 Feng Zhu1†
Feng Zhu1† Yuanlin Zhou
Yuanlin Zhou- 1Department of Neurology, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Taizhou, China
- 2Zhejiang Key Laboratory of Digital Technology in Medical Diagnostics, Dian Diagnostics Group Co., Ltd., Hangzhou, China
- 3Supply-Room, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Taizhou, China
Charcot-Marie-Tooth disease (CMT) is a hereditary peripheral neuropathy involving approximately 80 pathogenic genes. Whole-exome sequencing (WES) and confirmatory Sanger sequencing analysis was applied to identify the disease-causing mutations in a Chinese patient with lower limb weakness. We present an 18-year-old male with a 2.5-year history of progressive lower limb weakness and an unsteady gait. Upon admission, a physical examination revealed hands tremulousness, bilateral calf muscle wasting and weakness, pes cavus, and elevated serum creatine kinase (CK) levels. Electromyography demonstrated axonal neuropathy affecting both upper and lower limbs. A de novo heterozygous missense mutation was identified in the MORC2 gene, NM_001303256.3: c.1199A>G, NP_001290186.1: p.Gln400Arg. Consequently, these clinical and genetic findings suggested a diagnosis of hereditary peripheral neuropathy, CMT type 2Z. Oral mecobalamin and coenzyme Q10 was initiated as subsequent treatment. Our study firstly reports the MORC2 c.1199A>G mutation occurring de novo, highlighting its causal association with CMT2Z, and prompting its reclassification as likely pathogenic. Oral mecobalamin and coenzyme Q10 might be a potential treatment approach for early-stage CMT2Z. We recommend genetic testing for CMT patients to identify the genetic etiology, thereby improving clinical management and facilitating genetic counseling.
1 Introduction
Charcot-Marie-Tooth disease (CMT) is the most prevalent inherited peripheral neuropathy, with an incidence of 1 in 2,500 individuals (Fridman and Saporta, 2021). It is characterized by chronic motor and sensory polyneuropathy that manifests progressive, symmetrical distal muscle weakness and atrophy, diminished ankle dorsiflexion strength, depressed tendon reflexes, and pes cavus. CMT has significant genetic and clinical heterogeneity (Juneja et al., 2019; Volodarsky et al., 2021). Presently, over 80 genes have been implicated in various CMT subtypes (Stavrou and Kleopa, 2023). Although CMT can occur at any age, it predominantly arises between the first and third decades, severely impairing patients’ social participation and life quality from an early stage (Stavrou and Kleopa, 2023; Hertzog and Jacob, 2023). Therefore, elucidating the genetic etiology of CMT is critical for precision diagnosis, classification of subtypes, timely intervention, and genetic counseling for subsequent generations.
MORC2, encoding the microcheilia CW-type zinc finger protein 2, has been identified as the causative gene for CMT type 2Z (CMT2Z, MIM: #616688). CMT2Z is an autosomal dominant axonal peripheral neuropathy that initially presents with weakness and sensory impairment in the distal lower limb muscle and progressively involves the upper limbs and proximal muscle, leading to severe disability (Sevilla et al., 2016; Ando et al., 2017). However, the association between specific MORC2 mutations and the manifestation of CMT2Z remains controversial due to the limited patient population and the absence of functional assay. Further research is required to clarify this association.
In this study, we investigated a patient presenting with peripheral neuropathy. Utilizing whole-exome sequencing (WES) followed by Sanger sequencing, we identified a de novo MORC2 c.1199A>G mutation in the proband, leading to an upgrade in its mutation classification. Additionally, we detailed the proband’s disease progression and treatment approach, providing a comprehensive view on the clinical management of CMT2Z.
2 Case description
2.1 Case presentation
An 18-year-old Chinese Han male was admitted to Taizhou Hospital for a 2.5-year history of lower limb weakness and an unsteady gait. The patient presented with a generally lean body. Physical examination revealed decreased tendon reflex with a negative Babinski sign and intact cranial nerve function. Tremulousness was observed when extending his hand. Wasting of the bilateral calf muscle was observed, with muscle strength in the distal lower limbs rated at 4/5 (see Table 1; Figure 1A). Strength in the neck muscles, both proximal and distal upper limbs muscle, and proximal lower limbs muscle was normal and rated at 5/5. He was unable to perform heel walking due to bilateral dorsiflexion weakness of the feet, and a significant high arch was observed (Figure 1B). The patient didn’t report any hearing loss, decreased vision, or significant limb paresthesia. Notably, he experienced low physical achievement and a slower running speed since childhood (Supplementary Figure S1). His physical and intellectual development were normal, with a height of 1.68 m, a weight of 45 kg, and moderate academic performance in university. All immediate family members, including his parents (Ⅰ-1 and Ⅰ-2, aged 42) and younger sister (Ⅱ-2, aged 7), did not report any limb weakness. The physical assessments of the three family members revealed normal motor skills, characterized by an unimpaired gait and running posture, as well as normal arches and tendon reflexes, indicating their overall health. Given their unimpaired motor competencies, the family members declined further examinations.
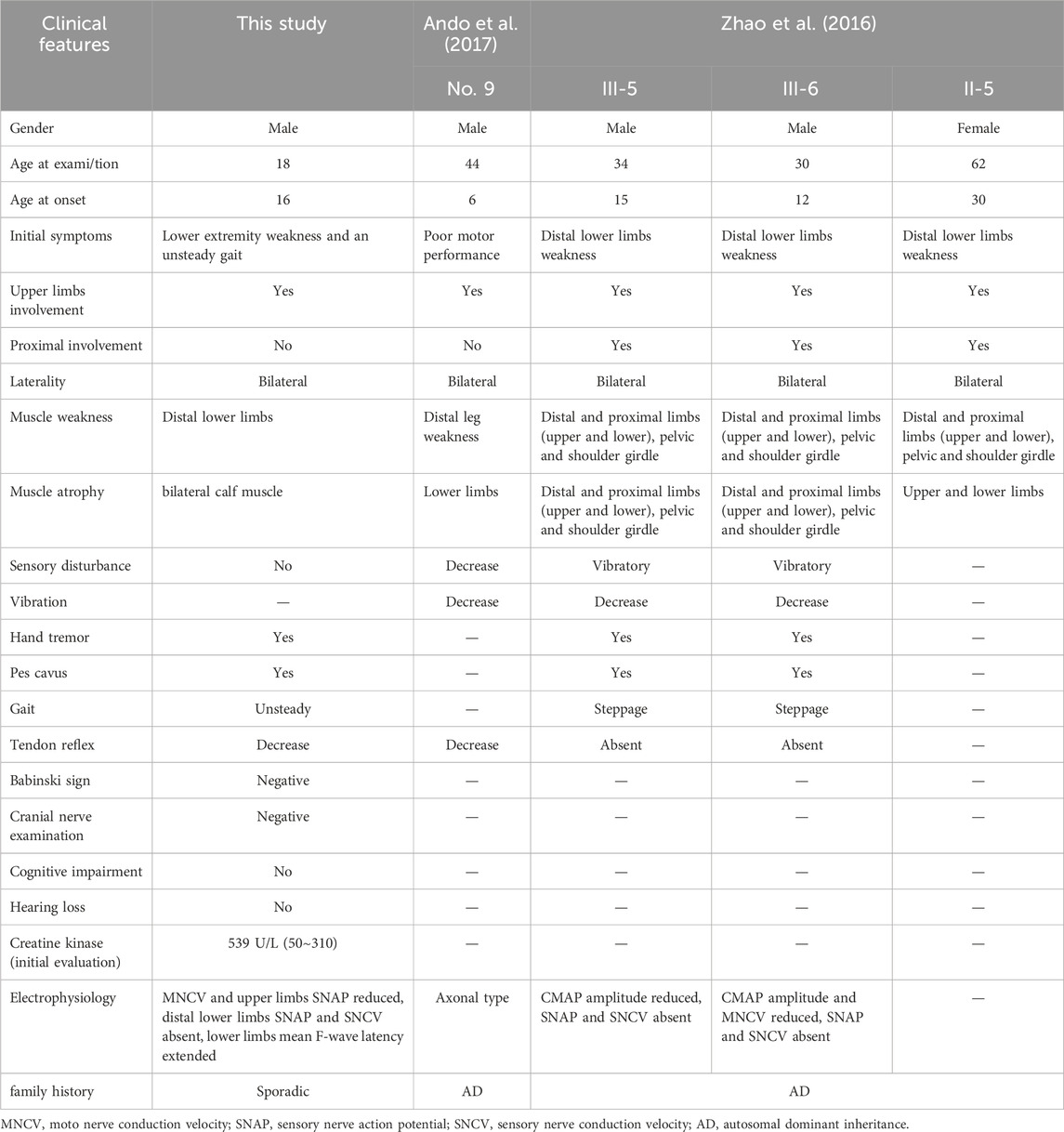
Table 1. Summarized clinical features of patients carrying MORC2 c.1199A>G.
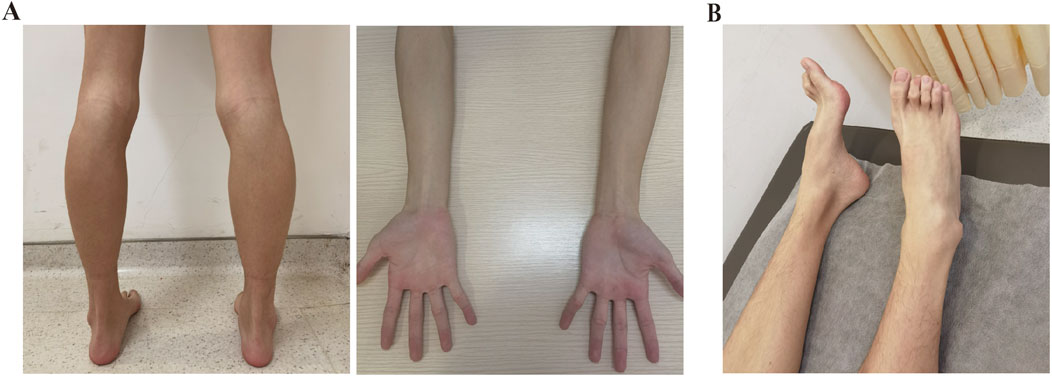
Figure 1. Clinical manifestations of the patient. (A) Physical examination revealed bilateral calf muscle wasting and slender arms. (B) Pes cavus was observed in the patient.
The patient showed an elevated serum CK at 539 U/L, which is above the normal range of 50 to 310 U/L. Electromyopraphy (EMG) study performed using Keypoint 9033A07 showed peripheral nerve axonopathy in both the upper and lower limbs, evidenced by a decreased motor nerve conduction rate and reduced amplitude of sensory nerve potentials in the upper limbs (see Table 2). Additionally, there was an absence of potential amplitude in the bilateral superficial peroneal and sural nerve, along with an extended mean F-wave latency in the bilateral tibial nerve (see Table 2). All these findings collectively indicate peripheral neuropathy in the patient.
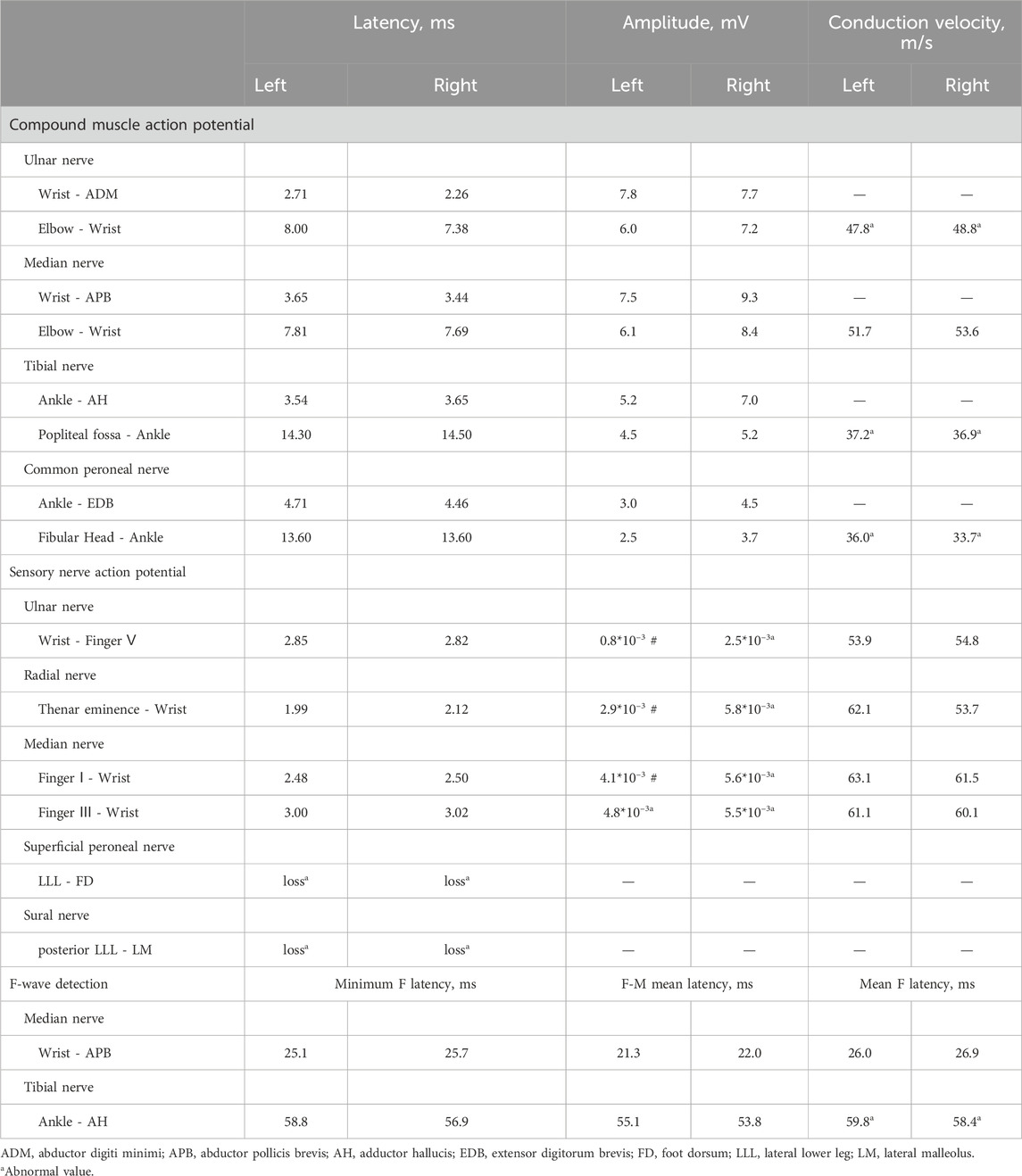
Table 2. Electrophysiological data of the proband.
To determine the genetic cause of peripheral neuropathy in this patient, we conducted WES on peripheral blood samples collected from the patient (II2). We identified a heterozygous missense mutation, MORC2 NM_001303256.3: c.1199A>G, p.Gln400Arg, located within the 13th exon of the MORC2 gene. Subsequent Sanger sequencing of the parents revealed that MORC2 c.1199A>G was a de novo mutation (meet ACMG evidence PS2) (Figures 2A, B). This mutation was not recorded in the Genome Aggregation Database (GnomAD, http://gnomad-sg.org/) (meet ACMG evidence PM2_supporting), while it has been previously identified in two independent probands diagnosed with autosomal dominant CMT2, being classified as of uncertain significance in the ClinVar database (also recorded as NM_014941.1: c.1013A>G, p.Q388R) (meet ACMG evidence PS4_Moderate) (Ando et al., 2017; Zhao et al., 2016). Predictions from computational programs such as REVEL, ClinPred, and MAGPIE were applied (Liu et al., 2024). The REVEL score was 0.411 (benign supporting), while the ClinPred and MAGPIE scores were 0.991 and 0.807, respectively (both indicating pathogenicity support). SIFT and Polyphen2 indicated that MORC2 p.Q400R is damaging and possibly damaging, respectively. An evolutionary conservation analysis showed that the MORC2 p.Q400 site is highly conserved (Figure 2D). 3D structure modeling suggested that MORC2 p.Q400R causes a change in the amino acid residue involved in the hydrogen bonds with 400 Glu residue, potentially impacting the Morc2 protein structure and stability (Figure 2C). Considering these results and following the ACMG guidelines (Richards et al., 2015), MORC2 c.1199A>G mutation’s classification can be upgraded to likely pathogenetic (PS2+PS4_Moderate + PM2_supporting). No additional rare pathogenic variants were detected in any other genes associated with neuromuscular disease.
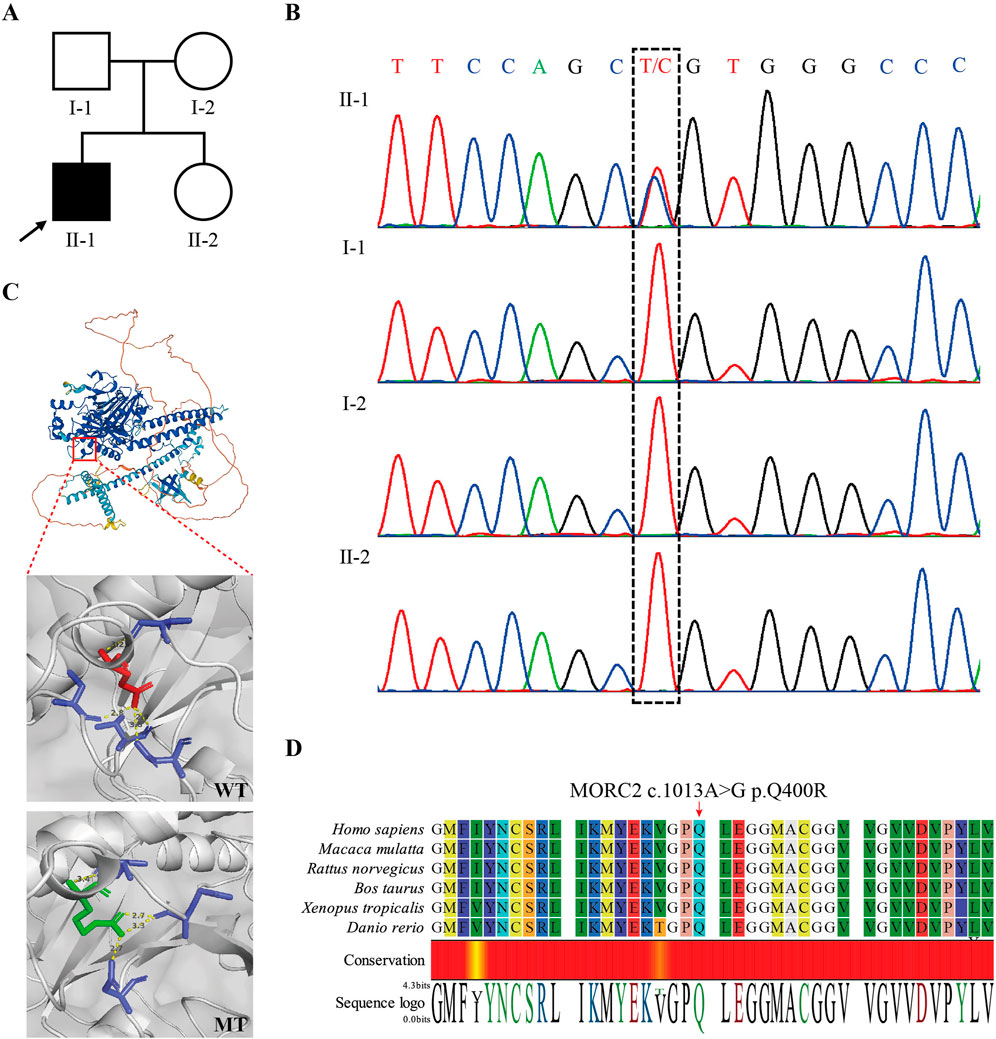
Figure 2. Identification of MORC2 NM_001303256.3: (C)1199A>G, p.Q400R in the patient. (A) Pedigree of the presented family. The arrow indicates the proband. (B) Sanger sequencing confirmed the de novo MORC2 c.1199A>G mutation in the proband. (C) 3D structure modeling shows the location of MORC2 p.Q400R. The MORC2 p.Q400R mutation alters the location and length of hydrogen bonds at the 400 residue. The wildtype Morc2 is predicted to form hydrogen bonds with the 380 Asp, 397 Val, 407 Cys, and 410 Val residues at the 400 Glu residue, while the mutant Morc2 is predicted to form hydrogen bonds with the 380 Asp, 395 Glu, and 397 Val residues at the 400 Arg residue. The Red amino acid indicates the Glu and green amino acid indicates the Arg. WT, wide type. MT, mutant type. (D) Evolutionary conservation of amino acid position p.Q400R in Morc2 protein.
Therefore, the combination of clinical manifestations and genetic analysis supported a diagnosis of CMT2Z in this patient, which is associated with the MORC2 c.1199A>G mutation. The patient had been prescribed vitamin B1 at a dose of 10 mg three times daily for a short period but discontinued it upon hospital admission. Considering that CMT2Z is pathophysiologically caused by axonal lesions, oral methylcobalamin and coenzyme Q10 (CoQ10) were initiated for subsequent treatment, at doses of 0.5 mg three times daily and 10 mg three times daily, respectively. As of the latest update 7 months post-diagnosis, the patient’s CK levels have normalized to 227 U/L, and he has not reported any further physical symptoms. Monitoring of the patient’s CK level and motor abilities will continue to determine the long-term effectiveness of the treatment and to manage his condition.
Furthermore, the proband’s younger sister doesn’t carry the mutation, suggesting a low risk of her developing similar symptoms. Nonetheless, prenatal diagnosis is recommended for future pregnancies in this family to prevent the potential transmission of a mutated allele, especially considering the possibility of gonadal mosaicism in a parent. Additionally, the proband could benefit from third-generation in vitro fertilization before pregnancy to prevent the inheritance of this disease.
2.2 Genetic testing methodology
Genomic DNA was exacted from 5 mL of patient’s peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Germany) and was subsequently fragmented into 180–280 bp segments with a Covaris LE220R-plus (Covaris, USA). A sequencing library was constructed, including end repair, phosphorylation, A-tailing, and adapters ligation. Then the exome enrichment was performed using the xGen Exome Research Panel v1.0 Kit (Integrated DNA Technologies, USA). The sequencing library underwent PCR amplification followed by sequencing on the NovaSeq 6000 platform (Illumina, USA). The overall average sequencing depth is 511×, with 99.1% of variants were covered at a depth greater than 20×.
The raw sequence data were firstly filtered to remove adapter sequences and low-quality reads. The remaining high-quality clean data were aligned to the GRCh37/hg19 human reference genome using the Burrows-Wheeler Aligner (BWA) tool (Li and Durbin, 2010). The variants calling was performed with the Genome Analysis Toolkit (GATK) (McKenna et al., 2010), followed by annotated according to the 1000 Genomes Project, GnomAD, ClinVar, and dbNSFP databases. Candidate mutations, were filtered based on allele frequency (AF < 1%), phenotypic concordance with the MIM database, inheritance patterns, and pathogenicity classification (pathogenic and likely pathogenic) according to ACMG guidelines (Richards et al., 2015).
Candidate variants were validated using Sanger sequencing. The primer sequences for MORC2 c.1199A>G were as follows: forward 5′-AGCTCTCCGGCGTTTGTAAC-3′; reverse 5′-ACATTGAACACCGGGATCTGG-3′. PCR amplicons were subsequently sequenced on an ABI 3130 Genetic Analyzer (Applied Biosystems, California).
3 Discussion
With the advancement of genome sequencing technology, an increasing number of CMT causal genes have been discovered. MORC2 was initially identified in a clinical cohort with axonal lesions and was reported to be associated with CMT in 2016 (Sevilla et al., 2016). The MORC2 gene is located on chromosome 22q12.2 in humans and encodes a DNA-dependent ATPase consisting of 26 exons and 1,032 amino acids (aa). The protein contains three functional domains: Histidine kinase/HSP90-like ATPase superfamily (23∼278aa), Morc S5 domain 2-like (330∼451aa), and CW-type zinc finger (490∼544aa), and is involved in epigenetic silencing, genome homeostasis, transcriptional regulation, and lipid metabolism (Guillen Sacoto et al., 2020; Sanchez-Solana et al., 2014; Li et al., 2012; Douse et al., 2018). To date, 15 MORC2 mutations have been identified in CMT2Z patients (Jacquier et al., 2022). Some of the functional impacts of these mutations have been elucidated. For instance, the MORC2 p.R252W mutation has been reported to hyper-activates Human Silencing Hub (HUSH)-mediated transcriptional repression and perturbs ATPase dimerization dynamics in neuronal cells (Douse et al., 2018; Tchasovnikarova et al., 2017). Another hotspot mutation, MORC2 p.S87L, has been shown to impair the proliferation of induced pluripotent stem cells (iPSCs) by inhibiting the MAPK/ERK and PI3K/Akt pathways, along with profiles of differentially expressed genes (DEGs) between p.S87L and p.Q400R (Zeng et al., 2024). Based on the above published transcriptomics data (Zeng et al., 2024; Mu et al., 2024), we investigated the DEGs between healthy controls and p.Q400R (Supplementary Figure S2). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed significant enrichment of the MAPK signaling pathway in moto neuron precursor cells (MNPs) (Supplementary Figure S3), a pathway also enriched in iPSCs harboring the p.S87L mutation. However, no significant enrichment of pathways associated with cell proliferation and differentiation was observed in iPSCs carrying the p.Q400R. This suggests that MORC2 p.Q400R mutation may dysregulate the intracellular and extracellular signaling pathways, such as MAPK signaling cascade, adversely affecting motor neuron function post-differentiation and ultimately contributing to the development of CMT2Z. Despite these findings, further research is still required to clarify the molecular mechanisms by which MORC2 mutations lead to neuronal cell dysfunction and the onset of CMT.
MORC2 exhibits significant phenotypic heterogeneity and a wide mutation spectrum. MORC2-associated disorders range from CMT2Z to symptoms resembling spinal muscular atrophy (SMA), with early onset and predominantly proximal muscle involvement. More severe neurodevelopmental conditions, characterized by developmental delay, impaired growth, dysmorphic facies, and axonal neuropathy (known as DIGFAN, MIM: #619090), have also been reported (Guillen Sacoto et al., 2020; Duan et al., 2021a; Lassuthova et al., 2016). It is noteworthy that all reported MORC2 mutations are missense, with a high missense Z score of 3.23 (Stafki et al., 2023; Sivera et al., 2021). One possible explanation for the predominance of missense mutations may be the high conservation and dosage sensitivity of the Morc2 protein, where more deleterious alterations such as nonsense or frameshift mutations, and exon deletions could result in embryonic lethality, thus making them unobservable in live births. According to GnomAD, MORC2 has a loss-of-function intolerant pLI score of 1 and a low observed/expected (oe) ratio of 0.1 (90% CI 0.06∼0.2), suggesting a high deleterious of virulent mutations. To date, no MORC2-knockout animal models have been developed, except for a mouse model harboring a Morc2a Ser87-to-Leu (p.S87L) knockin mutation (Lee et al., 2021). Additionally, the distribution of the MORC2 mutations does not exhibit a specific pattern correlating with symptom severities, mainly located on the ATPase, coiled-coil domain 1, and ribosomal protein S5 domain at the N-terminus (Jacquier et al., 2022; Duan et al., 2021a). However, mutations at different positions have varying effects on protein function. For instance, the MORC2 p.S87L and p.R252W mutations decrease Morc2 ATPase activity and hyperactivated HUSH-mediated epigenetic silencing, whereas the MORC2 p.T424R mutation has the opposite effect (Tchasovnikarova et al., 2017; Duan et al., 2021a). Furthermore, whether the mutation is inherited or de novo appears to influence phenotypes. A de novo MORC2 mutation is frequently associated with more severe manifestations, often involving central nervous system (CNS) disorder, while it is less common in CMT2Z patients. This discrepancy may be attributed to physiological or social infertility that naturally blocks the propagation of severely symptomatic MORC2 mutations to subsequent generations. Therefore, it is essential to pay close attention to disease progression for patients identified de novo MORC2 mutation, to facilitate timely disease-modifying interventions. Collectively, MORC2 has a broad mutation spectrum and phenotypic variability, rendering the disease subtype classification and clinical management of patients challenging.
In this study, we report for the first time a heterozygous missense, MORC2 c.1199A>G, occurring de novo in a young male with peripheral nerve axonopathy. Our study prompts a reclassification of this mutation from VUS to Likely Pathogenic, supporting the goal set by the National Human Genome Research Institute (NHGRI) to resolve all VUS in coding regions of the human Genome by 2030 (Green et al., 2020). Additionally, this mutation, also recorded as NM_014941.3: c.1013A>G, p.Gln338Arg, has been previously identified in four CMT-afflicted patients, three from a Chinese family and one from a Japanese family (see Table 1) (Ando et al., 2017; Zhao et al., 2016). Our study corroborates that individuals harboring MORC2 c.1199A>G mutations predominantly exhibit peripheral neuropathy symptoms with a low risk of CNS involvement. Furthermore, our study, together with previous reports of de novo MORC2 mutations, highlights the high mutation rate of the MORC2 gene. While the recurrence of the MORC2 c.1199A>G mutation suggests a potential hotspot within the East Asian CMT2Z cohort, further investigation such as haplotype analysis, is required to confirm this hypothesis.
Compared with previously reported four patients carrying MORC2 c.1199A>G mutation, we conducted a detailed study of the clinical features of this mutation (see Table 1) (Ando et al., 2017; Zhao et al., 2016). Patients typically develop bilateral lower limb weakness and muscle atrophy in childhood or adolescence, with subsequent progression to the proximal lower limbs and eventually the upper limbs. This progression is often accompanied by pes cavus, a pathologic gait, sensory disturbance, and areflexia. However, variability in clinical manifestations was also observed, particularly in gender differences. The female patient presented milder manifestations than her brothers, including a later onset at age 30 and fewer involved muscle groups. Similar gender-specific differences in symptom severity have also been observed in other neuromuscular diseases, such as SCN4A mutation-associated hypokalemic periodic paralysis, suggesting a potential area for further investigation of CMT2Z (Li et al., 2012). Notably, a previous study has reported elevated serum CK levels in MORC2-associated CMT(24). Similarly, our patient also showed a significant elevation in CK levels during the initial evaluation, approximately twice the upper limit of the normal range. Since muscle atrophy is a common progression in CMT2Z, we suggest longitudinal monitoring of CK levels might reflect muscle status to a certain extent, particularly during therapeutic and rehabilitative interventions. However, it is worth noting that using CK alone to assess disease progression and treatment efficacy is limited, as it can be affected by many factors, such as physical activities, alcohol abuse, and intramuscular injections.
Notably, EMG revealed an absent sensory nerve action potential (SNAP) amplitude of the superficial peroneal nerve and sural nerve, while the patient denied any sensory abnormalities. This discrepancy may be attributed to the high degree of clinically heterogeneity in CMT disease. In a cohort study involving 465 unrelated Chinese CMT patients, four patients exhibited absent sensory nerve action potential (SNAP) amplitude and conduction velocity of the sural nerve, ulnar nerve, and median nerve, while three of them still retained decreased sensory vibration and the other one retained decreased sensory pinprick (Duan et al., 2021b). In another study, a CMT patient denied any sensation symptoms despite neurophysiological examination showing slight abnormalities in EMG and impairment of sensory fibers in the peripheral nerves of all four limbs (Cao et al., 2023). Overall, electrophysiological results may significantly mismatch with clinical manifestations in CMT disease. Considering this findings, the patient’s tolerance to partial sensory loss may also contribute to the discrepancy between the EMG results and clinical presentation in our study. The difficulty in obtaining biopsy materials also prevents us from further verifying the specific damage to the sensory nerves.
Current therapeutic approaches for CMT are symptomatic and focus on rehabilitation such as strength training of proximal and core muscles, the application of foot orthoses, and anticonvulsants for managing positive sensory symptoms (Yiu et al., 2022; Corrado et al., 2016; Magy et al., 2018). Reports suggest that low-dose methylprednisolone tablets and energy supplements can help to reduce serum CK levels, while vincristine may induce neurotoxicity in patients with CMT2Z (Vujovic et al., 2021; Wang et al., 2022). In our study, Oral mecobalamin and CoQ10 were administered for subsequent treatment. Mecobalamin, an active form of vitamin B12, has been widely used as a supportive intervention in various peripheral neuropathies due to its capacity for neuronal preservation and facilitation of injured nerve regeneration (Xiong et al., 2022; Sawangjit et al., 2020). CoQ10, a small lipid-soluble benzoquinone, is a crucial source of indispensable endogenous antioxidants. CoQ10 has demonstrated protective properties against vincristine-induced peripheral neuropathy and assists in managing COQ7 gene mutation-associated distal hereditary motor neuropathy (Elshamy et al., 2022; Jacquier et al., 2023). Both mecobalamin and CoQ10 have been substantiated for safety (Sawangjit et al., 2020; Hathcock and Shao, 2006). After 7 months, the serum CK level normalized from 539 U/L to 227 U/L, suggesting a potentially treatment approach for early-stage CMT2Z. However, whether the decreased serum CK is due to this treatment is less clear and needs long-term follow-up and multiple perspectives assessment. Novel therapies, including gene therapy and targeted pharmaceuticals such as NRG-1 axis inhibitors and modulators of unfold protein response (UPR) and histone deacetylase (HDAC) enzyme family, are currently in development and promise to provide more precise treatment for CMT patients in the near future (Stavrou and Kleopa, 2023; Hertzog and Jacob, 2023; Beloribi-Djefaflia and Attarian, 2023). We will continue to monitor the proband’s disease progression and remain updated about the latest treatment advancements to improve his quality of life.
In summary, our study describes a male adolescent patient with peripheral neuropathy carrying a de novo heterozygous missense mutation, MORC2 c.1199A>G. Our findings further support the causal association between this mutation and CMT2Z, upgrading its mutation classification to likely pathogenic. Oral mecobalamin and CoQ10 appear to be a potential treatment option for early-stage CMT2Z patients but the efficacy of this therapy needs further evaluation. We emphasize the importance of genetic testing for individuals with CMT to determine the genetic etiology, which can significantly improve clinical management and provide valuable insights for genetic counseling.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of Taizhou Hospital of Zhejiang Province. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual (s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
FZ: Data curation, Resources, Writing–review and editing. CG: Formal Analysis, Visualization, Writing–original draft. XZ: Investigation, Validation, Writing–original draft. HJ: Investigation, Validation, Writing–original draft. MH: Methodology, Writing–review and editing. YZ: Conceptualization, Methodology, Project administration, Writing–review and editing.
Funding
The authors declare that no financial support was received for the research, authorship, and/or published of this article.
Acknowledgments
We would like to express our gratitude to the family, who made this study possible. Additionally, we thank the Clinical Genomics Center of Dian Diagnostics Group Co., Ltd. for conducting the genetic testing.
Conflict of interest
CG is employed by Dian Diagnostics Group Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1400906/full#supplementary-material
References
Ando, M., Okamoto, Y., Yoshimura, A., Yuan, J. H., Hiramatsu, Y., Higuchi, Y., et al. (2017). Clinical and mutational spectrum of Charcot-Marie-Tooth disease type 2Z caused by MORC2 variants in Japan. Eur. J. Neurol. 24 (10), 1274–1282. doi:10.1111/ene.13360
Beloribi-Djefaflia, S., and Attarian, S. (2023). Treatment of charcot-marie-tooth neuropathies. Rev. Neurol. Paris. 179 (1-2), 35–48. doi:10.1016/j.neurol.2022.11.006
Cao, L., Yang, J., Zhang, X., Wang, X., Chen, Z., Tan, S., et al. (2023). Clinical, neurophysiological evaluation and genetic features of axonal Charcot-Marie-Tooth disease in a Chinese family. Front. Neurol. 14, 1337065. doi:10.3389/fneur.2023.1337065
Corrado, B., Ciardi, G., and Bargigli, C. (2016). Rehabilitation management of the charcot-marie-tooth syndrome: a systematic review of the literature. Med. Baltim. 95 (17), e3278. doi:10.1097/MD.0000000000003278
Douse, C. H., Bloor, S., Liu, Y., Shamin, M., Tchasovnikarova, I. A., Timms, R. T., et al. (2018). Neuropathic MORC2 mutations perturb GHKL ATPase dimerization dynamics and epigenetic silencing by multiple structural mechanisms. Nat. Commun. 9 (1), 651. doi:10.1038/s41467-018-03045-x
Duan, X., Liu, X., Wang, G., Gu, W., Xu, M., Hao, Y., et al. (2021a). Characterization of genotype-phenotype correlation with MORC2 mutated Axonal Charcot-Marie-Tooth disease in a cohort of Chinese patients. Orphanet J. Rare Dis. 16 (1), 244. doi:10.1186/s13023-021-01881-7
Duan, X., Ma, Y., Fan, D., and Liu, X. (2021b). Characteristics of clinical and electrophysiological pattern in a large cohort of Chinese patients with charcot-marie-tooth 4C. Front. Neurol. 12, 598168. doi:10.3389/fneur.2021.598168
Elshamy, A. M., Salem, O. M., Safa, M. A. E., Barhoma, R. A. E., Eltabaa, E. F., Shalaby, A. M., et al. (2022). Possible protective effects of CO Q10 against vincristine-induced peripheral neuropathy: targeting oxidative stress, inflammation, and sarmoptosis. J. Biochem. Mol. Toxicol. 36 (3), e22976. doi:10.1002/jbt.22976
Fridman, V., and Saporta, M. A. (2021). Mechanisms and treatments in demyelinating CMT. Neurotherapeutics 18 (4), 2236–2268. doi:10.1007/s13311-021-01145-z
Green, E. D., Gunter, C., Biesecker, L. G., Di Francesco, V., Easter, C. L., Feingold, E. A., et al. (2020). Strategic vision for improving human health at the Forefront of Genomics. Nature 586 (7831), 683–692. doi:10.1038/s41586-020-2817-4
Guillen Sacoto, M. J., Tchasovnikarova, I. A., Torti, E., Forster, C., Andrew, E. H., Anselm, I., et al. (2020). De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial dysmorphism. Am. J. Hum. Genet. 107 (2), 352–363. doi:10.1016/j.ajhg.2020.06.013
Hathcock, J. N., and Shao, A. (2006). Risk assessment for coenzyme Q10 (Ubiquinone). Regul. Toxicol. Pharmacol. 45 (3), 282–288. doi:10.1016/j.yrtph.2006.05.006
Hertzog, N., and Jacob, C. (2023). Mechanisms and treatment strategies of demyelinating and dysmyelinating Charcot-Marie-Tooth disease. Neural Regen. Res. 18 (9), 1931–1939. doi:10.4103/1673-5374.367834
Jacquier, A., Roubille, S., Lomonte, P., and Schaeffer, L. (2022). Microrchidia CW-type zinc finger 2, a chromatin modifier in a spectrum of peripheral neuropathies. Front. Cell Neurosci. 16, 896854. doi:10.3389/fncel.2022.896854
Jacquier, A., Theuriet, J., Fontaine, F., Mosbach, V., Lacoste, N., Ribault, S., et al. (2023). Homozygous COQ7 mutation: a new cause of potentially treatable distal hereditary motor neuropathy. Brain 146 (8), 3470–3483. doi:10.1093/brain/awac453
Juneja, M., Burns, J., Saporta, M. A., and Timmerman, V. (2019). Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatry 90 (1), 58–67. doi:10.1136/jnnp-2018-318834
Lassuthova, P., Safka Brozkova, D., Krutova, M., Mazanec, R., Zuchner, S., Gonzalez, M. A., et al. (2016). Severe axonal Charcot-Marie-Tooth disease with proximal weakness caused by de novo mutation in the MORC2 gene. Brain 139 (Pt 4), e26. doi:10.1093/brain/awv411
Lee, G. S., Kwak, G., Bae, J. H., Han, J. P., Nam, S. H., Lee, J. H., et al. (2021). Morc2a p.S87L mutant mice develop peripheral and central neuropathies associated with neuronal DNA damage and apoptosis. Dis. Model Mech. 14 (10), dmm049123. doi:10.1242/dmm.049123
Li, D. Q., Nair, S. S., Ohshiro, K., Kumar, A., Nair, V. S., Pakala, S. B., et al. (2012). MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep. 2 (6), 1657–1669. doi:10.1016/j.celrep.2012.11.018
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 (5), 589–595. doi:10.1093/bioinformatics/btp698
Liu, Y., Zhang, T., You, N., Wu, S., and Shen, N. (2024). MAGPIE: accurate pathogenic prediction for multiple variant types using machine learning approach. Genome Med. 16 (1), 3. doi:10.1186/s13073-023-01274-4
Magy, L., Mathis, S., Le Masson, G., Goizet, C., Tazir, M., and Vallat, J. M. (2018). Updating the classification of inherited neuropathies: results of an international survey. Neurology 90 (10), e870–e876. doi:10.1212/WNL.0000000000005074
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
Mu, H., Chen, J., Huang, W., Huang, G., Deng, M., Hong, S., et al. (2024). OmicShare tools: a zero-code interactive online platform for biological data analysis and visualization. iMeta. e228. doi:10.1002/imt2.228
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Sanchez-Solana, B., Li, D. Q., and Kumar, R. (2014). Cytosolic functions of MORC2 in lipogenesis and adipogenesis. Biochim. Biophys. Acta 1843 (2), 316–326. doi:10.1016/j.bbamcr.2013.11.012
Sawangjit, R., Thongphui, S., Chaichompu, W., and Phumart, P. (2020). Efficacy and safety of mecobalamin on peripheral neuropathy: a systematic review and meta-analysis of randomized controlled trials. J. Altern. Complement. Med. 26 (12), 1117–1129. doi:10.1089/acm.2020.0068
Sevilla, T., Lupo, V., Martinez-Rubio, D., Sancho, P., Sivera, R., Chumillas, M. J., et al. (2016). Mutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease. Brain 139 (Pt 1), 62–72. doi:10.1093/brain/awv311
Sivera, R., Lupo, V., Frasquet, M., Argente-Escrig, H., Alonso-Perez, J., Diaz-Manera, J., et al. (2021). Charcot-Marie-Tooth disease due to MORC2 mutations in Spain. Eur. J. Neurol. 28 (9), 3001–3011. doi:10.1111/ene.15001
Stafki, S. A., Turner, J., Littel, H. R., Bruels, C. C., Truong, D., Knirsch, U., et al. (2023). The spectrum of MORC2-related disorders: a potential link to cockayne syndrome. Pediatr. Neurol. 141, 79–86. doi:10.1016/j.pediatrneurol.2023.01.011
Stavrou, M., and Kleopa, K. A. (2023). CMT1A current gene therapy approaches and promising biomarkers. Neural Regen. Res. 18 (7), 1434–1440. doi:10.4103/1673-5374.361538
Tchasovnikarova, I. A., Timms, R. T., Douse, C. H., Roberts, R. C., Dougan, G., Kingston, R. E., et al. (2017). Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nat. Genet. 49 (7), 1035–1044. doi:10.1038/ng.3878
Volodarsky, M., Kerkhof, J., Stuart, A., Levy, M., Brady, L. I., Tarnopolsky, M., et al. (2021). Comprehensive genetic sequence and copy number analysis for Charcot-Marie-Tooth disease in a Canadian cohort of 2517 patients. J. Med. Genet. 58 (4), 284–288. doi:10.1136/jmedgenet-2019-106641
Vujovic, D., Cornblath, D. R., and Scherer, S. S. (2021). A recurrent MORC2 mutation causes Charcot-Marie-Tooth disease type 2Z. J. Peripher Nerv. Syst. 26 (2), 184–186. doi:10.1111/jns.12443
Wang, Y., Han, J., Zhang, J., Wu, Y., and Song, X. (2022). MORC2 p.R252W mutant axonal charcot–marie–tooth disease causes peripheral neuropathies and pathological myofiber destruction. Balk. Med. J. 39 (3), 222–223. doi:10.4274/balkanmedj.galenos.2022.2021-12-97
Xiong, Z. L., Wang, Y., Ma, X. L., Zhou, C., Jiang, X. W., and Yu, W. H. (2022). Based on proteomics to explore the mechanism of mecobalamin promoting the repair of injured peripheral nerves. Can. J. Physiol. Pharmacol. 100 (6), 562–572. doi:10.1139/cjpp-2021-0692
Yiu, E. M., Bray, P., Baets, J., Baker, S. K., Barisic, N., de Valle, K., et al. (2022). Clinical practice guideline for the management of paediatric Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 93 (5), 530–538. doi:10.1136/jnnp-2021-328483
Zeng, S., Yang, H., Wang, B., Xie, Y., Xu, K., Liu, L., et al. (2024). The MORC2 p.S87L mutation reduces proliferation of pluripotent stem cells derived from a patient with the spinal muscular atrophy-like phenotype by inhibiting proliferation-related signaling pathways. Neural Regen. Res. 19 (1), 205–211. doi:10.4103/1673-5374.375347
Keywords: MORC2, lower limb weakness, charcot-marie-tooth disease, mecobalamin, coenzyme Q10, case report
Citation: Zhu F, Gao C, Zhu X, Jiang H, Huang M and Zhou Y (2024) Case Report: Charcot-marie-tooth disease caused by a de novo MORC2 gene mutation - novel insights into pathogenicity and treatment. Front. Genet. 15:1400906. doi: 10.3389/fgene.2024.1400906
Received: 14 March 2024; Accepted: 27 September 2024;
Published: 11 October 2024.
Edited by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesCopyright © 2024 Zhu, Gao, Zhu, Jiang, Huang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanlin Zhou, enlsQGVuemVtZWQuY29t; Mingchun Huang, aHVhbmdtY0BlbnplbWVkLmNvbQ==
†These authors have contributed equally to this work