 Fangli Gu1
Fangli Gu1 Wenwu Zhang
Wenwu Zhang Cheng Song
Cheng Song- 1Anhui Engineering Laboratory for Conservation and Sustainable Utilization of Traditional Chinese Medicine Resources, College of Biological and Pharmaceutical Engineering, West Anhui University, Lu’an, China
- 2College of Life and Health Sciences, Anhui Science and Technology University, Fengyang, China
- 3The First Affiliated Hospital, College of Clinical Medicine of Henan University of Science and Technology, Luoyang, China
Introduction: DNA-binding with one finger (Dof) transcription factors (TFs) are a unique family of TFs found in higher plants that regulate plant responses to light, hormones, and abiotic stresses. The specific involvement of Dof genes in the response to environmental stresses remains unknown in D. huoshanense.
Methods: A total of 22 Dof family genes were identified from the D. huoshanense genome.
Results: Chromosome location analysis showed that DhDof genes were distributed on 12 chromosomes, with the largest number of Dof genes located on chromosome 8. The phylogenetic tree revealed that DhDofs could be categorized into 11 distinct subgroups. In addition to the common groups, DhDof4, DhDof5, DhDof17, and the AtDof1.4 ortholog were clustered into the B3 subgroup. Group E was a newly identified branch, among which DhDof6, DhDof7, DhDof8, and DhDof9 were in an independent branch. The conserved motifs and gene structure revealed the differences in motif number and composition of DhDofs. The dof domain near the N-terminus was highly conserved and contained a C2-C2-type zinc finger structure linked with four cysteines. Microsynteny and interspecies collinearity revealed gene duplication events and phylogenetic tree among DhDofs. Large-scale gene duplication had not occurred among the DhDofs genes and only in one pair of genes on chromosome 13. Synteny blocks were found more often between D. huoshanense and its relatives and less often between Oryza sativa and Arabidopsis thaliana. Selection pressure analysis indicated that DhDof genes were subject to purifying selection. Expression profiles and correlation analyses revealed that the Dof gene under hormone treatments showed several different expression patterns. DhDof20 and DhDof21 had the highest expression levels and were co-expressed under MeJA induction. The cis-acting element analysis revealed that each DhDof had several regulatory elements involved in plant growth as well as abiotic stresses. qRT-PCR analysis demonstrated that DhDof2 was the main ABA-responsive gene and DhDof7 was the main cold stress-related gene. IAA suppressed the expression of some Dof candidates, and SA inhibited most of the candidate genes.
Discussion: Our results may provide new insights for the further investigation of the Dof genes and the screening of the core stress-resistance genes.
Introduction
Dof is a type of plant-specific TFs that regulate gene expression by binding to promoters or interacting with specific proteins, and play a crucial role in regulating a wide range of plant physiological functions (Gupta et al., 2018). The N-terminus of Dof protein shares a highly conserved Dof domain consisting of 52 amino acids. The core motif is covalently combined with Zn2+ to form a single zinc finger structure, which specifically binds to promoter sequences with the core [T/AAAAG] motif in the downstream gene (Gupta et al., 2015). The C-terminus harbors a transcriptional regulatory domain with diverse functionalities, enabling its interaction with various regulatory proteins and selective activation of gene expression. The amino acids in the domain are poorly conserved and vary greatly between different Dof members, which in turn leads to differences in Dof protein functions (Ma et al., 2015). Many Dof members have been found in higher plants and ZmDof1 was first found in Zea mays (Lijavetzky et al., 2003; Cai et al., 2013; Ma et al., 2015; Song et al., 2016; Song et al., 2022). In monocot plants, Eleusine coracana (48 individuals), Musa acuminata (74 individuals), and Setaria italica (35 individuals) had been identified (Dong et al., 2016; Zhang et al., 2017; Gupta et al., 2018). In dicotyledonous plants, A. thaliana (39 individuals), i (60 individuals), Populus trichocarpa (41 individuals), and Betula platyphylla (26 individuals) had been identified (Le Hir and Bellini, 2013; Wang et al., 2017; Zhang et al., 2017; Sun et al., 2021).
Dof TFs are involved in light response (Park et al., 2003; Ward et al., 2005), photoperiod regulation (Fornara et al., 2009; Corrales et al., 2014), sugar metabolism (Tanaka et al., 2009), nitrogen metabolism (Yanagisawa et al., 2004), seed development (Papi et al., 2002), cell cycle regulation (Xu et al., 2016), abiotic stresses (Zang et al., 2017), and other complex physiological processes (Zhuo et al., 2020). DAG1 (DOFAFFECTING GERMINATION 1) mutant was sensitive to red light and regulated by phytochrome B. It could reduce the red light and GA synthesis during seed germination in A. thaliana (Gabriele et al., 2010). OBP1 (OCS element binding factor binding protein 1) could shorten cell division cycle and cause dwarf plants (Skirycz et al., 2008). AtOBP3 was affected by SA and auxin. As a downstream regulator of phytochrome B, AtOBP3 is regulated by cryptochrome 1 (Ward et al., 2005). AtDof5.4/OBP4 acts as a negative regulator to regulate cell expansion and cell cycle progression. AtOBP4 inhibited cell growth and proliferation and caused obvious defects such as dwarfing growth and fewer flowers (Luo et al., 2022). CDFs (CYCLING DOF FACTOR) are widely involved in photoperiod regulation, and overexpression of the AtCDF1 gene showed an early flowering phenotype (Goralogia et al., 2017). SlCDF was not only involved in the regulation of photoperiod in tomato but also enhanced plant tolerance under abiotic stresses such as drought and salinity (Xu et al., 2021). AtDof2.4 and AtDof5.8 were involved in the formation of rosette leaf veins and flower bud vascularity (Konishi and Yanagisawa, 2007; Noguero et al., 2013). AtDof6 negatively regulated seed germination in the ABA hypersensitivity plants and increased the expressions of ABA1 and associated genes. AtDof5.6/HCA2 participated in the formation of the interfascicular cambium and vascular tissue development. AtDOF4.7 regulates abscission by directly controlling the transcription of cell wall hydrolases. AtDof4.7 gene was highly expressed in the siliques and inner layers of A. thaliana. AtDof2.1 sped up JA-stimulated senescence via the MYC2-AtDof2.1-MYC2 feed-forward loop, and promoted leaf senescence (Zhang et al., 2022).
Numerous studies have demonstrated that Dof TFs have a role in plant resistance responses to abiotic stress (Song et al., 2024). The expression of AtDof1.1 was stimulated by MeJA, resulting in a 2-3-fold increment in expression level (Skirycz et al., 2006). AtDof5.8 regulated the plasma membrane-bound NAC gene ANAC069, which contributed to the response to salt stress (He et al., 2015). High salt, drought, high temperature, and ABA all increased the expression of the AtCDF3 gene. Overexpression of AtCDF3 improved drought, low temperature, osmotic stress and shortened flowering time in transgenic A. thaliana. SlCDF1, the CDF homolog of A. thaliana, increased in expression to different levels when exposed to drought, salt, heat, and low temperatures. Overexpression of SICDF1 and SICDF3 in Arabidopsis improved the drought resistance of transgenic plants (Liu et al., 2023). The transgenic cotton overexpressing GhDof1 exhibited much greater salt and cold tolerances compared to the wild-type plants. Salt stress promoted the growth of the root system in GhDof1-overexpressing plants. The expressions of GhP5CS, GhSOD, and GhMYB in the transgenic lines was upregulated to varying degrees (Su et al., 2017). In Tamarix bristle, ThDof1.4 significantly improved the abiotic stress tolerance of transgenic plants by increasing proline content, ROS scavenging, and the expression of ThSOD and ThP5CS genes (Li et al., 2022). TaDofs were involved in wheat grain development and abiotic stress responses. TaDof16, TaDof26 and TaDof96 were significantly upregulated under drought stress (Liu et al., 2020). Thirty-three Dof genes were identified in pepper. The temporal and pathogen-specific differences under biotic stress were discovered in CaDofs, which demonstrated the functional diversity of CaDofs (Kang et al., 2016).
Dof transcription factors play a certain role in the regulation of primary metabolism and secondary metabolism. During carbon metabolism, Dof transcription factor regulates the expression of its related genes. In maize, ZmDof1 can bind to the AAAG motif in the promoter region of OsCS4PPDK to increase the expression of the C4 phosphoenolpyruvate carboxylase and pyruvate kinase genes in the cytoplasm; however, ZmDof2 inhibits the C4 phosphoenolpyruvate carboxylase gene expression (Yanagisawa, 2002). In sweet potatoes, overexpression of the SRF1 gene in the roots significantly reduces the transcription of the βfruct2 gene, thereby reducing the accumulation of sucrose invertase. This leads to a reduction in the concentration of monosaccharides and increases the starch content in the tubers, thereby regulating carbon metabolism (Tanaka et al., 2009). Dof protein can not only regulate carbon metabolism but also improve plant nitrogen utilization and increase nitrogen content. Once overexpressing the ZmDof1 gene in Arabidopsis, the nitrogen content of positive plants increased, and they were able to grow well under low-nitrogen conditions. Overexpression of OsDof25 in Arabidopsis thaliana promotes the expression of high-affinity and low-affinity ammonium transporter AtAMT1.1 and AtAMT2.1 and inhibits the expression of high-affinity nitrate transporter AtNRT2.1. The expression of kinase, phosphoenolpyruvate carboxylase, NADP-dependent and NAD-dependent isocitrate dehydrogenase genes is increased. Dof transcription factor is related to two secondary metabolic processes: the phenylpropionic acid synthesis pathway and the flavonoid synthesis pathway. Overexpression of AtDof4.2 can increase the sensitivity of plants to light at low temperatures. At low temperatures and strong light, AtDof4.2 can negatively regulate the synthesis of flavonoids and positively regulate the synthesis of cinnamic acid (Song et al., 2022). Dof transcription factors can also regulate lipid synthesis and control fatty acid content. Acetyl-CoA carboxylase and long-chain fatty acyl-CoA synthase are two key enzymes in the lipid synthesis process. GmDof4 and GmDof11 specifically bind to the promoters of the acetyl CoA carboxylase and long-chain CoA synthase genes, respectively (Dong et al., 2016).
Wild varieties of D. huoshanense have been plundered in large quantities over the past decade, and the original species is now threatened (Song et al., 2020). Semi-shade, moisture, and a particular environment around stones and moss are required for their fast growth (Song et al., 2021). There are lots of TFs that have been found to regulate abiotic stress and secondary metabolism in Dendrobium spp. (Song et al., 2022). bHLH TFs play a positive role in JA signaling cascade and abiotic stresses (Jiao et al., 2022). A genome-wide identification and analysis of WRKY TFs was employed and screened out several hormone- and cold stress-responsive genes (Zhang et al., 2023). Here, the identification, comparative genomics, and expression analysis of Dof genes were conducted in D. huoshanense. A total of 22 Dof genes were identified, which can be divided into 11 subgroups based on the homologous genes. Conserved motif and gene structure analysis indicated that these Dof proteins shared a common Dof domain, which contained a representative zinc finger structure through multiple sequence alignments. Dof members on the same evolutionary branch have similar motif compositions, but the structural composition of exons and introns is quite different. Microsynteny analysis revealed that the generation of DhDofs were not derived from large-scale gene duplication but mainly came from proximal and dispersed duplication, and these DhDof genes have undergone purifying selection. Comparative transcriptome analysis revealed that DhDof20 and DhDof21 are the main JA-responsive genes. The cis-acting element analysis revealed that the promoter of DhDof has many elements related to hormone response, abiotic stress, photoperiod, and vegetative growth. Expression pattern analysis showed that DhDof2 is the key ABA-responsive gene and DhDof7 is the main cold stress-related gene.
Materials and methods
Sample collection and conditions
D. Huoshanense materials were collected at the Plant Cell Engineering Center of West Anhui University (Luan, China). The culture conditions were 25°C ± 2°C, with a 12 h day and 12 h dark cycle (Zhang et al., 2023). MS medium (without hormone addition) was used for the tissue culture of seedlings. When the seedlings grow to a height of 5–8 cm in subculture, hormone treatments with different concentrations are added. A 100 mol/L ABA solution was supplied to the medium, and the expression levels of Dof genes were measured on days 0, 2, and 4. The samples were treated with a 50 mol/L MeJA solution and collected at 0, 2, and 8 days. The expression level of the Dof genes was measured on days 0, 2, and 4 after adding the 0.1 mg/L IAA solution and the 100 mol/L SA to the medium. After the application of the 0.5 mmol/L GA solution to the plants, samples were collected on days 0, 3, and 6 to measure the expression of Dof genes. During the low-temperature treatment, the samples were first stored at a temperature of 4°C in a refrigerator. To measure the expression of Dof genes, samples were obtained on days 0, 1, and 4.
Identification, physical properties and chromosomal location analysis of Dof gene
The genome sequence and annotation of D. huoshanense were obtained from the National Center for Biotechnology Information database (accession: PRJNA597621). The Hidden Markov Model (HMM) of the Dof domain (PF02701) is downloaded from the InterPro database. The Arabidopsis Dof homologs were downloaded from the Arabidopsis Information Resource (https://www.arabidopsis.org/), and the Blastp method was used for sequence alignment to obtain non-redundant Dof genes. Then, Pfam (http://pfam-legacy.xfam.org/), InterPro (http://www.ebi.ac.uk/interpro/), and SMART (https://smart.embl.de/) were applied for the verification of the Dof candidates. Using genome and gff annotations, chromosomal location analysis of 22 DhDof genes was completed, and then TBtools software was used to perform visual analysis of Dof genes (Chen et al., 2020). By using the Expasy web service (http://expasy.org/), it was possible to predict the molecular weight, isoelectric points, instability index, aliphatic index, and hydropathicity. The Plant-mPLoc plug-in of Cell-PLoc 2.0 software (http://www.csbio.sjtu.edu.cn/bioinf/Cell-PLoc-2/) was used for the subcellular localization prediction.
Phylogenetic tree analysis of Dof proteins
To determine the composition and classification of DhDof, a phylogenetic tree of the two species was constructed using the Arabidopsis nomenclature. MEGA (v.6.0.6) software was used to construct the evolutionary tree using the neighbor-joining method. First, ClustalW software was used to perform protein sequence alignment with the Poisson model and pairwise deletion mode. The bootstrap test was set at 1,000 replicates. IQ-TREE (v.1.6.12) software was used to construct the evolutionary tree using the maximum likelihood method. First, IQ-TREE was used to calculate the optimal model of these Dof sequence alignment files. “VT + F + R5” was chosen as the best-fit model under the Bayesian information criterion. Then, using this model and the “iqtree.exe -s./bidui.fas -m VT + F + R5 -bb 1000 -alrt 1000 -nt AUTO” command, we constructed an unrooted consistency evolutionary tree. The two evolutionary trees passed the identity (>40%) and SH-aLRT support (>80%)/ultrafast bootstrap support (>95%) thresholds to be manually classified.
Conserved domain, motif and exon/intron analysis of Dof genes
The conserved domain database (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) was used for domain searches of Dof proteins. The MEME-suite web service (http://meme.nbcr.net/meme/cgi-bin/meme.cgi) was applied for motif searching and the result was visualized using TBtools software (https://github.com/CJ-Chen/TBtools). Motifs identified by MEME were further retrieved in the InterPro database (http://www.ebi.ac.uk/interpro/). The gene structure was performed using Gene Structure Display Server (http://gsds.cbi.pku.edu.cn/) and visualized by TBtools.
Multiple sequence alignment and amino acid composition analysis of Dof proteins
ClustalW was used to align DhDof protein sequences first, and then the alignment files were imported into GeneDoc software (http://psc.edu/biomed/genedoc) for visualization of protein sequences. Black background, gray background, and white background represent different matching values of amino acids. WebLogo 3 software (https://weblogo.threeplusone.com/create.cgi) was used to visualize the sequence logo of the Dof domain.
Comparative genomic analysis of Dof genes
The MCScanX software (https://github.com/wyp1125/MCScanX) was applied to examine collinearity between D. huoshanense and the other four species. The genome sequences and annotations of D. nobile, D. chrysotoxum, O. sativa and A. thaliana are downloaded from NCBI (accession: PRJNA725550), NCBI (accession: PRJNA664445), Ensembl Plants (genome assembly: IRGSP-1.0), and TAIR (genome assembly: TAIR10). The microsynteny circus of the gene duplication event was visualized by TBtools software. Then, each Dof sequence of D. huoshanense, D. nobile, D. chrysotoxum, A. thaliana, and O. sativa was aligned against itself using BLASTp with an E-value threshold of e−10 to obtain the syntenic blocks between each two species.
Cis-acting element analysis of Dof genes
The 2 kilobase (kb) regions flanking the promoters were obtained from the genomic sequences. The cis-acting elements in Dof promoters were obtained from the PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). TBtools software was utilized to visualize the basic elements associated with growth, phytohormones, and environmental responses.
Expression profiling of Dof genes
The previous transcriptome datasets (https://bigd.big.ac.cn/gsa/browse/CRA006607) were used to compute and normalize the expression of Dof genes as fragments per kilobase per million mapped fragments (FPKM). The sample reads were aligned to the D. huoshanense reference genome using the HISAT software, which could be available at https://daehwankimlab.github.io/hisat2/. The FPKM values for mRNA expression analysis were computed using StringTie (https://github.com/gpertea/stringtie). Salmon was employed to quantify expression levels from RNA-seq data (https://combine-lab.github.io/salmon/). The heatmap construction utilizes the normalized FPKM values (log2 (FPKM)) of the unigenes.
qRT-PCR analysis of Dof genes
RNA was isolated and extracted using the RNA extraction reagent (QIAGEN, Germany) according to the manufacturer’s protocol. The genomic DNA (gDNA) was cleaved using the RNAse-Free DNase Set from QIAGEN, a company based in Germany. The RNA samples were assessed for purity using the NanoDrop 2000c (Thermo Fisher Scientific, United States) and gel electrophoresis. The reverse transcription PCR was performed using the PrimeScriptTM II 1st Strand cDNA Synthesis Kit from TaKaRa, a company based in China. The SYBR Premix Ex TaqTM II, manufactured by TaKaRa in China, was employed for quantitative reverse transcription polymerase chain reaction (qRT-PCR) on a 7,500 series real-time fluorescence quantitative cycler manufactured by Bio-RAD in the United States of America. The primers for the qRT-PCR test were prepared using the Primer Premier 5.0 software. The ACTIN gene served as the reference gene (Song et al., 2021). The primers utilized for qRT-PCR analysis are listed in Supplementary Table S2. Each experiment was repeated three times in triplicate, and a total of three biological replicates were undertaken. The gene expression levels were determined using the 2-△△CT method.
Results
Chromosome location and phylogenetic analysis of Dof genes
A total of 22 Dof genes were screened from D. huoshanense genome, and their conserved domains were further compared and verified through Pfam, InterPro, and SMART databases. Using the genome and annotation files, the chromosomal locations of these Dof genes were determined. The original Dofs were renamed based on their order and location on different chromosomes. The results showed that 22 Dof genes were dispersed on 12 chromosomes (Figure 1). The Dof genes anchored on chromosome 8 genetically formed a cluster at a physical distance. However, there was only one Dof gene on chromosomes 11, 12, 13, 15, 16, and 19. To clarify the evolutionary relationships and classification of these DhDofs, the phylogenetic tree was initially built using the neighbor-joining method (Figure 2A). According to the branch value and the default classification, DhDof proteins were categorized into 5 groups and 11 subgroups, namely, A, B1, B2, B3, C1, C2.1, C2.2, C3, D1, D2, and E. Among them, DhDof4, DhDof5, DhDof17 and AtDof1.4 were on the B3 subgroup, which was different from the previous result. There were no DhDofs that belonged to the C3 subgroup. Group E is a new branch identified for the first time. The four genes DhDof6, DhDof7, DhDof8, and DhDof9 are independent from branches A and D2. In addition, the ML phylogenetic tree of the two species was also constructed (Figure 2B). The only difference between two phylogenetic trees was that AtDof1.7 and AtDof3.1 were classified in the D2 subgroup. The properties and subcellular localization of Dof genes were further predicted (Table 1). Subcellular localization prediction showed that all the DhDofs were localized in the nucleus.
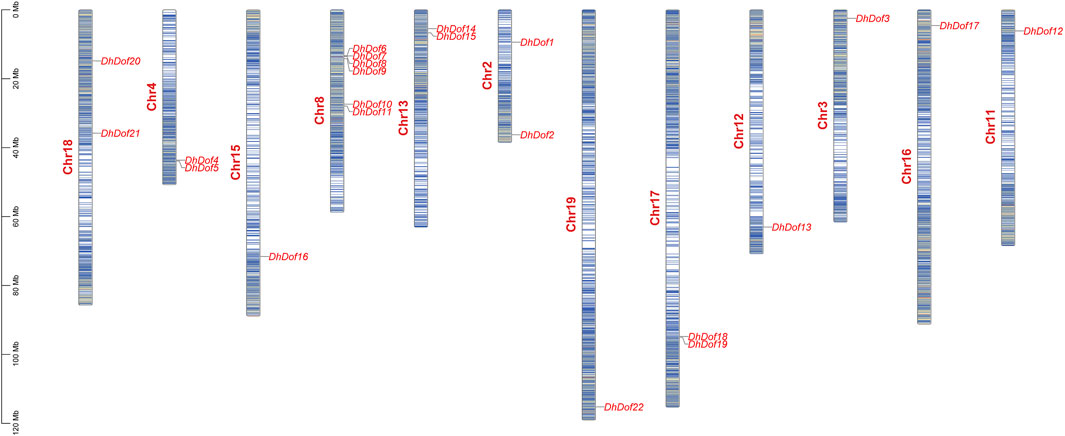
Figure 1. Chromosomal location analysis of the DhDof genes.
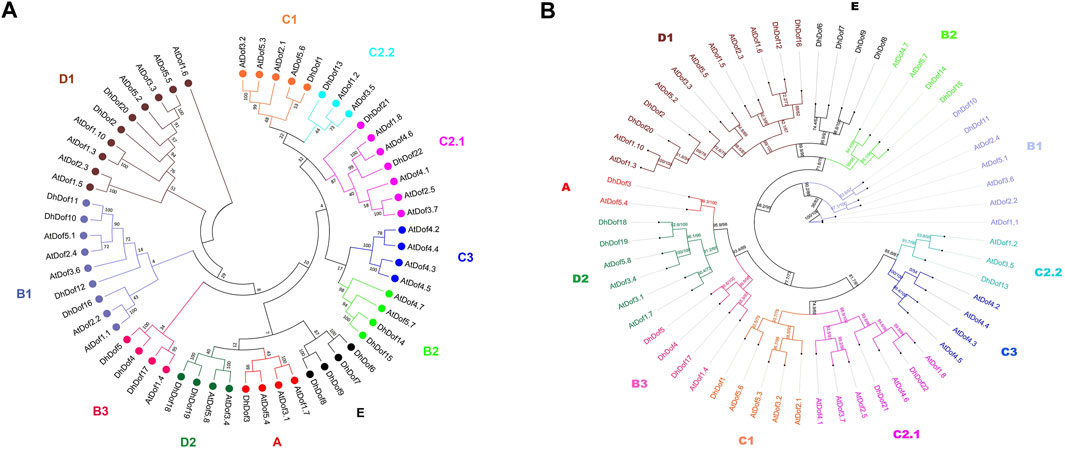
Figure 2. Phylogenetic tree of Dof proteins from D. huoshanense and A. thaliana. (A) Neighbor-joining method; (B) Maximum-likelihood method.
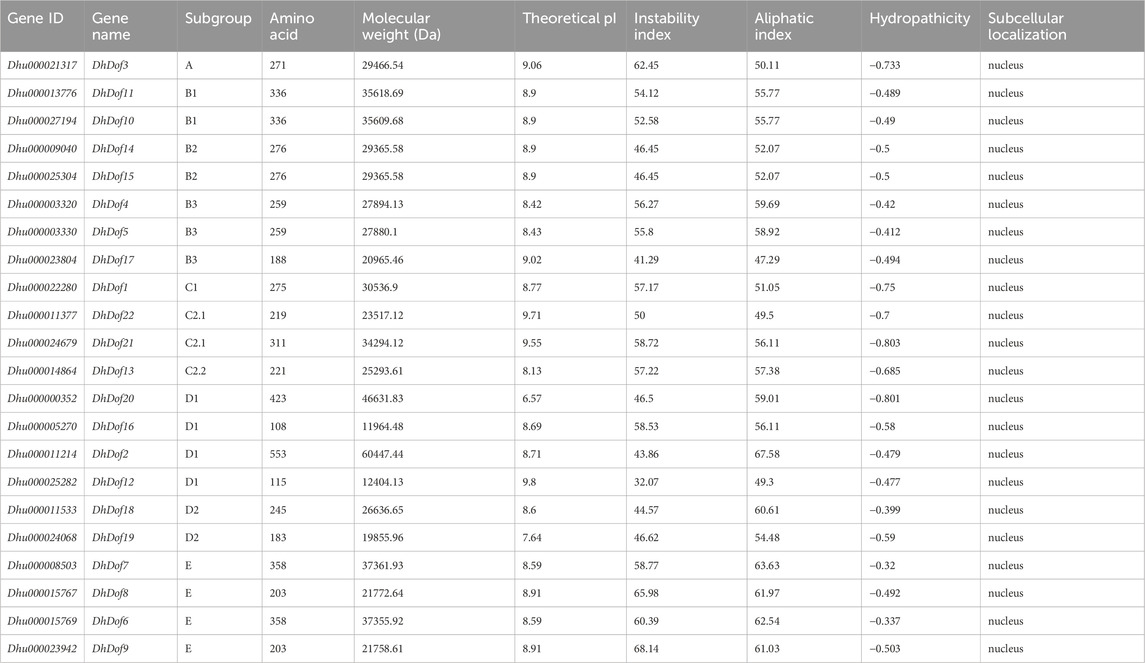
Table 1. The classification and physical properties of Dof genes in D. huoshansense
Conserved domain, conserved motif and exon/intron composition of Dof genes
To clarify the gene structure and composition of different types of Dof genes, the number and composition of motifs of DhDofs were first analyzed. Motif 1 was a conserved zinc-finger motif common to all Dof genes (Figure 3A). For the same subgroup, their conserved motif composition was the same, like DhDof10 and DhDof11, which both contained motif 1, motif 3 and motif 9. DhDof14 and DhDof15 included motif 1, motif 2, motif 8 and motif 10. The conserved domain database search results showed that all DhDofs contained the zf-Dof superfamily domains, while the N-ternimus of DhDof4, DhDof5 and DhDof21 also contained an InsA superfamily domain (Figure 3B). Gene structure analysis revealed that the number and composition of introns and exons in different DhDof genes varied greatly. Some DhDofs (such as DhDof2, DhDof20, DhDof10 and DhDof1) also contained 3’ UTR regions (Figure 3C). There was a long intron insertion between the two exons of DhDof18. The zinc finger structure in Dof domain is required to recognize AAAG/CTTT elements. A sequence comparison of the Dof proteins revealed that DhDofs contained a conserved CPRC(X)21CK(X)1C motif, which formed part of the Dof domain (Figure 4).
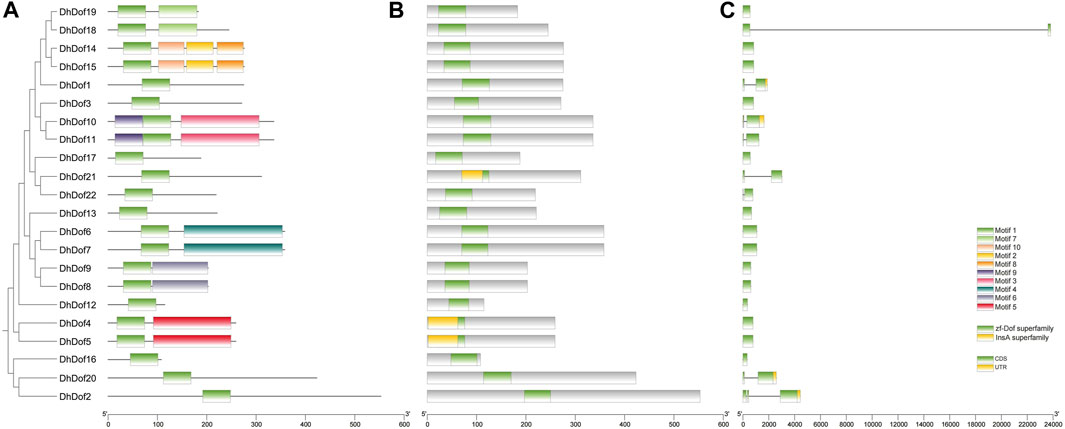
Figure 3. Conserved motif and exon/intron analysis of the DhDof gene. (A) Conserved motif; (B) conserved domain; (C) cds, introns and UTR.

Figure 4. Multiple sequence alignments of the Dof protein. “C” stands for cysteine. The dashed line represents the zinc-finger motif of the Dof domain.
Comparative genomic analysis of Dof genes
Comparative genomics analysis provided clues for the evolutionary relationships among species of D. spp. To clarify the genetic relationship of Dof genes between D. huoshanense and its closely related species, a collinearity analysis of Dof genes was employed. Microsynteny analysis showed that out of all the DhDof genes, only one pair of genes (Dhu000025304 and Dhu000009040) had collinearity, and dispersed and proximal duplication contributed to the expansion of DhDof genes. This implied that the formation of DhDof homologs was not due to whole genome duplication (Figure 5A). Synteny analysis from interspecies reveals that D. huoshanense and D. nobile exhibited the highest level of collinearity, with a total of 21 gene pairs, followed by D. huoshanense and D. chrysotoxum. However, there were only 7 and 6 syntenic blocks compared with O. sativa and A. thaliana, respectively. This was in accordance with the species’ relationship (Figure 5B). Artificial selection pressure provides the driving force for genome evolution and domestication. Through calculating Ka and Ks values, the Ka/Ks ratios of all DhDof genes were shown less than 1, which meant that DhDof genes were subject to purifying selection (Supplementary Table S1).
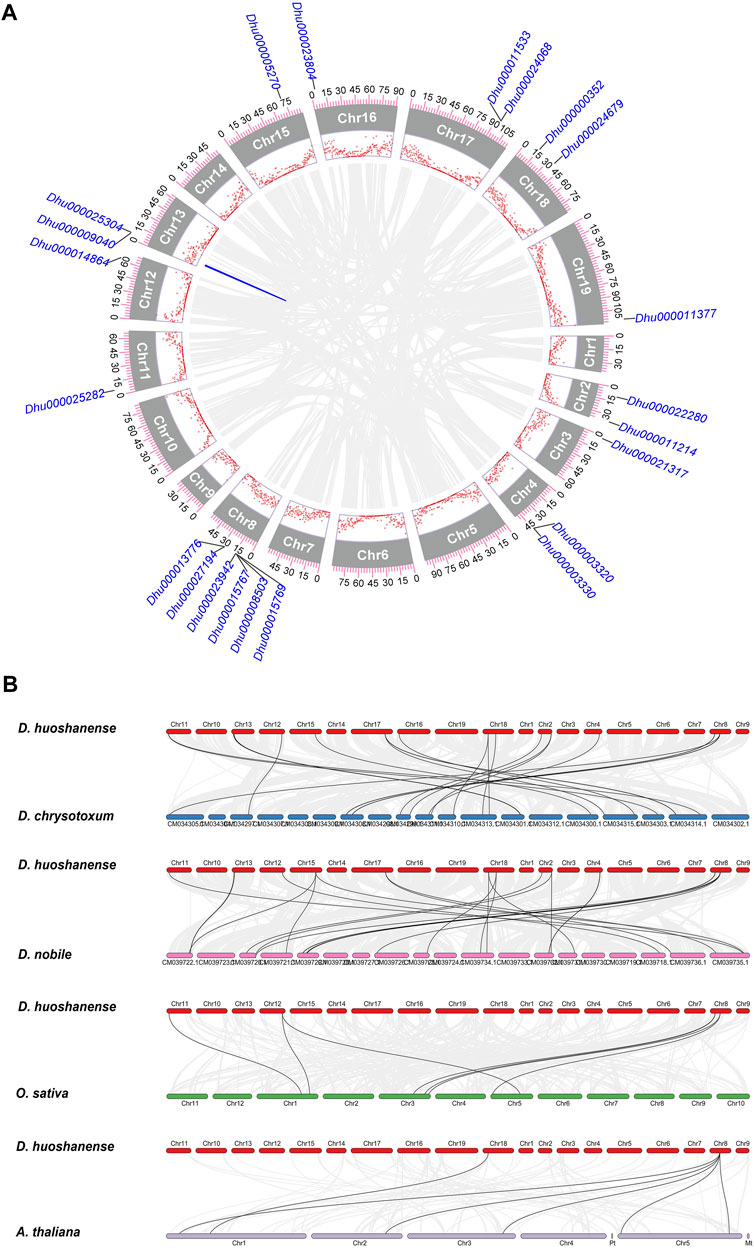
Figure 5. Comparative genomic analysis of Dof genes. (A) Microsynteny of DhDof genes; (B) Collinearity of D. huoshanense with D. nobile, D. chrysotoxum, O. sativa and A. thaliana. The ticks and scatter points are the genome scale and gene density, respectively. Gray lines represent shared gene pairs.
Expression profile and correlation analysis of Dof genes
Based on the transcriptome sequencing and annotation results, the expression profile of Dofs was examined under MeJA treatment. Almost half of the DhDof genes were not expressed, and only a few genes were highly expressed after treatment (Figure 6A). DhDof20 and DhDof21 were more sensitive to MeJA stimulation and showed high expression throughout the entire period. This suggests that they are the core JA-responsive genes and participate in the downstream regulation of JA signaling. The expression of DhDof2 continued to increase with treatment time. The expression level reached its maximum at Time 7, showing a delayed JA-induced effect. On the contrary, the expression of DhDof17 was the highest at the beginning and decreased significantly after MeJA treatment, which suggested that DhDof17 was a negative effector gene of JA signaling. The expressions of DhDof6, DhDof8, and DhDof13 were highest at Time 3 with MeJA treatment and then gradually decreased, indicating that these DhDofs were early-responsive genes in JA signaling. To find out which Dof genes are co-expressed, a correlation analysis was performed based on their expression levels. DhDof6 showed a significantly positive correlation with DhDof8 and DhDof13. DhDof8 and DhDof13 also showed a significantly positive correlation, followed by DhDof15. DhDof6, DhDof8, and DhDof13 were co-expressed during MeJA treatment (Figure 6B). In addition, the expression of DhDof21 and DhDof17 showed a significant negative correlation, which was consistent with the previous result.
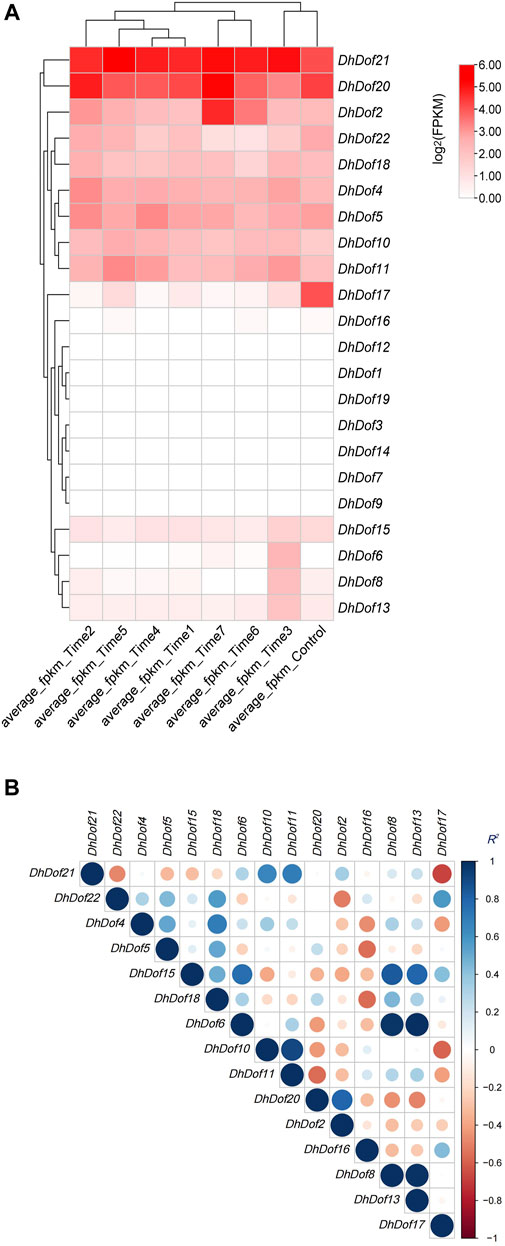
Figure 6. Expression profile and correlation analysis of the DhDof gene. (A) Expression profile after MeJA treatment; (B) correlation analysis of DhDof genes. Mean_Control, Mean_T1, Mean_T2, Mean_T3, Mean_T4, Mean_T5, Mean_T6, and Mean_T7 represent the groups treated with MeJA after 0, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 16.0 h, respectively.
Cis-acting element analysis of Dof genes
To determine which kind of environmental stresses these Dof genes could respond to, the composition of cis-acting elements was analyzed. The DhDof gene contained numerous types of regulatory elements. DhDofs from the same branch or subgroup contained similar cis-acting elements but differed in the distribution of seldom elements. For instance, DhDof18 contained a wound-induced element at the 5′end, but not in DhDof19. DhDof19 contained a GA-responsive element at the 5′end, but not in DhDof18. A total of 37 functional classifications were present here, which were mainly involved in growth, hormone induction, and abiotic stress response (Figure 7A). The most distributed element was related to light responsiveness, which was present in all DhDof genes. Among them, DhDof2, DhDof14, and DhDof21 were Dof genes that contained more functional categories. The second most common component was the MYB-binding element. DhDof1, DhDof12, and DhDof16 contained a large number of these elements. The third common component is the MYC-binding element. A total of 76 different cis-acting elements were discovered, with Myb, MYC, and Box4 elements being the most prominent (Figure 7B). The regulatory elements related to plant growth and development mainly included GCN4_motif, circadian, A-box, CAT-box, CCGTCC motif, re2f-1, HD-Zip 1 and RY-element. Cis-acting elements involved in hormone responses were deferentially distributed in the DhDof genes. DhDof2 and DhDof17 were the key genes involved in ABA responsiveness. DhDof21 was the key gene involved in GA responsiveness. DhDof2 and DhDof13 were the key genes involved in MeJA responsiveness. DhDof10 and DhDof11 were the key genes involved in SA responsiveness. DhDof22 was the key gene involved in ethylene responsiveness. The cis-acting elements involved in abiotic stress mainly included ARE, GC-motif, TC-rich repeats, MBS, DRE1, DRE core, LTR, STRE, WUN-motif, and WRE3, which were relevant to drought, low temperature, defense stresses, anaerobic induction, mechanical injury, etc. (Figure 7C).
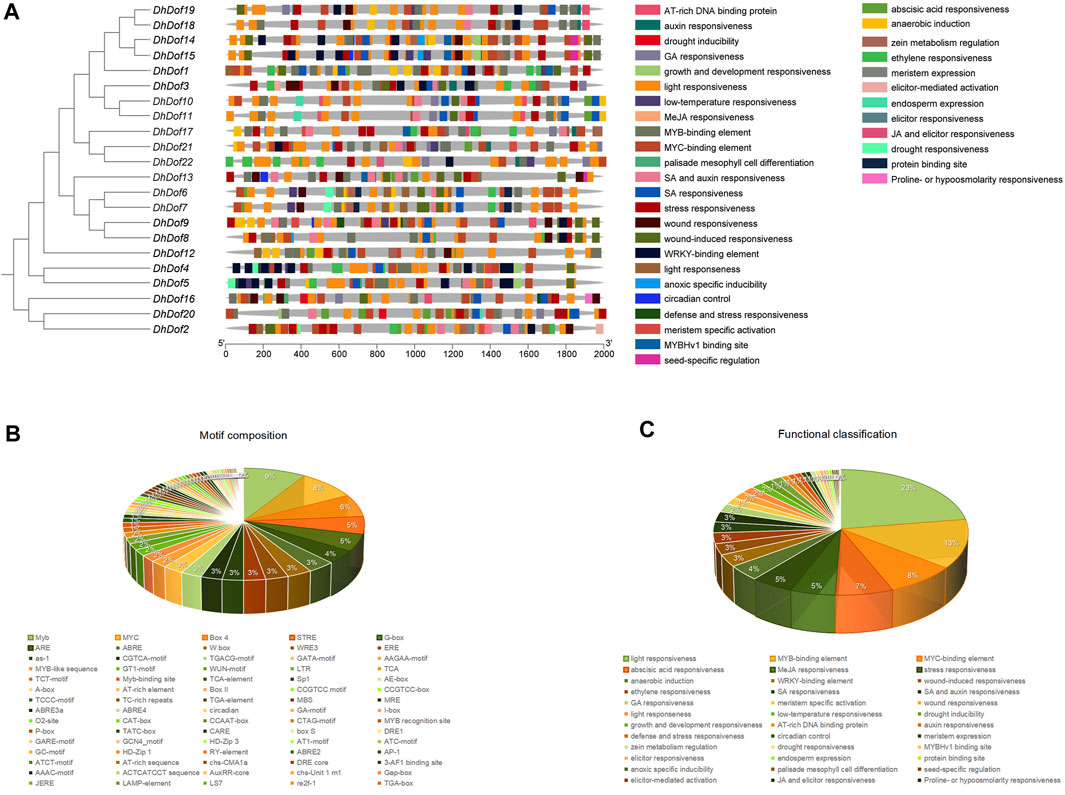
Figure 7. Analysis of cis-acting elements of the DhDof gene. (A) Distribution and composition of elements on the promoter of the DhDof gene; (B) Number and proportion of elements in the DhDof gene; (C) Functional classification and proportion of the DhDof gene.
Expression pattern analysis of Dof genes
The expression pattern of some of Dofs was confirmed by qRT-PCR method. The results showed that on the second and fourth days of ABA treatment, the expression of DhDof2 dramatically increased by approximately 92-fold and 49-fold compared with the control, respectively. DhDof2 was the main ABA-responsive gene and could be strongly induced. The expression levels of DhDof1 and DhDof12 exhibited an initial increase followed by a later drop under ABA treatment. The expression of DhDof4 decreased significantly after ABA treatment (Figure 8A). The expression level of DhDof13 decreased significantly at the 2nd hour after GA treatment and remained at a low level at 8 h. The expressions of DhDof21 and DhDof16 were also inhibited by GA. However, the expression of DhDof11 and DhDof17 decreased sharply at the 2nd hour of GA treatment but basically returned to the initial level at the 8th hour (Figure 8B). IAA treatment could significantly inhibit the expression of all DhDof candidates, and this inhibitory effect persisted until day 4. TGA-element (AACGAC) and AuxRR-core element (GGTCCAT) in the promoter region of DhDof were the negative auxin-responsive elements (Figure 8C).
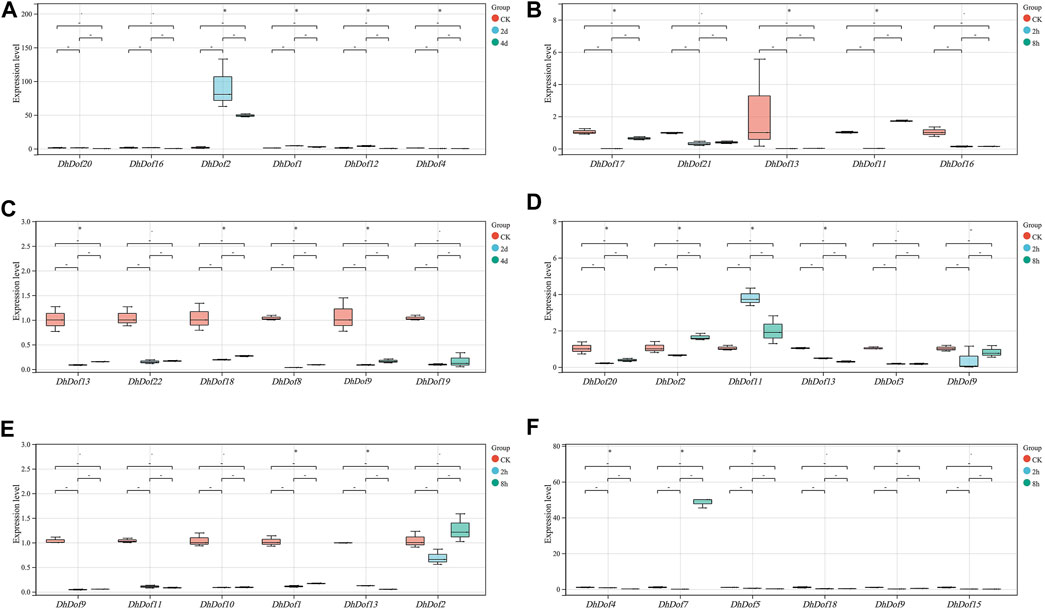
Figure 8. qRT-PCR analysis of the DhDof gene. (A) ABA treatment; (B) GA treatment; (C) IAA treatment; (D) MeJA treatment; (E) SA treatment; (F) low temperature treatment.
Discussion
Molecular characterization and evolution of DhDof genes
Dof TFs are ubiquitously found across the full spectrum of the plant kingdom, encompassing both lower plants, such as unicellular green algae, and higher plants, like angiosperms and gymnosperms (Yu et al., 2020; Song et al., 2024). D. huoshanense did not have as many Dof genes as other angiosperms, which meant it probably had not experienced large-scale genome duplication (Melgar and Zelada, 2021). DhDofs were unevenly distributed on 12 pseudochromosomes and not equivalent to chromosome size (Figure 1). We infer this result from the unequal gene duplications of chromosomal segments. Microsynteny analysis revealed that only one pair of DhDof genes had collinearity, which proved that other duplication may be relevant (Figure 5). In addition to the Dof gene, some other transcription factors, like bHLH and WRKY, have been shown shared syntenic blocks among D. huoshanense, D. chrysotoxum, and D. nobile. This provided evidence for their close genetic relationship (He et al., 2023; Pan et al., 2023). D. huoshanense and D. nobile had the most collinear gene pairs, followed by D. huoshanense and D. chrysotoxum (Figure 5). This result was consistent with the other monocots, such as Z. mays, Brachypodium distachyon, Saccharum officinarum, etc. (Zou and Sun, 2023).
Phylogenetic tree, exon/intron structure and conserved motif analysis of DhDof genes
Phylogenetic analysis helps us understand the evolution and genetic relationship between Dof genes and Dof in other species. Based on the composition of domains and motifs, Dof genes are usually divided into four groups: A, B, C, and D. The B, C, and D groups can be further divided into several subgroups. There are also some studies that classify Dof genes into groups I-VII, which are obtained through domains and motifs. For example, 30 Dof proteins of O. sativa were divided into four groups and further subdivided into seven subgroups by constructing an unrooted phylogenetic tree with Arabidopsis, sorghum, and maize (Khan et al., 2021). Dof gene family was divided into six subfamilies (Group A-F) in sorghum, of which Group B contained only AtDof4.2, AtDof4.3, AtDof4.4, and AtDof4.5. 96 Dof genes from the wheat genome were divided into five subfamilies (Groups A-E), but only four AtDofs were categorized in Group A. Our results indicated that these four AtDofs were classified into the C3 subgroup, and no DhDof was assigned to this subgroup (Figure 2). Based on the phylogenetic tree, DhDof genes were divided into 11 subgroups, among which B3 and E subgroups are new branches identified (Figure 2). In addition to the E group, the D1 subgroup, also known as the CDF subfamily, contains the most Dof genes, including DhDof2, DhDof20, DhDof12, and DhDof16. AtDof1.4 was classified into Group IV or B2 subgroups in previous studies (Yanagisawa, 2002; Lijavetzky et al., 2003; Zhang et al., 2023). However, the phylogenetic trees in our study indicated that AtDof1.4 did not group with AtDof1.7, AtDof3.1, AtDof3.4, and AtDof5.8 in the D2 subgroup but as an independent subgroup. Three DhDof genes also belong to this subgroup (Figure 2). Remarkably, the E group, to which DhDof6/7/8/9 belonged, was a new branch that originated from the paralogs in D1. All the DhDof proteins possessed the highly conserved zinc-finger Dof domain (Figures 3, 4). Gene structure analysis revealed notable variations among distinct subgroups, whereas similar structures were observed in the common subgroup, as demonstrated in the motif analysis (Figure 3). Some subgroups contained their own unique motifs. Studies indicated that the introns of IbDof genes were relatively small, and most genes had only one intron or even no intron. Such intron-free genes may play a role in the accelerated stress response.
Expression profiling and functional prediction of DhDof genes
The involvement of Dof genes in response to biotic stress has been well documented. However, the regulatory function of Dof on abiotic stress responses was only reported in a limited number of plants (Zhang et al., 2018). RcDOFs expressed at two distinct levels varied in response to ABA (Waschburger et al., 2022). CmDOFs had a role in the response to ABA and SA, which contributed to differential expression patterns. Exogenous ABA specifically dramatically increased the expression of CmDOF12 and CmDOF20. OsDOF15 mediated the growth of the main root in rice when exposed to high salinity by releasing ethylene (Qin et al., 2019). The expressions of OsDof1 and OsDof19 increased at low temperatures in the cold-tolerant cultivar. Overexpression of OsDof1 resulted in a greater seed setting rate (Liu et al., 2021). An investigation and analysis of DhDofs were conducted in several abiotic stresses. Expression profiles showed the spatiotemporal profile of DhDofs after MeJA induction. DhDof20 and DhDof21 were identified as the main responsive genes of JA signaling, and DhDof17 was considered to be an early-responsive gene (Figure 6). DhDofs showed distinct expression patterns under different abiotic stresses. Under a series of abiotic stresses, the expression patterns of the DhDofs were determined at different phases. DhDof2 was strongly induced by ABA, while DhDof7 was significantly induced by low temperatures. The expression of most DhDof genes showed an inhibitory state after treatments. Most genes were inhibited after IAA and SA treatment, which were probably related to the cis-acting elements in their promoter (Figures 7, 8). Our results revealed how Dof genes work and how Dendrobium plants interact and coordinate with the external environment.
Conclusion
Dof TFs play multiple roles in physiological processes that involved in biotic or abiotic stresses. Twenty-two Dof genes were dispersed on twelve pseudochromosomes. Microsynteny analysis revealed that DhDof genes were not generated from large-scale gene duplication. Interspecies synteny analysis suggested that these Dof genes originated from a common ancestral homolog (Song et al., 2023). The syntenic blocks between D. huoshanense, D. chrysotoxum, and D. nobile were larger. Selection pressure analysis further revealed DhDof genes underwent purifying selection. Multiple sequence alignments showed that all Dof genes contain highly conserved zinc finger motif. The phylogenetic analysis divided 22 DhDof into 11 subgroups. The E group is a new branch assigned and contained four genes, including DhDof6, DhDof7, DhDof8 and DhDof9. In addition to the zf-Dof subfamily domain, a few Dof genes also contained the Ins A subfamily domain, which was thought to be related to specific regulatory functions. However, the exon/intron analysis reflected that even Dof genes from the same subgroup would have widely differing intron and 3′or 5′UTR. Expression profiling analysis showed that DhDof21 and DhDof22 were the main JA-responsive genes. The promoter of Dof genes has numerous hormone response-related elements, such as the CGTCA-motif, the TGACG-motif, as-1, TCA, CARE, P-box, and others. This makes them better able to sense changes in external hormones. qRT-PCR analysis showed that DhDof genes had different expression patterns in response to different abiotic stresses. As there is some variation in the number of Dof genes among various species, the potential functional redundancy of these genes needs to be validated by developing several knockout lines and other approaches. Dofs with the potential to raise crop yields can provide promising opportunities for the advancement of the food manufacturing and biofuel production.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
FG: Formal Analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. WZ: Formal Analysis, Investigation, Software, Validation, Visualization, Writing–original draft. TW: Formal Analysis, Investigation, Software, Writing–original draft. XH: Formal Analysis, Funding acquisition, Investigation, Supervision, Writing–review and editing. NC: Project administration, Resources, Supervision, Writing–review and editing. YZ: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Visualization, Writing–review and editing. CS: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Software, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is supported by Demonstration Experiment Training Center of Anhui Provincial Department of Education (2022sysx033), Startup fund for high-level talents of West Anhui University (WGKQ2022025, WGKQ2023010), Anhui Provincial Department of Education Excellent Top Talents Cultivation Project for Universities (gxgnfx2021144, gxgnfx2022151) and Joint Construction Project of Henan Provincial Medical Science and Technology Research Program (LHGJ20190550).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1394790/full#supplementary-material
References
Cai, X., Zhang, Y., Zhang, C., Zhang, T., Hu, T., Ye, J., et al. (2013). Genome-wide analysis of plant-specific dof transcription factor family in tomato. J. Integr. Plant Biol. 55, 552–566. doi:10.1111/jipb.12043
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi:10.1016/j.molp.2020.06.009
Corrales, A. R., Nebauer, S. G., Carrillo, L., Fernández-Nohales, P., Marqués, J., Renau-Morata, B., et al. (2014). Characterization of tomato Cycling Dof Factors reveals conserved and new functions in the control of flowering time and abiotic stress responses. J. Exp. Bot. 65, 995–1012. doi:10.1093/jxb/ert451
Dong, C., Hu, H., and Xie, J. (2016). Genome-wide analysis of the DNA-binding with one zinc finger (Dof) transcription factor family in bananas. Genome 59, 1085–1100. doi:10.1139/gen-2016-0081
Fornara, F., Panigrahi, K. C. S., Gissot, L., Sauerbrunn, N., Rühl, M., Jarillo, J. A., et al. (2009). Arabidopsis DOF transcription factors act redundantly to reduce CONSTANS expression and are essential for a photoperiodic flowering response. Dev. Cell 17, 75–86. doi:10.1016/j.devcel.2009.06.015
Gabriele, S., Rizza, A., Martone, J., Circelli, P., Costantino, P., and Vittorioso, P. (2010). The Dof protein DAG1 mediates PIL5 activity on seed germination by negatively regulating GA biosynthetic gene AtGA3ox1. Plant J. 61, 312–323. doi:10.1111/j.1365-313X.2009.04055.x
Goralogia, G. S., Liu, T. K., Zhao, L., Panipinto, P. M., Groover, E. D., Bains, Y. S., et al. (2017). CYCLING DOF FACTOR 1 represses transcription through the TOPLESS co-repressor to control photoperiodic flowering in Arabidopsis. Plant J. 92, 244–262. doi:10.1111/tpj.13649
Gupta, S., Malviya, N., Kushwaha, H., Nasim, J., Bisht, N. C., Singh, V. K., et al. (2015). Insights into structural and functional diversity of Dof (DNA binding with one finger) transcription factor. Planta 241, 549–562. doi:10.1007/s00425-014-2239-3
Gupta, S., Pathak, R. K., Gupta, S. M., Gaur, V. S., Singh, N. K., and Kumar, A. (2018). Identification and molecular characterization of Dof transcription factor gene family preferentially expressed in developing spikes of Eleusine coracana L. 3 Biotech. 8, 82–13. doi:10.1007/s13205-017-1068-z
He, L., Su, C., Wang, Y., and Wei, Z. (2015). ATDOF5.8 protein is the upstream regulator of ANAC069 and is responsive to abiotic stress. Biochimie 110, 17–24. doi:10.1016/j.biochi.2014.12.017
He, X., Zhang, W., Sabir, I. A., Jiao, C., Li, G., Wang, Y., et al. (2023). The spatiotemporal profile of Dendrobium huoshanense and functional identification of bHLH genes under exogenous MeJA using comparative transcriptomics and genomics. Front. Plant Sci. 14, 1–18. doi:10.3389/fpls.2023.1169386
Jiao, C., Wei, M., Fan, H., Song, C., Wang, Z., Cai, Y., et al. (2022). Transcriptomic analysis of genes related to alkaloid biosynthesis and the regulation mechanism under precursor and methyl jasmonate treatment in Dendrobium officinale. Front. Plant Sci. 13, 941231–941317. doi:10.3389/fpls.2022.941231
Kang, W. H., Kim, S., Lee, H. A., Choi, D., and Yeom, S. I. (2016). Genome-wide analysis of Dof transcription factors reveals functional characteristics during development and response to biotic stresses in pepper. Sci. Rep. 6, 33332–33412. doi:10.1038/srep33332
Khan, I., Khan, S., Zhang, Y., and Zhou, J. (2021). Genome-wide analysis and functional characterization of the Dof transcription factor family in rice (Oryza sativa L.). Planta 253, 101–114. doi:10.1007/s00425-021-03627-y
Konishi, M., and Yanagisawa, S. (2007). Sequential activation of two Dof transcription factor gene promoters during vascular development in Arabidopsis thaliana. Plant Physiol. Biochem. 45, 623–629. doi:10.1016/j.plaphy.2007.05.001
Le Hir, R., and Bellini, C. (2013). The plant-specific Dof transcription factors family: new players involved in vascular system development and functioning in Arabidopsis. Front. Plant Sci. 4, 164. doi:10.3389/fpls.2013.00164
Li, J., Jia, X., Yang, Y., Chen, Y., Wang, L., Liu, L., et al. (2022). Genome-wide identification of the DOF gene family involved in fruitlet abscission in areca catechu L. Int. J. Mol. Sci. 23, 11768. doi:10.3390/ijms231911768
Lijavetzky, D., Carbonero, P., and Vicente-Carbajosa, J. (2003). Genome-wide comparative phylogenetic analysis of the rice and Arabidopsis Dof gene families. BMC Evol. Biol. 3, 17–11. doi:10.1186/1471-2148-3-17
Liu, J., Meng, Q., Xiang, H., Shi, F., Ma, L., Li, Y., et al. (2021). Genome-wide analysis of Dof transcription factors and their response to cold stress in rice (Oryza sativa L.). BMC Genomics 22, 800–812. doi:10.1186/s12864-021-08104-0
Liu, W., Ren, W., Liu, X., He, L., Qin, C., Wang, P., et al. (2023). Identification and characterization of Dof genes in Cerasus humilis. Front. Plant Sci. 14, 1152685–1152712. doi:10.3389/fpls.2023.1152685
Liu, Y., Liu, N., Deng, X., Liu, D., Li, M., Cui, D., et al. (2020). Genome-wide analysis of wheat DNA-binding with one finger (Dof) transcription factor genes: evolutionary characteristics and diverse abiotic stress responses. BMC Genomics 21, 276–318. doi:10.1186/s12864-020-6691-0
Luo, T., Song, Y., Gao, H., Wang, M., Cui, H., Ji, C., et al. (2022). Genome-wide identification and functional analysis of Dof transcription factor family in Camelina sativa. BMC Genomics 23, 812–817. doi:10.1186/s12864-022-09056-9
Ma, J., Li, M. Y., Wang, F., Tang, J., and Xiong, A. S. (2015). Genome-wide analysis of Dof family transcription factors and their responses to abiotic stresses in Chinese cabbage. BMC Genomics 16, 33. doi:10.1186/s12864-015-1242-9
Melgar, A. E., and Zelada, A. M. (2021). Evolutionary analysis of angiosperm dehydrin gene family reveals three orthologues groups associated to specific protein domains. Sci. Rep. 11, 23869–23912. doi:10.1038/s41598-021-03066-5
Noguero, M., Atif, R. M., Ochatt, S., and Thompson, R. D. (2013). The role of the DNA-binding One Zinc Finger (DOF) transcription factor family in plants. Plant Sci. 209, 32–45. doi:10.1016/j.plantsci.2013.03.016
Pan, H., Chen, Y., Zhao, J., Huang, J., Shu, N., Deng, H., et al. (2023). In-depth analysis of large-scale screening of WRKY members based on genome-wide identification. Front. Genet. 13, 1104968–1105014. doi:10.3389/fgene.2022.1104968
Papi, M., Sabatini, S., Altamura, M. M., Hennig, L., Schäfer, E., Costantino, P., et al. (2002). Inactivation of the phloem-specific Dof zinc finger gene DAG1 affects response to light and integrity of the testa of Arabidopsis seeds. Plant Physiol. 128, 411–417. doi:10.1104/pp.010488
Park, D. H., Lim, P. O., Kim, J. S., Cho, D. S., Hong, S. H., and Nam, H. G. (2003). The Arabidopsis COG1 gene encodes a Dof domain transcription factor and negatively regulates phytochrome signaling. Plant J. 34, 161–171. doi:10.1046/j.1365-313X.2003.01710.x
Qin, H., Wang, J., Chen, X., Wang, F., Peng, P., Zhou, Y., et al. (2019). Rice OsDOF15 contributes to ethylene-inhibited primary root elongation under salt stress. New Phytol. 223, 798–813. doi:10.1111/nph.15824
Skirycz, A., Radziejwoski, A., Busch, W., Hannah, M. A., Czeszejko, J., Kwaśniewski, M., et al. (2008). The DOF transcription factor OBP1 is involved in cell cycle regulation in Arabidopsis thaliana. Plant J. 56, 779–792. doi:10.1111/j.1365-313X.2008.03641.x
Skirycz, A., Reichelt, M., Burow, M., Birkemeyer, C., Rolcik, J., Kopka, J., et al. (2006). DOF transcription factor AtDof1.1 (OBP2) is part of a regulatory network controlling glucosinolate biosynthesis in Arabidopsis. Plant J. 47, 10–24. doi:10.1111/j.1365-313X.2006.02767.x
Song, A., Gao, T., Li, P., Chen, S., Guan, Z., Wu, D., et al. (2016). Transcriptome-wide identification and expression profiling of the DOF transcription factor gene family in Chrysanthemum morifolium. Front. Plant Sci. 7, 199. doi:10.3389/fpls.2016.00199
Song, C., Cao, Y., Dai, J., Manzoor, M. A., Chen, C., et al. (2022). The multifaceted roles of MYC2 in plants: toward transcriptional reprogramming and stress tolerance by jasmonate signaling. Front. Plant Sci. 13, 868874–868914. doi:10.3389/fpls.2022.868874
Song, C., Jiao, C., Jin, Q., Chen, C., Cai, Y., and Lin, Y. (2020). Metabolomics analysis of nitrogen-containing metabolites between two Dendrobium plants. Physiol. Mol. Biol. Plants 26, 1425–1435. doi:10.1007/s12298-020-00822-1
Song, C., Li, G., Dai, J., and Deng, H. (2021). Genome-wide analysis of PEBP genes in Dendrobium huoshanense: unveiling the antagonistic functions of FT/TFL1 in flowering time. Front. Genet. 12, 687689–687711. doi:10.3389/fgene.2021.687689
Song, C., Wang, Y., Manzoor, M. A., Mao, D., Wei, P., Cao, Y., et al. (2022). In-depth analysis of genomes and functional genomics of orchid using cutting-edge high-throughput sequencing. Front. Plant Sci. 13, 1018029–1018115. doi:10.3389/fpls.2022.1018029
Song, C., Zhang, Y., Chen, R., Zhu, F., Wei, P., Pan, H., et al. (2022). Label-free quantitative proteomics unravel the impacts of salt stress on Dendrobium huoshanense. Front. Plant Sci. 13, 874579–874612. doi:10.3389/fpls.2022.874579
Song, C., Zhang, Y., Zhang, W., Manzoor, M. A., Deng, H., and Han, B. (2023). The potential roles of acid invertase family in Dendrobium huoshanense: identification, evolution, and expression analyses under abiotic stress. Int. J. Biol. Macromol. 253, 127599. doi:10.1016/j.ijbiomac.2023.127599
Song, H., Ji, X., Wang, M., Li, J., Wang, X., Meng, L., et al. (2024). Genome-wide identification and expression analysis of the Dof gene family reveals their involvement in hormone response and abiotic stresses in sunflower (Helianthus annuus L.). Gene 910, 148336. doi:10.1016/j.gene.2024.148336
Su, Y., Liang, W., Liu, Z., Wang, Y., Zhao, Y., Ijaz, B., et al. (2017). Overexpression of GhDof1 improved salt and cold tolerance and seed oil content in Gossypium hirsutum. J. Plant Physiol. 218, 222–234. doi:10.1016/j.jplph.2017.07.017
Sun, S., Wang, B., Jiang, Q., Li, Z., Jia, S., Wang, Y., et al. (2021). Genome-wide analysis of BpDof genes and the tolerance to drought stress in birch (Betula platyphylla). PeerJ 9, 119388–e12024. doi:10.7717/peerj.11938
Tanaka, M., Takahata, Y., Nakayama, H., Nakatani, M., and Tahara, M. (2009). Altered carbohydrate metabolism in the storage roots of sweetpotato plants overexpressing the SRF1 gene, which encodes a Dof zinc finger transcription factor. Planta 230, 737–746. doi:10.1007/s00425-009-0979-2
Wang, H., Zhao, S., Gao, Y., and Yang, J. (2017). Characterization of dof transcription factors and their responses to osmotic stress in poplar (populus trichocarpa). PLoS One 12, e0170210–e0170219. doi:10.1371/journal.pone.0170210
Ward, J. M., Cufr, C. A., Denzel, M. A., and Neff, M. M. (2005). Erratum: the Dof transcription factor OBP3 modulates phytochrome and cryptochrome signaling in Arabidopsis. Plant Cell 17, 2614. doi:10.1105/tpc.105.170960
Waschburger, E. L., Guzman, F., and Turchetto-Zolet, A. C. (2022). Genome-wide identification and analysis of DOF gene family in eugenia uniflora L. (Myrtaceae). Genes (Basel) 13, 2235. doi:10.3390/genes13122235
Xu, D., Li, X., Wu, X., Meng, L., Zou, Z., Bao, E., et al. (2021). Tomato SlCDF3 delays flowering time by regulating different FT-like genes under long-day and short-day conditions. Front. Plant Sci. 12, 650068–650069. doi:10.3389/fpls.2021.650068
Xu, P., Chen, H., Ying, L., and Cai, W. (2016). AtDOF5.4/OBP4, a DOF transcription factor gene that negatively regulates cell cycle progression and cell expansion in Arabidopsis thaliana. Sci. Rep. 6, 27705–27713. doi:10.1038/srep27705
Yanagisawa, S. (2002). The Dof family of plant transcription factors. Trends Plant Sci. 7, 555–560. doi:10.1016/S1360-1385(02)02362-2
Yanagisawa, S., Akiyama, A., Kisaka, H., Uchimiya, H., and Miwa, T. (2004). Metabolic engineering with Dof1 transcription factor in plants: improved nitrogen assimilation and growth under low-nitrogen conditions. Proc. Natl. Acad. Sci. U. S. A. 101, 7833–7838. doi:10.1073/pnas.0402267101
Yu, Q., Li, C., Zhang, J., Tian, Y., Wang, H., Zhang, Y., et al. (2020). Genome-wide identification and expression analysis of the Dof gene family under drought stress in tea (Camellia sinensis). PeerJ 8, e9269. doi:10.7717/peerj.9269
Zang, D., Wang, L., Zhang, Y., Zhao, H., and Wang, Y. (2017). ThDof1.4 and ThZFP1 constitute a transcriptional regulatory cascade involved in salt or osmotic stress in Tamarix hispida. Plant Mol. Biol. 94, 495–507. doi:10.1007/s11103-017-0620-x
Zhang, A., Matsuoka, K., Kareem, A., Robert, M., Roszak, P., Blob, B., et al. (2022). Cell-wall damage activates DOF transcription factors to promote wound healing and tissue regeneration in Arabidopsis thaliana. Curr. Biol. 32, 1883–1894.e7. doi:10.1016/j.cub.2022.02.069
Zhang, C., Dong, T., Yu, J., Hong, H., Liu, S., Guo, F., et al. (2023). Genome-wide survey and expression analysis of Dof transcription factor family in sweetpotato shed light on their promising functions in stress tolerance. Front. Plant Sci. 14, 1140727–1140816. doi:10.3389/fpls.2023.1140727
Zhang, L., Liu, B., Zheng, G., Zhang, A., and Li, R. (2017). Genome-wide characterization of the SiDof gene family in foxtail millet (Setaria italica). BioSystems 151, 27–33. doi:10.1016/j.biosystems.2016.11.007
Zhang, Y., Zhang, W., Manzoor, M. A., Sabir, I. A., Zhang, P., Cao, Y., et al. (2023). Differential involvement of WRKY genes in abiotic stress tolerance of Dendrobium huoshanense. Ind. Crops Prod. 204, 117295. doi:10.1016/j.indcrop.2023.117295
Zhang, Z., Yuan, L., Liu, X., Chen, X., and Wang, X. (2018). Evolution analysis of Dof transcription factor family and their expression in response to multiple abiotic stresses in Malus domestica. Gene 639, 137–148. doi:10.1016/j.gene.2017.09.039
Zhuo, M., Sakuraba, Y., and Yanagisawa, S. (2020). A jasmonate-activated MYC2-dof2.1-MYC2 transcriptional loop promotes leaf senescence in Arabidopsis. Plant Cell 32, 242–262. doi:10.1105/tpc.19.00297
Keywords: Dendrobium huoshanense, DNA binding with one finger, phylogeny, abiotic stress, bioinformactics analysis
Citation: Gu F, Zhang W, Wang T, He X, Chen N, Zhang Y and Song C (2024) Identification of Dof transcription factors in Dendrobium huoshanense and expression pattern under abiotic stresses. Front. Genet. 15:1394790. doi: 10.3389/fgene.2024.1394790
Received: 02 March 2024; Accepted: 09 April 2024;
Published: 22 April 2024.
Edited by:
Yang Yang, Shanxi Agricultural University, ChinaReviewed by:
Ake Liu, Changzhi University, ChinaIrfan Ali Sabir, South China Agricultural University, China
Muhammad Waheed Riaz, Zhejiang Agriculture and Forestry University, China
Copyright © 2024 Gu, Zhang, Wang, He, Chen, Zhang and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng Song, bGFubmlhbzgxMjMyOTIxOEAxNjMuY29t; Yingyu Zhang, emhhbmd5aW5neXUwNjEzQDE2My5jb20=