 Dalal A. Al-Mutairi
Dalal A. Al-Mutairi Basel H. Alsabah2
Basel H. Alsabah2 Petra Pennekamp
Petra Pennekamp Heymut Omran
Heymut Omran- 1Department of Pathology, Faculty of Medicine, Kuwait University, Kuwait City, Kuwait
- 2Zain Hospital for Ear, Nose and Throat, Kuwait City, Kuwait
- 3Department of Pediatrics, University Hospital Muenster, Muenster, Germany
Introduction: Primary ciliary dyskinesia (PCD) is caused by the dysfunction of motile cilia resulting in insufficient mucociliary clearance of the lungs. This study aimed to map novel PCD variants and determine their pathogenicity in PCD patients in Kuwait.
Methods: Herein, we present five PCD individuals belonging to a cohort of 105 PCD individuals recruited from different hospitals in Kuwait. Genomic DNAs from the family members were analysed to screen for pathogenic PCD variants. Transmission electron microscopy (TEM) and immunofluorescence (IF) analyses were performed on the nasal biopsies to detect specific structural abnormalities within the ciliated cells.
Results: Genetic screening and functional analyses confirmed that the five PCD individuals carried novel pathogenic variants of DNAH5 causing PCD in three Arabic families. Of these, one multiplex family with two affected individuals showed two novel homozygous missense variants in DNAH5 causing PCD with situs inversus; another multiplex family with two affected individuals showed two newly identified compound heterozygous variants in DNAH5 causing PCD with situs solitus. In addition, novel heterozygous variants were identified in a child with PCD and situs solitus from a singleton family with unrelated parents. TEM analysis demonstrated the lack of outer dynein arms (ODAs) in all analysed samples, and IF analysis confirmed the absence of the dynein arm component of DNAH5 from the ciliary axoneme.
Conclusion: The newly identified pathogenic variants of DNAH5 are associated with PCD as well as variable pulmonary clinical manifestations in Arabic families.
Introduction
Primary ciliary dyskinesia (PCD; OMIM: 244400) is a clinically and genetically heterogeneous group of disorders of the motile cilia that results in abnormal mucociliary clearance and pulmonary dysfunction. PCD-affected individuals have a history of neonatal respiratory distress and suffer from lifelong symptoms of wet cough, rhinosinusitis, and otitis media. Recurrent chest infections are also common, which eventually lead to bronchiectasis and progressive decline in pulmonary functions (Ratjen et al., 2016; Davis et al., 2019; Al-Mutairi et al., 2022). Almost 50% of PCD affected individuals also have laterality defects, mainly situs inversus totalis and other situs abnormalities, owing to dysmotility of the monocilia of the left-right organiser (LRO). Male infertility and female subfertility are also associated with PCD (Zariwala et al., 2007; Leigh et al., 2009; Zariwala et al., 2011; Al-Mutairi et al., 2022). A recent study showed that the global incidence of PCD is 1 in 7,550 and that the frequency of PCD is higher in isolated populations, such as individuals of African descent, than in most other populations (Hannah et al., 2022; Al-Mutairi et al., 2023). Geographically isolated populations may also exhibit increased incidence of PCD owing to the practise of consanguineous marriages over multiple generations (Zariwala et al., 2007; Al-Mutairi et al., 2022; Hannah et al., 2022; Al-Mutairi et al., 2023), which is typical in the Kuwaiti population. Despite recent improvements in PCD diagnosis since the first reported case at the beginning of the 20th century, only a limited number of individuals have been accurately diagnosed with PCD, reflecting the limited capability of PCD diagnosis. Almost one-third of such individuals with well-characterised PCD phenotypes have no detectable genetic variants corresponding to known, published PCD-causing genetic variants (Knowles et al., 2013; Horani et al., 2016; Leigh et al., 2019; Wheway et al., 2021; Al-Mutairi et al., 2023). The diagnosis of PCD is therefore quite challenging, and a large proportion of PCD individuals are either misdiagnosed or receive delayed diagnoses since there is no ‘optimum PCD diagnostic approach’. In addition, some PCD-causing variants, especially compound heterozygous variants in PCD individuals with situs solitus, escape inclusion in the current diagnostic criteria for confirmed PCD diagnosis (Dalrymple and Kenia, 2019; Muller et al., 2021; Al-Mutairi et al., 2022; Goutaki and Shoemark, 2022; Al-Mutairi et al., 2023).
Currently, more than 53 PCD genes (OMIM: PS244400) have been identified that mostly encode structural components of the axonemes of motile cilia and sperm flagella (Al-Mutairi et al., 2022). In general, there are six structural genes that encode the dynein components reported to cause PCD and are associated with outer dynein arm (ODA) defects. Three ciliary structural genes, namely, DNAH5 (Olbrich et al., 2002; Hornef et al., 2006; Failly et al., 2009), DNAH9 (Loges et al., 2018), and DNAH11 (Schwabe et al., 2008; Pifferi et al., 2010; Knowles et al., 2012), encode the axonemal ODA heavy chains. In addition, there are two ciliary structural genes DNAI1 (Zariwala et al., 2006) and DNAI2 (Loges et al., 2008; Al-Mutairi et al., 2022) that encode the axonemal ODA intermediate chains. Only one ciliary structural gene is known to encode the axonemal ODA light chains: DNAL1 (Mazor et al., 2011). Dynein axonemal heavy chain 5 (DNAH5) (MIM: 603335) was the first reported gene to cause PCD associated with the absence of the ODAs; it is also the most common defective gene in PCD-affected Caucasians (Failly et al., 2009).
The purpose of this study was to determine the genetic causes and ciliary abnormalities of the respiratory system in Arabic PCD patients in Kuwait. This report summarises the genetic and functional analyses of ciliated respiratory epithelial cells from five PCD individuals with variable laterality defects who inherited novel pathogenic variants of DNAH5. Immunofluorescence (IF) and transmission electron microscopy (TEM) analyses show the complete absence of the dynein arm protein component DNAH5 as well as the absence of the axonemal ODAs in all five PCD individuals. This confirms that these newly reported variants of DNAH5 are pathogenic and associated with PCD.
Methods
Inclusion criteria for the selected human subjects
Ethical approval for this project was obtained from the Ministry of Health Research Ethics Committee (Ethics ID: 62/2013). This study was carried out with the permission of all participating family members; informed written consent was obtained from the adult participants and from the parents/guardians/next-of-kin of children for collection of blood samples from the patients and non-affected parents as well as nasal biopsies.
Patients with a clinical picture of PCD were referred to clinics run by the Kuwait Ministry of Health for genetic assessment in collaboration with the Kuwait University Faculty of Medicine. The ethnic background of the patients included in this study include Asian Arabs descended from different tribes originating from the Arabian Peninsula (Al-Mutairi et al., 2022; Al-Mutairi et al., 2023). The inclusion criteria were clinical features typical of the hallmark disease of PCD (MIM: PS244400), including neonatal respiratory distress, chronic respiratory disease with symptoms of chronic airway infections, rhinosinusitis, otitis media, and bronchiectasis. Most patients were suspected to carry recessive disease-causing hereditary variants as more than one individual in their families exhibited similar clinical phenotypes.
Pedigrees for the families were constructed using Cyrillic version 2.1 (http://www.cyrillicsoftware.com/) as previously published (Al-Mutairi et al., 2022; Al-Mutairi et al., 2023). All radiological examinations of the patients in this study were performed at Sabah Hospital in Kuwait. The genomic DNA extractions and exome sequencing protocols were performed as described previously (Al-Mutairi et al., 2022; Al-Mutairi et al., 2023).
Autozygosity mapping and variant screening
Genetic screening using autozygosity mapping was performed as described previously (Carr et al., 2013; Al-Mutairi et al., 2022; Al-Mutairi et al., 2023). AgileMultiIdeogram software was used to visualise the homozygous intervals using exome data for linkage, in which all the homozygous intervals were displayed against a circular ideogram for the 22 autosomal chromosomes for the related patients (Carr et al., 2013; Al-Mutairi et al., 2022; Al-Mutairi et al., 2023). Linkage analysis using autozygosity mapping was performed for all the affected individuals belonging to the same family, such as family KU-5 and family KU-9 together in one setting. This linkage was also performed for the patient belonging to family KU-2 separately. The reference genome annotation used was the Human Genome Build hg19 (UCSC genome browser) (Al-Mutairi et al., 2022; Al-Mutairi et al., 2023).
Primers for variant confirmation and segregation analyses were designed using the Primer3 software (http://frodo.wi.mit.edu/primer3/). Exon sequences were obtained from Genome Browser of the University of California, Santa Cruz (http://genome.ucsc.edu/) and RefSNP Report—dbSNP—NCBI (nih.gov) for all exons of DNAH5 as follows. For the first heterozygous variant rs761622153 in family KU-2 exon 34, the forward primer sequence used was 5′-AATGAGAAACGTGACTTCAACA-3´ and reverse primer sequence used was 5′-ACTGGGTAAATGCAGATAGTGTC-3´. For the second heterozygous variant in family KU-2 exon 72, the forward primer sequence used was 5′-TTTGACCTGGATTAATTTAAAATGG-3´ and reverse primer sequence used was 5′-CACAAAAGAGGACCATCACC-3´. For the first homozygous variant in family KU-5 exon 73, the forward primer sequence used was 5′-TCAGGGATTTACAAAAATGATTACT-3´ and reverse primer sequence used was 5′-TACGCCAAAGCTAGGAGGTC-3´. For the second homozygous variant in family KU-5 exon 75, the forward primer sequence used was 5′-TGAGTTTGTAGCTTGACACCAA-3´ and reverse primer sequence used was 5′-TCTCCCAACCGAAGATATGC-3´. For the first heterozygous variant in family KU-9 exon 10, the forward primer sequence used was 5′-ATCAAAGGGTTATTGGTGGAT-3´ and reverse primer sequence used was 5′-TCCCAAGGAGGGTACAAAAA-3´. For the second heterozygous variant in family KU-9 exon 77, the forward primer sequence used was 5′-AAACACTTACGCCAGAATCCA-3´ and reverse primer sequence used was 5′-AGTCGAGCTAGCAGCCTGAG-3´. The Sanger sequencing protocol was performed as described previously (Al-Mutairi et al., 2022; Al-Mutairi et al., 2023), and the chromatograms were analysed using the GeneScreen software (Carr et al., 2011a; Al-Mutairi et al., 2022; Al-Mutairi et al., 2024). The allele frequencies of the two homozygous missense variants of DNAH5 in the Arab population were estimated using the allelic discrimination test described previously (Al-Mutairi et al., 2022; Al-Mutairi et al., 2023).
In silico analyses of the two novel pathogenic variants in family KU-5
The algorithms used to estimate the pathogenicity of the two missense variants, DNAH5:c.12614G>A and DNAH5:c.12947T>C, in Tables 1, 2 are derived from the extended information in the MutPred database, which estimates the pathogenicities of the identified variants based on the protein sequences and model changes with regard to the structural features and functional sites between the wild-type and mutant sequences (Li et al., 2009). All pathogenicity scores mentioned in Table 3 are from the dbNSFP v4.1a database, which is a human database of aggregated scores from a large number of precalculated scores and algorithms that have also been augmented with custom rank scores to improve comparability. The dbNSFP v4.1a database focuses on non-synonymous single-nucleotide variants (nsSNVs) (Liu et al., 2020).
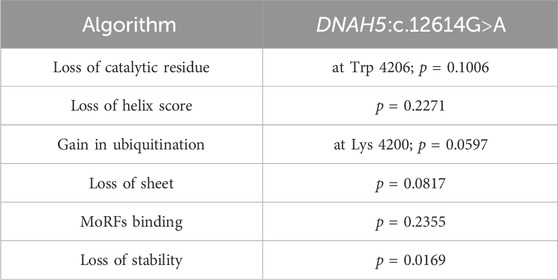
Table 1. Summaries of the in silico analyses for DNAH5:c.12614G>A showing the prediction tools with the most significant pathogenicity scores.
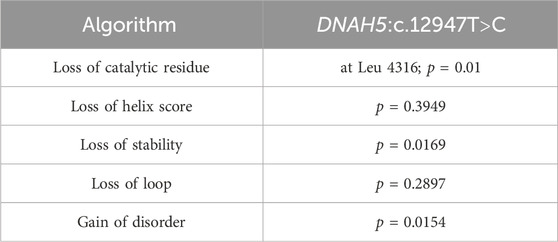
Table 2. Summaries of the in silico analyses for DNAH5:c.12947T>C showing the prediction tools with the most significant pathogenicity scores.
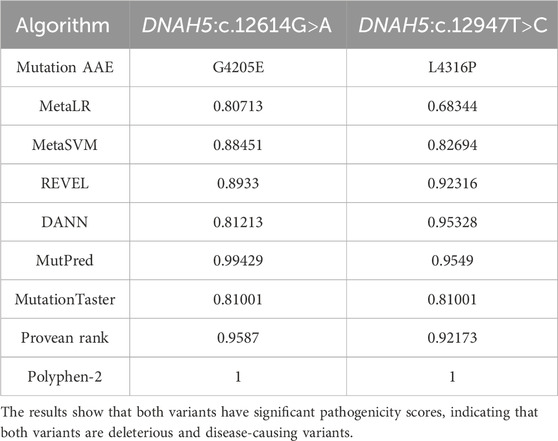
Table 3. Summaries of the in silico analyses using nine meta-tools available in the dbNSFP database for the two newly identified missense variants DNAH5:c.12614G>A and DNAH5:c.12947T>C.
IF analyses of the nasal biopsies
Respiratory epithelial cells were obtained from the subjects by nasal brush biopsy and suspended in a cell culture medium. Samples were then spread onto glass slides, air dried, and stored at −80°C until use. The respiratory epithelial cells from the healthy control and five PCD-affected individuals highlighted in the pedigrees (Figures 1A, 3A, 6A) were dual labelled for IF analysis with antibodies against axonemal components (visualised with red-fluorescent secondary antibody) and antibodies to acetylated anti-tubulin (visualised with green-fluorescent secondary antibody) as the ciliary markers. The nuclei were stained with Hoechst 33342 (blue fluorescent) (Wallmeier et al., 2014; Al-Mutairi et al., 2023). The IF images were acquired with a Zeiss LSM 800 confocal microscope and processed using ZEN and ImageJ software (Al-Mutairi et al., 2022; Al-Mutairi et al., 2023).
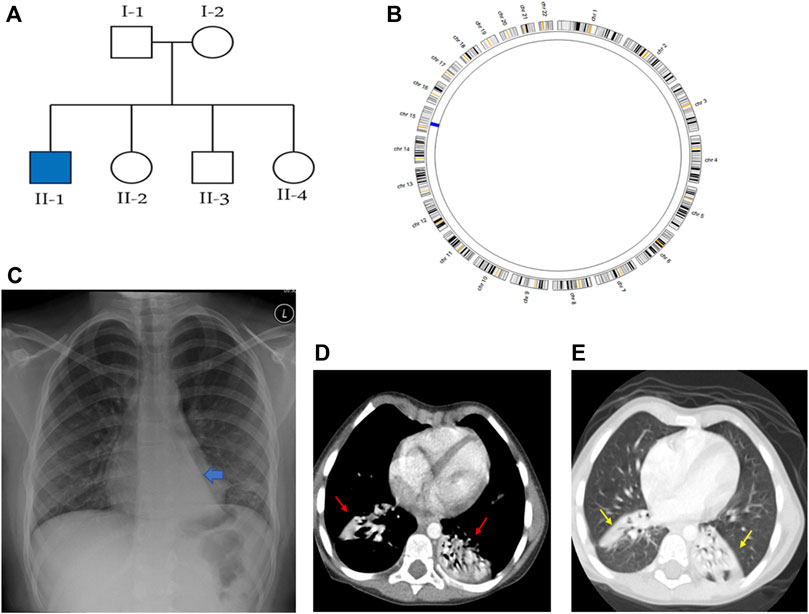
Figure 1. Summaries of the genetic and radiological data of family KU-2, where (A) shows the pedigree of family KU-2 and (B) shows the AgileMultiIdeogram for the KU-2.II.1 patient indicating that there was a single region of homozygosity (ROH) at chromosome 15. (C) Shows the chest X-ray for the KU-2.II.1 patient demonstrating a triangular retrocardiac opacity with a ‘flat-waist sign’ suggesting left lower lobe collapse. (D, E) Selected axial sections of the CT chest study of the same patient in the soft-tissue and lung windows showing subsegmental right lower lobe collapse in addition to the left lower lobe collapse (arrows).
The panel of primary antibodies used for the initial screening included mouse monoclonal anti-tubulin (acetylated) antibody (Sigma Aldrich, T6793) for staining the ciliary microtubules and rabbit polyclonal antibody anti-DNAH5 (1:300) (HPA037470, Atlas Antibodies). The secondary antibodies used were highly cross absorbed, including Alexa Fluor 488-conjugated goat antibodies to mouse (1:1,000) (A11029, Molecular Probes, Invitrogen) and Alexa Fluor 546-conjugated goat antibodies to rabbit (1:1,000) (A11035, Molecular Probes, Invitrogen) (Al-Mutairi et al., 2022; Al-Mutairi et al., 2023).
TEM analyses of the nasal biopsies
For the TEM analyses, nasal biopsies were obtained from the middle turbinate for the same five affected individuals. The ciliated cells were first fixed with glutaraldehyde (2.5%) in Sorensen’s phosphate buffer (pH 7.4), following which the samples were treated with osmium tetroxide (1.3%), dehydrated using graded ethanol series, and immersed in hexamethyldisilazane as the chemical drying reagent. The samples were next embedded overnight in 1,2-epoxypropan-epon- mixture (1:1) at 4°C. After polymerisation, several sections were retrieved onto copper grids and were stained with Reynold’s lead citrate. TEM was then performed using the Philips CM10 device (Wallmeier et al., 2014; Al-Mutairi et al., 2022; Al-Mutairi et al., 2023).
Results
We present the genetic and functional analyses for the five PCD individuals belonging to three families KU-2, KU-5, and KU-9 who were independently diagnosed with PCD from a cohort of 105 PCD individuals recruited from different hospitals in Kuwait. According to the medical reports of the patients, the paediatric pulmonologists performed differential diagnoses for all five patients and ruled out other conditions, such as cystic fibrosis, asthma, immunodeficiencies, and anatomic anomalies. These patients showed similar signs and symptoms as other healthy children, except for recurrent chest infections. The five patients were hospitalised during infancy for recurrent mild respiratory distress and required oxygen administration. None of the patients in this study required surgical intervention for bronchiectasis.
Clinical and genetic findings for the KU-2.II.1 patient
The KU-2 family had one affected male child KU-2.II.1 who met the inclusion criteria for PCD symptoms with situs solitus (Figure 1A); the child underwent spirometry testing at various ages and showed declining pulmonary functions with both obstructive and restrictive patterns at the age of 18 years. His diagnosis of PCD was confirmed by genetic and in vitro functional analyses of the airway cilia. The selected chest X-ray for KU-2.II.1 shows a triangular retrocardiac opacity with ‘flat-waist sign’ suggesting left lower lobe collapse (Figure 1C). Selected axial sections of the computed tomography (CT) chest study for this patient from the soft-tissue and lung windows show subsegmental right lower lobe collapse in addition to the left lower lobe collapse (arrows in Figures 1D, E).
Exome data reveals novel deleterious heterozygous variant of DNAH5 in family KU-2
Autozygosity mapping for KU-2.II.1 revealed a single region of homozygosity (ROH) summation at chromosome 15, as shown in Figure 1B. Filtering the variants in the exome data across this interval did not show any pathogenic variants; furthermore, since the parents are not related, this indicates that the PCD-causing variants are more likely inherited in compound heterozygous form. The analysis of exome data for deleterious variants in the known 53 PCD genes revealed that the patient carried two deleterious variants of DNAH5 inherited in the compound heterozygous fashion. The first heterozygous variant was a heterozygous non-sense mutation NM_001369.2(DNAH5):c.5503C>T; (p.Gln 1835*); rs761622153 in exon 34 from paternal inheritance; RS = 5.8 and RVIS = 0.18 indicate that this is significantly damaging. This was recently reported as a PCD-causing variant inherited in a consanguineous family from United Arab Emirates (Alsamri et al., 2021). The second heterozygous variant was a novel frameshift deletion NM_001369.2(DNAH5):c.12357_12358delTG; (p.Glu4120Alafs*4) resulting in premature termination of translation from maternal inheritance. Segregation analysis confirmed that the two heterozygous variants of DNAH5 were segregated with the disease phenotype, consistent with both being the PCD-causing variants in the KU-2.II.1 patient (Figures 2A–C).
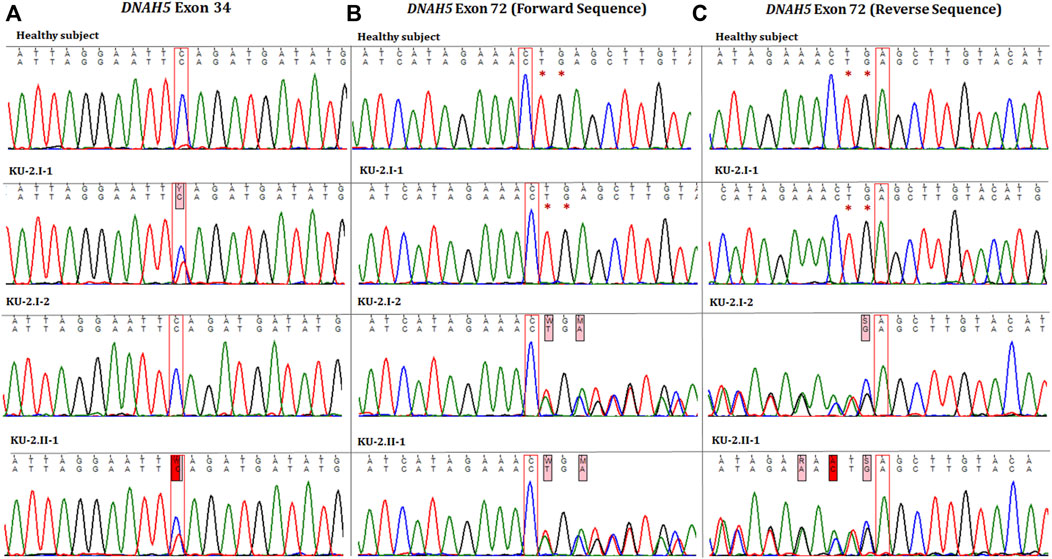
Figure 2. Chromatographs of the DNAH5 variants for the members of family KU-2. (A) Represents the sequences of the healthy subject, parents (KU-2.I.1 and KU-2.I.2), and KU-2.II.1 patient, respectively. The KU-2.II.1 patient carried a heterozygous loss of function (LOF) variant (c.5503C>T; p.Gln 1835*; rs761622153) in exon 34 of DNAH5 that predicted premature termination of translation. Segregation analysis demonstrated that the rs761622153 variant had a paternal inheritance. (B, C) Represent the forward and reverse sequences for the healthy subject, parents (KU-2.I.1 and KU-2.I.2), and KU-2.II.1 patient, respectively. The KU-2.II.1 patient carried a heterozygous novel LOF TG deletion mutation (c.12357_12358delTG; p.Glu4120Alafs*4) in exon 72 of DNAH5 that also predicted premature termination of translation. Segregation analysis demonstrated that the variant (c.12357_12358delTG) had a maternal inheritance. This set of data indicates that the KU-2.II.1 patient has PCD due to inheritance of two LOF heterozygous pathogenic variants (non-sense and truncated).
Clinical and genetic findings in the two PCD siblings of family KU-5
The KU-5 family was a multiplex family with two affected females, KU-5.IV.1 and KU-5.IV.3, both of whom presented with the following symptoms: chronic productive cough, recurrent pneumonias, wheezing dyspnea, and constant nasal mucopurulent secretions with situs inverses totalis (Figure 3A). Confirmed diagnoses of PCD were made at the ages of 7 and 5 years, with both subjects having situs inversus totalis. According to the KU-5.IV.1 patient’s questionnaire, the symptoms became gradually progressive with age, and the results of spirometry tests showed that the patient had declining pulmonary function, with both obstructive and restrictive patterns at the age of 12 years. The radiological findings for the two affected siblings (KU-5.IV.1 and KU-5.IV.3) can be summarised as follows: chest X-rays demonstrate right-sided heart apex (arrow) and gastric air bubbles (arrowhead), consistent with situs inversus totalis (Figures 4A, B). In addition, KU-5.IV.1 had mild diffuse bronchiectatic changes that were visible in the medial aspects of both lower zones, as highlighted by the thin yellow arrows (Figure 4A). The main radiological findings from the high-resolution CT (HRCT) chest examination of KU-5.IV.1 were situs inversus and subsegmental atelectasis in both lungs with mild underlying bronchiectatic changes (arrows in Figures 4C, D). Furthermore, selected axial and coronal reconstruction images of the CT of the paranasal sinus (CT-PNS) revealed complete opacification of the bilateral paranasal sinuses, consistent with chronic inflammatory pansinusitis (highlighted by arrows in Figures 4E, F).
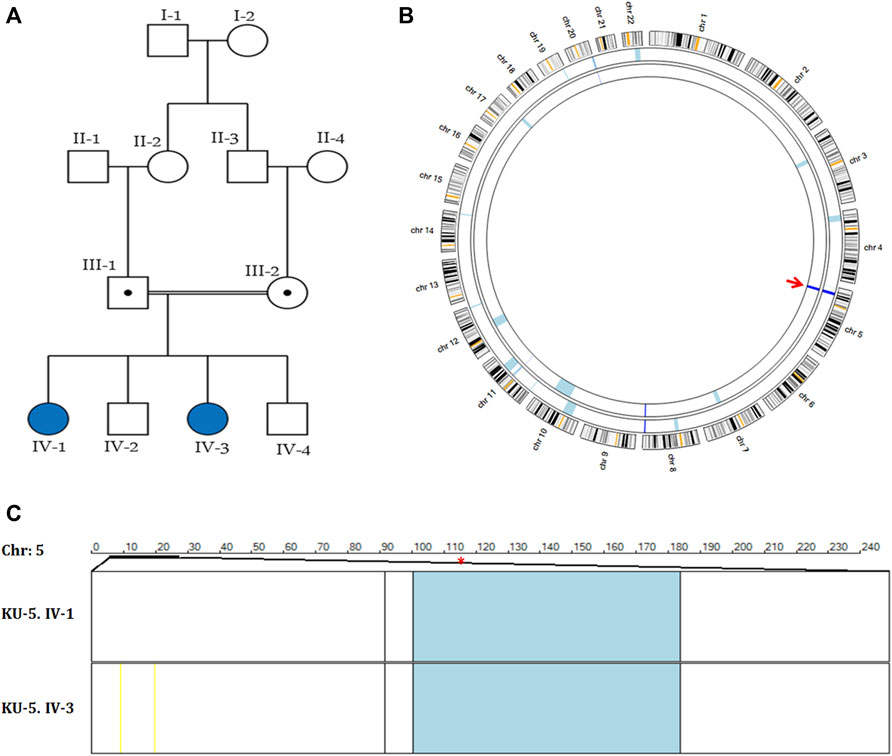
Figure 3. Genetic linkage data of family KU-5. (A) Pedigree of family KU-5 showing that the family has two affected females. (B) AgileMultiIdeogram showing the exclusive ROHs for KU-5.IV.1 and KU-5.IV.3 PCD individuals. The two PCD individuals had shared regions of autozygosity (dark blue) across the DNAH5 locus on chromosome 5, as indicated by the red arrow. (C) Multiple linkage analysis at chromosome 5 for the two affected siblings using AgileVCFMapper software. The shared IBD interval at chromosome 5 was estimated to be (chr5:11,427,782-21,860,252), with a length of almost 10.5 kB. The physical location of DNAH5 is at chr5:13,690,328-13,944,688 (within the shared IBD interval) and is highlighted by the red asterisk. As seen, each row represents the genotypes for chromosome 5 for one affected individual. The homozygous discordant alleles are represented as yellow lines, concordant autozygous alleles are represented as blue lines, and rare concordant autozygous alleles are represented as black lines.
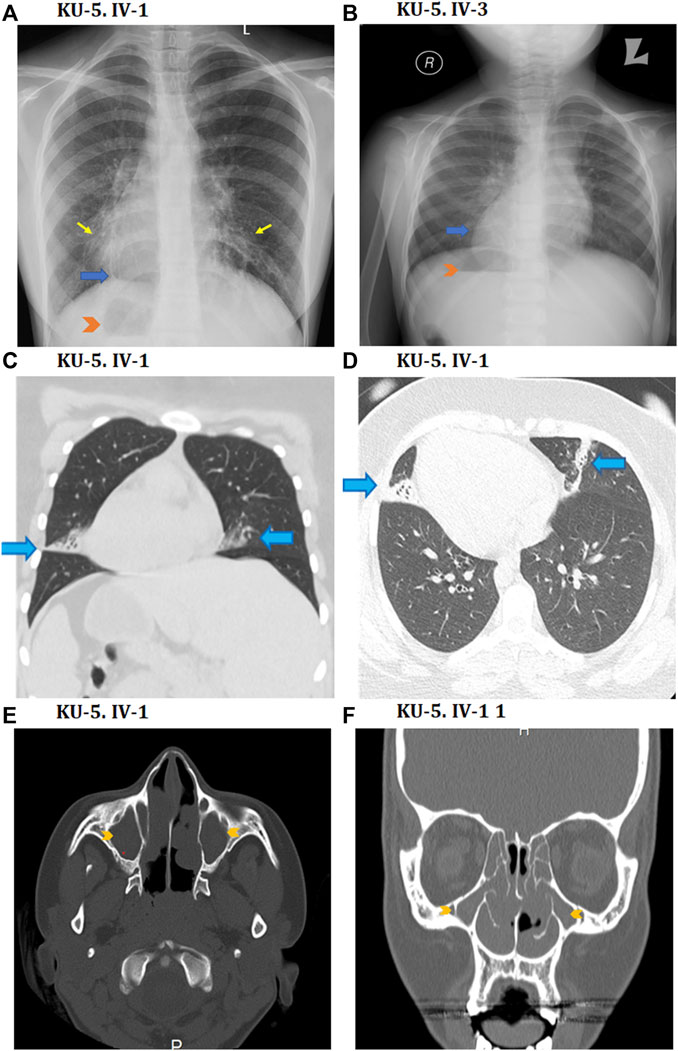
Figure 4. Radiological findings for the two PCD individuals of family KU-5. (A, B) Show chest X-rays for the KU-5.IV.1 and KU-5.IV.3 patients demonstrating right-sided heart apices (blue arrows) and gastric air bubbles (arrowheads) in conformity with situs inversus totalis. In addition, (A) shows mild diffuse bronchiectatic changes in the medial aspects of both lower zones (yellow arrows) for the KU-5.IV.1 patient. Selected coronal CT image in the lung window (C) as well as axial image of the lung window (D) from the CT chest study of a patient reveal situs inversus and subsegmental atelectasis in both lungs with mild underlying bronchiectatic changes (arrows). Selected axial (E) and coronal (F) reconstruction images of the CT-PNS study on the KU-5.IV.1 patient reveal complete opacification of the bilateral paranasal sinuses (arrowheads) in conformity with chronic inflammatory pansinusitis.
Autozygosity mapping reveals two novel homozygous variants of DNAH5 in family KU-5
Autozygosity mappings for the two PCD individuals of family KU-5 showed a single shared interval that was identical by descent (IBD) at chromosome 5, as seen in Figure 3B. The estimated shared homozygous intervals were precisely determined using AgileVCFMapper software (chr5:11,427,782-21,860,252) and found to be almost 10.5 kB (Figure 3C). The DNAH5 location at the shared IBD interval (chr5:13,690,328-13,944,688) is highlighted with a red asterisk in Figure 3C. The exome data for the two affected siblings were filtered at the shared IBD interval at chromosome 5 for screening of the deleterious variants of DNAH5.
Two novel homozygous missense variants of DNAH5 were identified in both patients by whole exome sequencing. The first homozygous missense variant NM_001369.2(DNAH5):c.12614G>A; (p.Gly4205Glu) was identified in exon 73, as shown in Figure 5A. The second homozygous missense mutation NM_001369.2(DNAH5):c.12947T>C; (p.Leu4316Pro) was identified in exon 75, as shown in Figure 5B. Both variants have no RefSNPs (rs) numbers to date in either Ensembl or the UCSC genome browsers. Following linkage and sequencing analyses, segregation analysis was performed to confirm that both variants identified in DNAH5 were segregated with the disease phenotype. The results of segregation analyses confirmed that the parents, KU-5.III.1 and KU-5.III-2, were heterozygous carriers for both missense variants (Figures 5A, B). This implies that either of these variants could be responsible for PCD in family KU-5.
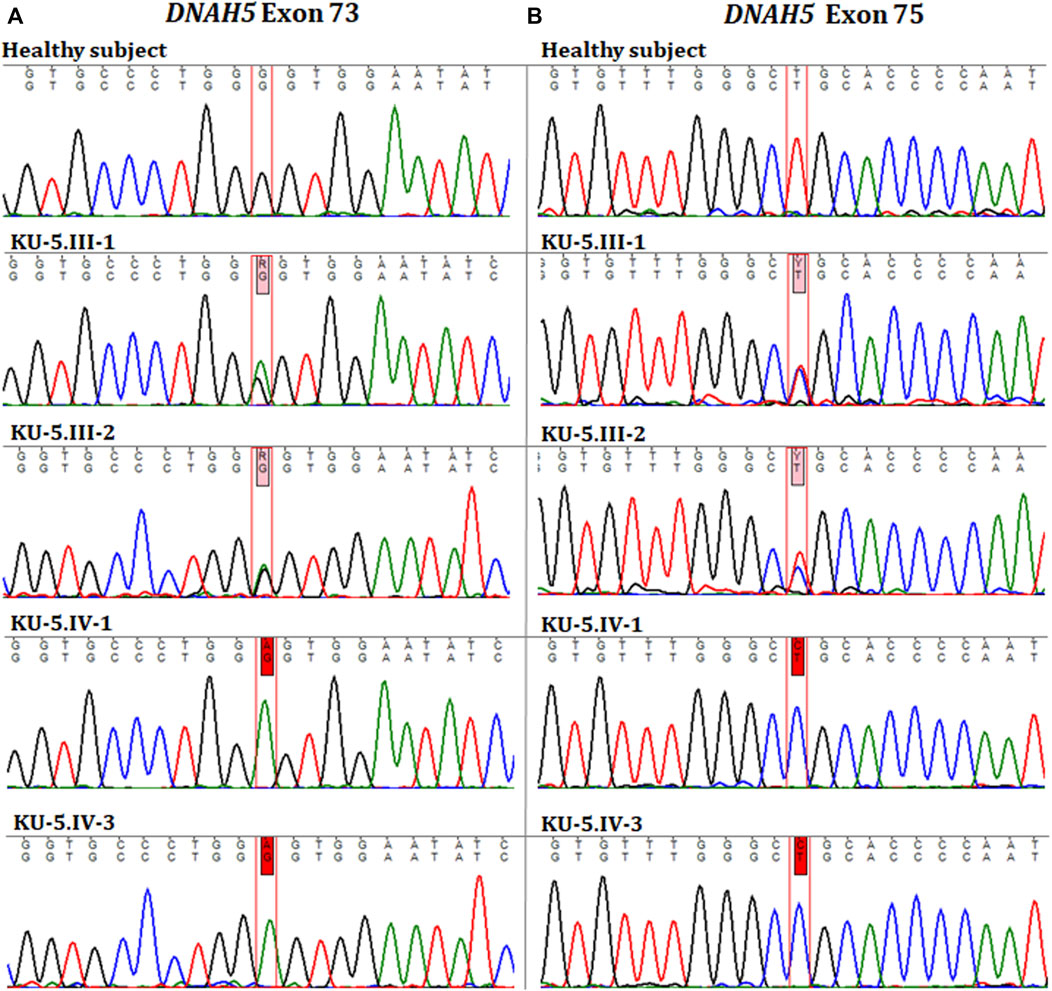
Figure 5. Chromatographs of the DNAH5 variants in the members of family KU-5. (A) Represents the sequences for the healthy subject, parents (KU-5.III.1 and KU-5.III.2), and two PCD individuals (KU-5.IV.1 and KU-5.IV.3), respectively. The two PCD individuals inherited a novel homozygous missense variant in DNAH5 (c.12614G>A; p.Gly4205Glu) located within exon 73 that results in a change of the amino acid glycine to glutamic acid. This variant has no RS number to date. The parents KU-5.III.1 and KU-5.III.2 were both carriers for this variant, consistent with it being a segregating and PCD-causing variant. (B) Represents the sequences for the healthy subject, parents (KU-5.III.1 and KU-5.III.2), and two PCD individuals (KU-5.IV.1 and KU-5.IV.3), respectively. Interestingly, the two PCD individuals inherited a second novel homozygous missense variant in DNAH5 (c.12947T>C; p.Leu4316Pro) located within exon 75, which results in leucine to proline change. The second variant also has no RS number to date. The parents KU-5.III.1 and KU-5.III.2 were both carriers for the second variant, consistent with it being a segregating and PCD-causing variant. Genetic screening suggests that the two novel homozygous missense variants are responsible for developing PCD with situs inversus in family KU-5.
The two variants were assessed using different algorithms and statistical measurements to predict their pathogenicity. The first of these was rejected substitutions (RSs), defined as the number of substitutions expected under a neutral model minus the number ‘observed’ (estimated) for a particular alignment (Pazour et al., 2006; Al-Mutairi et al., 2022; Al-Mutairi et al., 2024); the second was polymorphism phenotyping (PolyPhen), which is a tool to predict the possible impacts of an amino acid substitution on the structure and function of a human protein (Adzhubei et al., 2013; Al-Mutairi et al., 2022; Al-Mutairi et al., 2024). The missense variant DNAH5:c.12614G>A resulting in an amino acid exchange (p.Gly4205Glu) had RS = 5.2, PPH = 3, MedPred = 81, and Varsome: likely pathogenic, all of which indicate that the variant is highly damaging. The missense variant DNAH5:c.12947T>C resulting in an amino acid exchange (p.Leu4316Pro) had RS = 4.6, PPH = 3, and MedPred = 83, all of which indicate that the second variant is also highly damaging. Together, these data for family KU-5 are unique and interesting since this is the only family in the study cohort that has two novel homozygous missense variants in DNAH5 that are not reported previously in any database.
We extended our in silico analyses to predict the pathogenicities of the DNAH5:c.12614G>A and DNAH5:c.12947T>C variants. Our findings reveal noteworthy scores using mutation prediction tools designed to assess the pathogenicity of the identified mutations, as outlined in Tables 1–3. First, the loss of the catalytic residue is critical for ciliary targeting, and the loss of integrity of the catalytic domain in multi-ciliated respiratory cell mutants is directly related to ciliary mistargeting. This involves the targeting of cargo-containing membrane carriers or vesicles to the periciliary membrane (Lu and Madugula, 2018; Al-Mutairi et al., 2022; Cilleros-Rodriguez et al., 2022; Al-Mutairi et al., 2024). Loss of catalytic residues for the two missense variants were significantly low and were considered pathogenic (Tables 1, 2). In addition, the two variants DNAH5:c.12614G>A and DNAH5:c.12947T>C had significant loss of helix scores, indicating that both variants were highly damaging (Romero Romero et al., 2018). The missense variant DNAH5:c.12614G>A had significant scores for gain in ubiquitination at Lys 4200, loss of sheet, and loss of molecular recognition feature (MoRF) binding, all of which indicate that the variant is highly pathogenic (Table 1).
Furthermore, the missense variant DNAH5:c.12947T>C causes severe loss of the catalytic residue at Leu 4316, which is also directly associated with cilia mistargeting. The DNAH5:c.12947T>C variant leads to significant loss of stability, which is a common and widely used predictive tool for estimating the pathogenicity of missense mutations and disease development. The predicted pathogenicity from single amino acid changes affecting the thermodynamic stabilities of proteins highlights the role of protein stability in genetic diseases (Blaabjerg et al., 2023; Al-Mutairi et al., 2024). Furthermore, the DNAH5:c.12947T>C variant has a significant loss of loop score, which estimates the pathogenicity of the missense variant on a phosphate-binding loop (P-loop) motif that is necessary for ATP binding (Romero Romero et al., 2018; Al-Mutairi et al., 2024). It is well known that axonemal dyneins are directly involved in driving ciliary movements by providing the sliding forces for the microtubules within the cilia, which require energy from ATP hydrolysis within the heavy chain domains of axonemal dyneins (Mitchison and Mitchison, 2010). The DNAH5:c.12947T>C variant has a significant gain of disorder score supporting the previous pathogenic value of this amino acid substitution (Table 2).
In addition, we utilised nine meta-tools available in the dbNSFP database for annotation and functional predictions of all potential nsSNVs in the human genome (Liu et al., 2011; Kopanos et al., 2019; Liu et al., 2020; Al-Mutairi et al., 2022; Al-Mutairi et al., 2024). All of these determine the pathogenicity based on combined evidence from several other in silico predictors, and all of them predict both DNAH5:c.12614G>A and DNAH5:c.12947T>C to be deleterious/pathogenic and disease-causing missense variants, as seen in Table 3. Thus, the in silico analyses indicate that each variant is potentially highly pathogenic and PCD-causing, i.e. the presence of each variant alone may be sufficient for the development of PCD.
According to the ACMG criteria, the missense variant DNAH5:c.12614G>A has been annotated as likely pathogenic and missense variant DNAH5:c.12947T>C has been annotated to have uncertain significance despite their high pathogenicity prediction scores. Subsequently, allelic discrimination assays were performed on the two homozygous missense DNAH5 variants using RT-PCR to estimate their carrier rate frequencies (DNAH5:c.12614G>A in Supplementary Figure S1 and DNAH5:c.12947T>C in Supplementary Figure S2) using a control panel composed of 100 DNA samples from normal individuals belonging to different Arabian tribes. As shown in Supplementary Figure S1, the genotype for the two PCD individuals is homozygous for the missense variant (A), and their parents are all heterozygous carriers for the wild-type allele (G) and defective allele (A), consistent with the Sanger sequencing data. Remarkably, the genotypes for the Arab control DNA panel show that all healthy individuals have the two wild-type alleles (G). Regarding the second variant, the genotype for the two PCD individuals is also homozygous for the second missense variant (C), and their parents are heterozygous carriers for the wild-type allele (T) and defective allele (C), which is again consistent with the Sanger sequencing data (Supplementary Figure S2).
Clinical and genetic findings in the two PCD siblings of family KU-9
The KU-9 family is a multiplex family with two affected siblings KU-9.II.1 and KU-9.II.2 (Figure 6A) who met the inclusion criteria for PCD with situs solitus but showed only obstructive spirometry patterns. The male patient KU-9.II.1 was a smoker, so it is difficult to predict whether the pulmonary dysfunction was due to PCD or smoking; the patient was also substerile and had conceived two children by IVF. The female patient KU-9.II.2 had typical PCD symptoms, and her physician referred her to a genetic clinic for a confirmed diagnosis; she also experienced fertility problems and conceived a child by IVF after 5 years of marriage. The confirmed diagnoses of PCD were made through genetic and ciliary functional analyses of the two affected siblings KU-9.II.1 and KU-9.II.2 at the ages of 32 and 27 years, respectively. The main radiological findings from the HRCT chest investigation for KU-9.II.2 demonstrate situs solitus morphology with moderate cystic bronchiectatic changes involving the lateral segment of the right middle lobe. No other abnormalities or signs of active infection were observed (Figures 6C–E).
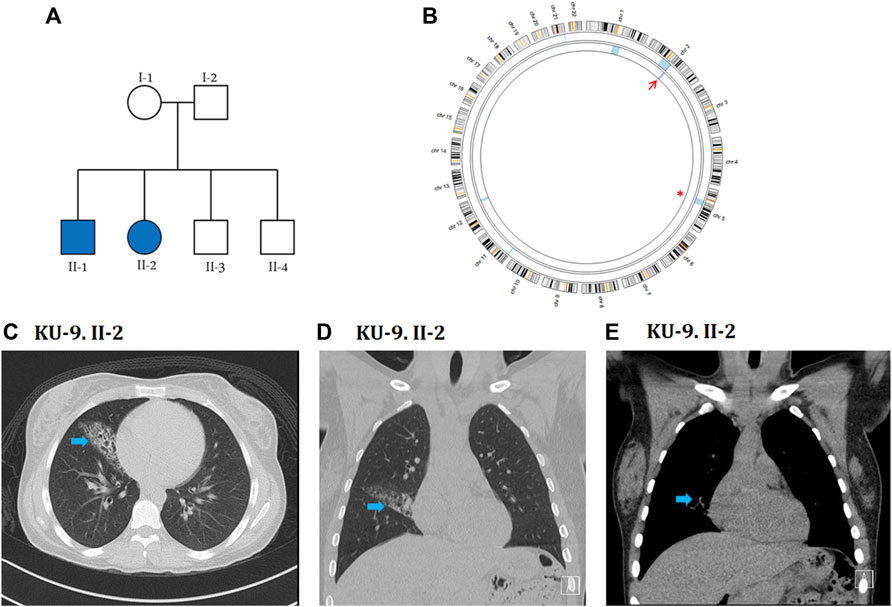
Figure 6. Genetic linkage and radiological data of family KU-9. (A) Shows the pedigree of family KU-9 that has two affected siblings. (B) AgileMultiIdeogram showing the exclusive ROH for the KU-9.II.2 patient (light blue) across the DNAH5 locus at chromosome 5, highlighted by a red asterisk. There is a detectable shared IBD in chromosome 2 for the two siblings KU-9.II.2 and KU-9.II.1, as highlighted by the red arrow. Radiological data for the KU-9.II.2 individual are summarised in (C–E); selected axial (C) and coronal (D, E) images of a HRCT study demonstrate situs solitus morphology with a wedge-shaped area of chronic inflammatory changes with bronchiectasis in the lateral segment of the right middle lobe (arrow). No other abnormalities or signs of active infection were noted. These abnormalities were stable in comparison to multiple previous studies over the years.
Exome data reveals novel deleterious heterozygous variants of DNAH5 in family KU-9
Although family KU-9 is a multiplex family with two affected siblings, the parents were not close relatives; it is very rarely the case that a family with two affected individuals has unrelated parents. However, exome sequencing of the two siblings indicated that the family was highly inbred, with the ROH summation being 220 and 117 for KU-9.II.1 and KU-9.II.2, respectively. The ROH summation cutoff value for inbreeding is 100, so the individuals theoretically belong to a highly inbred family. It is worth noting that family KU-9 belongs to a tribe that has practised consanguineous marriages among members of the tribe for generations and is considered to be particularly isolated, i.e. they have never married outside the tribe for historical and/or cultural reasons. Linkage analysis for the two affected siblings showed multiple ROH segments consistent with the ROH scores. However, none of these were shared between the two affected siblings, except for a minute interval at chromosome 2 (Figure 6B). Screening of this interval does not show any pathogenic variants. Subsequently, two interesting ROH intervals at chromosome 1 for patient KU-9.II.1 and chromosome 5 for patient KU-9.II.2 were prioritised for screening of the pathogenic variants. The ROH highlighted with a red asterisk (Figure 6B) is across the DNAH5 locus at chromosome 5 for patient KU-9.II.2. The other ROH interval at chromosome 1 did not harbour any pathogenic variants. Interestingly, the exome data for the two affected siblings showed that both had two pathogenic heterozygous variants of DNAH5. Theoretically, autozygosity mapping can only map an IBD interval harbouring homozygous variants, and since we mapped the compound heterozygous variants in DNAH5 in the two patients of family KU-9, this might explain the reason for not detecting the shared IBD using exome data across the DNAH5 locus in the two affected siblings. The set of genetic data for family KU-9 is also unique and interesting since this multiplex family has two affected individuals who have inherited two novel compound heterozygous variants of DNAH5 causing PCD with situs solitus, which has not been reported previously.
Exome and Sanger sequencings show that the two PCD individuals shared compound heterozygous variants; they were both carriers for a missense variant leading to amino acid exchange in exon 10 of DNAH5, i.e. NM_001369.2(DNAH5):c.1206 T>A; (p.Asn402Lys); rs140782270 (Figure 7A). The allele frequency of T was 100% for all ethnic backgrounds in the Ensembl genome browser with uncertain significance. They were also carriers for a second non-sense heterozygous variant in exon 77 of DNAH5, namely, NM_001369.2(DNAH5):c.13465C>T; (p. Gln4489*), that led to premature termination of translation and the variant having no RS number till date (Figure 7B). Segregation analyses for the two heterozygous variants showed that the heterozygous missense variant c.1206 T>A had a paternal inheritance. The heterozygous non-sense variant c.13465C>T had a maternal inheritance. This confirms that the presence of two heterozygous variants in the two PCD siblings of family KU-9 is most likely responsible for developing the PCD phenotype in the compound heterozygous mode of inheritance (Figures 7A, B). In conclusion, family KU-9 is the first multiplex family in which we failed to detect a shared IBD interval between the two affected individuals; nevertheless, both PCD individuals harboured the same heterozygous pathogenic variants. In conclusion, the variants mapped for DNAH5 were pathogenic and associated with PCD. We also summarise the variants in the three Arabic families from Kuwait in Table 4.
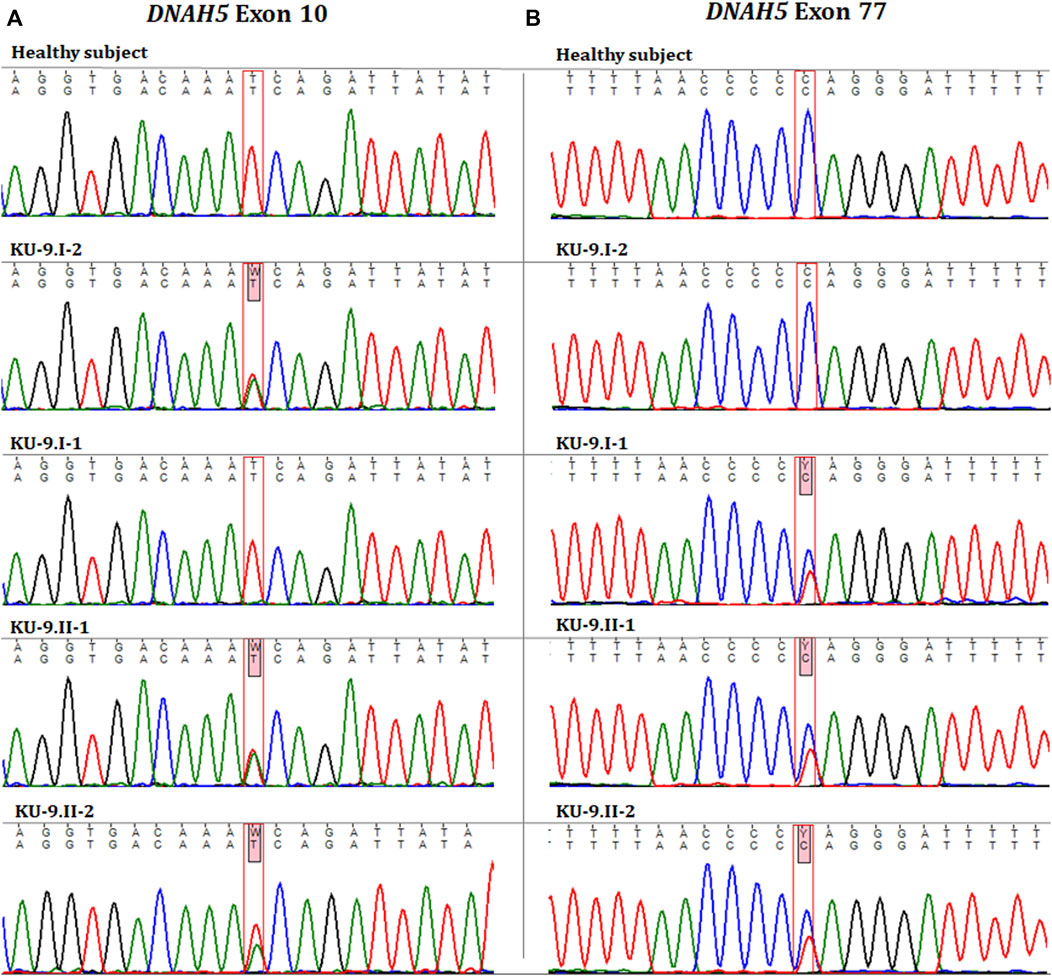
Figure 7. Chromatographs for the DNAH5 variants in the members of family KU-9. Sequences for the healthy subject, parents (KU-9.I.1 and KU-9.I.2), and two PCD individuals (KU-9.II.1 and KU-9.II.2) are shown. The two PCD individuals inherited two compound heterozygous variants of DNAH5. (A) Shows that both PCD individuals were carriers for a missense variant leading to amino acid exchange (c.1206 T>A; p.Asn402Lys; rs140782270) in exon 10 of DNAH5 that had a paternal inheritance. (B) Shows that both PCD individuals were carriers for a non-sense variant (c.13465C>T; p. Gln4489*) in exon 77 of DNAH5 that leads to premature termination of translation and had a maternal inheritance. These data suggest that the two PCD individuals in family KU-9 had PCD due to inheritance of two heterozygous (missense and non-sense) pathogenic variants.
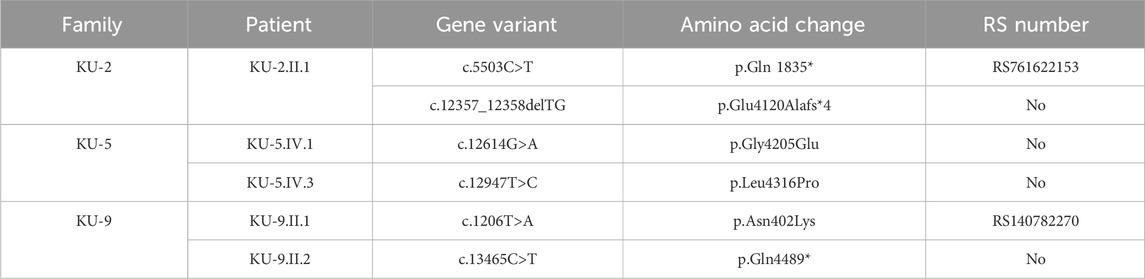
Table 4. Summary of the DNAH5 variants reported in this study in the three Arabic families in Kuwait.
Detection of ultrastructural defects of the motile cilia in the pulmonary system
IF and TEM analyses were performed in this study on the nasal biopsies derived from five PCD individuals with different variants of DNAH5 to identify the ultrastructural defects of the axoneme of the multi-ciliated respiratory epithelial cells. IF analyses of the primary airway epithelial cells using monoclonal antibodies showed staining for acetylated α-tubulin, which acts as an axonemal marker protein, in the five PCD individuals and in the healthy control, as seen in panel (a) of Figure 8. However, IF analysis using polyclonal antibodies directed against the ODA heavy chain DNAH5 showed complete lack of staining due to the pathogenic variants of DNAH5, resulting in loss of DNAH5 from the ciliary axoneme, as demonstrated by the absence of the DNAH5 signal in panel (b) of Figure 8 as well as absence of yellow co-staining in panel (c) of Figure 8 compared to the healthy subject who showed DNAH5 staining across the ciliary axoneme. In addition, TEM analyses confirmed the lack of ODAs (Figure 9), consistent with previously published reports (Olbrich et al., 2002; Hornef et al., 2006; Al-Mutairi et al., 2022). These analyses demonstrated that DNAH5 was absent from the ciliary axonemes of the affected individuals, thus supporting our genetic findings.
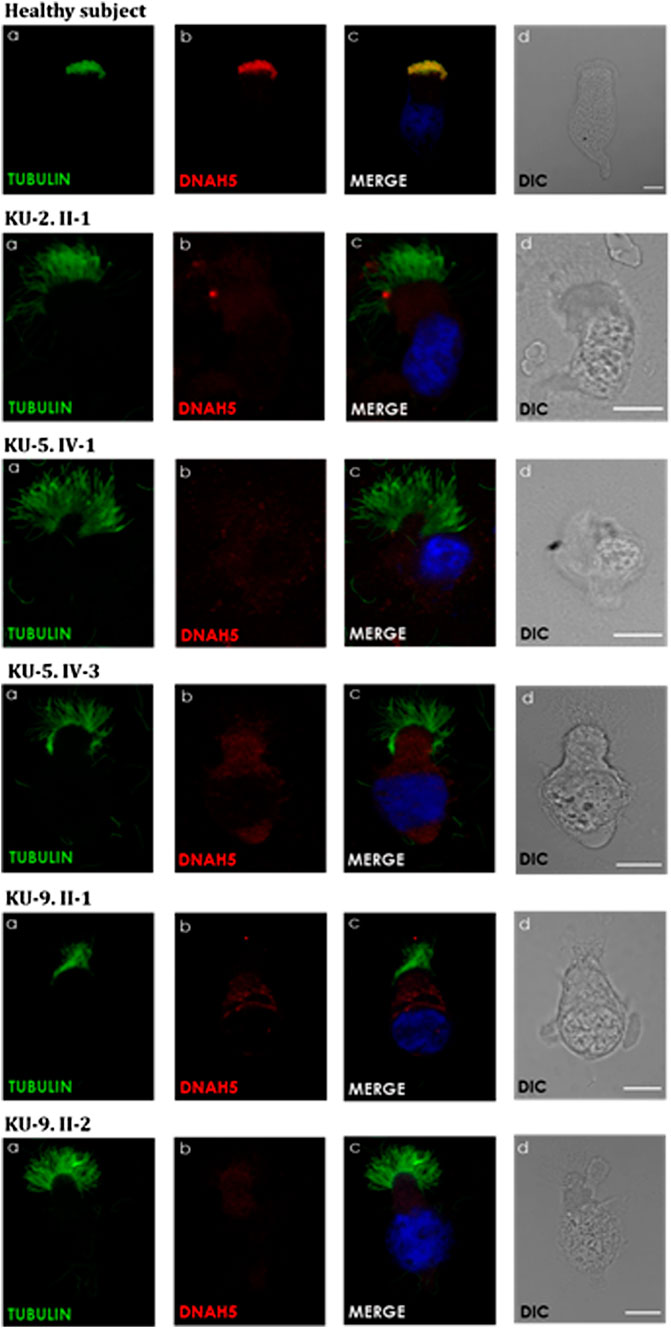
Figure 8. Immunofluorescence (IF) images of the primary airway epithelial cells of the PCD subjects. IF was performed using monoclonal anti-acetylated α-tubulin (a) and polyclonal anti-DNAH5 (b) antibodies for the five PCD individuals versus healthy controls. As seen in the controls, the merged images (c) show yellow co-staining within the ciliary axonemes, indicating that both proteins were co-localised within the airway cilia, compared to the images of the PCD individuals that show the absence of anti-DNAH5 staining. (d) shows differential interference contrast figures (DIC) for analyzed cells. Scale bar is 10 µm.

Figure 9. TEM sections of the ciliary axonemes for PCD individuals versus control. The TEM results for the ciliary axonemes of the ciliated epithelial cells of the respiratory system show that the PCD patients do not have ODAs as compared to the control, which is mainly seen in patients with defects in the genes encoding dynein arm components, consistent with their genotypes.
Discussion
Our study presents novel findings regarding genetic variants within DNAH5 inherited in both homozygous and compound heterozygous patterns, resulting in PCD with variable laterality defects. By employing an autozygosity mapping approach (Lander and Botstein, 1987; Abecasis et al., 2002; Carr et al., 2011b; Al-Mutairi et al., 2022; Al-Mutairi et al., 2023), we identified two novel homozygous missense variants within DNAH5 in two PCD individuals from family KU-5 as well as compound heterozygous variants of DNAH5 in PCD individuals from families KU-2 and KU-9, all of which were responsible for the development of PCD.
Previous studies on human DNAH5 have characterised it as a structural component of the ODAs that are responsible for the main dynamic motility forces of the ciliary beats (Fowkes and Mitchell, 1998; Hornef et al., 2006; Al-Mutairi et al., 2022). The main structural components of the ODAs and inner dynein arms (IDAs) are motor heterotrimeric proteins that are preassembled in the cytoplasm prior to transportation into and assembly in the ciliary axonemes (Fowkes and Mitchell, 1998; Al-Mutairi et al., 2022). Human respiratory cilia have two distinct ODA heavy chains (HCs): ODA type 1, which is the component of the proximal part of the axoneme consisting of γ-HC DNAH5 and β-HC DNAH11; ODA type 2, which is the component of the distal part of the axoneme consisting of γ-HC DNAH5 and β-HC DNAH9 (Hornef et al., 2006; Loges et al., 2018; Al-Mutairi et al., 2022).
IF studies of the respiratory cilia show that DNAH5 has subcellular localisation along the entire lengths of the ciliary axonemes in both ODA type 1 and type 2 complexes. In addition, previous IF analyses of the airway cilia from PCD individuals with biallelic pathogenic DNAH5 variants showed complete absence of not only DNAH5 but also DNAH11 (the ODA type 1 component) and DNAH9 (the ODA type 2 component). This shows that DNAH5 is required for accurate assembly of both type 1 and type 2 ODAs (Fliegauf et al., 2005; Schwabe et al., 2008; Pifferi et al., 2010; Knowles et al., 2012; Al-Mutairi et al., 2022).
Applying IF and TEM as two well-established in vitro functional analyses revealed the complete absence of the dynein arm protein component DNAH5 in all affected individuals in this study, indicating compromised axonemal assembly of both the proximal and distal localised ODA types 1 and 2 as well as lack of ODAs as the main ultrastructural defects in these individuals presenting with DNAH5 pathogenic variants. These data support our genetic findings that previously unpublished homozygous and compound heterozygous DNAH5 variants are the cause of PCD in these families and that the identified DNAH5 variants are pathogenic.
Conclusion
In previous reports, we described founder variants in RSPH9 in eleven individuals shared by several unrelated Arabic families in our Kuwaiti PCD cohort (Al-Mutairi et al., 2023); we also reported novel founder variants in DNAI2 in seven individuals (Al-Mutairi et al., 2022). Both of these are very rare PCD-causing genes in the global population. Herein, we identified the pathogenic variants of DNAH5 in only five PCD individuals in our cohort of 105 Arabic PCD individuals recruited from different hospitals in Kuwait. Thus, in contrast to other ethnicities that have an incidence of almost 50%, DNAH5 is not the most common PCD gene in the Kuwaiti population.
Summary
In this study, we present our findings on five individuals having PCD belonging to three Arabic families, two of which are multiplex consanguineous families with two affected siblings each. All five PCD individuals had a history of neonatal respiratory distress, lifelong symptoms of wet cough, rhinosinusitis, otitis media, and bronchiectasis. Genetic screening revealed that these individuals had different pathogenic variants of DNAH5 inherited in both homozygous and compound heterozygous forms. Molecular functional assessments using TEM and IF analyses confirmed the absence of the ODAs and missing DNAH5 components from the ciliary axonemes. In conclusion, DNAH5 harbours rare novel pathogenic variants causing PCD in the Arab population of Kuwait.
Author’s note
The author contributions in detail: D.A-M. designed the study, provided the clinical data of the PCD individuals, collected DNA from all members of the PCD families, performed autozygosity mapping, variant screening and validation including in silico analyses, segregation analyses, IF analysis and interpretation, prepared all figures and wrote the manuscript. B.H.A. performed nasal brushing of all PCD subjects in the study. P.P. performed the TEM analysis. P.P. and H.O. reviewed the manuscript.
Data availability statement
The raw sequence data is available on request, as it contains potentially identifiable information.
Ethics statement
The studies involving humans were approved by protocol no. 62/2013 of the Medical and Health Research Coordination Committee, Ministry of Health, Kuwait. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was also obtained from the individual(s) and minor(s)’ legal guardians/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
DA-M: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. BA: Investigation, Data curation, Writing–review and editing. PP: Investigation, Writing–review and editing. HO: Validation, Writing–review and editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the Kuwait Foundation for the Advancement of Sciences (KFAS) (grant no. P114-13MG-01) as well as the General Facility Grant by the Health Science Center of Kuwait University (grant no. SRUL02/13).
Acknowledgments
The authors are very grateful to all the family members who agreed to participate in this study. The authors are grateful to Dr. Wasif Mohammed, radiologist, at Sabah Hospital, Kuwait, for his assistance in providing and interpreting the radiological data. The authors also thank Prof. Trevor Batten at the University of Leeds, United Kingdom, for his invaluable comments on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, editors, and reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1396797/full#supplementary-material
References
Abecasis, G. R., Cherny, S. S., Cookson, W. O., and Cardon, L. R. (2002). Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101. doi:10.1038/ng786
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter 7, doi:10.1002/0471142905.hg0720s76
Al-Mutairi, D. A., Alsabah, B. H., Alkhaledi, B. A., Pennekamp, P., and Omran, H. (2022). Identification of a novel founder variant in DNAI2 cause primary ciliary dyskinesia in five consanguineous families derived from a single tribe descendant of Arabian Peninsula. Front. Genet. 13, 1017280. doi:10.3389/fgene.2022.1017280
Al-Mutairi, D. A., Alsabah, B. H., Pennekamp, P., and Omran, H. (2023). Mapping the most common founder variant in RSPH9 that causes primary ciliary dyskinesia in multiple consanguineous families of bedouin arabs. J. Clin. Med. 12, 6505. doi:10.3390/jcm12206505
Al-Mutairi, D. A., Jarragh, A. A., Alsabah, B. H., Wein, M. N., Mohammed, W., and Alkharafi, L. (2024). A homozygous SP7/OSX mutation causes osteogenesis and dentinogenesis imperfecta with craniofacial anomalies. JBMR Plus 8, ziae026. doi:10.1093/jbmrpl/ziae026
Alsamri, M. T., Alabdouli, A., Iram, D., Alkalbani, A. M., Almarzooqi, A. S., Souid, A. K., et al. (2021). A study on the genetics of primary ciliary dyskinesia. J. Clin. Med. 10, 5102. doi:10.3390/jcm10215102
Blaabjerg, L. M., Kassem, M. M., Good, L. L., Jonsson, N., Cagiada, M., Johansson, K. E., et al. (2023). Rapid protein stability prediction using deep learning representations. Elife 12, e82593. doi:10.7554/eLife.82593
Carr, I. M., Camm, N., Taylor, G. R., Charlton, R., Ellard, S., Sheridan, E. G., et al. (2011a). GeneScreen: a program for high-throughput mutation detection in DNA sequence electropherograms. J. Med. Genet. 48, 123–130. doi:10.1136/jmg.2010.082081
Carr, I. M., Markham, S. A., and Pena, S. D. (2011b). Estimating the degree of identity by descent in consanguineous couples. Hum. Mutat. 32, 1350–1358. doi:10.1002/humu.21584
Carr, I. M., Morgan, J., Watson, C., Melnik, S., Diggle, C. P., Logan, C. V., et al. (2013). Simple and efficient identification of rare recessive pathologically important sequence variants from next generation exome sequence data. Hum. Mutat. 34, 945–952. doi:10.1002/humu.22322
Cilleros-Rodriguez, D., Martin-Morales, R., Barbeito, P., Deb Roy, A., Loukil, A., Sierra-Rodero, B., et al. (2022). Multiple ciliary localization signals control INPP5E ciliary targeting. Elife 11, e78383. doi:10.7554/eLife.78383
Dalrymple, R. A., and Kenia, P. (2019). European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia: a guideline review. Arch. Dis. Child. Educ. Pract. Ed. 104, 265–269. doi:10.1136/archdischild-2017-312902
Davis, S. D., Rosenfeld, M., Lee, H. S., Ferkol, T. W., Sagel, S. D., Dell, S. D., et al. (2019). Primary ciliary dyskinesia: longitudinal study of lung disease by ultrastructure defect and genotype. Am. J. Respir. Crit. Care Med. 199, 190–198. doi:10.1164/rccm.201803-0548OC
Failly, M., Bartoloni, L., Letourneau, A., Munoz, A., Falconnet, E., Rossier, C., et al. (2009). Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia. J. Med. Genet. 46, 281–286. doi:10.1136/jmg.2008.061176
Fliegauf, M., Olbrich, H., Horvath, J., Wildhaber, J. H., Zariwala, M. A., Kennedy, M., et al. (2005). Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 171, 1343–1349. doi:10.1164/rccm.200411-1583OC
Fowkes, M. E., and Mitchell, D. R. (1998). The role of preassembled cytoplasmic complexes in assembly of flagellar dynein subunits. Mol. Biol. Cell 9, 2337–2347. doi:10.1091/mbc.9.9.2337
Goutaki, M., and Shoemark, A. (2022). Diagnosis of primary ciliary dyskinesia. Clin. Chest Med. 43, 127–140. doi:10.1016/j.ccm.2021.11.008
Hannah, W. B., Seifert, B. A., Truty, R., Zariwala, M. A., Ameel, K., Zhao, Y., et al. (2022). The global prevalence and ethnic heterogeneity of primary ciliary dyskinesia gene variants: a genetic database analysis. Lancet Respir. Med. 10, 459–468. doi:10.1016/S2213-2600(21)00453-7
Horani, A., Ferkol, T. W., Dutcher, S. K., and Brody, S. L. (2016). Genetics and biology of primary ciliary dyskinesia. Paediatr. Respir. Rev. 18, 18–24. doi:10.1016/j.prrv.2015.09.001
Hornef, N., Olbrich, H., Horvath, J., Zariwala, M. A., Fliegauf, M., Loges, N. T., et al. (2006). DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 174, 120–126. doi:10.1164/rccm.200601-084OC
Knowles, M. R., Daniels, L. A., Davis, S. D., Zariwala, M. A., and Leigh, M. W. (2013). Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am. J. Respir. Crit. Care Med. 188, 913–922. doi:10.1164/rccm.201301-0059CI
Knowles, M. R., Leigh, M. W., Carson, J. L., Davis, S. D., Dell, S. D., Ferkol, T. W., et al. (2012). Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax 67, 433–441. doi:10.1136/thoraxjnl-2011-200301
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: the human genomic variant search engine. Bioinformatics 35, 1978–1980. doi:10.1093/bioinformatics/bty897
Lander, E. S., and Botstein, D. (1987). Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 236, 1567–1570. doi:10.1126/science.2884728
Leigh, M. W., Horani, A., Kinghorn, B., O'Connor, M. G., Zariwala, M. A., and Knowles, M. R. (2019). Primary Ciliary Dyskinesia (PCD): a genetic disorder of motile cilia. Transl. Sci. Rare Dis. 4, 51–75. doi:10.3233/TRD-190036
Leigh, M. W., Pittman, J. E., Carson, J. L., Ferkol, T. W., Dell, S. D., Davis, S. D., et al. (2009). Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 11, 473–487. doi:10.1097/GIM.0b013e3181a53562
Li, B., Krishnan, V. G., Mort, M. E., Xin, F., Kamati, K. K., Cooper, D. N., et al. (2009). Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744–2750. doi:10.1093/bioinformatics/btp528
Liu, X., Jian, X., and Boerwinkle, E. (2011). dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899. doi:10.1002/humu.21517
Liu, X., Li, C., Mou, C., Dong, Y., and Tu, Y. (2020). dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 12, 103. doi:10.1186/s13073-020-00803-9
Loges, N. T., Antony, D., Maver, A., Deardorff, M. A., Gulec, E. Y., Gezdirici, A., et al. (2018). Recessive DNAH9 loss-of-function mutations cause laterality defects and subtle respiratory ciliary-beating defects. Am. J. Hum. Genet. 103, 995–1008. doi:10.1016/j.ajhg.2018.10.020
Loges, N. T., Olbrich, H., Fenske, L., Mussaffi, H., Horvath, J., Fliegauf, M., et al. (2008). DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 83, 547–558. doi:10.1016/j.ajhg.2008.10.001
Lu, L., and Madugula, V. (2018). Mechanisms of ciliary targeting: entering importins and Rabs. Cell Mol. Life Sci. 75, 597–606. doi:10.1007/s00018-017-2629-3
Mazor, M., Alkrinawi, S., Chalifa-Caspi, V., Manor, E., Sheffield, V. C., Aviram, M., et al. (2011). Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 88, 599–607. doi:10.1016/j.ajhg.2011.03.018
Mitchison, T. J., and Mitchison, H. M. (2010). Cell biology: how cilia beat. Nature 463, 308–309. doi:10.1038/463308a
Muller, L., Savas, S. T., Tschanz, S. A., Stokes, A., Escher, A., Nussbaumer, M., et al. (2021). A comprehensive approach for the diagnosis of primary ciliary dyskinesia-experiences from the first 100 patients of the PCD-UNIBE diagnostic center. Diagn. (Basel) 11, 1540. doi:10.3390/diagnostics11091540
Olbrich, H., Haffner, K., Kispert, A., Volkel, A., Volz, A., Sasmaz, G., et al. (2002). Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 30, 143–144. doi:10.1038/ng817
Pazour, G. J., Agrin, N., Walker, B. L., and Witman, G. B. (2006). Identification of predicted human outer dynein arm genes: candidates for primary ciliary dyskinesia genes. J. Med. Genet. 43, 62–73. doi:10.1136/jmg.2005.033001
Pifferi, M., Michelucci, A., Conidi, M. E., Cangiotti, A. M., Simi, P., Macchia, P., et al. (2010). New DNAH11 mutations in primary ciliary dyskinesia with normal axonemal ultrastructure. Eur. Respir. J. 35, 1413–1416. doi:10.1183/09031936.00186209
Ratjen, F., Waters, V., Klingel, M., McDonald, N., Dell, S., Leahy, T. R., et al. (2016). Changes in airway inflammation during pulmonary exacerbations in patients with cystic fibrosis and primary ciliary dyskinesia. Eur. Respir. J. 47, 829–836. doi:10.1183/13993003.01390-2015
Romero Romero, M. L., Yang, F., Lin, Y. R., Toth-Petroczy, A., Berezovsky, I. N., Goncearenco, A., et al. (2018). Simple yet functional phosphate-loop proteins. Proc. Natl. Acad. Sci. U. S. A. 115, E11943–E11950. doi:10.1073/pnas.1812400115
Schwabe, G. C., Hoffmann, K., Loges, N. T., Birker, D., Rossier, C., de Santi, M. M., et al. (2008). Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum. Mutat. 29, 289–298. doi:10.1002/humu.20656
Wallmeier, J., Al-Mutairi, D. A., Chen, C. T., Loges, N. T., Pennekamp, P., Menchen, T., et al. (2014). Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat. Genet. 46, 646–651. doi:10.1038/ng.2961
Wheway, G., Thomas, N. S., Carroll, M., Coles, J., Doherty, R., Genomics England Research, C., et al. (2021). Whole genome sequencing in the diagnosis of primary ciliary dyskinesia. BMC Med. Genomics 14, 234. doi:10.1186/s12920-021-01084-w
Zariwala, M. A., Knowles, M. R., and Omran, H. (2007). Genetic defects in ciliary structure and function. Annu. Rev. Physiol. 69, 423–450. doi:10.1146/annurev.physiol.69.040705.141301
Zariwala, M. A., Leigh, M. W., Ceppa, F., Kennedy, M. P., Noone, P. G., Carson, J. L., et al. (2006). Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. Am. J. Respir. Crit. Care Med. 174, 858–866. doi:10.1164/rccm.200603-370OC
Keywords: primary ciliary dyskinesia, pulmonary diseases, genetics of ciliopathy, DNAH5 gene, laterality defects
Citation: Al-Mutairi DA, Alsabah BH, Pennekamp P and Omran H (2024) Novel pathogenic variants of DNAH5 associated with clinical and genetic spectra of primary ciliary dyskinesia in an Arab population. Front. Genet. 15:1396797. doi: 10.3389/fgene.2024.1396797
Received: 06 March 2024; Accepted: 20 May 2024;
Published: 10 July 2024.
Edited by:
Ahmed Rebai, Centre of Biotechnology of Sfax, TunisiaReviewed by:
Antonio Martinez-Gimeno, Complejo Hospitalario de Toledo, SpainSteven Pei, Yale University, United States
Copyright © 2024 Al-Mutairi, Alsabah, Pennekamp and Omran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dalal A. Al-Mutairi, ZGFsYWwuYWxtdXRhaXJpQGt1LmVkdS5rdw==