromosome maintenance complex component 3 associated protein (MCM3AP) is a gene in which mutations can result in autosomal recessive peripheral neuropathy with or without impaired intellectual development. The MCM3AP genotype-phenotype correlation and prognosis remain unclear. The aim of this study was to explore the genotype–phenotype correlations pertaining to MCM3AP.
Methods: Whole-exome sequencing (WES) combined with copy number variation sequencing (CNV-seq) were performed on the genomic DNA isolated from a Chinese family, and Sanger sequencing, quantitative PCR and cDNA analyses were performed to examine the mutations. The retrospective study was conducted on 28 individuals with biallelic MCM3AP mutation-related diseases, including features such as mutations, motor development impairment, intellectual disability, weakness/atrophy, and cerebral magnetic resonance imaging abnormalities.
Results: Sequencing identified novel compound heterozygous mutations in MCM3AP, namely, a paternal variant c.1_5426del (loss of exons 1–25) and a maternal splicing variant c.1858 + 3A>G. Functional studies revealed that the variant c.1858 + 3A>G resulted in the heterozygous deletion of exon 5, thereby affecting splicing functionality. Furthermore, the compound heterozygous mutation may affect the functionality of the protein domain. Retrospective analysis revealed different genotype–phenotype correlations for the pathogenic variants in biallelic MCM3AP: all individuals (100%) with mutations outside the Sac3 domain exhibited early-onset symptoms, motor developmental delays, and cognitive abnormalities, conversely, the proportions of individuals carrying mutations within the domain were 26.7% (motor delays) and 46.7% (cognitive abnormalities).
Conclusion: Our findings further expand the genetic mutation spectrum of biallelic MCM3AP and highlight the genotype-phenotype associations. Additionally, we elaborate on the importance of rehabilitation intervention.
1 Introduction
Biallelic mutations in minichromosome maintenance complex component 3 associated protein (MCM3AP, MIM*603294) can result in autosomal recessive peripheral neuropathy with or without impaired intellectual development (PNRIID, MIM#618124). This early-onset neurologic syndrome is often progressive and results in distal motor impairment and gait difficulties, frequently accompanied by loss of ambulation and difficulties with manual dexterity. Furthermore, most of the affected individuals present with intellectual disability (ID) or learning difficulties (Schuurs-Hoeijmakers et al., 2013; Mert et al., 2017; Ylikallio et al., 2017; Kennerson et al., 2018; Woldegebriel et al., 2020). Eye movement abnormalities, characteristic hand and foot deformities, and scoliosis are some additional features.
The minichromosome maintenance (MCM) family is highly conserved in vertebrates and comprises MCM2–MCM10. These proteins play essential roles in DNA replication and cell cycle progression. MCM3AP interacts with the MCM3 protein, which is involved in the initiation of DNA replication (Abe et al., 2000; Masai et al., 2005). Germinal center-associated nuclear protein (GANP) is a 218 kDa multidomain protein encoded by 28 exons of MCM3AP in chromosome 21q22.3; it is ubiquitously expressed and predominantly localized in the nucleus (Abe et al., 2000; Wickramasinghe et al., 2010). It contains an N-terminal domain comprising phenylalanine-glycine repeats, a C-terminal acetyltransferase domain, DNA primase region, suppressor of actin 3 (Sac3) mRNA-binding domain, and the Cdc31-interaction domain (CID) in the middle (Takei et al., 2001; Takei et al., 2002; Ylikallio et al., 2017). However, the overall function of mammalian GANP has not been determined. Functionally, GANP serves as an mRNA export factor; its depletion leads to mRNA accumulation in the nucleus and subsequent cellular degeneration, which is associated with various neurodegenerative disorders (Wickramasinghe et al., 2010; Mert et al., 2017; Ylikallio et al., 2017; Nussbacher et al., 2019). In flies, GANP can suppress TDP-43-mediated degeneration of motor neurons (Sreedharan et al., 2015). Overall, recessive mutations in MCM3AP are associated with neuropathy and ID as well as functional impairment of the GANP protein (Ylikallio et al., 2017; Woldegebriel et al., 2020).
Biallelic MCM3AP-associated disease has been reported previously; however, the genotype-phenotype correlation pertaining to MCM3AP remains unclear, and the prognosis has not been investigated. In this study, we identified a novel compound heterozygous variant of MCM3AP and investigated the efficacy of rehabilitation in a Chinese family. Furthermore, we reviewed several previous studies to explore the genetic spectrum of MCM3AP and its relationship with the PNRIID phenotype.
2 Methods
2.1 Clinical evaluation
In this study, patients were recruited from the Department of Rehabilitation, the Second Affiliated Hospital of Wenzhou Medical University. The signs and symptoms of the disease in the proband and his family members were documented. The Ethics Committee of the Second Affiliated Hospital of Wenzhou Medical University approved this study (2021-K-127-02). All procedures were conducted according to the principles outlined in the Declaration of Helsinki (World Medical Association, 2013). Legal guardians provided written informed consent.
2.2 Whole-exome sequencing (WES) and copy number variant sequencing (CNV-seq)
Trio-(WES&CNV-seq) were performed to identify the gene mutations in the proband and his parents, and the proband's sister verified the mutations. Ethylenediaminetetraacetic acid (EDTA)-anticoagulated blood was collected, followed by genomic DNA (gDNA) extraction using a blood column medium extraction kit (Kangweishiji, China). The Qubit 2.0 fluorometer and 1.0% agarose gel electrophoresis were utilized to measure DNA quality and concentration, respectively. Subsequently, the gDNA was fragmented, end-repaired, ligated, and subjected to polymerase chain reaction (PCR) to construct a sequencing library. Then, IDT xGen® Exome Research Panel v2.0 was used to hybridize the resulting target DNA fragments for enrichment to generate a whole exome library. The BGI DNBSEQ-T7 platform was used to perform sequencing. Burrows–Wheeler Aligner (BWA) was used to align the sequencing data against the Ensemble reference genome GRCh37/hg19. Thereafter, single nucleotide polymorphisms and indels were analyzed using GATK. Structural variations (SVs) and copy number variations (CNVs) were annotated and ranked by AnnotSV. Chigene (www.chigene.org), an independently developed mutation annotation software, was used to annotate these high-quality mutations and analyze their potential hazards.
2.3 Quantitative PCR (qPCR)
Blood samples were anticoagulated with EDTA and subjected to DNA extraction using the magnetic bead method (TIANGEN, China). Specific primers were designed based on the target region. The system was configured using the SYBR qPCR kit (Sigma-Aldrich, United States). qPCR experiments were performed using a two-step method, and the 2−△△Ct method was utilized (Table 1).
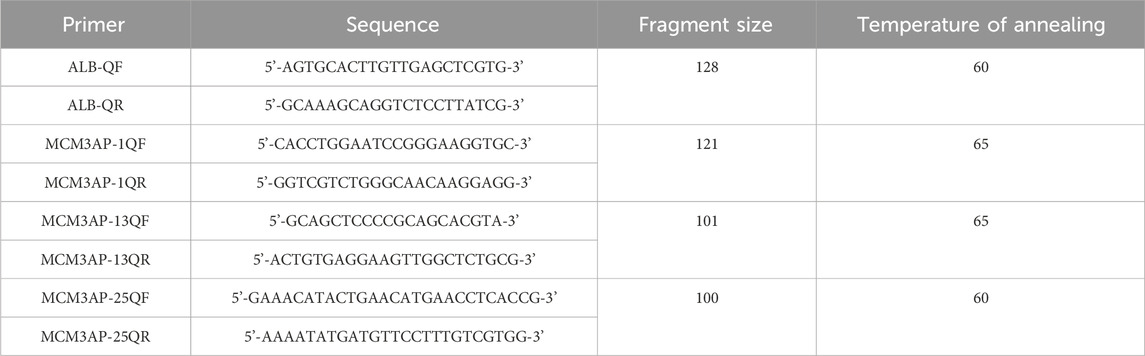
Table 1. qPCR experimental procedure.
2.4 cDNA analysis
Blood samples were collected from the proband and anticoagulated with EDTA. Then, RNA was extracted using the Blood RNA Extraction Kit (BioTek, China), followed by reverse transcription with random hexamers (Vazyme, China) to obtain cDNA. Based on MCM3AP (NM_003906.5 → NP_003897.2), c.1858 + 3(IVS5)A>G-specific primers were designed (Table 2). PCR was performed to amplify the target sequences, followed by 1.0% agarose gel electrophoresis and Sanger sequencing using the ABI3730 sequencer for verification. The results were analyzed using Chromas software.

Table 2. Primer sequences used for cDNA sequencing.
2.5 Clinical data collection and literature review
Databases such as PubMed, the Web of Science, and Google Scholar were searched using the term “MCM3AP” from inception until February 2024 without language restrictions to retrieve relevant human mutation cases. The clinical and genetic characteristics of the patients were collected.
3 Results
3.1 Clinical manifestations
This study outlines the cases of two affected siblings aged 14 and 5 years (sister, II-1; brother, II-2) who were born to nonconsanguineous parents (Figure 1A). They had experienced delayed motor milestones during infancy and had a history of global developmental delay. Patient II-2 presented with mouth twitching and lip cyanosis from the first day of birth, raising the suspicion of convulsions. Patient II-1 could independently sit at 8–9 months of age but was unable to independently stand at 3 years of age. In contrast, patient II-2 was able to independently sit at 14 months of age and independently stand at 3 years of age. Patient II-2 underwent surgery for a right foot deformity at the age of 4 years; however, abnormal posture remained despite independent walking ability. Moreover, both siblings exhibited impaired fine motor skills. After starting rehabilitation at the age of 7 months, patient II-2 exhibited improvements in overall movement. At present, he can walk independently, climb stairs with orthopedic shoes, perform complete gripping and other actions (except for slight atrophy of the thenar muscles and poor thumb movement), and has better cognition than patient II-1. In patient II-1, rehabilitation was initiated at 3.5 years of age; she achieved maximum motor status at 4 years of age and was able to independently walk with aid for short distances. However, she exhibited inflexibility in grasping objects and could not climb stairs. Moreover, upon treatment cessation, her mobility significantly regressed, resulting in an inability to sit independently, stand, and grasp objects. These symptoms were accompanied by weakness and muscle atrophy, which primarily affected the distal region of the limbs, with hand and foot deformities and scoliosis. Patient II-1 also continued to exhibit issues with urinary incontinence. The right lower limb of patient II-2 was thinner than the left lower limb. Both siblings exhibited ID and dysarthria but no paresthesia, hearing impairment, or eye movement disorders. In both cases, right pes cavus and left crural valgus were observed (Figure 1B). Tendon reflexes were absent. Electromyography revealed motor axonal neuropathy, whereas cerebral magnetic resonance imaging (MRI) revealed abnormal signals in the bilateral lateral ventricles. Lastly, electroencephalography revealed epileptic wave discharges in both individuals; however, patient II-1 did not experience seizure attacks.
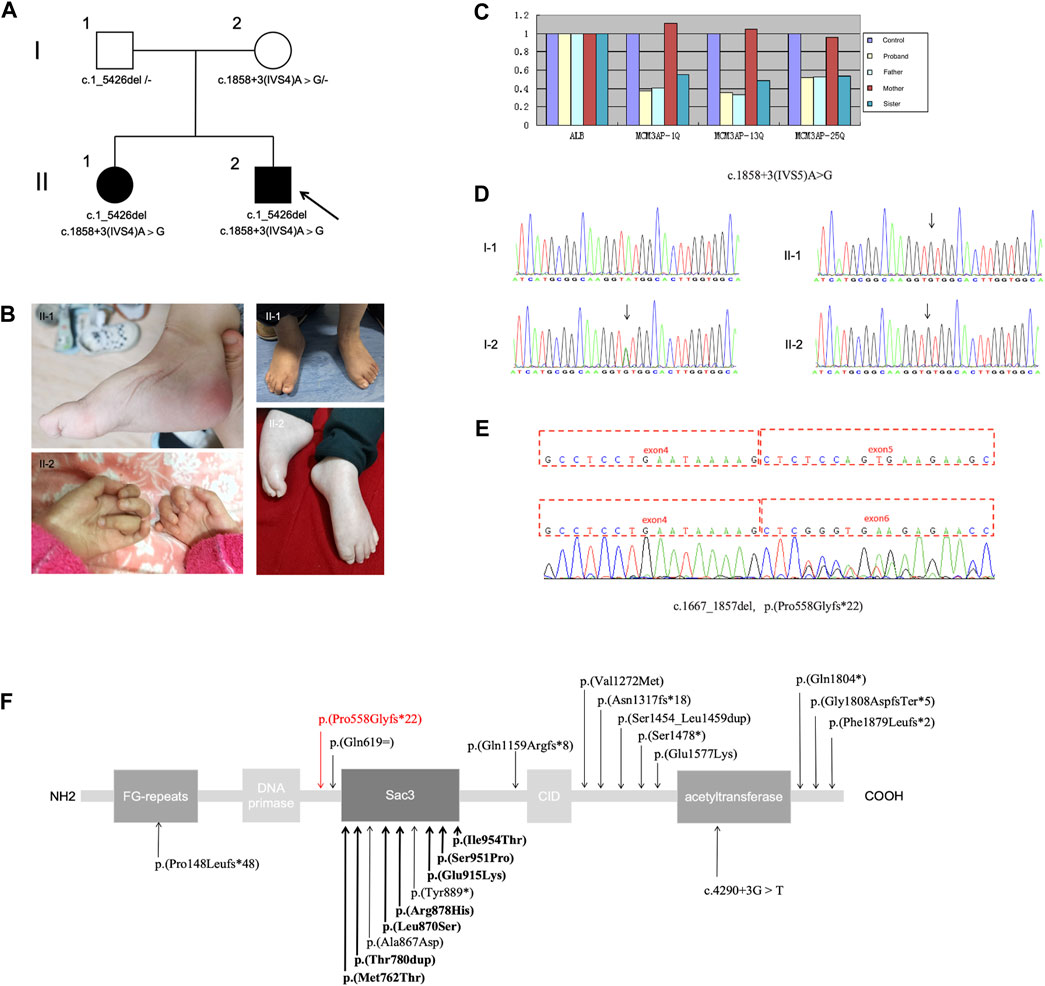
Figure 1. (A) Pedigree of the family; the arrow indicates the proband. (B) Distal muscle atrophy and contractures accompanied by pes cavus and claw hand. (C–E) Presence of the compound heterozygous variants c.1_5426del (loss of exons 1–25) and c.1858 + 3A>G in MCM3AP. (C) Copy number analysis of exons 1–25 of MCM3AP in the affected siblings and parents. (D) Chromatograms of the c.1858 + 3A>G variant. (E) Functional experiments showing that the splice variant c.1858 + 3A>G (p.Pro558Glyfs*22) results in the deletion of exon 5. (F) Schematic illustrating the structure of the GANP protein along with the MCM3AP variants; the novel variant is indicated using a red arrow, whereas previously reported variants are indicated in black (bold indicates homozygous variants).
3.2 Genetic findings
Through diagnostic WES combined with CNV-seq, the compound heterozygous mutation c.1_5426del (loss of exons 1–25) and splicing variant c.1858 + 3A>G in MCM3AP were identified. Sanger sequencing validated the findings. The mutations observed in MCM3AP of the patients were inherited from their parents (both heterozygous; Figure 1A). The identified variants were not observed in databases such as 1,000 Genomes, Exome Aggregation Consortium, dbSNP 150, or the Genome Aggregation Database. The phenotype of the patients was consistent with that of biallelic MCM3AP-associated diseases.
Next, qPCR was performed to analyze the copy numbers of exons 1–25 in the target gene MCM3AP (Figure 1C). Compared with normal controls, the copy number ratio of exons 1–25 in MCM3AP was approximately 0.5 in both siblings and their father, indicating a loss of heterozygosity for exons 1–25. In contrast, the ratio was approximately 1 in the mother, suggesting a normal copy number. The paternally inherited variant potentially resulted in the loss of gene function and was not detected in the normal control population database. Therefore, according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015), this variant was classified as a likely pathogenic variant (PVS1 + PM2).
Splicing functional experiments revealed that the maternal variant c.1858 + 3A>G (p.Pro558Glyfs*22) in MCM3AP can affect splicing, leading to a transcript without exon 5, which, in turn, results in the domain deletion of Sac3, CID, and acetyltransferase (Figures 1D–F). Subsequently, based on the ACMG standards and guidelines, this variant was classified as PS3 + PM2 + PP3.
Co-segregation studies confirmed that both parents were carriers of MCM3AP mutations without any evidence of neurologic symptoms or signs. These findings suggest an association between MCM3AP variations and the phenotypic manifestations observed in the affected individuals.
3.3 Clinical and genetic characteristics of the patients carrying biallelic mutations in MCM3AP
Eight studies reported 26 cases of biallelic MCM3AP-associated diseases in 15 families. Two families (patients #19, #20, #21, and #26) were affected by multiple sclerosis (MS). Including our variants, 23 homozygous or compound heterozygous mutations have been identified. Table 3 summarizes the biallelic MCM3AP mutations and associated clinical characteristics of the 28 affected individuals.
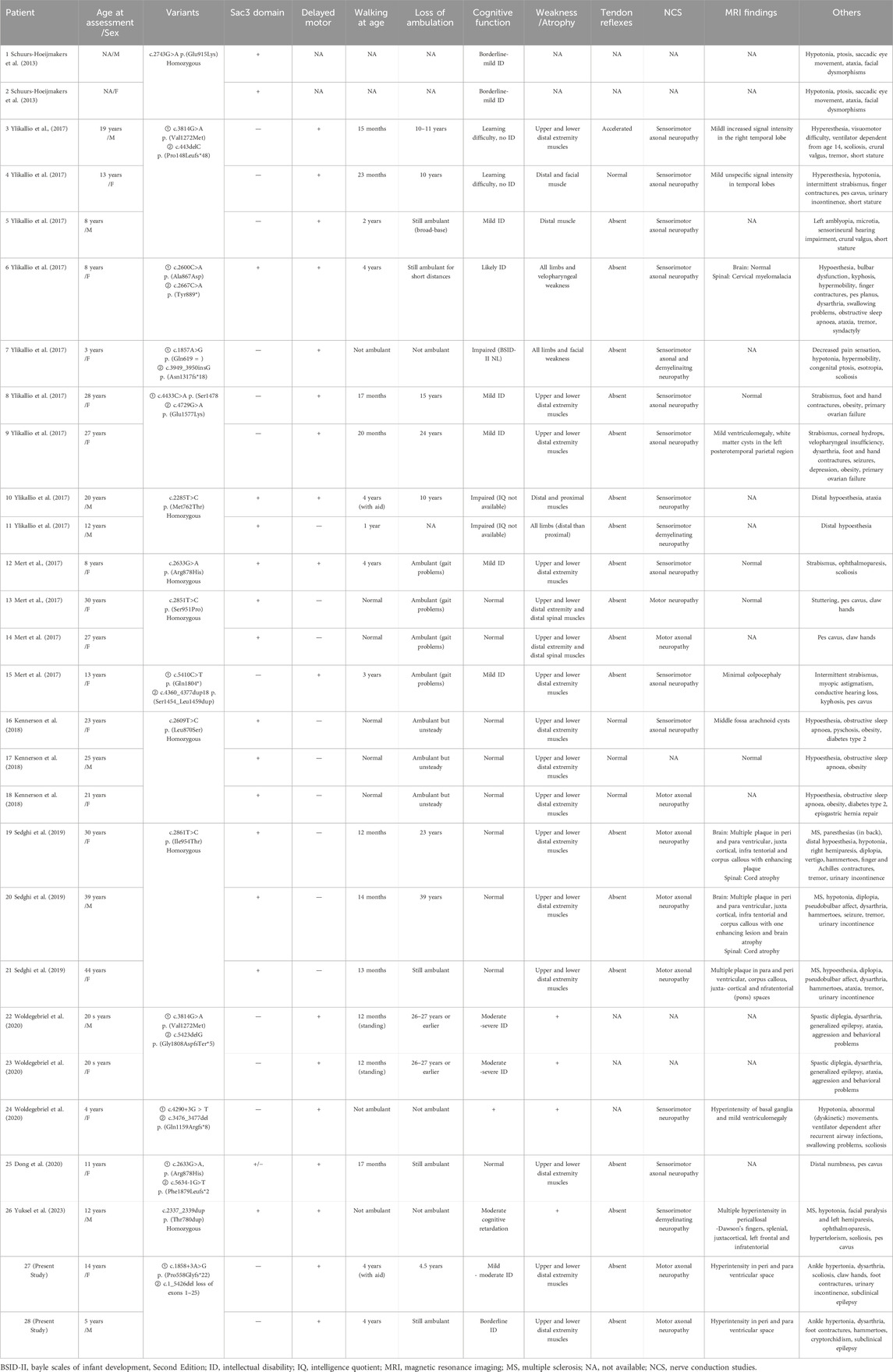
Table 3. Clinical characteristics of the patients included in this study and previous studies.
Among the 16 families, seven exhibited homozygous mutations (all within the Sac3 domain), and nine exhibited compound heterozygous mutations (seven outside the Sac3 domain, one within the Sac3 domain, and one was a mixed mutation). In the literature cohort, early-onset symptoms accompanied by motor developmental delays were observed in 100.0% (12/12) of individuals carrying mutations outside the Sac3 domain of GANP, and in 26.7% (4/15) of individuals carrying mutations within the Sac3 domain. ID was observed in 17 out of 28 individuals, with two individuals experiencing learning difficulties specifically associated with this condition. Among them, 100% (12/12) of the individuals carrying mutations outside the Sac3 domain presented with cognitive abnormalities, compared to 46.7% (7/15) of those with mutations within the domain.
4 Discussion
In this study, we summarized the clinical findings of two patients from a Chinese family with PNRIID who exhibited typical symptoms. The affected individuals presented with slow-progressive distal motor neuropathy, resulting in weakness and atrophy of the foot and/or hand muscles as well as joint deformities, accompanied by ID, dysarthria, urinary incontinence, and cryptorchidism. WES combined with CNV-seq revealed compound heterozygous variants in MCM3AP. These variants segregated with the disease phenotype, and the affected individuals carried both variants, whereas their father was a heterozygous carrier of a large fragment deletion, c.1_5426del (loss of exons 1–25), and their mother was heterozygous for c.1858 + 3A>G, a novel splicing variant resulting in the heterozygous deletion of exon 5. Coincidentally, these mutations were passed on to the siblings, eventually leading to the homozygous deletion of exon 5.
GANP, encoded by MCM3AP, is a protein associated with neurologic disorders in humans; defects in this protein have been described as causative agents of autosomal recessive Charcot–Marie–Tooth disease with ID (Ylikallio et al., 2017). The disease is characterized by progressive symmetrical distal limb muscle weakness and atrophy, with or without hypoesthesia, and weakened or absent tendon reflexes in the limbs (Mert et al., 2017). The affected individuals primarily exhibited developmental delay from infancy or experienced developmental regression following early normal motor development. To date, 23 pathogenic variants of MCM3AP have been identified in 16 families. The vast majority are missense and frameshift mutations. In our study, CNV-seq combined with WES enabled us to identify a novel variant c.1_5426del (loss of exons 1–25) in MCM3AP gene, a large pathogenic fragment deletion. These results suggest that CNV-seq or multiplex ligation-dependent probe amplification (MLPA) may be optimal for further evaluation in patients with highly suspected genetic motor/ID disorders who remain undiagnosed after gene panel testing or WES. Disease-associated MCM3AP variations can be broadly divided into two classes: those occurring within or outside the Sac3 domain of GANP (Woldegebriel et al., 2020). Notably, the Sac3 domain exhibits high homology across vertebrates (Jones et al., 2000; Jani et al., 2009; Kopytova et al., 2010), where it plays a vital role in exporting mRNAs via nuclear pores into the cytoplasm (Wickramasinghe et al., 2010). The effect of Sac3 on actin and microtubule functions may be a pathological mechanism underlying motor neuron degeneration (Bauer and Kölling, 1996). Among the 16 families, most mutations within the Sac3 domain are homozygous. Literature analysis (Mert et al., 2017; Ylikallio et al., 2017; Kennerson et al., 2018; Ylikallio et al., 2018) revealed the genotype–phenotype correlation of the pathogenic variation in MCM3AP, with a tendency toward a milder and slower-progressing phenotype in individuals with homozygous mutations within the Sac3 domain. In contrast, compound heterozygous mutations outside the Sac3 domain lead to early-onset and severe disease courses. In our study, we identified a homozygous exon 5 deletion, which was located outside the Sac3 domain; this deletion possibly affected mRNA stability and resulted in early-onset symptoms with rapid progression. Statistical analysis revealed that all the affected individuals carrying mutations outside the Sac3 domain presented early-onset symptoms accompanied by motor developmental delays. Notably, one severely affected individual carrying mutations outside the Sac3 domain experienced loss of independent ambulation during adolescence and required ventilator support (Ylikallio et al., 2017). Furthermore, missense variants within the Sac3 domain did not lead to GANP protein depletion; however, compound heterozygous MCM3AP variants outside this domain drastically decreased GANP protein levels in the nuclear envelope (Woldegebriel et al., 2020). GANP protein depletion and/or loss of function impair the transcription and/or export of mRNAs, resulting in the distinct neurologic disease phenotypes associated with MCM3AP mutations.
Previous studies have revealed MCM3AP expression in the brain and neuronal tissues (Schuurs-Hoeijmakers et al., 2013). In addition to peripheral neuropathy, ID was also observed as another prominent feature, presenting in 60.7% (17/28) patients. Furthermore, mutations outside the Sac3 domain may predispose individuals to ID. Some affected individuals exhibited other central nervous system (CNS) signs, including seizures (6/28, 21.4%), ataxia (7/28, 25.0%), tremors (5/28, 17.9%), and psychopsychic abnormalities (4/28, 14.3%). In the present study, electroencephalography revealed frequent epileptiform activity in both siblings, with the suspicion of convulsions in patient II-2. Woldegebriel et al. (2020) reported the case of a family, carrying mutations outside the Sac3 domain, that displayed a motor phenotype with progressive encephalopathy as an additional feature. According to the literature, cerebral MRI abnormalities have been observed in 70.6% (12/17) of the cases studied to date. Moreover, GANP expression has been detected in the brain (Abe et al., 2000), with suggestions of its involvement in CNS-originating neoplasms (Ohta et al., 2009). Collectively, these findings suggest the emerging role of GANP, not only in peripheral nerves but also within the CNS. Furthermore, the progression of MCM3AP-associated disease to peripheral neuropathy and MS with white matter lesions in the CNS has been described in two families (Sedghi et al., 2019; Yuksel et al., 2023). It has been reported that variants in MCM3AP encoding GANP can also cause complex phenotypes such as immunodeficiency, genomic instability, and skin changes (Gatz et al., 2016). The pathogenic mechanism of MS is considered to be immunologically mediated, involving demyelination, with B cells contributing to the immune inflammatory response. The role of GANP in B cell antibody maturation has been suggested (Kuwahara et al., 2004; Singh et al., 2013). The pathogenic mechanism of MCM3AP variants may involve neuroimmune damage. Further studies in additional families are needed to clarify these associations and explore the potential pathogenesis further.
The pathogenic variation in MCM3AP primarily results in motor neuropathy, with a less pronounced effect on sensory function; however, approximately 42.9% (12/28) of the patients revealed sensory disorders clinically, which primarily manifested as decreased or absent sensation of the cuff and sock distribution. Other potentially associated symptoms included eye movement disorders (10/28, 35.7%), dysarthria (8/28, 28.6%), respiratory dysfunction (6/28, 21.4%), obesity (5/28, 17.9%), and ovarian failure (2/28, 7.1%). In addition, cryptorchidism, which may be a novel PNRIID phenotype, was observed in our case. Studies (Ylikallio et al., 2017; Zhou et al., 2023) have shown that MCM3AP is involved in the DNA replication process, and this association may result in a phenotype of steroid hormonal dysregulation.
Rehabilitation, orthotics, and supportive care are some of the currently available treatment modalities. Evidence suggests that mild to moderate exercise is effective and safe (Chetlin et al., 2004; Young et al., 2008). In the present study, both patients exhibited improvements in limb strength and walking ability during rehabilitation; however, patient II-1 experienced functional degeneration as well as joint contracture upon rehabilitation cessation. As the disease progresses, most patients develop joint deformities, with foot and/or hand deformities (16/28, 57.1%) and scoliosis (8/28, 28.6%) being observed in the affected individuals. Surgery can be performed to treat skeletal deformities, including foot or hand deformities or scoliosis, to relieve pain and improve function (Karol and Elerson, 2007). Furthermore, orthotic and assistive devices can be helpful. Patient II-2 underwent plantar fasciotomy to correct the cavus deformity, which improved gait abnormalities and allowed better navigation of stairs while wearing orthopedic shoes. Our study findings highlight the efficacy of rehabilitation interventions for two siblings exhibiting similar clinical manifestations. Patient II-1 missed the rehabilitation period, resulting in functional regression; in contrast, the function (including cognitive) of patient II-2 continued to improve during convalescence. Due to an insufficient sample size, we cannot be certain about the efficacy of rehabilitation; therefore, an increase in the number of reported cases will help clarify this. In addition, PNRIID represents a type of progressive neuropathy. Ongoing research on upstream biological disorders and neurotrophic factors holds promise for mitigating neurodegeneration and instilling optimism among patients.
5 Conclusion
We presented the findings of a Chinese family with PNRIID harboring a novel compound heterozygous mutation in MCM3AP. We identified a large fragment deletion in MCM3AP and confirmed that the splicing variant c.1858 + 3A>G causes the deletion of exon 5. Furthermore, the genotype–phenotype correlation analysis of reported cases of biallelic MCM3AP-associated disease suggests that compound heterozygous mutations outside the Sac3 domain may lead to ID and more severe peripheral nerve phenotypes compared to homozygous mutations within the Sac3 domain. The molecular sub-regional location of variants and genotypes helps explain the phenotypic heterogeneity of patients with MCM3AP variants. More cases will help clarify genotype–phenotype correlations. Finally, our findings highlight the importance of rehabilitation interventions.
Data availability statement
The data presented in the study are deposited in the Genome Variantion Map repository, accession number GVM000827.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the Second Affiliated Hospital of Wenzhou Medical University (2021-K-127-02). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
CL: Conceptualization, Data curation, Investigation, Writing–original draft, Writing–review and editing. QX: Methodology, Writing–review and editing. QH: Data curation, Investigation, Writing–original draft. BX: Data curation, Investigation, Validation, Writing–original draft. KZ: Supervision, Writing–review and editing. XC: Conceptualization, Writing–original draft. FZ: Conceptualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Scientific Research Fund of the Zhejiang Provincial Education Department (No. Y202148132).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abe, E., Kuwahara, K., Yoshida, M., Suzuki, M., Terasaki, H., Matsuo, Y., et al. (2000). Structure, expression, and chromosomal localization of the human gene encoding a germinal center-associated nuclear protein (GANP) that associates with MCM3 involved in the initiation of DNA replication. Gene 255 (2), 219–227. doi:10.1016/s0378-1119(00)00336-x
Bauer, A., and Kölling, R. (1996). The SAC3 gene encodes a nuclear protein required for normal progression of mitosis. J. Cell. Sci. 109 (Pt 6), 1575–1583. doi:10.1242/jcs.109.6.1575
Chetlin, R. D., Gutmann, L., Tarnopolsky, M. A., Ullrich, I. H., and Yeater, R. A. (2004). Resistance training exercise and creatine in patients with Charcot-Marie-Tooth disease. Muscle Nerve 30 (1), 69–76. doi:10.1002/mus.20078
Dong, H. L., Wei, Q., Li, J. Q., Li, H. F., Bai, G., Ma, H., et al. (2020). Genetic spectrum of MCM3AP and its relationship with phenotype of Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 25 (2), 107–111. doi:10.1111/jns.12377
Gatz, S. A., Salles, D., Jacobsen, E. M., Dörk, T., Rausch, T., Aydin, S., et al. (2016). MCM3AP and POMP mutations cause a DNA-repair and DNA-damage-signaling defect in an immunodeficient child. Hum. Mutat. 37 (3), 257–268. doi:10.1002/humu.22939
Jani, D., Lutz, S., Marshall, N. J., Fischer, T., Köhler, A., Ellisdon, A. M., et al. (2009). Sus1, Cdc31, and the Sac3 CID region form a conserved interaction platform that promotes nuclear pore association and mRNA export. Mol. Cell 33 (6), 727–737. doi:10.1016/j.molcel.2009.01.033
Jones, A. L., Quimby, B. B., Hood, J. K., Ferrigno, P., Keshava, P. H., Silver, P. A., et al. (2000). SAC3 may link nuclear protein export to cell cycle progression. Proc. Natl. Acad. Sci. U.S.A. 97 (7), 3224–3229. doi:10.1073/pnas.97.7.3224
Karol, L. A., and Elerson, E. (2007). Scoliosis in patients with Charcot-Marie-Tooth disease. J. Bone Jt. Surg. Am. 89 (7), 1504–1510. doi:10.2106/JBJS.F.01161
Kennerson, M. L., Corbett, A. C., Ellis, M., Perez-Siles, G., and Nicholson, G. A. (2018). A novel MCM3AP mutation in a Lebanese family with recessive Charcot-Marie-Tooth neuropathy. Brain 141 (9), e66. doi:10.1093/brain/awy184
Kopytova, D. V., Orlova, A. V., Krasnov, A. N., Gurskiy, D. Y., Nikolenko, J. V., Nabirochkina, E. N., et al. (2010). Multifunctional factor ENY2 is associated with the THO complex and promotes its recruitment onto nascent mRNA. Genes Dev. 24 (1), 86–96. doi:10.1101/gad.550010
Kuwahara, K., Fujimura, S., Takahashi, Y., Nakagata, N., Takemori, T., Aizawa, S., et al. (2004). Germinal center-associated nuclear protein contributes to affinity maturation of B cell antigen receptor in T cell-dependent responses. Proc. Natl. Acad. Sci. U.S.A. 101 (4), 1010–1015. doi:10.1073/pnas.0307609100
Masai, H., You, Z., and Arai, K. (2005). Control of DNA replication: regulation and activation of eukaryotic replicative helicase, MCM. IUBMB Life 57 (4-5), 323–335. doi:10.1080/15216540500092419
Mert, K., Neda, M., Ipek, P., Diana, B., Sandra, D., Reza, M., et al. (2017). Biallelic MCM3AP mutations cause Charcot-Marie-Tooth neuropathy with variable clinical presentation. Brain 140e65. doi:10.1093/brain/awx222
Nussbacher, J. K., Tabet, R., Yeo, G. W., and Lagier-Tourenne, C. (2019). Disruption of RNA metabolism in neurological diseases and emerging therapeutic interventions. Neuron 102 (2), 294–320. doi:10.1016/j.neuron.2019.03.014
Ohta, K., Kuwahara, K., Zhang, Z., Makino, K., Komohara, Y., Nakamura, H., et al. (2009). Decreased expression of germinal center-associated nuclear protein is involved in chromosomal instability in malignant gliomas. Cancer Sci. 100 (11), 2069–2076. doi:10.1111/j.1349-7006.2009.01293.x
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schuurs-Hoeijmakers, J. H., Vulto-van Silfhout, A. T., Vissers, L. E., van de Vondervoort, I. I., van Bon, B. W., de Ligt, J., et al. (2013). Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. J. Med. Genet. 50 (12), 802–811. doi:10.1136/jmedgenet-2013-101644
Sedghi, M., Moslemi, A. R., Cabrera-Serrano, M., Ansari, B., Ghasemi, M., Baktashian, M., et al. (2019). Recessive Charcot-Marie-Tooth and multiple sclerosis associated with a variant in MCM3AP. Brain Commun. 1 (1), fcz011. doi:10.1093/braincomms/fcz011
Singh, S. K., Maeda, K., Eid, M. M., Almofty, S. A., Ono, M., Pham, P., et al. (2013). GANP regulates recruitment of AID to immunoglobulin variable regions by modulating transcription and nucleosome occupancy. Nat. Commun. 41830, 1830. doi:10.1038/ncomms2823
Sreedharan, J., Neukomm, L. J., Brown, R. H., and Freeman, M. R. (2015). Age-dependent TDP-43-mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr. Biol. 25 (16), 2130–2136. doi:10.1016/j.cub.2015.06.045
Takei, Y., Assenberg, M., Tsujimoto, G., and Laskey, R. (2002). The MCM3 acetylase MCM3AP inhibits initiation, but not elongation, of DNA replication via interaction with MCM3. J. Biol. Chem. 277 (45), 43121–43125. doi:10.1074/jbc.C200442200
Takei, Y., Swietlik, M., Tanoue, A., Tsujimoto, G., Kouzarides, T., and Laskey, R. (2001). MCM3AP, a novel acetyltransferase that acetylates replication protein MCM3. EMBO Rep. 2 (2), 119–123. doi:10.1093/embo-reports/kve026
Wickramasinghe, V. O., McMurtrie, P. I., Mills, A. D., Takei, Y., Penrhyn-Lowe, S., Amagase, Y., et al. (2010). mRNA export from mammalian cell nuclei is dependent on GANP. Curr. Biol. 20 (1), 25–31. doi:10.1016/j.cub.2009.10.078
Woldegebriel, R., Kvist, J., Andersson, N., Õunap, K., Reinson, K., Wojcik, M. H., et al. (2020). Distinct effects on mRNA export factor GANP underlie neurological disease phenotypes and alter gene expression depending on intron content. Hum. Mol. Genet. 29 (9), 1426–1439. doi:10.1093/hmg/ddaa051
World Medical Association (2013). World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 310 (20), 2191–2194. doi:10.1001/jama.2013.281053
Ylikallio, E., Woldegebriel, R., Tumiati, M., Isohanni, P., Ryan, M. M., Stark, Z., et al. (2017). MCM3AP in recessive Charcot-Marie-Tooth neuropathy and mild intellectual disability. Brain 140 (8), 2093–2103. doi:10.1093/brain/awx138
Ylikallio, E., Woldegebriel, R., and Tyynismaa, H. (2018). Reply: a novel MCM3AP mutation in a Lebanese family with recessive Charcot-Marie-Tooth neuropathy. Brain 141 (9), e67. doi:10.1093/brain/awy185
Young, P., De Jonghe, P., Stögbauer, F., and Butterfass-Bahloul, T. (2008). Treatment for charcot-marie-tooth disease. Cochrane Database Syst. Rev. 2008 (1), CD006052. doi:10.1002/14651858.CD006052.pub2
Yuksel, D., Gocmen, R., Temucin, C., and Lafci, N. (2023) “Recessive Charcot-Marie-Tooth and multiple sclerosis associated with a variant in MCM3AP: a case report,” in 28th international annual Congress of the world-muscle-society S99. doi:10.1016/j.nmd.2023.07.135
Keywords: peripheral neuropathy, intellectual disability, MCM3AP, GANP, biallelic mutations
Citation: Liu C, Xie Q, Hu Q, Xiang B, Zhao K, Chen X and Zheng F (2024) Identification of biallelic mutations in MCM3AP and comprehensive literature analysis. Front. Genet. 15:1405644. doi: 10.3389/fgene.2024.1405644
Received: 23 March 2024; Accepted: 31 July 2024;
Published: 20 August 2024.
Edited by:
Jayesh Sheth, Foundation for Research In Genetics and Endocrinology, IndiaReviewed by:
Maoqiang Tian, Affiliated Hospital of Zunyi Medical University, ChinaJessie Poquérusse, The University of Auckland, New Zealand
Copyright © 2024 Liu, Xie, Hu, Xiang, Zhao, Chen and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Chen, Y2hlbnhpYW5nZmV5QDEyNi5jb20=; Feixia Zheng, emhmeGlhMDZAMTYzLmNvbQ==