
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 07 June 2024
Sec. Livestock Genomics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1404031
 Hua Yang1*†
Hua Yang1*† Mengting Zhu1,2†
Mengting Zhu1,2† Mingyuan Wang1,3
Mingyuan Wang1,3 Huaqian Zhou1,3Jingjing Zheng1,3Lixia Qiu1Wenhua Fan1,3Jinghui Yang1Qian Yu1Yonglin Yang1Wenzhe Zhang1
Huaqian Zhou1,3Jingjing Zheng1,3Lixia Qiu1Wenhua Fan1,3Jinghui Yang1Qian Yu1Yonglin Yang1Wenzhe Zhang1The identification of genome-wide selection signatures can reveal the potential genetic mechanisms involved in the generation of new breeds through natural or artificial selection. In this study, we screened the genome-wide selection signatures of prolific Suffolk sheep, a new strain of multiparous mutton sheep, to identify candidate genes for reproduction traits and unravel the germplasm characteristics and population genetic evolution of this new strain of Suffolk sheep. Whole-genome resequencing was performed at an effective sequencing depth of 20× for genomic diversity and population structure analysis. Additionally, selection signatures were investigated in prolific Suffolk sheep, Suffolk sheep, and Hu sheep using fixation index (FST) and heterozygosity H) analysis. A total of 5,236.338 Gb of high-quality genomic data and 28,767,952 SNPs were obtained for prolific Suffolk sheep. Moreover, 99 selection signals spanning candidate genes were identified. Twenty-three genes were significantly associated with KEGG pathway and Gene Ontology terms related to reproduction, growth, immunity, and metabolism. Through selective signal analysis, genes such as ARHGEF4, CATIP, and CCDC115 were found to be significantly correlated with reproductive traits in prolific Suffolk sheep and were highly associated with the mTOR signaling pathway, the melanogenic pathway, and the Hippo signaling pathways, among others. These results contribute to the understanding of the evolution of artificial selection in prolific Suffolk sheep and provide candidate reproduction-related genes that may be beneficial for the establishment of new sheep breeds.
China has the largest sheep flock and is the largest producer of sheep meat worldwide. According to the “National Breed List of Livestock and Poultry Genetic Resources” (2021 edition), there are 89 sheep breeds in China, including 44 indigenous breeds, 32 improved breeds, and 13 introduced breeds. Developing sheep breeds with high prolificacy has become a key goal in livestock breeding (Cottle, 2010; Miao et al., 2016; Gowane et al., 2017). Many new sheep breeds have been established in China in the past decade, such as Luxi Black Head sheep (Liu et al., 2022), Huang-huai sheep (Quan et al., 2020), and a new breed of prolific Suffolk sheep (Wang et al., 2020), which was established through grading hybridization between Suffolk sheep and Hu sheep over 12 years of breeding. Suffolk sheep make excellent male parents for terminal crosses while Hu sheep are known for their high fertility, and are widely used in sheep farming in Xinjiang, China. Thus, prolific Suffolk sheep have the advantages of high-quality meat from Suffolk sheep and prolificacy characteristics from Hu sheep.
Selection signature analysis can help identify the genomic imprint of livestock resulting from the process of domestication or artificial selection. Based on this strategy, researchers can scan the regions that are associated with important economic traits that have been subjected to selection during the domestication of livestock, locate the selected genes or genetic markers, and identify the genetic mutations associated with these traits, so as to achieve variety improvement and new germplasm creation (Horscroft et al., 2019). Additionally, population-based resequencing can reveal the evolutionary relationships between populations, help identify the excellent genetic resources of each breed, and contribute to the understanding of the genetic diversity between populations; this provides strong support for the selection of new breeds and the promotion of the development of animal husbandry. Li et al. conducted 25.7× whole-genome resequencing of wild and domestic sheep, which revealed the genetic mechanism underlying various agricultural traits in domesticated sheep (e.g., reproduction, wool production), thereby providing valuable genomic resources for research on sheep genetics (Li et al., 2020). Similarly, Zhang et al. employed genome resequencing technology to uncover the natural selection molecular imprinting of wild and domesticated sheep. They identified IFI44, PNK2, and RNF24 as being related to the immunity of sheep, thereby providing insights into the molecular mechanism underpinning the origins of phenotypic variation induced by sheep domestication and improvement (Zhang et al., 2022). Sweet-Jones et al. used whole-genome resequencing to perform high-depth scanning for selection signatures linked to the adaptability of Welsh sheep (Sweet-Jones et al., 2021). The authors reported that the RNF24, PANK2, and MUC15 genes had strong selection signals, with potential functions in the environmental adaptability of local Welsh breeds. Furthermore, Wang et al. (2017) sequenced mixed pools of multiple-lamb and single-lamb Duolang sheep populations, and identified six genes related to reproductive performance, including INHBA, NCOA1, INGS, BMPR-IB, ARNT, and KLHL1 (Sui, 2017). Zhang et al. used fixation index (FST) analysis to detect genome-wide selection signals in five sheep breeds, and found that RXFP2, GHR, and ASIP were related to the shape, growth, and lipid metabolism of horns (Zhang et al., 2013). Therefore, we used whole genome resequencing technology to screen genes related to important economic traits, reveal the genetic basis of breeding breeds, and provide a basis for the selection and breeding of multiple new breeds of mutton sheep.
In this study, we resequenced the whole genomes of 90 Hu sheep, Suffolk sheep, and prolific Suffolk sheep to explore their genetic structure and the genetic variance among the breeds as well as identify candidate regions and genes related to reproductive traits. Additionally, FST and H analysis was used to identify selection signals unique to prolific Suffolk sheep, while functional enrichment analysis was undertaken to identify major genes closely related to reproductive traits. Our aim was to provide a theoretical basis for the breeding of prolific Suffolk sheep breeds as well as offer further insights into the selection of local sheep breeds in China.
In this study, based on pedigree information, 50 healthy and unrelated prolific Suffolk sheep of the same age (26 rams and 24 ewes with 196% average lambing rate) were selected. The 24 ewes were divided into two groups, namely, a multi-lamb group, consisting of 12 ewes with a 275% lambing rate (Figure 1A), and a single-lamb group, comprising 12 ewes with a lambing rate of 117% (Figure 1B). Twenty Hu sheep with a 230% lambing rate (Figure 1C) and 20 Suffolk sheep with a 140% lambing rate (Figure 1D) were also used (Supplementary Table S6). All the sheep came from the sheep farm of Xinjiang Academy of Agricultural and Reclamation Science. Blood samples (5 mL) collected from the jugular vein were placed in EDTA-Na2 and stored at −20 °C until further processing.

Figure 1. Sheep breeds. (A) Prolific Suffolk sheep in the multi-lamb group. (B) Prolific Suffolk sheep in the single-lamb group. (C) Hu sheep. (D) Suffolk sheep.
DNA was extracted from blood using the TIANamp Blood DNA Kit (TIANGEN, China) according to the manufacturer’s instructions. The quality of the genomic DNA was assessed by 1% agarose gel electrophoresis while its concentration and purity were evaluated using a NanoDrop 2000 spectrophotometer. DNA libraries were prepared using the TruSeq Library Construction Kit. The DNA was randomly fragmented to an average size of 350 bp using ultrasonication and sequencing libraries were constructed following the manufacturer’s instructions (Illumina, San Diego, CA, USA). The libraries were paired-end sequenced at ∼20× coverage on the Illumina HiSeq2500 Platform (Illumina Inc.) by Beijing Compass Biotechnology Co., Ltd (Beijing, China). To provide reliable data for subsequent analysis, the original sequencing data were filtered to remove reads with linker sequences, sequences with more than 10% N content, and low-quality data (Q-value ≤5). Subsequent analyses were based on these clean data.
The effective sequencing data were compared to the sheep reference genome (Oar_v4.0) through BWA (v.0.7.12) with the parameters “mem -t 4 -k 32 -M” (Li and Durbin, 2009). Individual SNPs were detected with SAMtools (v.1.2) using the parameters “mpileup -m 2 -F 0.002 -d 1,000” (Li et al., 2009). The filtering criteria for minimizing variant calling errors were as follows: variant sites with QD < 2.0, MQ < 20, and FS > 60.0 were discarded and the remaining variants were annotated using ANNOVAR v.21-Jun-2013.
Based on the neighbor-joining (NJ) method, a phylogenetic tree was constructed with the set of quality-filtered SNPs using the Phylogeny Inference Package (PHYLIP) (Felsenstein, 1989). Cluster analysis for elucidating population structure was performed using ADMIXTURE (v.1.3.0) with the following parameters: “for K in 2 3, do admixture --cv sheep.bed $K | tee log${K }.out, done” with a maximum of 10,000 iterations. Principal Component Analysis (PCA) of the 90 samples was performed using the EIGENSOFT package, v.7.2.1 (Price et al., 2006).
In this study, H and the FST were calculated with VCFtools v.0.1.14 using a sliding window approach (100-kb windows with a 50-kb step size) (Zhang et al., 2021). The parameters for the VCFtools program were as follows: “--fst-window -size 100,000 --fst-window-step 50,000”. The top 5% FST and H values were selected as the threshold to map the selected loci on autosomes and identify differences between any two populations. The intersection candidate area was considered as the selection signal in the test.
The FST calculation formulas:
The H calculation formulas:
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed to identify clusters of functionally related genes (Harris et al., 2004; Jiao et al., 2012). GO term analysis included the biological process (BP), molecular function (MF), and cellular component (CC) categories. A significance threshold of <0.05 was used to determine GO term enrichment in a set of genes. KEGG pathway enrichment analysis was performed using KOBAS 2.0 (http://kobas.cbi.pku.edu.cn/) and a corrected p-value of <0.05 was set as the threshold for significance. In order to avoid false positive results, the enrichment analysis result P was corrected by multiple tests (False Discovery Rate, FDR).
The formula of FDR is:
A total of 5,245.855 Gb of raw data for 90 individuals were obtained by resequencing on the Illumina HiSeq 2,500 platform, with 5,236.338 Gb of clean reads remaining after filtering. The Q20 value of the clean reads was ≥95.73% while the Q30 value was ≥89.69%. The GC content ranged between 43.09% and 46.62%. The genome mapping rate relative to the sheep reference genome (Oar_v4.0) ranged between 98.65% and 99.32%. The average coverage depth was approximately 17.39× for all three sheep breeds, the 1 × average coverage was more than 98.06%, and the 4 × average coverage was more than 94.29%, indicating that the data were accurate and reliable (Supplementary Table S1). SAMTools was used to collect summary information from the input binary alignment/map (BAM) files, compute the likelihood of each genotype, and then convert the information into binary variant call format (BCF). ANNOVAR software was used for the functional annotation of gene mutations and for converting the data into variant call format (VCF) for subsequent analysis.
Additionally, 84,505,591 SNPs in all three sheep breeds and 28,767,952 SNPs in prolific Suffolk sheep were annotated with SAMTools v.0.1.19. In the prolific Suffolk sheep population, 204,304 exonic SNPs, 1,520,657 non-synonymous mutations (5.29%), and 2,101,043 synonymous mutations were identified. In the Hu sheep population, a total of 29,080,742 SNPs were annotated, 207,059 of which were located in exons; 620,064 non-synonymous mutations (2.13%) and 866,996 synonymous mutations were also identified. In Suffolk sheep, a total of 26,656,897 SNPs were annotated; 187,316 of these SNPs were located in exons, while 606,196 non-synonymous mutations (2.27%) and 838,500 synonymous mutations were identified. The non-synonymous/synonymous ratio was 0.72 in all three sheep breeds (Supplementary Table S2).
To investigate the genetic relationship among the three sheep breeds, a genetic distance matrix was calculated based on the SNPs after whole-genome quality control, and a phylogenetic tree of the three populations was constructed using the NJ method. The NJ tree was constructed based on the JTT + G model with 1,000 bootstrap replicates. NJ tree analysis showed that the three sheep varieties were separated into three independent genetic groups (Figure 2A).
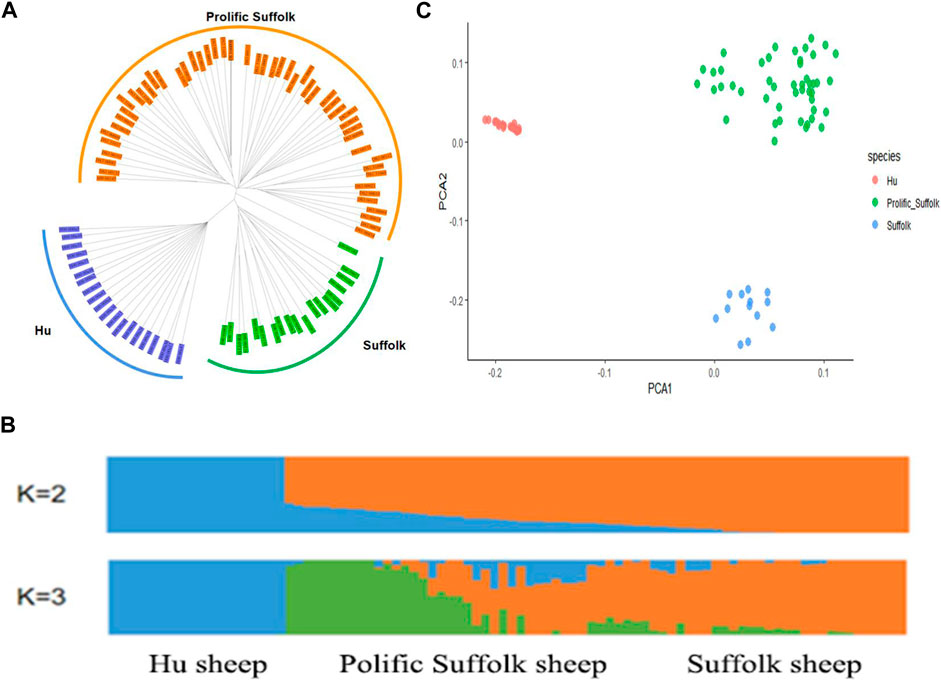
Figure 2. Population genetics analysis. (A) Neighbor-joining phylogenetic tree. (B) Population structure based on 90 individuals as determined using ADMIXTURE with K = 2, 3. (C) The results of the Principal Component Analysis (PCA) for the three sheep breeds.
As shown in Figure 2B, at K = 2, Hu sheep clustered into one type, prolific Suffolk sheep and Suffolk sheep clustered into another type, and there was gene flow within them. At K = 3, prolific Suffolk sheep were clearly separated from both Hu and Suffolk sheep. Prolific Suffolk sheep and Suffolk sheep were clustered together. Hu sheep had a long genetic distance from the other two populations. Also, a close genetic relationship was discovered between prolific Suffolk sheep and Suffolk sheep, which was also consistent with the breeding process of the former.
To examine the genetic relationship among and within the three varieties, we conducted a PCA. The first Eigenvector clearly distinguished Hu sheep from Prolific Suffolk sheep and Suffolk sheep, while the second Eigenvector distinguished prolific Suffolk sheep from Suffolk sheep (Figure 2C). As expected, the results of the PCA were similar to those obtained for the phylogenetic tree and population genetic structure analysis, showing that the selected samples had good consistency.
To accurately identify the biological markers associated with the germplasm characteristics of prolific Suffolk sheep, the sliding window method (window size: 100 kb, step size: 50 kb) was used to scan the selection signals on autosomes. The top 5% FST and H values were selected as the threshold to map the selected loci on autosomes and identify differences between any two populations. A total of 137 selected regions were scanned and 154 candidate genes were mapped in the comparison between prolific Suffolk sheep and Hu sheep (FST > 0.249001 and H < 0.224707) (Figures 3A,C; Supplementary Tables S3 and S4). For prolific Suffolk sheep versus Suffolk sheep, a total of 99 selected regions were screened and 59 candidate genes were mapped (FST > 0.178916 and H < 0.224707) (Figures 3B,C; Supplementary Tables S3 and S4). Repeats were removed from the 213 candidate genes in the two comparison groups. Finally, 190 candidate genes were screened, 14 of which were related to reproduction traits, including WNT10A, SENP2, and WNT6 (Table 1). Furthermore, 23 genes, including ARHGEF4, CATIP, CCDC115, and CDK5R2, were found to be unique to prolific Suffolk sheep (Figure 3D; Supplementary Table S5). These genes may represent the germplasm-specific genetic information retained during prolific Suffolk sheep breeding and selection (Table 2).
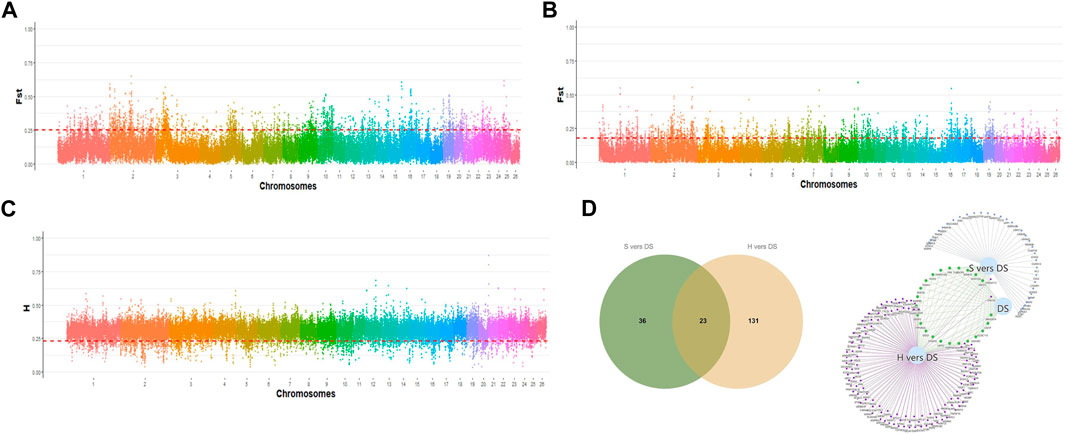
Figure 3. (A) The distribution of FST values on autosomes in prolific Suffolk sheep and Hu sheep. (B) The distribution of FST values on autosomes in prolific Suffolk sheep and Suffolk sheep. (C) The distribution of heterozygosity (H) on autosomes in prolific Suffolk sheep. (D) Venn diagram of the unique genes in prolific Suffolk sheep.
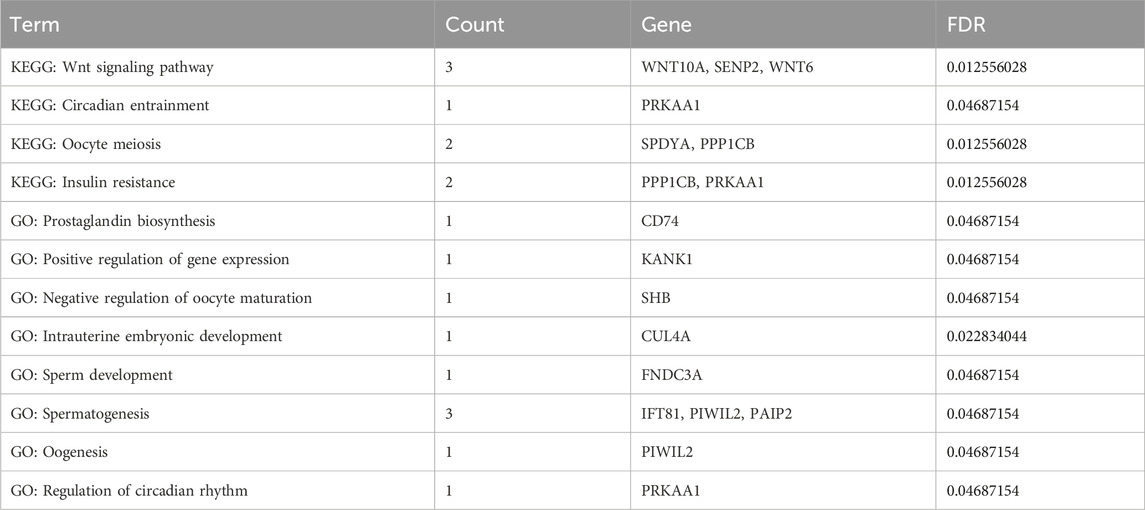
Table 1. Enriched items and genes related to reproduction in prolific Suffolk sheep.
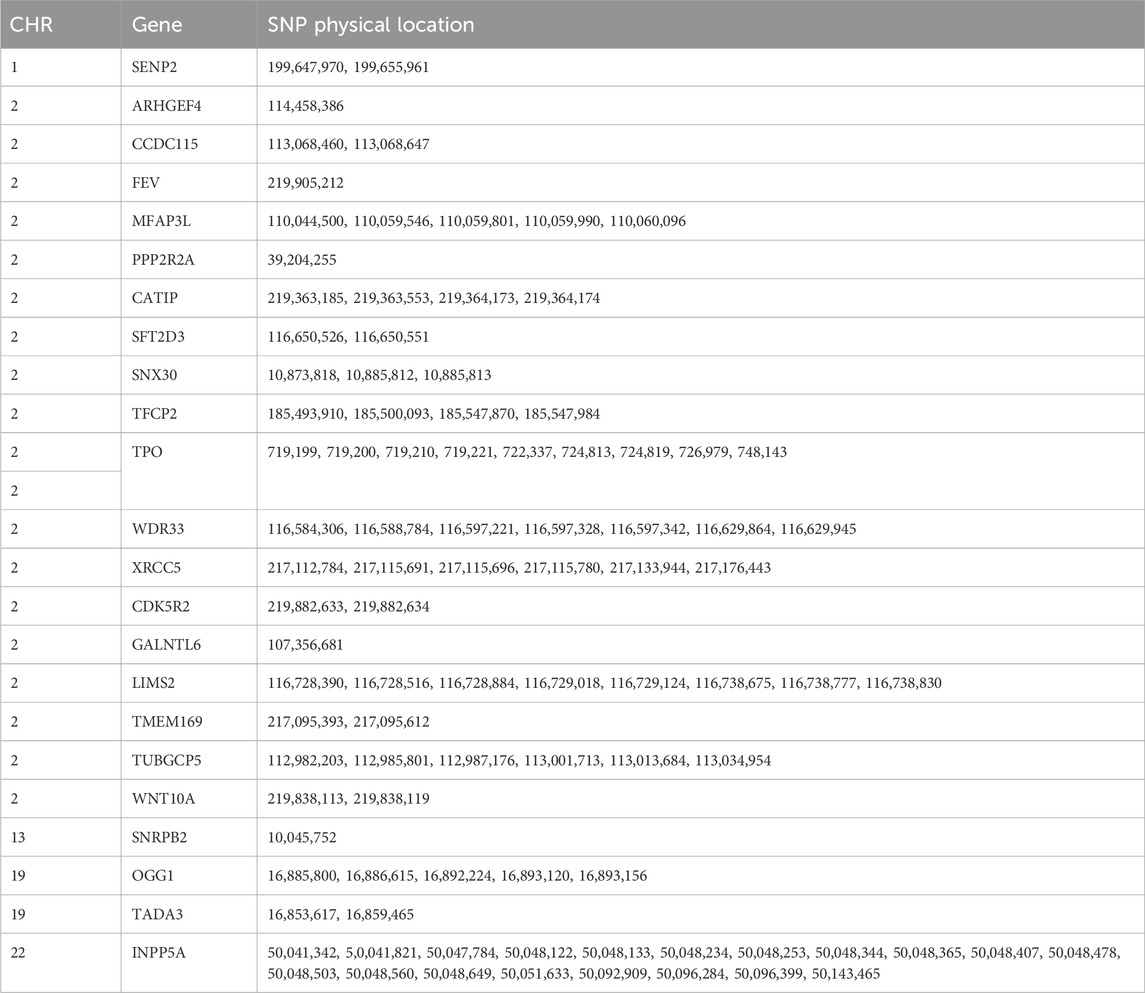
Table 2. Genetic variation information for prolific Suffolk sheep.
Additionally, Gene Ontology (GO) enrichment analysis revealed key biological processes associated with germplasm-specific genes in prolific Suffolk sheep. Candidate genes were enriched in positive regulation of defense response to virus by host, and G protein-coupled purinergic nucleotide receptor activity and other processes and were mainly related to signal transduction. Meanwhile, KEGG pathway enrichment analysis indicated that the candidate genes WNT10A, PPP2R2A, and WDR33 were enriched in the mTOR signaling pathway, melanogenesis, and the Hippo signaling pathway (p < 0.05) (Figure 4).
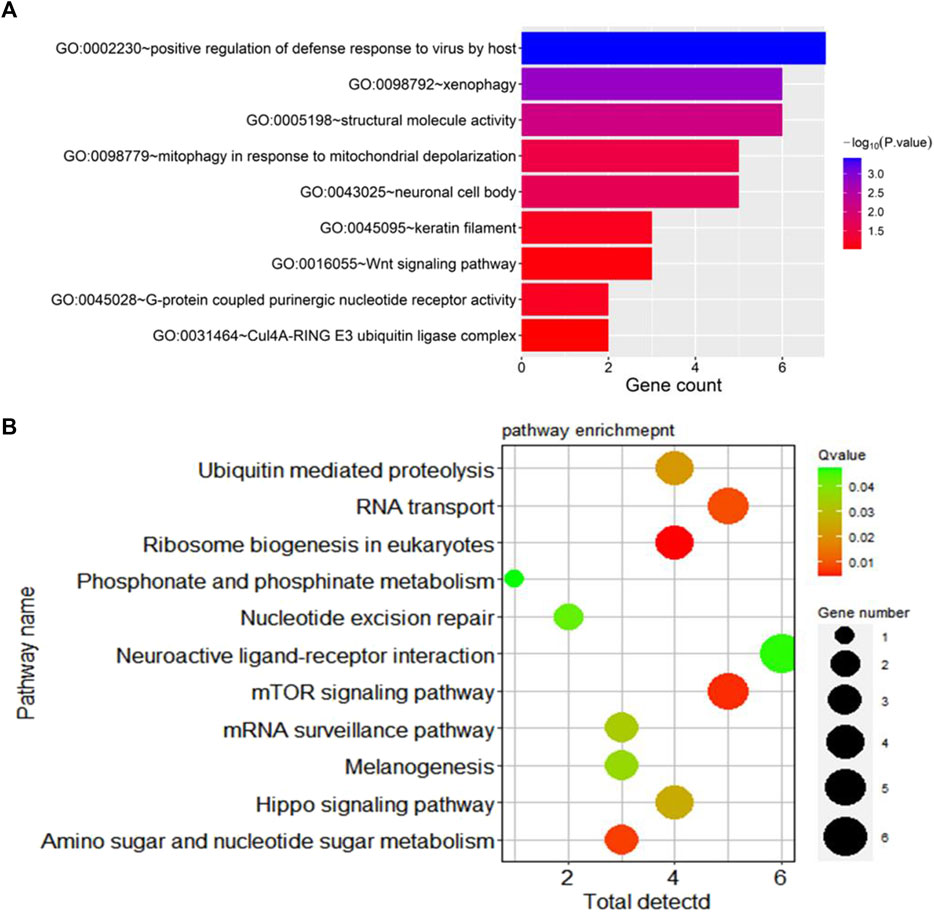
Figure 4. Enrichment results for specific genes in prolific Suffolk sheep. (A) GO term enrichment. (B) KEGG pathway enrichment.
Next, we explored the selective imprints of reproduction traits in prolific Suffolk sheep resulting from artificial selection. For this, selection signals were compared between the multi-lamb and single-lamb groups using 100-kb sliding windows with a step size of 50 kb across the genome. A combination of both FST and H analysis methods was also employed to scan for selection signals on autosomes. We identified 29 selected regions and 24 candidate genes (Figures 5A,B; Table 3). Genes related to reproductive traits, such as MTNR1A, ITSN1, and GBE1, among others, were subjected to GO and KEGG enrichment analysis and were found to be associated with litter size. Furthermore, the identified genes were mainly enriched in circadian entrainment, MAPK signaling pathway, AMPK signaling pathway, and mTOR signaling pathway (Figures 5C,D).
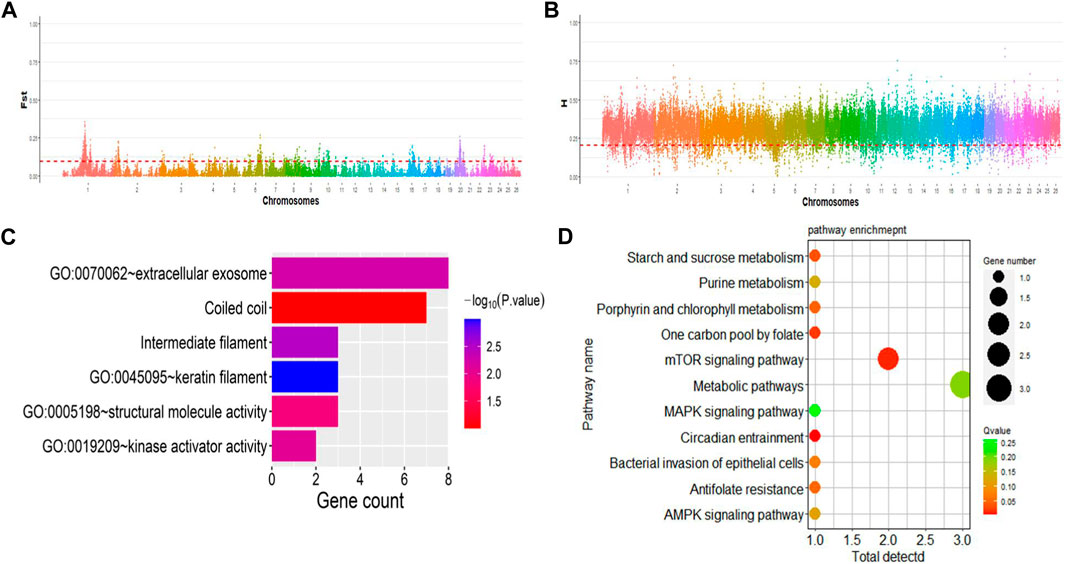
Figure 5. Selective imprints of the reproductive traits in multi-lamb and single-lamb groups. (A) Selection signals in the multi-lamb and single-lamb groups. (B) Distribution of heterozygosity (H) on the autosome of the multi-lamb group. (C) GO term enrichment analysis for genes related to reproductive traits. (D) KEGG pathway enrichment analysis for genes related to reproductive traits.
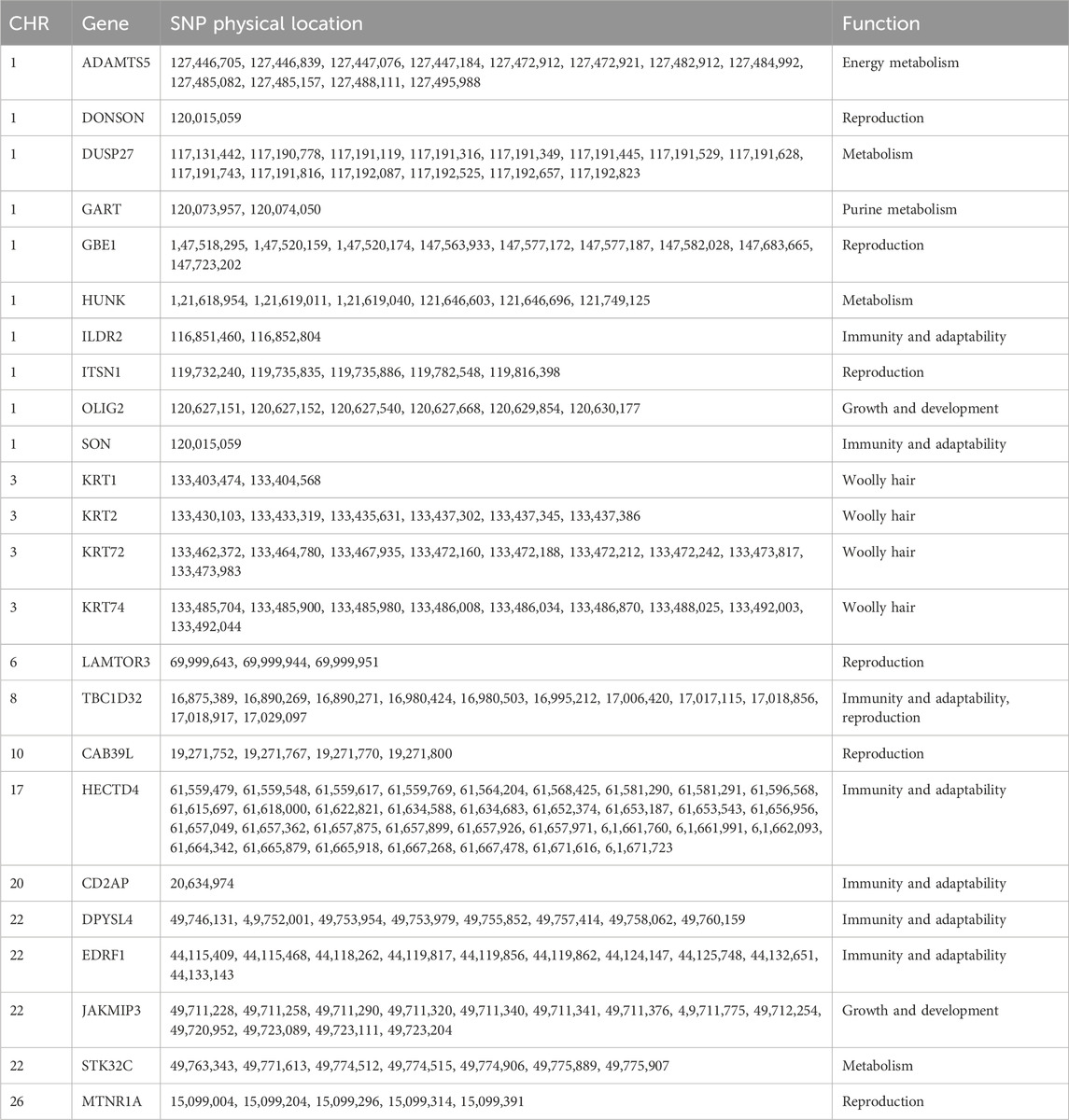
Table 3. Variation information for genes in prolific Suffolk sheep.
Sheep farming plays an important role in the global animal husbandry industry, supplying a wide range of products such as wool, meat, milk, and skin. In China, sheep farming has been an important part of the agricultural economy and rural life for many centuries. Over recent years, the meat sheep breeding industry has undergone rapid development, characterized by an increasing abundance of germplasm resources, a gradually improving breeding system, and a steadily increasing level of sheep production (Meadows and Kijas, 2010). However, overall, there is still a large gap between China and developed countries in this respect, a limitation that can mostly be explained by the low fecundity of most Chinese breeds (Han et al., 2015). Litter size is a complex trait that is influenced by many factors, including genetic background, nutritional level, and feeding management. It is the main trait in sheep reproductive performance and one of the main selection objectives in meat sheep breeding programs (Baelden et al., 2005). Increasing litter size proved to be an effective means of improving the economic benefits of the mutton industry (Dong et al., 2013). Therefore, in this study, we performed genome-wide resequencing to analyze germplasm-specific genes, genetic variation, and the genome map of self-bred prolific Suffolk sheep. Furthermore, we identified the main genes and SNPs related to their reproductive traits. Our findings contribute to unraveling the germplasm characteristics and population genetic evolution of this new strain of Suffolk sheep and provide a basis for the creation of new breeds of prolific meat sheep in China.
Prolific Suffolk sheep is a new strain developed by our group through 12 years of breeding in Xinjiang, China. Hu sheep served as the female parents and Suffolk sheep as the male parents in hybrid breeding. The breeding process involved hybridization, fixation in a two-way crossbreed closed flock, and herd propagation, resulting in a breed with high prolificacy and high-quality meat performance. The new strain is a stabilized composite breed made up of 87.5% Suffolk blood proportion and 12.5% Hu blood proportion (Yang et al., 2021a). In this study, we found that the Hu sheep breed was highly distinct from the other two breeds, exhibiting a large genetic distance from both, which was also consistent with the breeding process of prolific Suffolk sheep. Through selective signal analysis, the WNT10A, PPP2R2A, and WDR33 genes were found to be significantly related to reproductive traits in prolific Suffolk sheep and were highly associated with the mTOR signaling pathway, the melanogenic pathway, and the Hippo signaling pathway, among others. mTOR is a highly conserved serine-threonine protein kinase that forms two distinct complexes, mTORC1 and mTORC2 (Sui et al., 2021). mTOR complexes are sensitive to growth factor, amino acid, and oxidative stress stimulation and are involved in multiple biological processes, such as lipid metabolism, autophagy, protein synthesis, and nucleosome biosynthesis (Yao et al., 2021). The melanogenesis pathway involves a complex series of enzymatically and chemically catalyzed reactions (Pillaiyar et al., 2017). The MC1 receptor (MC1R) and its ligand melanocortin are important positive regulators of melanin production (Dnyane and Gadgil, 2020). MC1R activates cyclic AMP (cAMP) response element binding protein (CREB). Tyrosinase (TYR) is the rate-limiting enzyme in melanin synthesis, which occurs in specialized cell organelles, called melanosomes, which are transferred to keratinocytes through mechanisms that have not been fully characterized (Varghese et al., 2021). The results of the present study indicated that phenotypic uniformity was achieved in prolific Suffolk sheep during the 12 years of breeding. The appearance of pure black coat color on the head and limbs is one of the characteristics of the breed, indicating that differences in melanin production may underlie this phenotype. Additionally, the Hippo and WNT signaling pathways are closely related. The Hippo signaling pathway comprises a group of conserved kinases that can inhibit the normal growth of cells and participates in the regulation of organ and tissue size (Heallen et al., 2011; Yang et al., 2021b). The phosphorylation of the protein kinase Warts, a constituent of the Hippo signaling pathway, results in its activation, leading to a series of changes in signaling pathways associated with reproduction traits in female livestock (Vitezslav and Vladimir, 2018; Hidayah et al., 2019). The WNT10A gene has been shown to act in the canonical Wnt/β-catenin signaling pathway. It is expressed in epithelial and mesenchymal cells throughout tooth development and plays an important role in this process (Kanchanasevee et al., 2020; Zeng et al., 2021). In this study, we found that the selected genes were enriched in the WNT and mTOR signaling pathways, suggesting that WNT10A may have contributed to changes in tooth development, hair follicle growth, and reproductive performance during the breeding of prolific Suffolk sheep. Similarly, the PPP2R2A gene is mainly involved in signal transduction and may participate in the growth and development of mammals (Zhang et al., 2020). Cui et al. showed that the downregulation of PPP2R2A by CRISPR/Cas9-mediated deletion inhibits the growth of non-small cell lung cancer cells (Cui et al., 2020), which was similar to the results of this study. This suggests that the WNT10A and PPP2R2A genes may have played a role in the breeding process of prolific Suffolk sheep, affecting their growth and development, hair follicle growth, and reproductive performance.
Furthermore, selective imprint analysis indicated that the MTNR1A and GBE1 genes were mainly involved in circadian entrainment, metabolism, and signal transduction, and were closely related to the high fecundity of prolific Suffolk sheep. In addition, MTNR1A has been reported to be related to sheep litter size (Luridiana et al., 2020; Stari et al., 2020). MTNR1A is a G protein-coupled seven-transmembrane receptor that mediates the effects of melatonin on mammalian circadian rhythms, thereby affecting mammalian reproductive function (Cosso et al., 2021). Notably, the MTNR1A gene is a key regulator of the reproductive traits of sheep and may be closely related to the litter size trait in prolific Suffolk sheep. He et al. analyzed the association between MTNR1A gene polymorphism and litter size in Small Tail Han sheep and showed that the mutation (TT) in the g.15118756C > T locus significantly increased litter size (He et al., 2012). Additionally, Starič et al. investigated the correlation between MTNR1A gene polymorphism and litter size in Slovenian sheep and similarly found that the g.17355452 locus had a significant effect on litter size (Stari et al., 2020). These results were similar to those recorded in this study. The protein encoded by the GBE1 gene is a glycogen-branching enzyme that may be involved in energy regulation during animal growth (Li et al., 2018). UROIIIS, encoded by the UROS gene, may be involved in metabolic regulation during growth (Blouin et al., 2021). Our results suggested that the GBE1 and UROS genes may regulate the growth, development, and metabolism of prolific Suffolk sheep.
Through selective signature analysis, we identified the genes that were specifically differentially expressed in prolific Suffolk sheep, a new strain developed by crossing Hu sheep and Suffolk sheep. These genes were mainly enriched in pathways related to reproduction, immunity, growth, energy metabolism, and sugar metabolism. Our findings provide both a basis for the molecular breeding of new breeds of prolific meat sheep as well as target genes and functional sites for the establishment of other new sheep breeds.
In conclusion, this study provided a comprehensive insight into the germplasm characteristics of prolific Suffolk sheep. We identified several germplasm-specific candidate genes and markers under selection in prolific Suffolk sheep, Suffolk sheep, and Hu sheep. These genes play essential roles in reproduction, growth and development, among other economic traits. The large number of genetic variants identified in the study represents an opportunity for further exploring the genetic diversity and the associated phenotypic variation in prolific Suffolk sheep. Our results contribute to the understanding of the genetic makeup of prolific Suffolk sheep and provide valuable information for future development and improvement of new breeds.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal studies were approved by The study was conducted in accordance with the Experimental Animal Care and Use Committee of Xinjiang Academy of Agricultural and Reclamation Sciences (protocol code XJNKKXY-2020-34, 30 December 2020). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
HY: Writing–original draft, Writing–review and editing, Project administration, Resources. MZ: Writing–original draft, Writing–review and editing, Formal Analysis. MW: Formal Analysis, Writing–review and editing. HZ: Writing–review and editing. JZ: Writing–review and editing. LQ: Writing–review and editing. WF: Writing–review and editing. JY: Writing–review and editing. QY: Writing–review and editing. YY: Writing–review and editing. WZ: Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by Youth Science and Technology Innovation Talent of Tianshan Talent Training Program in Xinjiang; Bingtuan Talent Training Program in Xinjiang; the National Natural Science Foundation of China, grant number 32360818; Agricultural Science and Technology Innovation Project of the Xinjiang Production and Construction Corps, grant number NCG202211 and Major Scientific and Technological Project of the Xinjiang Production and Construction Corps, grant number 2017AA006, China.
The authors would like to express their thanks for help with various aspects of the study. We are grateful for the support of Youth Science and Technology Innovation Talent of Tianshan Talent Training Program in Xinjiang; Bingtuan Talent Training Program in Xinjiang; the National Natural Science Foundation of China, grant number 32360818; Agricultural Science and Technology Innovation Project of the Xinjiang Production and Construction Corps, grant number NCG202211 and Major Scientific and Technological Project of the Xinjiang Production and Construction Corps, grant number 2017AA006, China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1404031/full#supplementary-material
BP, Biological process; CC, Cellular component; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MF, Molecular function; NJ, Neighbor-joining; PCA, Principal Component Analysis; VCF, Variant call format.
Baelden, M., Tiphine, L., Poivey, J. P., Bouix, J., Bibé, B., Robert-Granié, C., et al. (2005). Estimation of genetic parameters for litter size after natural and hormone-induced oestrus in sheep. Livest. Prod. Sci. 97 (2-3), 275–281. doi:10.1016/j.livprodsci.2005.05.003
Blouin, J. M., Ged, C., Bernardo-Seisdedos, G., Cabantous, T., Richard, E., Poli, A., et al. (2021). Identification of novel UROS mutations in a patient with congenital erythropoietic porphyria and efficient treatment by phlebotomy. Mol. Genet. Metabolism Rep. 27 (Jan 9), 100722. doi:10.1016/j.ymgmr.2021.100722
Cosso, G., Nehme, M., Luridiana, S., Pulinas, L., Mura, M. C., Hosri, C., et al. (2021). Detection of polymorphisms in the MTNR1A gene and their association with reproductive performance in awassi ewes. Animals 11 (2), 583. doi:10.3390/ani11020583
Cui, R., Jiang, N., Zhang, M., Du, S., Zhang, J., Ge, R., et al. (2020). AMOTL2 inhibits JUN Thr239 dephosphorylation by binding PPP2R2A to suppress the proliferation in non-small cell lung cancer cells. Biochimica Biophysica Acta (BBA) - Mol. Cell. Res. 1868, 118858. doi:10.1016/j.bbamcr.2020.118858
Dnyane, P., and Gadgil, C. (2020). Boolean model for melanogenesis. Phys. Biol. 18, 026004. doi:10.1088/1478-3975/abd3dd
Dong, Y., Xie, M., Jiang, Y., Xiao, N., Du, X., Zhang, W., et al. (2013). Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 31 (2), 135–141. doi:10.1038/nbt.2478
Felsenstein, J. (1989). PHYLIP-phylogeny inference package (version 3.2). Cladistics-the Int. J. Willi Hennig Soc. 5, 164–166.
Gowane, G. R., Gadekar, Y. P., Prakash, V., Kadam, V., Chopra, A., and Prince, L. L. L. (2017). “Climate change impact on sheep production: growth, milk, wool, and meat,” in Sheep production adapting to climate change, Germany, Springer, 31–69.
Han, Y., Chen, H., Shen, B., Jia, Y., Sheng, Z., and Zeng, X. C. (2015). Differential gene expression in ovaries of qira black sheep and hetian sheep using RNA-seq technique. PLOS ONE 10 (3), e0120170. doi:10.1371/journal.pone.0120170
Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., et al. (2004). The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261. doi:10.1093/nar/gkh036
He, J. N., Zhang, B. Y., Chu, M. X., Wang, P. Q., Fen, T., Cao, G. L., et al. (2012). Polymorphism of insulin-like growth factor 1 gene and its association with litter size in Small Tail Han sheep. Mol. Biol. Rep. 39 (10), 9801–9807. doi:10.1007/s11033-012-1846-y
Heallen, T., Zhang, M., Wang, J., Bontlla-Claudio, M., Klysik, E., Ohnson, R. L., et al. (2011). Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332 (6028), 458–461. doi:10.1126/science.1199010
Hidayah, N., Kritsiligkou, P., and Grant, C. M. (2019) Loss of mrna surveillance pathways results in widespread protein aggregation open.
Horscroft, C., Ennis, S., Pengelly, R. J., Sluckin, T. J., and A, C. (2019). Sequencing era methods for identifying signatures of selection in the genome. Brief. Bioinform 20 (6), 1997–2008. doi:10.1093/bib/bby064
Jiao, X., Sherman, B. T., Huang da, W., Stephens, R., Baseler, M. W., Lane, H. C., et al. (2012). DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 28 (13), 1805–1806. doi:10.1093/bioinformatics/bts251
Kanchanasevee, C., Sriwattanapong, K., Theerapanon, T., Thaweesapphithak, S., Shotelersuk, V., Porntaveetus, T., et al. (2020). Phenotypic and genotypic features of Thai patients with nonsyndromic tooth agenesis and WNT10A variants. Front. Physiology 11, 573214. doi:10.3389/fphys.2020.573214
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, W., Wong, C. C., Zhang, X., Kang, W., Nakatsu, G., Zhao, Q., et al. (2018). CAB39L elicited an anti-Warburg effect via a LKB1-AMPK-PGC1α axis to inhibit gastric tumorigenesis. Oncogene 37 (50), 6383–6398. doi:10.1038/s41388-018-0402-1
Li, X., Yang, J., Shen, M., Xie, X. L., Liu, G. J., Xu, Y. X., et al. (2020). Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 11 (1), 2815. doi:10.1038/s41467-020-16485-1
Liu, Z., Tan, X., Wang, J., Jin, Q., Meng, X., Cai, Z., et al. (2022). Whole genome sequencing of Luxi Black Head sheep for screening selection signatures associated with important traits. Anim. Biosci. 35 (9), 1340–1350. doi:10.5713/ab.21.0533
Luridiana, S., Cosso, G., Pulinas, L., Stefano, M., Mura, M. C., Carcangiu, V., et al. (2020). New polymorphisms at MTNR1A gene and their association with reproductive resumption in sarda breed sheep. Theriogenology 158, 438–444. doi:10.1016/j.theriogenology.2020.10.006
Meadows, J. R. S., and Kijas, J. W. (2010). Re-sequencing regions of the ovine Y chromosome in domestic and wild sheep reveals novel paternal haplotypes. Anim. Genet. 40 (1), 119–123. doi:10.1111/j.1365-2052.2008.01799.x
Miao, X., Luo, Q., Zhao, H., and Qin, X. (2016). Ovarian proteomic study reveals the possible molecular mechanism for hyperprolificacy of Small Tail Han sheep. Rep 6, 27606. doi:10.1038/srep27606
Pillaiyar, T., Manickam, M., and Jung, S. H. (2017). Recent development of signaling pathways inhibitors of melanogenesis. Cell. Signal. 99.
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38 (8), 904–909. doi:10.1038/ng1847
Quan, K., Li, J., Han, H., Wei, H., Zhao, J., Si, H. A., et al. (2020). Review of Huang-huai sheep, a new multiparous mutton sheep breed first identified in China. Trop. Anim. Health Prod. 53 (1), 35. doi:10.1007/s11250-020-02453-w
Stari, J., Farci, F., Luridiana, S., Mura, M. C., Carcangiu, V., Cosso, G., et al. (2020). Reproductive performance in three Slovenian sheep breeds with different alleles for the MTNR1A gene. Animal Reproduction Sci. 216, 106352. doi:10.1016/j.anireprosci.2020.106352
Sui, X., Han, X., Chen, P., Feng, J., Duan, T., Liu, S., et al. (2021). Baicalin induces apoptosis and suppresses the cell cycle progression of lung cancer cells through downregulating akt/mTOR signaling pathway. Soc. Sci. Electron. Publ. 16, 20–28. doi:10.3389/fmolb.2020.602282
Sui, Z (2017) Re-sequencing strategy for mining multiple birth genes in Duolang sheep and analysis of candidate genes and litter size. China: Shihezi University.
Sweet-Jones, J., Lenis, V. P., Yurchenko, A. A., Yudin, N. S., Swain, M., and Larkin, D. M. (2021). Genotyping and whole-genome resequencing of Welsh sheep breeds reveal candidate genes and variants for adaptation to local environment and socioeconomic traits. Front. Genet. 12, 612492. doi:10.3389/fgene.2021.612492
Varghese, P. K., Abuasab, M., Dimitriadis, E. K., Dolinska, M. B., Morcos, G. P., and Sergeev, Y. V. (2021). Tyrosinase nanoparticles: understanding the melanogenesis pathway by isolating the products of tyrosinase enzymatic reaction. Int. J. Mol. Sci. 22 (2), 734. doi:10.3390/ijms22020734
Vitezslav, K., and Vladimir, K. (2018). Wnt, RSPO and Hippo signalling in the intestine and intestinal stem cells. Genes. 9 (1), 20. doi:10.3390/genes9010020
Wang, Z., Guo, J., Guo, Y., Yang, Y., Yang, H., Yu, Q., et al. (2020). Genome-wide detection of CNVs and association with body weight in sheep based on 600K SNP arrays. Front. Genet. 11, 558. doi:10.3389/fgene.2020.00558
Yang, H., Yang, Y. L., Li, G. Q., Yu, Q., and Yang, J. (2021a). Identifications of immune-responsive genes for adaptative traits by comparative transcriptome analysis of spleen tissue from Kazakh and Suffolk sheep. Sci. Rep. 11 (1), 3157. doi:10.1038/s41598-021-82878-x
Yang, M., Han, Y., Han, Q., Rong, X., Liu, X., and Ln, X. (2021b). KCTD11 inhibits progression of lung cancer by binding to β-catenin to regulate the activity of the Wnt and Hippo pathways. J. Cell. Mol. Med. 25 (19), 9411–9426. doi:10.1111/jcmm.16883
Yao, J., Wang, Z., Cheng, Y., Ma, C., Li, Z., Xiao, Y., et al. (2021). M2 macrophage-derived exosomal microRNAs inhibit cell migration and invasion in gliomas through PI3K/AKT/mTOR signaling pathway. J. Transl. Med. 19 (1), 99. doi:10.1186/s12967-021-02766-w
Zeng, Y., Baugh, E., Akyalcin, S., and Letra, A. (2021). Functional effects of WNT10A rare variants associated with tooth agenesis. J. Dent. Res. 100 (3), 302–309. doi:10.1177/0022034520962728
Zhang, D. Y., Zhang, X. X., Li, F. D., Yuan, L. F., Li, X. L., Zhang, Y. K., et al. (2022). Whole-genome resequencing reveals molecular imprints of anthropogenic and natural selection in wild and domesticated sheep. Zool. Res. 43 (5), 695–705. doi:10.24272/j.issn.2095-8137.2022.124
Zhang, L., Mousel, M. R., Wu, X., Michal, J. J., Xiang, Z., Bo, D., et al. (2013). Genome-wide genetic diversity and differentially selected regions among Suffolk, rambouillet, columbia, polypay, and targhee sheep. PLOS ONE 8, e65942. doi:10.1371/journal.pone.0065942
Zhang, M., Wang, S., Yi, A., and Qiao, Y. (2020). microRNA-665 is down-regulated in gastric cancer and inhibits proliferation, invasion, and EMT by targeting PPP2R2A. Cell. Biochem. Funct. 38 (4), 409–418. doi:10.1002/cbf.3485
Keywords: selection signatures, fst, reproduction traits, candidate genes, sheep
Citation: Yang H, Zhu M, Wang M, Zhou H, Zheng J, Qiu L, Fan W, Yang J, Yu Q, Yang Y and Zhang W (2024) Genome-wide comparative analysis reveals selection signatures for reproduction traits in prolific Suffolk sheep. Front. Genet. 15:1404031. doi: 10.3389/fgene.2024.1404031
Received: 20 March 2024; Accepted: 20 May 2024;
Published: 07 June 2024.
Edited by:
Ran Di, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Zhipeng Wang, Northeast Agricultural University, ChinaCopyright © 2024 Yang, Zhu, Wang, Zhou, Zheng, Qiu, Fan, Yang, Yu, Yang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Yang, eWh4amNuQHNpbmEuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.