 Huihui Sun
Huihui Sun Geng Zhang2
Geng Zhang2- 1Department of Paediatrics, Beijing Jishuitan Hospital, Capital Medical University, Beijing, China
- 2Beijing Chigene Translational Medical Research Center Company, Beijing, China
- 3Department of Radiology, Beijing Jishuitan Hospital, Capital Medical University, Beijing, China
Background: Short stature is a complex disorder with phenotypic and genetic heterogeneity. This study aimed to investigate clinical phenotypes and molecular basis of a cohort of patients with short stature.
Methods: Trio whole-exome sequencing (Trio-WES) was performed to explore the genetic aetiology and obtain a molecular diagnosis in twenty Chinese probands with syndromic and isolated short stature.
Results: Of the twenty probands, six (6/20, 30%) patients with syndromic short stature obtained a molecular diagnosis. One novel COMP pathogenic variant c.1359delC, p.N453fs*62 and one LZTR1 likely pathogenic variant c.509G>A, p.R170Q were identified in a patient with short stature and skeletal dysplasia. One novel de novo NAA15 pathogenic variant c.63T>G, p.Y21X and one novel de novo KMT2A pathogenic variant c.3516T>A, p.N1172K was identified in two probands with short stature, intellectual disability and abnormal behaviours, respectively. One patient with short stature, cataract, and muscle weakness had a de novo POLG pathogenic variant c.2863 T>C, p.Y955H. One PHEX pathogenic variant c.1104G>A, p.W368X was identified in a patient with short stature and rickets. Maternal uniparental disomy 7 (mUPD7) was pathogenic in a patient with pre and postnatal growth retardation, wide forehead, triangular face, micrognathia and clinodactyly. Thirteen patients with isolated short stature had negative results.
Conclusion: Trio-WES is an important strategy for identifying genetic variants and UPD in patients with syndromic short stature, in which dual genetic variants are existent in some individuals. It is important to differentiate between syndromic and isolated short stature. Genetic testing has a high yield for syndromic patients but low for isolated patients.
Introduction
Short stature is a common disorder for referral to paediatric endocrinologists (Collett-Solberg et al., 2019a). The clinical diagnosis of short stature is challenging based mainly on birth weight, growth pattern, clinical manifestations, family history, and laboratory tests. It is important to differentiate between isolated short stature and syndromic short stature, which is concurrent with facial dysmorphism, skeletal dysplasia and other malformations (Jee et al., 2018). Nevertheless, some clinical manifestations are unspecific and mild, especially in young children. With the rapid development of molecular technology, recent studies have demonstrated that the disorder is mainly caused by various genetic aberrations, including polygenic inheritance, chromosomal abnormalities, copy number variants (CNVs), uniparental disomy of chromosome (UPD), nuclear genetic variants, mitochondrial genetic variants, and methylation defects (Wit et al., 2016; Grunauer and Jorge, 2018; Homma et al., 2018). Treatment with recombinant human growth hormone (rhGH) is an effective approach for improving heights of children with short stature; however, not all of these patients can be treated with rhGH, and neither the patient’s response to rhGH nor the adverse effects in the context of some genetic disorders are well understood (Murray et al., 2018; Collett-Solberg et al., 2019a).
A definitive molecular diagnosis provides important information about specific associated features, prognostic predictions, individualized therapeutic options and genetic counselling (Jee et al., 2018). Recent studies have demonstrated that isolated and syndromic short stature could be caused by monogenic defects in the growth hormone insulin-like growth factor 1 (GH-IGF-1) axis, cartilage growth plate, fundamental cellular processes and signalling pathway (Hu et al., 2016; Andrade et al., 2017; Homma et al., 2019; Stavber et al., 2022). Trio whole-exome sequencing (Trio-WES) is a diagnostic approach for identifying genetic variations in patients with syndromic short stature with unknown aetiology (Sun et al., 2021). Despite tremendous advances in the molecular genetics of growth disorders, the underlying molecular mechanisms remain unknown in a significant proportion of individuals with short stature (Zahnleiter et al., 2013). In this study, Trio-WES was performed to explore genetic causes in patients with isolated short stature and syndromic short stature (Miller et al., 2010; Wang et al., 2020). We describe detailed clinical manifestations and molecular analysis in these patients to guide individualized therapy.
Methods
Patients
This study was approved by Ethics Committee of Beijing Jishuitan Hospital, Capital Medical University (202104-07 and 202201-20-01). Patients were referred to the paediatric department of Beijing Jishuitan Hospital, Capital Medical University from January 2021 to December 2022. The diagnostic criteria of short stature were as follows (Siklar and Berberoglu, 2014; Grunauer and Jorge, 2018; Collett-Solberg et al., 2019b): height or predicted adult height (PAH) measuring 1.88 standard deviations (SD) below the mean for individuals of the same age and sex among healthy Chinese children aged 1–15 years. The exclusion criteria were chronic systematic diseases and comorbidities that could affect growth, such as malnutrition, psychological disturbances, hypothyroidism, and known syndromes. Patients were classified into two groups: isolated short stature and syndromic short stature, which was concurrent with multiple malformations including facial dysmorphism, intellectual disability, skeletal dysplasia and other abnormalities (Collett-Solberg et al., 2019b). Twenty Chinese trios of probands with short stature were recruited in the study. Written informed consent was obtained from their legal guardians and parents.
Trio-WES
Genomic DNA was extracted from peripheral blood lymphocytes of the probands and their available family members using the Blood Genome Column Medium Extraction Kit (Kangweishiji, China). Trio-WES was performed in the entire cohort by Beijing Chigene Translational Medical Research Center. Exome capture was carried out using xGen Exome Research Panel v2.0 (IDT, Iowa, United States), which consists of 429,826 individually synthesized and quality-controlled probes, targeting a 39 Mb protein-coding region (19,396 genes) of the human genome and covering 51 Mb of end to-end tiled probe space. High-throughput sequencing (100×) was performed by MGI DNBSEQ-T7 sequencing instruments (PE150) and the average coverage of target sequence was above 99%.The online system independently developed by Chigene (www.chigene.org) was used to annotate database-based minor allele frequencies (MAFs) and American college of medical genetics (ACMG) practice guideline-based pathogenicity of genetic variants. Bioinformatics software tools, including VarCards (http://varcards.biols.ac.cn/) and InterVar (https://wintervar.wglab.org/), were both applied for clinical interpretation of each genetic variant according to the ACMG and the Association for Molecular Pathology (ACMG/AMP) 2015 guidelines (Richards et al., 2015). Genotype-phenotype correlations were analysed by crossreferencing with Online Mendelian Inheritance in Man (OMIM, https://www.omim.org/), ClinVar databases and the relevant references. The identified genetic variants were classified as “benign”, “likely benign”, “variants of uncertain significance (VUS)”, “likely pathogenic” or “pathogenic”. Sanger sequencing was further used to confirm the candidate variants.
Results
Clinical features and Trio-WES results of twenty probands are summarized in Table 1. In this cohort, thirteen patients (13/20, 65%) were diagnosed as isolated short stature including twelve patients with idiopathic short stature (ISS) and one patient born small for gestational age (SGA). Seven probands (7/20, 35%) including one with SGA were diagnosed with syndromic short stature. These abnormal features included skeletal dysplasia (n = 6, P4, P6, P8, P9, P14, and P19), facial dysmorphism (n = 4, P4, P8, P9, and P19), intellectual disability (n = 2, P8, and P9), and visual impairment (n = 2, P9, and P13).
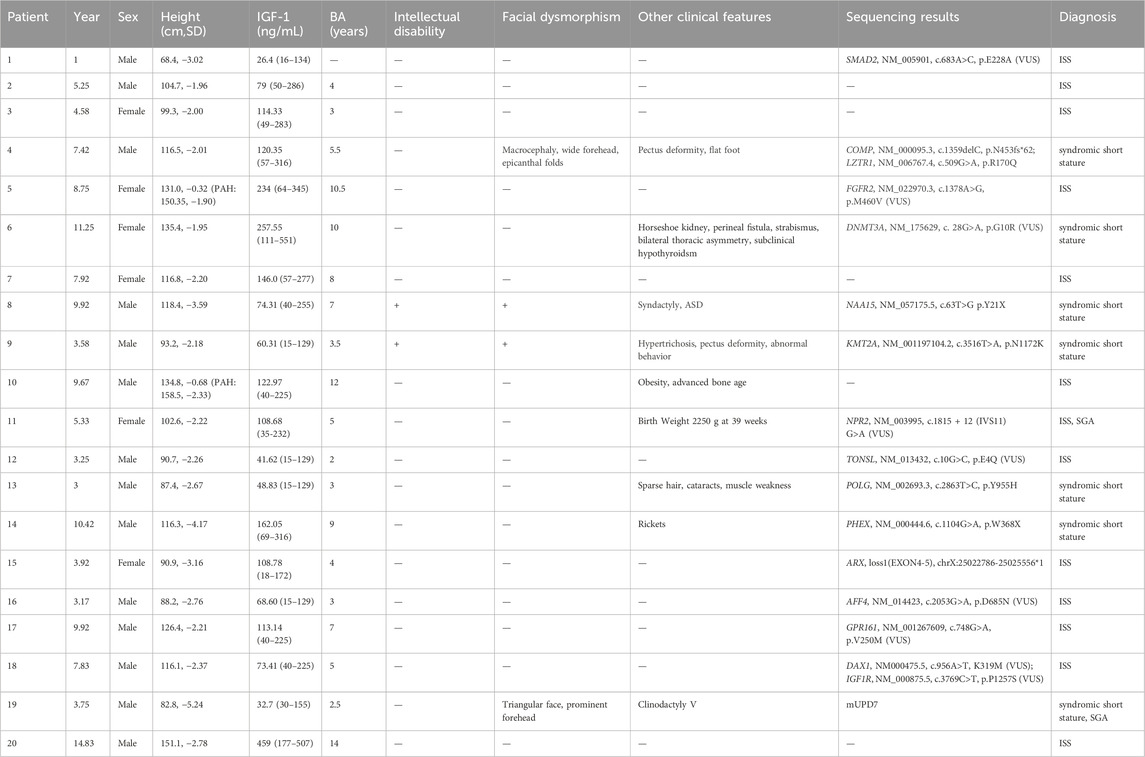
Table 1. Clinical phenotypes and genetic analysis in patients with short stature.
Of the twenty probands, six (6/20, 30%) patients with syndromic short stature obtained molecular diagnosis. Thirteen patients with isolated short stature had negative results. One novel COMP pathogenic variant c.1359delC, p.N453fs*62 and one LZTR1 likely pathogenic variant c.509G>A, p.R170Q were identified in a patient with short stature and skeletal dysplasia. One novel de novo NAA15 pathogenic variant c.63T>G, p.Y21X and one novel de novo KMT2A pathogenic variant c.3516T>A, p.N1172K was identified in two probands with short stature, intellectual disability and abnormal behaviours, respectively. One patient with short stature, cataract, and muscle weakness had a de novo POLG pathogenic variant c.2863 T>C, p.Y955H. One PHEX pathogenic variant c.1104G>A, p.W368X was identified in a patient with short stature and rickets. The primers used by Sanger sequencing to confirm these variants are shown in the Supplementary Table. Maternal uniparental disomy 7 (mUPD7) was pathogenic in a patient with pre and postnatal growth retardation, wide forehead, triangular face, micrognathia and clinodactyly. Clinical manifestations of these patients are described in detail sequentially below.
COMP variant and LZTR1 variant
The fourth proband was a boy aged 7 years and 5 months. He showed signs of short stature after 2 years of age. The patient was born at full term by caesarean section. His birth weight was 3,600 g, and his birth length was 50 cm. He had no history of perinatal asphyxia. His height was 116.5 cm (−2.01 SD), weight 21.2 kg (−1.24 SD), and BMI 15.62 kg/m2 (−0.08 SD) at visit. He had normal intelligence. His head circumference was 54 cm. The upper segment was 62.5 cm, and lower segment was 54.0 cm. The upper/lower segment ratio was 1.16 (−0.82 SD). His arm span was 114 cm. The proband had macrocephaly, wide forehead, epicanthal folds, pectus deformity, wrinkled skin on the hands and feet, flat foot and café-au-lait skin pigmentation (Figures 1A–C). He had no short neck or webbed neck. Growth hormone stimulation tests showed peak concentrations of 8.14 ng/mL after combined treatment with levodopa and arginine. The proband had normal liver and kidney function, myocardial enzyme levels and thyroid function. The echocardiography and urological ultrasound findings were all normal. His bone age (BA) was 5.5 years, 2 years below his chronological age (CA). His father’s height, weight and BMI were 164.4 cm (−1.36 SD), 52.4 kg, and 19.39 kg/m2, respectively. He had facial dysmorphism, including a triangular face and prominent nasolabial folds. He had pectus deformity (Figure 1D), movement limitation of the lumber spine, and flatfoot. He had multiple lentigines on the face and trunk (Figure 1E), and wrinkled skin on the hands and feet. He had complained of ankle pain after long-distance walking beginning at the age of 30 years. The height, weight and BMI of the proband’s mother were 154.7 cm, 58.8 kg, and 24.57 kg/m2, respectively. She had no complaints. A COMP pathogenic variant c.1359delC p.N453fs*62 in exon 13 was identified in the proband and his father. There were five other members segregating with this variant including I-2 (153 cm, −1.41 SD), II-4 (154 cm, −1.22 SD), II-6 (155 cm, −1.04 SD), III-5 (123 cm, 0.04 SD), and III-7 (120 cm, −1.27 SD) confirmed by sanger sequencing. The pedigree tree is shown in Figure 1F. All adult members carrying the COMP variation had short stature or low–normal height and joint pain, including in the knees, wrist, ankle, and lumber vertebra. There was no complaint of joint pain among the young affected members (III-5, III-7 and III-9). A LZTR1 heterozygous variant c.509G>A p.R170Q (MAF 0.000014/2, GnomAD) in exon five was identified in the proband and his father. There were four other affected members with this variant, including II-2 (152 cm, −1.59 SD), III-3 (152 cm, −0.63 SD), II-4 (154 cm, −1.22 SD), and III-5 (123 cm, 0.04 SD). In silico analysis predicted that it was a likely pathogenic variant (InterVar, PM1, PM2, PP3 and PP5) according to clinical interpretation of genetic variants by ACMG/AMP 2015 guideline. Blood sample of the members I-1, II-1, II-5, III-1, III-2, III-6, and III-8 were unavailable, preventing molecular testing.
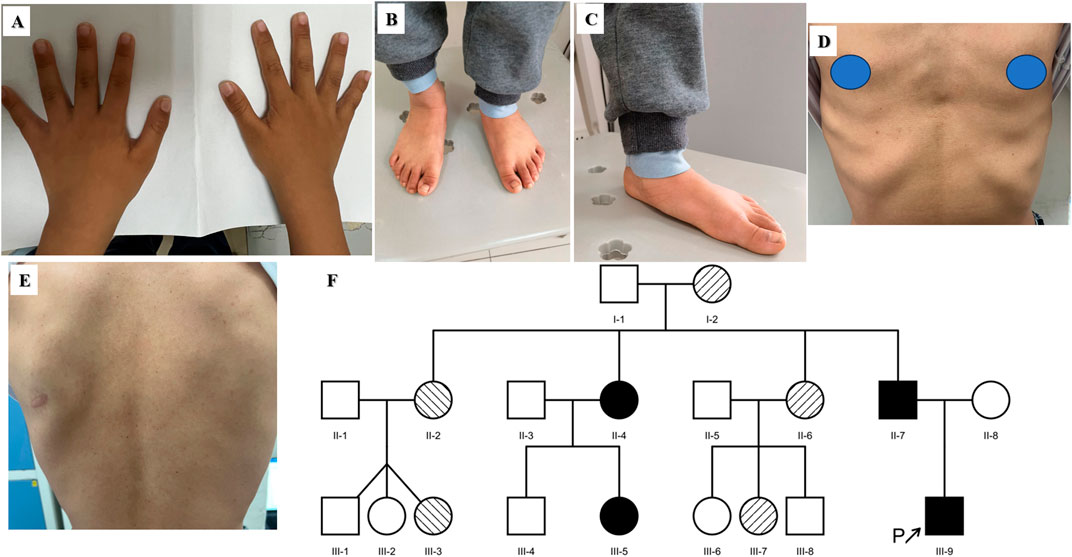
Figure 1. Clinical features of the fourth proband (A–C) and his father (D, E). (A) and (B) Wrinkled skin of the hands and feet. (C) Flatfoot. (D) Pectus deformity. (E) Multiple lentigines of the trunk. (F) Pedigree tree of the fourth proband. The black arrow represents the proband. The shaded symbols represent the affected individuals. The members I-2, II-6 and III-7 had the COMP variant (Right diagonal). The members II-2 and III-3 had the LZTR1 variant (Left diagonal). The members II-4, II-7, III-5 and III-9 had both the COMP variant and LZTR1 variant (Solid). The members II-3, II-8 and III-4 were normal. Blood sample of other members were unavailable.
NAA15
The eighth proband was a boy aged 9 years and 11 months. He showed signs of short stature. The patient was born at full term by vaginal delivery. His birth weight was 2,690 g, and his birth length was 49 cm. He had no history of perinatal asphyxia. His height, weight and BMI were 118.4 cm (−3.59 SD), 18.55 kg (−3.35 SD), and 13.23 kg/m2 (−2.14 SD), respectively. He had intellectual disability and language delay. He was diagnosed with autism spectrum disorder (ASD) at 5 years old. The proband had prominent eyebrows, epicanthus inversus, anteverted nares, multiple dental caries and low-set and posteriorly rotated ears. He had partial syndactyly between the third and fourth fingers in both hands since birth and had completed surgical management (Figure 2A). His growth hormone stimulation tests showed peak concentrations of 14.20 ng/mL after combination treatment with levodopa and arginine. His BA was 7 years, which was 3 years below CA (Figure 2B). His pituitary MRI showed a partial empty sella (Figure 2C). One novel de novo NAA15 variant c.63T>G, p.Y21X was identified in the proband.
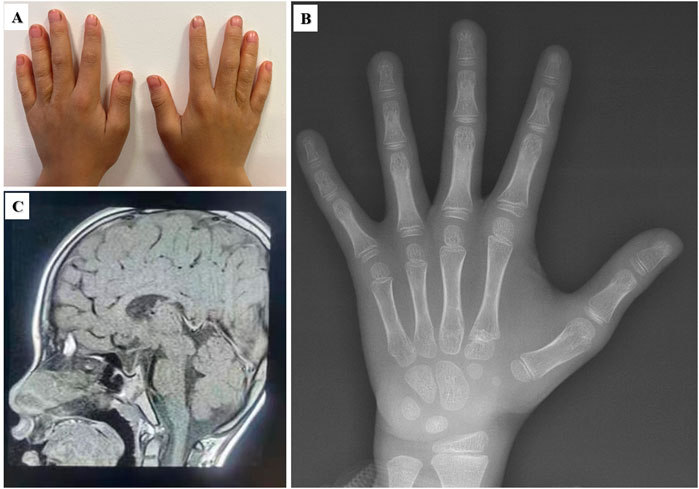
Figure 2. Clinical features of the eighth proband with the NAA15 variant. (A) Surgical scar of partial syndactyly in both hands. (B) Bone age radiograph. CA: 9/11/12 years; BA: 7 years (C) Partial empty sella of the pituitary gland.
KMT2A
The ninth proband was a boy aged 3 years and 7 months. He showed signs of short stature. The patient was born at full term by vaginal delivery. His birth weight was 3,850 g, and his birth length was 47 cm. He had no history of perinatal asphyxia. His height, weight and BMI were 93.2 cm (−2.18 SD), 12.1 kg (−2.51 SD), and 13.93 kg/m2 (−1.41 SD), respectively. His head circumference was 47 cm (−2.40 SD). He had intellectual disability, language delay, and abnormal hyperactivity behaviours. The proband had an abnormal facial appearance: flat face, thick eyebrows, epicanthus, ptosis, long eyelashes, downslanting and vertically narrow palpebral fissures, strabismus, wide nasal bridge, broad nasal tip and thin upper vermillion border. He had slim build, muscular hypotonia, pectus deformity, hypertrichosis on the upper arms and back, puffy hands and feet (Figure 3). He had chronic constipation and poor sleep since birth. His echocardiography and urological ultrasound were normal. His BA was 3.5 years. His father’s height was 176 cm, and his mother’s height was 160 cm, and their phenotypes were normal. Trio-WES identified a KMT2A de novo variant c.3516T>A, p.N1172K in the proband.

Figure 3. Clinical features of the nineth proband with KMT2A variant. (A) Pectus deformity. (B) and (C) Hypertrichosis on the upper arms and back. (D, E) Puffy hands and feet.
POLG
The thirteenth proband was a male patient aged 3 years. The child was born by breech delivery at term. His birth weight was 2,950 g. After birth, he had hypoxia asphyxia. At visit, his height, weight and BMI were 87.4 cm (−2.67 SD), 10.7 kg (−3.01 SD), and BMI 14.01 kg/m2 (−1.54SD), respectively. The proband had normal thyroid function, liver and kidney function, myocardial enzymes and blood electrolytes. His BA was equivalent to 3 years. Then he was diagnosed as isolated short stature. At the follow-up of 4 years and 1 month, he was diagnosed with bilateral cataracts in ophthalmic department. Then his height, weight and BMI were 91.9 cm (−3.29 SD), 11.95 kg (−3.14 SD), and BMI 14.15 kg/m2 (−1.06SD), respectively. His upper segment was 48.9 cm and his lower segment was 43.0 cm. The upper/lower segment ratio was 1.13 (−3.61 SD). His head circumference was 50 cm (−0.27 SD). Muscle weakness and exercise intolerance were apparent. His BA was about 4 years. He had elevated liver and muscle enzymes including AST 50.8 (14–44) U/L, GGT 19.4 (5–19) U/L, CG 4.48 (0–2.70) mg/L, ADA 26.3 (0–25) U/L, LDH 331 (110–295) U/L, and HBDH 283 (80–220) U/L. He had high TBA 13.48 (0–10) μmol/L, high TG 3.86 (0.4–1.7) mmol/L and low HDL 0.9 (1–1.55) mmol/L levels. A de novo POLG pathogenic variant c.2863T>C, p.Y955H was identified in the proband.
PHEX
The fourteenth proband was a male patient aged 10 years and 10 months. He showed signs of rickets. He manifested with severe lower extremity bowing, impaired mobility, and genu valgus. His height, weight and BMI were 116.3 cm (−4.39 SD), 24.3 kg (−2.23 SD), and 17.97 kg/m2 (0.20 SD), respectively. His upper segment was 67.0 cm (−2.70 SD), and his lower segment was 49.3 cm. The upper/lower segment ratio was 1.36 (4.04 SD). His pubertal stage was Tanner II. The child was born by caesarean section at 34 weeks’ gestational age. His birth weight was 2,800 g. The proband had normal liver and kidney function, myocardial enzymes and blood electrolytes. The findings for the indicators of bone metabolism were as follows: Ca 2.26 (2.25–2.67) mmol/L, P 0.67 (1.45–2.10) mmol/L, and ALP 673 (50–400) IU/L. His intact parathyroid hormone (iPTH) level was 63.9 (15–65) pg/mL. He had a low level of 25-(OH)-VD3 17.77 (30–100) ng/mL. His BA was equivalent to 9 years. He had normal thyroid function. His radiographic features were widened, frayed, and cupped metaphyses, pseudofractures, and osteoporosis in the femurs (Figure 4A). The younger brother of the proband was 7 years and 3 months old. His manifestations and radiographic features (Figure 4B) were similar to the proband. His height, weight and BMI were 105.1 cm (−4.16 SD), 18.9 kg (−1.95 SD), and 17.11 kg/m2 (0.77 SD), respectively. His upper segment was 63.5 cm (−1.95 SD), and his lower segment was 41.6 cm. The upper/lower segment ratio was 1.53 (5.48 SD). Bilateral cryptorchidism was present at birth. The findings for the indicators of bone metabolism were as follows: Ca 2.21 (2.25–2.67) mmol/L, P 0.58 (1.45–2.10) mmol/L, and ALP 870 (50–400) IU/L. His iPTH was 93.9 (15–65) pg/mL. He had a low level of 25-(OH)-VD3 18.16 (30–100) ng/mL. His BA was equivalent to 6 years. His father had a normal phenotype with a height of 170 cm. His mother had short stature and rickets. She had received surgical management. Her height was 141.4 cm (−3.55 SD). A PHEX pathogenic variant c.1104G>A, p.W368X in exon 10 was identified in the proband, his younger brother, and their mother.
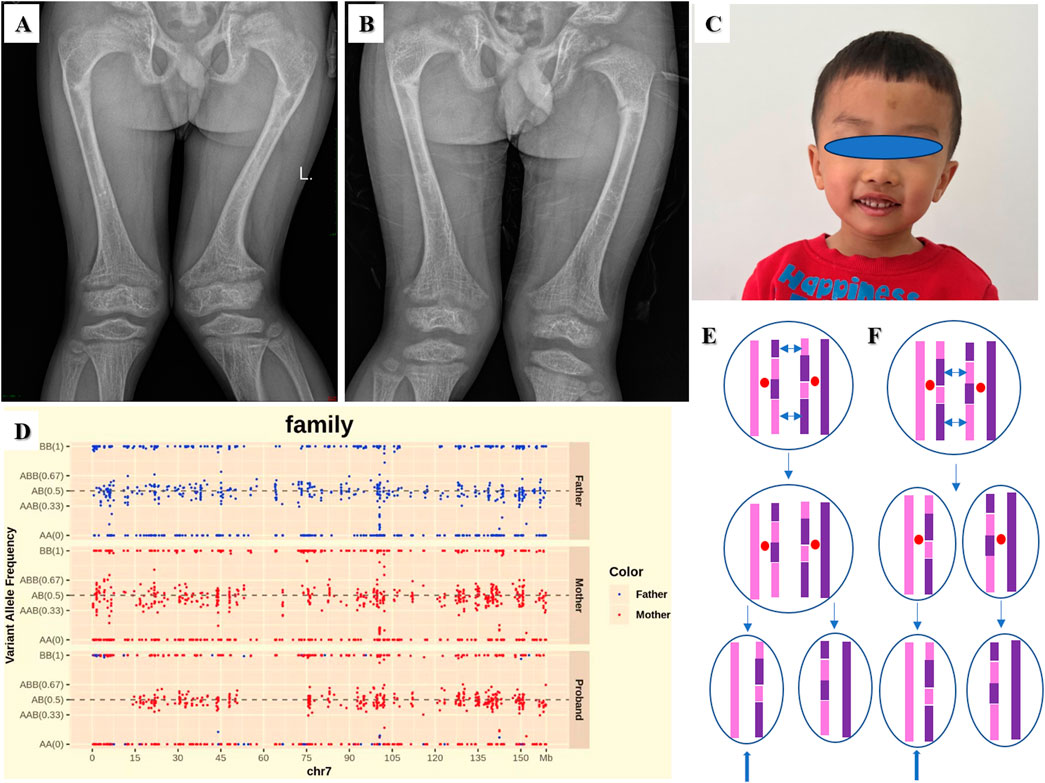
Figure 4. Radiographic features of the fourteenth proband (A) and his younger brother (B): frayed and cupped metaphyses, pseudofractures, and osteoporosis in the femurs. (C) The facial feature of the nineteenth proband. (D) SNPs analysis based on Trio-WES revealed a mixture of maternal isodisomy and heterodisomy of chromosome 7. The possible mechanism of the mixture pattern of uniparental disomy (UPD) is based on recombination of maternal homologs and maternal nondisjunction at Meiosis I (E) or Meiosis II (F). The double-headed arrows show crossing over of genetic material between homologous chromosomes during meiosis. The blue arrows show a mixture of maternal isodisomy and heterodisomy of chromosome 7.
UPD 7
The nineteenth proband was a boy aged 3 years and 9 months. The patient was born at full term by caesarean section. His birth weight was 2,100 g, and his birth length was 46 cm. He was diagnosed as SGA. He had feeding difficulties after birth. At visit his height was 82.8 cm (−5.24 SD), weight 8.8 kg (−5.24 SD), and BMI 12.84 kg/m2 (−2.62 SD). His head circumference was 46.2 cm (−2.90 SD). His upper segment was 44.8 cm, and his lower segment was 38.0 cm. The upper/lower segment ratio was 1.18 (−3.11SD). His arm span was 83.0 cm. The proband had slim build, wide forehead, triangular face, micrognathia and fifth-finger clinodactyly (Figure 4C). Growth hormone stimulation tests by levodopa showed peak concentrations of 13.03 ng/mL. The proband had normal liver and kidney function, myocardial enzyme levels and thyroid function. The echocardiography and urological ultrasound findings were normal. His BA was 2.5 years. His father’s and mother’s height was175 cm and 153 cm, respectively. Single nucleotide polymorphisms (SNPs) analysis based on Trio-WES revealed mUPD 7, maternal isodisomy at chr7:1-12727826 and chr7:63505837-75513216 and maternal heterodisomy at chr7:12727826-63505837 and chr7: 75513216-159138663 as shown in Figure 4D. The possible mechanism is based on recombination of maternal homologs and maternal nondisjunction at Meiosis I (Figure 4E) or Meiosis II (Figure 4F).
Discussion
We performed Trio-WES in a cohort of Chinese children with syndromic and isolated short stature. The diagnostic yield in the entire cohort was 30% (6/20), which was consistent with previous studies (Huang et al., 2018; Murray et al., 2018). Six pathogenic/likely pathogenic genetic variants were identified in five patients with syndromic short stature, including four monogenic pathogenic variants and one patient with dual pathogenic and likely pathogenic variants. Importantly, mUPD7 was identified in a patient with SGA and dysmorphic features.
Cartilage oligomeric matrix protein encoded by COMP (MIM *600310) is an extracellular matrix (ECM) glycoprotein and plays an important role in the structural integrity of cartilage and chondrocyte growth plate development (Posey et al., 2018; Caron et al., 2020). It has been demonstrated that COMP variants are associated with multiple epiphyseal dysplasia (MED, MIM#132400) and pseudoachondroplasia (PSACH, MIM #177170) with autosomal dominant inheritance (El-Lababidi et al., 2020). COMP variants could result in endoplasmic reticulum retention of COMP in chondrocytes and negative regulation of cartilage growth (Posey et al., 2018). MED is a genetic skeletal dysplasia characterized by delayed and irregular ossification of the epiphyses and early-onset osteoarthritis (Shao et al., 2020). The COMP pathogenic variant p.N453fs*62 segregated with short stature or low-normal height and early-onset osteoarthritis in the three-generation pedigree. The proband had short stature and delayed BA maturation. His father had low–normal height, movement limitation of the lumbar spine, and joint pain. All adult members carrying the COMP variation had a short height and various degrees of joint pain. This suggests that patients with short stature, delayed BA and a family history of early arthritis may have a COMP variant. Additionally, a LZTR1 likely pathogenic variant p.R170Q was identified in the proband. LZTR1 encoding leucine-zipper-like transcription regulator 1, which belongs to the BTB-POZ protein superfamily, is a tumour suppressor gene (Jacquinet et al., 2020). The variant p.R170Q localized in the KELCH 2 domain has been predicted to enhance RAS and MAPK signalling and identified in patients with Noonan syndrome and schwannomatosis (Piotrowski et al., 2014; Yamamoto et al., 2015; Johnston et al., 2018; Motta et al., 2019). Noonan syndrome (MIM #616564) called RASopathy syndrome is characterized by craniofacial malformations, cardiac abnormalities, short stature, cryptorchidism, and cognitive impairment (Roberts, 1993; Roberts et al., 2013; Noonan and Kappelgaard, 2015). The proband harbouring LZRT1 variant was characterized by short stature, facial abnormalities, and pectus deformity. His father had typical facial features, pectus deformity, and multiple lentigines. In a patient with Noonan syndrome carrying the LZTR1 variant c.850C>T, p.Arg284Cys, there were possible associations between rhGH treatment and late-onset oligoastrocytoma and ganglioblastoma (Jacquinet et al., 2020). We used rhGH in the proband with caution due to risks of nervous system disorders associated with LZTR1 variants (Piotrowski et al., 2014; Jacquinet et al., 2020). Genotype-phenotype analysis suggested that clinical manifestations of the proband and his father had features of dual disorders of MED and Noonan syndrome. The findings allowed us to identify a novel COMP pathogenic variant and a LZTR1 likely pathogenic variant segregating in the pedigree.
NAA15, located at 4q31.1, encodes for N-alpha-acetyltransferase 15, which is the auxiliary subunit of the N-terminal acetyltransferase (NatA) complex that is crucial for posttranslational modification of proteins (Stessman et al., 2017). NAA15 plays an important role in the generation and differentiation of neurons in development of the nervous system (Cheng et al., 2018; Zhao et al., 2018). This deleterious variant p.Y21X identified in our study is predicted to induce nonsense-mediated decay (NMD) and haploinsufficiency, affecting either NatA co-translational activity or complex stability (Cheng et al., 2018; Cheng et al., 2019; Lyon et al., 2023). NAA15 (OMIM *608000) variants have been identified in patients with intellectual disability, speech delay, ASD, facial dysmorphology and cardiac anomalies (MIM # 617787) (Stessman et al., 2017; Cheng et al., 2018). Clinical manifestations of the proband included short stature, intellectual disability, speech delay, ASD, facial abnormalities, and syndactyly. He had a partial empty sella but no growth hormone deficiency. In contrast to previous studies, we highlight the vital role of NAA15 in growth development based on short stature in the proband and the mechanism requires further functional study (Cheng et al., 2018; Zhao et al., 2018). The proband had a good response to rhGH during the follow-up period. Other pituitary hormones should be monitored (Ucciferro and Anastasopoulou, 2022).
KMT2A (OMIM *159555) encodes a DNA-binding histone methyltransferase that regulates chromatin-mediated transcription through histone H3K4 methylation and positively regulates expression of target genes (Milne et al., 2002). It has been identified as the pathogenic gene of Weidemann-Steiner syndrome (WSS, MIM #605130) (Jones et al., 2012). We identified a novel de novo KMT2A likely pathogenic variant p. N1172K in the proband with short stature, intellectual disability, facial malformations, ocular abnormalities, hypertrichosis and pectus deformity. His characteristics were consistent with a diagnosis of WSS, which is a disorder of phenotype heterogeneity. KMT2A is widely expressed in the central nervous system (CNS) including the retina and plays crucial role in CNS development (Brightman et al., 2018). The variant p.N1172K in the cysteine-rich CXXC domain (amino acids 1147-1204) is functionally conserved and crucial for binding nonmethylated CpG-containing DNA, contributing to severe symptoms of multisystem involvements (Xu et al., 2018; Luo et al., 2021). The mechanisms of histone modification for growth failure remains to be determined.
The patient with short stature, cataract, and muscle weakness had a de novo heterozygous POLG pathogenic variant p.Y955H. A previous study had identified another patient with the same variant had a severe early-onset multisystemic involvement with bilateral sensorineural hearing loss, cataract, myopathy, and liver failure (Siibak et al., 2017). This study has demonstrated that the variant p.Y955H in the critical polymerase catalytic domain caused severe mitochondrial DNA (mtDNA) polymerase dysfunction, affecting mtDNA replication with dominant negative effect on DNA synthesis (Bekheirnia et al., 2012; Siibak et al., 2017). Both the patients had similar clinical phenotypes, especially cataracts, suggesting an important role for POLG during lens development. The POLG-related disorders comprise a continuum of overlapping phenotypes based on biallelic POLG pathogenic variants except autosomal dominant progressive external ophthalmoplegia (adPEO,MIM #157640) (Cohen et al., 1993). The disorder is characterized by generalized myopathy, variable degrees of sensorineural hearing loss, axonal neuropathy, ataxia, depression, parkinsonism, hypogonadism and cataracts (Rahman and Copeland, 2019). The patient in our study had some, but not all, of the features of adPEO. Given his young age, his clinical phenotype requires long-term follow-up. Our findings expand the clinical spectrum associated with POLG-associated disorders (Bekheirnia et al., 2012). Because of milder phenotypic presentation in early childhood, it’s difficult to recognize and diagnose POLG-related disorders in some patients with short stature.
X-linked hypophosphatemic rickets (XLH, MIM #307800) is due to abnormalities of a phosphate-regulating gene homologous to endopeptidase on the X chromosome (PHEX) (Bitzan and Goodyer, 2019). PHEX, highly expressed in osteoblasts, osteocytes, and odontoblasts, is a local negative regulator of fibroblast growth factor 23 (FGF23). The PHEX variant p.W368X might cause PHEX inactivation leading to upregulation of FGF23. FGF23 excess increases inorganic phosphate (Pi) excretion and lowers serum Pi levels by downregulating the renal expression of type IIa and IIc sodium/Pi (Na+/Pi) cotransporters (NaPi-IIa and NaPi-IIc) (Yamazaki and Michigami, 2022). In addition, FGF23 reduces the production of 1,25(OH)2D, which can increase intestinal Pi absorption by upregulating the expression of the type IIb Na+/Pi cotransporter NaPi-IIb, leading to hypophosphatemia (Yamazaki and Michigami, 2022). Hypophosphatemia impairs skeletal mineralization and results in skeletal manifestations of rickets. The proband and his brother were characterized by disproportionate short stature with leg length significantly lower than the sitting height, osteoporosis, severe bone malformations and genu valgus. They had typical chemical findings of XLH: low serum phosphate concentration and elevated alkaline phosphatase. Based on the pathogenesis, XLH is a disorder of FGF23-related hypophosphatemic rickets.
WES has been routinely used for identifying pathogenic variants in genetic disorders and presents a higher diagnostic yield than short stature panels (Li et al., 2022). Recent studies have demonstrated that Trio-WES was an important approach to investigate UPD through analysis of SNP origin from trio sequencing data (Del Gaudio et al., 2020; Chen et al., 2021). Silver-Russell syndrome (SRS, MIM #180860) is a genetically heterogeneous disorder characterized by intrauterine growth retardation, postnatal growth failure, relative macrocephaly at birth, prominent forehead usually with frontal bossing, micrognathia with narrow chin, body asymmetry, clinodactyly, and feeding difficulties (Saal et al., 1993). The proband with SGA (P19) had pre and postnatal growth retardation, wide forehead, triangular face, micrognathia, clinodactyly and low BMI, however, characteristic body asymmetry was obscure. According to the Netchine-Harbison Clinical Scoring System (NH-CSS) clinical criteria, his score was four (Wakeling et al., 2017). Clinical diagnosis of SRS was confirmed by trio-WES, in which mUPD7 was identified as the pathogenic cause. Growth anomalies in mUPD7 were caused by two maternally overexpression of growth suppressor genes and lack of paternally expressed growth promoter genes (Butler, 2020; Zhang et al., 2020). Diagnosis of SRS was challenging as patients with mUPD7 are more likely to have less typical phenotypes than those with loss of methylation on chromosome 11p15 (Bernard et al., 1999; Wakeling, 2011; Wakeling, 2021). UPD evaluation was recommended in Trio-WES analysis for patients who manifested with short stature, especially SGA.
This study suggests that it is important to separate and differentiate between syndromic and isolated short stature patients. Genetic testing has high yield for syndromic patients but low for isolated patients. In this cohort, 70% (14/20) of these patients had negative Trio-WES results. The main reason was that 65% (13/20) patients had isolated short stature, which might be normal variants of growth caused by combination of hundreds of common variants associated with linear growth and environment effects (Wit et al., 2016; Collett-Solberg et al., 2019a; Homma et al., 2019). Comprehensive genetic analysis, such as whole-genome sequencing (WGS) and genome-wide methylation assays, is recommended to explore potential causal variants in deep introns and genetic methylation defects in patients with negative Trio-WES findings (Collett-Solberg et al., 2019a).
Our study highlights the importance of Trio-WES to identify genetic variants and UPD in the molecular diagnosis of syndromic short stature. Except for monogenic causes of short stature, our study identified dual genetic variants in a patient with short stature expanding our knowledge about complex aetiology of growth failure. These molecular defects cause abnormal functions of the cartilage extracellular matrix (COMP), paracrine signalling factors (PHEX, FGFR3-related rickets), transcriptional regulation (LZTR1), histone methylation (KMT2A), posttranslational modification (NAA15), and mtDNA replication and repair (POLG) (Andrade et al., 2017). The precise aetiology and signalling pathways of mUPD7 need to be elucidated in further studies. Molecular diagnosis of short stature provides new insights into personalized treatment of growth disorders beyond growth hormone replacement in these individuals.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author. The variants COMP c.1359delC (p.N453fs*62), LZTR1 c.509G>A (p.R170Q), NAA15 c.63T>G (p.Y21X), KMT2A c.3516T>A (p.N1172K), POLG c.2863T>C (p.Y955H), and PHEX c.1104G>A (p.W368X) were submitted to the ClinVar database (http://ncbi.nlm.nlh.nih.gov/clinvar) with the ClinVar variant ID SCV004805897, SCV004805898, SCV004808384, SCV004809195, SCV004809197, SCV004809198.
Ethics statement
The studies involving humans were approved by the study was approved by the Ethics Committee of Beijing Jishuitan Hospital, Capital Medical University. Written informed consent was obtained from the participants and parents of the patients in the study. The study was conducted according to the World Medical Association Declaration of Helsinki. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
HS: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. GZ: Data curation, Conceptualization, Formal Analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Writing–eview and editing. NL: Data curation, Formal Analysis, Conceptualization, Investigation, Methodology, Software, Supervision, Validation, Writing–review and editing. XB: Data curation, Formal Analysis, Investigation, Conceptualization, Resources, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by research grants from the Beijing Hospitals Authority Youth Programme (QML20210403) and Beijing JST Research Funding (ZR202114).
Acknowledgments
We would like to thank all members of the family participating in the study for agreeing to publish their available clinical data.
Conflict of interest
Author GZ was employed by Beijing Chigene Translational Medical Research Center Company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1399186/full#supplementary-material
References
Andrade, A. C., Jee, Y. H., and Nilsson, O. (2017). New genetic diagnoses of short stature provide insights into local regulation of childhood growth horm res paediatr. Horm. Res. Paediatr. 88 (1), 22–37. doi:10.1159/000455850
Bekheirnia, M. R., Zhang, W., Eble, T., Willis, A., Shaibani, A., Wong, L. J., et al. (2012). POLG mutation in a patient with cataracts, early-onset distal muscle weakness and atrophy, ovarian dysgenesis and 3-methylglutaconic aciduria. Gene 499 (1), 209–212. doi:10.1016/j.gene.2012.02.034
Bernard, L. E., Penaherrera, M. S., Van Allen, M. I., Wang, M. S., Yong, S. L., Gareis, F., et al. (1999). Clinical and molecular findings in two patients with russell-silver syndrome and UPD7: comparison with non-UPD7 cases. Am. J. Med. Genet. 87 (3), 230–236. doi:10.1002/(sici)1096-8628(19991126)87:3<230::aid-ajmg7>3.0.co;2-s
Bitzan, M., and Goodyer, P. R. (2019). Hypophosphatemic rickets. Pediatr. Clin. North Am. 66 (1), 179–207. doi:10.1016/j.pcl.2018.09.004
Brightman, D. S., Grant, R. L., Ruzycki, P. A., Suzuki, R., Hennig, A. K., and Chen, S. (2018). MLL1 is essential for retinal neurogenesis and horizontal inner neuron integrity. Sci. Rep. 8 (1), 11902. doi:10.1038/s41598-018-30355-3
Butler, M. G. (2020). Imprinting disorders in humans: a review. Curr. Opin. Pediatr. 32 (6), 719–729. doi:10.1097/MOP.0000000000000965
Caron, M. M. J., Janssen, M. P. F., Peeters, L., Haudenschild, D. R., Cremers, A., Surtel, D. A. M., et al. (2020). Aggrecan and COMP improve periosteal chondrogenesis by delaying chondrocyte hypertrophic maturation. Front. Bioeng. Biotechnol. 8, 1036. doi:10.3389/fbioe.2020.01036
Chen, W., Chen, F., Shen, Y., Yang, Z., and Qin, J. (2021). Case report: a case of epileptic disorder associated with a novel CNTN2 frameshift variant in homozygosity due to maternal uniparental disomy. Front. Genet. 12, 743833. doi:10.3389/fgene.2021.743833
Cheng, H., Dharmadhikari, A. V., Varland, S., Ma, N., Domingo, D., Kleyner, R., et al. (2018). Truncating variants in NAA15 are associated with variable levels of intellectual disability, autism spectrum disorder, and congenital anomalies. Am. J. Hum. Genet. 102 (5), 985–994. doi:10.1016/j.ajhg.2018.03.004
Cheng, H., Gottlieb, L., Marchi, E., Kleyner, R., Bhardwaj, P., Rope, A. F., et al. (2019). Phenotypic and biochemical analysis of an international cohort of individuals with variants in NAA10 and NAA15. Hum. Mol. Genet. 28 (17), 2900–2919. doi:10.1093/hmg/ddz111
Cohen, B. H., Chinnery, P. F., and Copeland, W. C. (1993). “POLG-related disorders,” in GeneReviews((R)). Editors M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Beanet al. (Seattle WA).
Collett-Solberg, P. F., Ambler, G., Backeljauw, P. F., Bidlingmaier, M., Biller, B. M. K., Boguszewski, M. C. S., et al. (2019a). Diagnosis, genetics, and therapy of short stature in children: a growth hormone research society international perspective. Horm. Res. Paediatr. 92 (1), 1–14. doi:10.1159/000502231
Collett-Solberg, P. F., Jorge, A. A. L., Boguszewski, M. C. S., Miller, B. S., Choong, C. S. Y., Cohen, P., et al. (2019b). Growth hormone therapy in children; research and practice - a review. Growth Horm. IGF Res. 44, 20–32. doi:10.1016/j.ghir.2018.12.004
Del Gaudio, D., Shinawi, M., Astbury, C., Tayeh, M. K., Deak, K. L., Raca, G., et al. (2020). Diagnostic testing for uniparental disomy: a points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 22 (7), 1133–1141. doi:10.1038/s41436-020-0782-9
El-Lababidi, N., Zikanova, M., Baxova, A., Noskova, L., Leiska, A., Lambert, L., et al. (2020). Age dependent progression of multiple epiphyseal dysplasia and pseudoachondroplasia due to heterozygous mutations in COMP gene. Prague Med. Rep. 121 (3), 153–162. doi:10.14712/23362936.2020.14
Grunauer, M., and Jorge, A. A. L. (2018). Genetic short stature. Growth Horm. IGF Res. 38, 29–33. doi:10.1016/j.ghir.2017.12.003
Homma, T. K., Freire, B. L., Honjo Kawahira, R. S., Dauber, A., Funari, M. F. A., Lerario, A. M., et al. (2019). Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. J. Pediatr. 215, 192–198. doi:10.1016/j.jpeds.2019.08.024
Homma, T. K., Krepischi, A. C. V., Furuya, T. K., Honjo, R. S., Malaquias, A. C., Bertola, D. R., et al. (2018). Recurrent copy number variants associated with syndromic short stature of unknown cause. Horm. Res. Paediatr. 89 (1), 13–21. doi:10.1159/000481777
Hu, G., Fan, Y., Wang, L., Yao, R. E., Huang, X., Shen, Y., et al. (2016). Copy number variations in 119 Chinese children with idiopathic short stature identified by the custom genome-wide microarray. Mol. Cytogenet 9, 16. doi:10.1186/s13039-016-0225-0
Huang, Z., Sun, Y., Fan, Y., Wang, L., Liu, H., Gong, Z., et al. (2018). Genetic evaluation of 114 Chinese short stature children in the next generation era: a single center study. Cell Physiol. Biochem. 49 (1), 295–305. doi:10.1159/000492879
Jacquinet, A., Bonnard, A., Capri, Y., Martin, D., Sadzot, B., Bianchi, E., et al. (2020). Oligo-astrocytoma in LZTR1-related noonan syndrome. Eur. J. Med. Genet. 63 (1), 103617. doi:10.1016/j.ejmg.2019.01.007
Jee, Y. H., Baron, J., and Nilsson, O. (2018). New developments in the genetic diagnosis of short stature. Curr. Opin. Pediatr. 30 (4), 541–547. doi:10.1097/MOP.0000000000000653
Johnston, J. J., van der Smagt, J. J., Rosenfeld, J. A., Pagnamenta, A. T., Alswaid, A., Baker, E. H., et al. (2018). Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet. Med. 20 (10), 1175–1185. doi:10.1038/gim.2017.249
Jones, W. D., Dafou, D., McEntagart, M., Woollard, W. J., Elmslie, F. V., Holder-Espinasse, M., et al. (2012). De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am. J. Hum. Genet. 91 (2), 358–364. doi:10.1016/j.ajhg.2012.06.008
Li, X., Yao, R., Chang, G., Li, Q., Song, C., Li, N., et al. (2022). Clinical profiles and genetic spectra of 814 Chinese children with short stature. J. Clin. Endocrinol. Metab. 107 (4), 972–985. doi:10.1210/clinem/dgab863
Luo, S., Bi, B., Zhang, W., Zhou, R., Chen, W., Zhao, P., et al. (2021). Three de novo variants in KMT2A (MLL) identified by whole exome sequencing in patients with Wiedemann-Steiner syndrome. Mol. Genet. Genomic Med. 9 (10), e1798. doi:10.1002/mgg3.1798
Lyon, G. J., Vedaie, M., Beisheim, T., Park, A., Marchi, E., Gottlieb, L., et al. (2023). Expanding the phenotypic spectrum of NAA10-related neurodevelopmental syndrome and NAA15-related neurodevelopmental syndrome. Eur. J. Hum. Genet. 31 (7), 824–833. doi:10.1038/s41431-023-01368-y
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86 (5), 749–764. doi:10.1016/j.ajhg.2010.04.006
Milne, T. A., Briggs, S. D., Brock, H. W., Martin, M. E., Gibbs, D., Allis, C. D., et al. (2002). MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 10 (5), 1107–1117. doi:10.1016/s1097-2765(02)00741-4
Motta, M., Fidan, M., Bellacchio, E., Pantaleoni, F., Schneider-Heieck, K., Coppola, S., et al. (2019). Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum. Mol. Genet. 28 (6), 1007–1022. doi:10.1093/hmg/ddy412
Murray, P. G., Clayton, P. E., and Chernausek, S. D. (2018). A genetic approach to evaluation of short stature of undetermined cause. Lancet Diabetes Endocrinol. 6 (7), 564–574. doi:10.1016/S2213-8587(18)30034-2
Noonan, J. A., and Kappelgaard, A. M. (2015). The efficacy and safety of growth hormone therapy in children with noonan syndrome: a review of the evidence. Horm. Res. Paediatr. 83 (3), 157–166. doi:10.1159/000369012
Piotrowski, A., Xie, J., Liu, Y. F., Poplawski, A. B., Gomes, A. R., Madanecki, P., et al. (2014). Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat. Genet. 46 (2), 182–187. doi:10.1038/ng.2855
Posey, K. L., Coustry, F., and Hecht, J. T. (2018). Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol. 71-72, 161–173. doi:10.1016/j.matbio.2018.02.023
Rahman, S., and Copeland, W. C. (2019). POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 15 (1), 40–52. doi:10.1038/s41582-018-0101-0
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Roberts, A. E. (1993). “Noonan syndrome,” in GeneReviews((R)). Editors M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Grippet al. (Seattle WA: University of Washington), 1993–2021.
Roberts, A. E., Allanson, J. E., Tartaglia, M., and Gelb, B. D. (2013). Noonan syndrome. Lancet 381 (9863), 333–342. doi:10.1016/S0140-6736(12)61023-X
Saal, H. M., Harbison, M. D., and Netchine, I. (1993) “Silver-Russell syndrome,” in GeneReviews. Seattle (WA).
Shao, J., Zhao, S., Yan, Z., Wang, L., Zhang, Y., Lin, M., et al. (2020). A novel COMP mutation in a Chinese family with multiple epiphyseal dysplasia. BMC Med. Genet. 21 (1), 115. doi:10.1186/s12881-020-01040-y
Siibak, T., Clemente, P., Bratic, A., Bruhn, H., Kauppila, T. E. S., Macao, B., et al. (2017). A multi-systemic mitochondrial disorder due to a dominant p.Y955H disease variant in DNA polymerase gamma. Hum. Mol. Genet. 26 (13), 2515–2525. doi:10.1093/hmg/ddx146
Siklar, Z., and Berberoglu, M. (2014). Syndromic disorders with short stature. J. Clin. Res. Pediatr. Endocrinol. 6 (1), 1–8. doi:10.4274/Jcrpe.1149
Stavber, L., Gaia, M. J., Hovnik, T., Bizjan, B. J., Debeljak, M., Kovac, J., et al. (2022). Heterozygous NPR2 variants in idiopathic short stature. Genes (Basel) 13 (6), 1065. doi:10.3390/genes13061065
Stessman, H. A., Xiong, B., Coe, B. P., Wang, T., Hoekzema, K., Fenckova, M., et al. (2017). Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49 (4), 515–526. doi:10.1038/ng.3792
Sun, H., Li, N., and Wan, N. (2021). Molecular genetic analysis and growth hormone response in patients with syndromic short stature. BMC Med. Genomics 14 (1), 261. doi:10.1186/s12920-021-01113-8
Wakeling, E. L. (2011). Silver-Russell syndrome. Arch. Dis. Child. 96 (12), 1156–1161. doi:10.1136/adc.2010.190165
Wakeling, E. L., Brioude, F., Lokulo-Sodipe, O., O'Connell, S. M., Salem, J., Bliek, J., et al. (2017). Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat. Rev. Endocrinol. 13 (2), 105–124. doi:10.1038/nrendo.2016.138
Wang, J., Wang, Y., Wang, L., Chen, W. Y., and Sheng, M. (2020). The diagnostic yield of intellectual disability: combined whole genome low-coverage sequencing and medical exome sequencing. BMC Med. Genomics 13 (1), 70. doi:10.1186/s12920-020-0726-x
Wit, J. M., Oostdijk, W., Losekoot, M., van Duyvenvoorde, H. A., Ruivenkamp, C. A., and Kant, S. G. (2016). Mechanisms in endocrinology: novel genetic causes of short stature. Eur. J. Endocrinol. 174 (4), R145–R173. doi:10.1530/EJE-15-0937
Xu, C., Liu, K., Lei, M., Yang, A., Li, Y., Hughes, T. R., et al. (2018). DNA sequence recognition of human CXXC domains and their structural determinants. Structure 26 (1), 85–95. doi:10.1016/j.str.2017.11.022
Yamamoto, G. L., Aguena, M., Gos, M., Hung, C., Pilch, J., Fahiminiya, S., et al. (2015). Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J. Med. Genet. 52 (6), 413–421. doi:10.1136/jmedgenet-2015-103018
Yamazaki, M., and Michigami, T. (2022). Osteocytes and the pathogenesis of hypophosphatemic rickets. Front. Endocrinol. (Lausanne) 13, 1005189. doi:10.3389/fendo.2022.1005189
Zahnleiter, D., Uebe, S., Ekici, A. B., Hoyer, J., Wiesener, A., Wieczorek, D., et al. (2013). Rare copy number variants are a common cause of short stature. PLoS Genet. 9 (3), e1003365. doi:10.1371/journal.pgen.1003365
Zhang, C., Hao, S., Zhang, Q., Liu, F., Zhou, B., Xuan, F., et al. (2020). Maternal UPD of chromosome 7 in a patient with Silver-Russell syndrome and Pendred syndrome. J. Clin. Lab. Anal. 34 (9), e23407. doi:10.1002/jcla.23407
Keywords: short stature, molecular diagnosis, trio whole-exome sequencing, variant, uniparental disomy
Citation: Sun H, Zhang G, Li N and Bu X (2024) Molecular diagnosis of patients with syndromic short stature identified by trio whole-exome sequencing. Front. Genet. 15:1399186. doi: 10.3389/fgene.2024.1399186
Received: 11 March 2024; Accepted: 16 September 2024;
Published: 02 October 2024.
Edited by:
Claudia Gonzaga-Jauregui, Universidad Nacional Autónoma de México, MexicoReviewed by:
Ludmila Matyakhina, GeneDx, United StatesYongchu Pan, Nanjing Medical University, China
Copyright © 2024 Sun, Zhang, Li and Bu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huihui Sun, c3VuaHVpaHVpMDEwQDEyNi5jb20=